
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced imino functionalizations of alkenes via intermolecular charge transfer†
Xiang-Xin
Zhang
ab,
Hao
Zheng
ab,
Yong-Kang
Mei
 ab,
Yan
Liu
ab,
Ying-Ying
Liu
ab,
Ding-Wei
Ji
a,
Boshun
Wan
a and
Qing-An
Chen
*ab
ab,
Yan
Liu
ab,
Ying-Ying
Liu
ab,
Ding-Wei
Ji
a,
Boshun
Wan
a and
Qing-An
Chen
*ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, People's Republic of China
bUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China. E-mail: qachen@dicp.ac.cn; Web: http://www.lbcs.dicp.ac.cn
First published on 25th September 2023
Abstract
A catalyst-free photosensitized strategy has been developed for regioselective imino functionalizations of alkenes via the formation of an EDA complex. This photo-induced protocol facilitates the construction of structurally diverse β-imino sulfones and vinyl sulfones in moderate to high yields. Mechanistic studies reveal that the reaction is initiated with an intermolecular charge transfer between oximes and sulfinates, followed by fragmentation to generate a persistent iminyl radical and transient sulfonyl radical. This catalyst-free protocol also features excellent regioselectivity, broad functional group tolerance and mild reaction conditions. The late stage functionalization of natural product derived compounds and total synthesis of some bioactive molecules have been demonstrated to highlight the utility of this protocol. Meanwhile, the compatibility of different donors has proved the generality of this strategy.
Introduction
Nitrogen-containing molecules are widely found in natural products and pharmaceuticals, and play an important role in organic synthesis.1 Particularly, β-functionalized amino compounds display unique properties in terms of pharmaceutical activity and ligand design.2 Given the availability and abundance of olefins, difunctionalization of alkenes has been recognized as a straightforward and powerful strategy for constructing complex molecules. Therefore, transition metal-catalyzed difunctionalization of olefins has been developed for the construction of a series of products containing β-functionalized amino structural units.3 However, this method faces problems such as relatively harsh reaction conditions and the participation of precious metals (Fig. 1A).3d–f,3h Considering the problem of heavy metal residues and the applicability of the reactions, searching for gentler and non-toxic methods becomes necessary. Recently, photoredox catalysis has been employed to construct β-functionalized amino compounds via multicomponent alkene difunctionalization (Fig. 1A).4 Nevertheless, unlike amino functionalizations of alkenes, olefin imino functionalization is still in the initial stage and mainly limited to intramolecular cyclization reactions.3d,4a,5 Additionally, most of these photoredox protocols require the participation of noble metal photocatalysts. Under energy transfer photocatalysis, various elegant bifunctional reagents have been developed to generate the required radical pairs for alkene difunctionalizations (Fig. 1B).6 Although significant progress has been made, this energy transfer catalytic strategy relies on the design and synthesis of bifunctional reagents, and often requires multi-step synthesis for the introduction of other functional groups. Therefore, it is of great interest to develop a flexible and variable olefin difunctionalization method under mild conditions.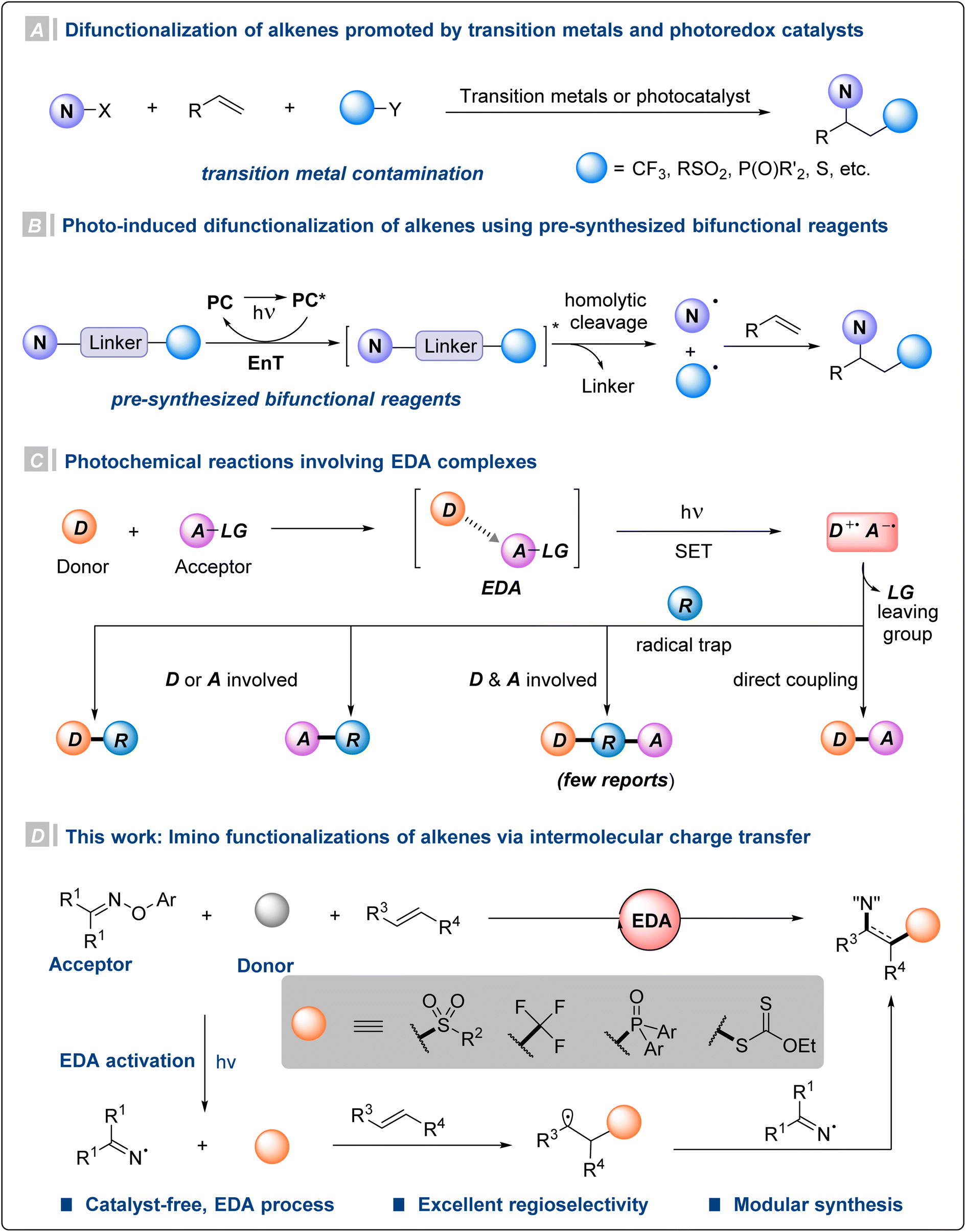 | ||
| Fig. 1 Imino functionalizations of alkenes. | ||
Considering the current problems, an alternative strategy is the direct photoexcitation of an electron donor–acceptor (EDA) complex. It is defined as a new molecular aggregate formed by an electron acceptor substrate A and a donor molecule D through non-bonding interactions.7 The two components A and D may not absorb visible light themselves, but their in situ formed EDA complex does. And subsequent intramolecular single-electron transfer (SET) is triggered to generate radical intermediates under the excitation of visible light.8 With regard to photochemical reactions involving EDA complexes, there are two reaction modes. On the one hand, the free radicals generated by the two substrates respectively could occur a direct radical–radical coupling reaction.9 On the other hand, the free radical intermediates produced by the EDA complex are reacted by free radical trapping reagents.5a,10 However, only a few protocols are able to incorporate the donor D and acceptor A motifs simultaneously onto the radical traps (Fig. 1C).4a,10g,11
Based on this point and our group's research on olefin functionalization,12 we wonder whether the two free radicals produced by the EDA complex can both be utilized with alkenes as trap reagents. The studies by Narasaka,13 Walton,14 Leonori,5 Yang6j and Glorius6a,d,6g,i have demonstrated oximes15 as good nitrogen radical precursors. The β-amino sulfones display unique pharmaceutical activities that render them as good candidates in drug discovery research. Then we speculated that the electron-poor O-aryl oximes and electron-rich sulfinates could form EDA complexes and enable access to sulfonyl radicals and iminyl radicals upon exposure to visible light. Herein, we report a catalyst-free intermolecular radical sulfonylimination of alkenes via photochemical EDA activation. Preliminary mechanistic studies suggest that the initial radical addition of the reactive transient sulfonyl radical with alkenes should be kinetically favored compared to that of the persistent iminyl radical. Meanwhile, this strategy can also be applied to other donors to construct structurally different β-amino compounds.
Results and discussion
To verify our hypothesis, O-aryl oxime 1a, styrene 2a, and sodium benzosulfonate 3a were chosen as model substrates to optimize the reaction (Table 1). After careful evaluation of reaction parameters, β-imino sulfone 4a was formed in 88% yield with 1a as the benzophenone iminyl radical precursor under blue light irradiation (entry 1). Control experiments confirmed the essential roles of visible light and the substituents of O-aryl oximes (entries 2 and 3). Meanwhile, screening of light sources revealed that the wavelength (390–456 nm) had little effect on the further improvement of the reaction yield (entries 4–6). The evaluation of solvents showed that MeCN and DMSO had better performance on yields (entries 7–10 and Table S2 in the ESI†). This result inspired us to test the mixed solvents and it was found that the MeCN/DMSO solvent system in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v was the optimal choice (entries 1, 11 and 12).
:1 v/v was the optimal choice (entries 1, 11 and 12).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 4a (%) |
|
a Conditions: 1a (0.12 mmol), 2a (0.10 mmol), 3a (0.18 mmol), MeCN/DMSO (9:1, 1.0 mL), rt, 18 h.
b Determined by HPLC with anthracene as the internal standard.
c Isolated yield.
|
||
| 1 | None | 88(82)c |
| 2 | Without light | — |
| 3 | R = Me or H | — |
| 4 | 390 nm | 76 |
| 5 | 427 nm | 78 |
| 6 | 456 nm | 65 |
| 7 | MeCN | 74 |
| 8 | DMSO | 46 |
| 9 | THF | 21 |
| 10 | DCM | 5 |
| 11 | MeCN/DMSO = 2:1 |
77 |
| 12 | MeCN/DCM = 9:1 |
73 |
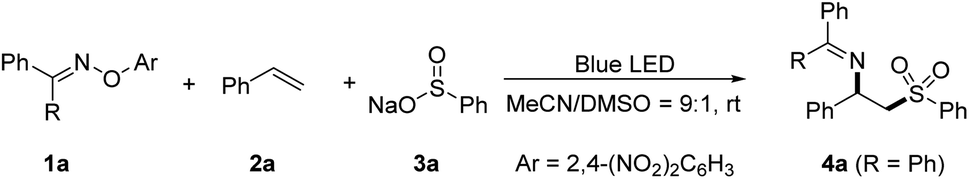
With the optimized conditions in hand, we systematically explored the substrate scope of this sulfonylimination approach (Fig. 2). A wide range of styrene derivatives all underwent the sulfonylimination reaction smoothly, delivering the corresponding β-imino sulfones with excellent regioselectivity. Styrenes bearing electron-donating or electron-withdrawing substituents at different positions of the aromatic rings were tolerated (4a–4j). Halogen and chloromethyl groups that are susceptible in an EDA reaction system could be well compatible in this protocol. Other terminal aryl alkenes, such as 2-vinylnaphthalene and 2-vinylpyridine could also be applicable (4k and 4l). Excitingly, 1,3-enyne could be easily accommodated, leaving the alkyne moiety untouched (4m). Next, a series of disubstituted and multi-substituted alkenes were investigated under our sulfonylimination conditions. 1,1-Disubstituted alkenes with different electronic properties and steric hindrance could provide β-imino sulfones with a tertiary alkylamine centre (4n–4r). The desired products (4s–4w) could also be obtained from the coupling of different 1,2-disubstituted and multi-substituted alkenes. Notably, high regio- and diastereoselectivities were obtained for unsymmetrical internal alkenes. The anti-geometries between sulfonyl and iminyl groups were confirmed by single crystal analysis of 4s (CCDC: 2224155) and 4t (CCDC: 2224158). Gratifyingly, two consecutive distinct quaternary carbon centers could be constructed simultaneously (4w). Subsequently, a series of sulfinates and O-aryl oximes were evaluated with 4-tert-butylstyrene. Moderate to excellent yields were obtained for phenylsulfinates with different substituents at the para position (5a–5g). This protocol was also compatible with other aromatic groups, such as naphthyl (5h), thienyl (5i), and pyridyl (5j). Besides, alkyl sulfinates also worked in this protocol with high reactivities and regioselectivities (5k–5m). It is worth noting that the cyclopropyl group could remain intact in the target product (5m). O-Aryl oximes derived from other diaryl ketones were also well accommodated in this sulfonylimination (5n and 5o).
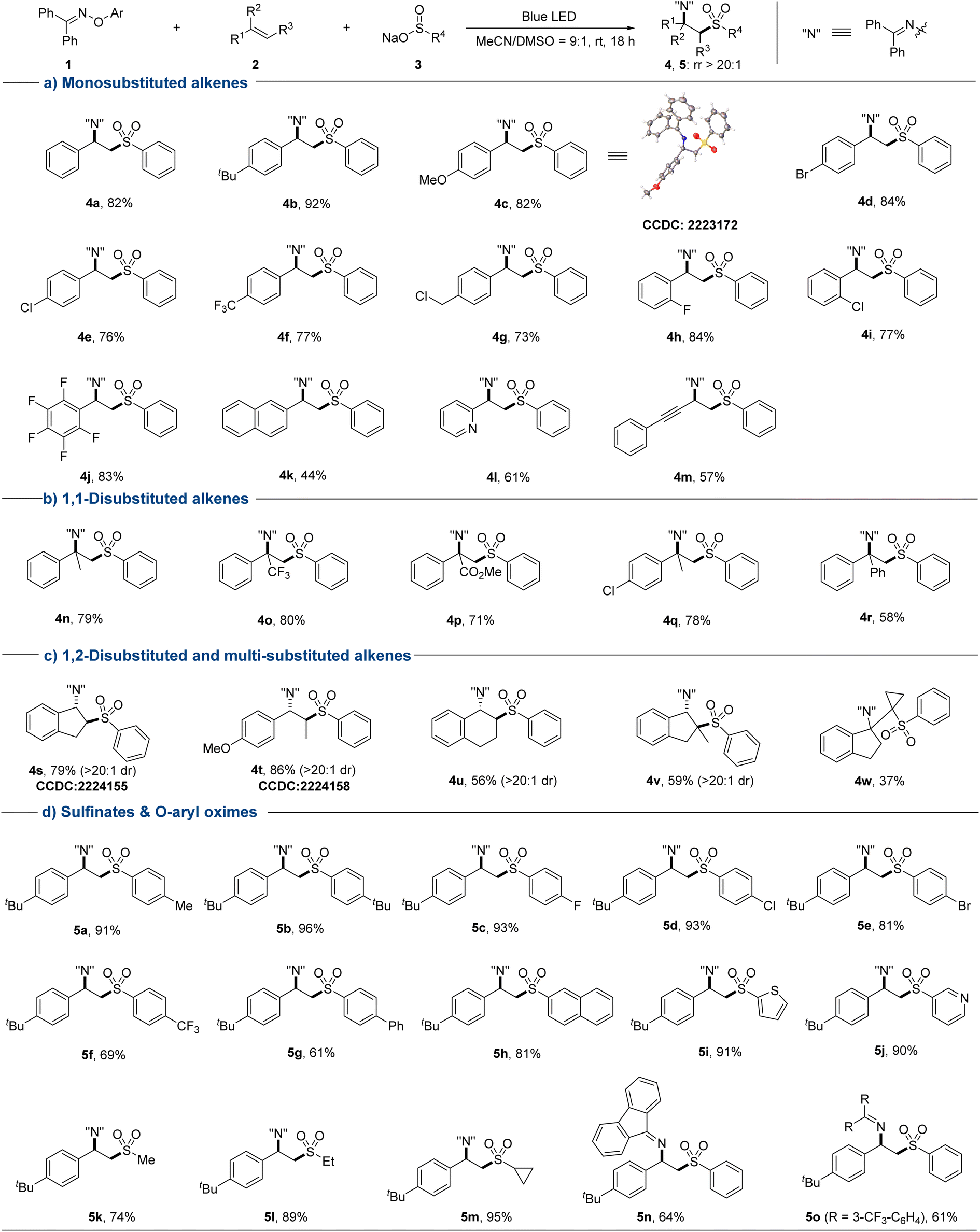 | ||
| Fig. 2 Substrate scope of sulfonylimination of alkenes. Conditions: 1 (0.12 mmol), 2 (0.10 mmol), 3 (0.18 mmol), MeCN/DMSO (9:1, 1.0 mL), rt, 18 h. Isolated yields are given in all cases. | ||
The synthetic utilization of this protocol in late-stage functionalization was further evaluated through direct sulfonylimination of a range of complex alkene substrates (Fig. 3a). Natural products and drug-derived alkenes reacted smoothly to deliver the desired products in good yields (6a–6d). The ester groups, cyclic olefin and amides of complex olefins could be well maintained in this protocol. Ultimately, the synthesis of pharmaceuticals has been conducted to further demonstrate its synthetic utility.
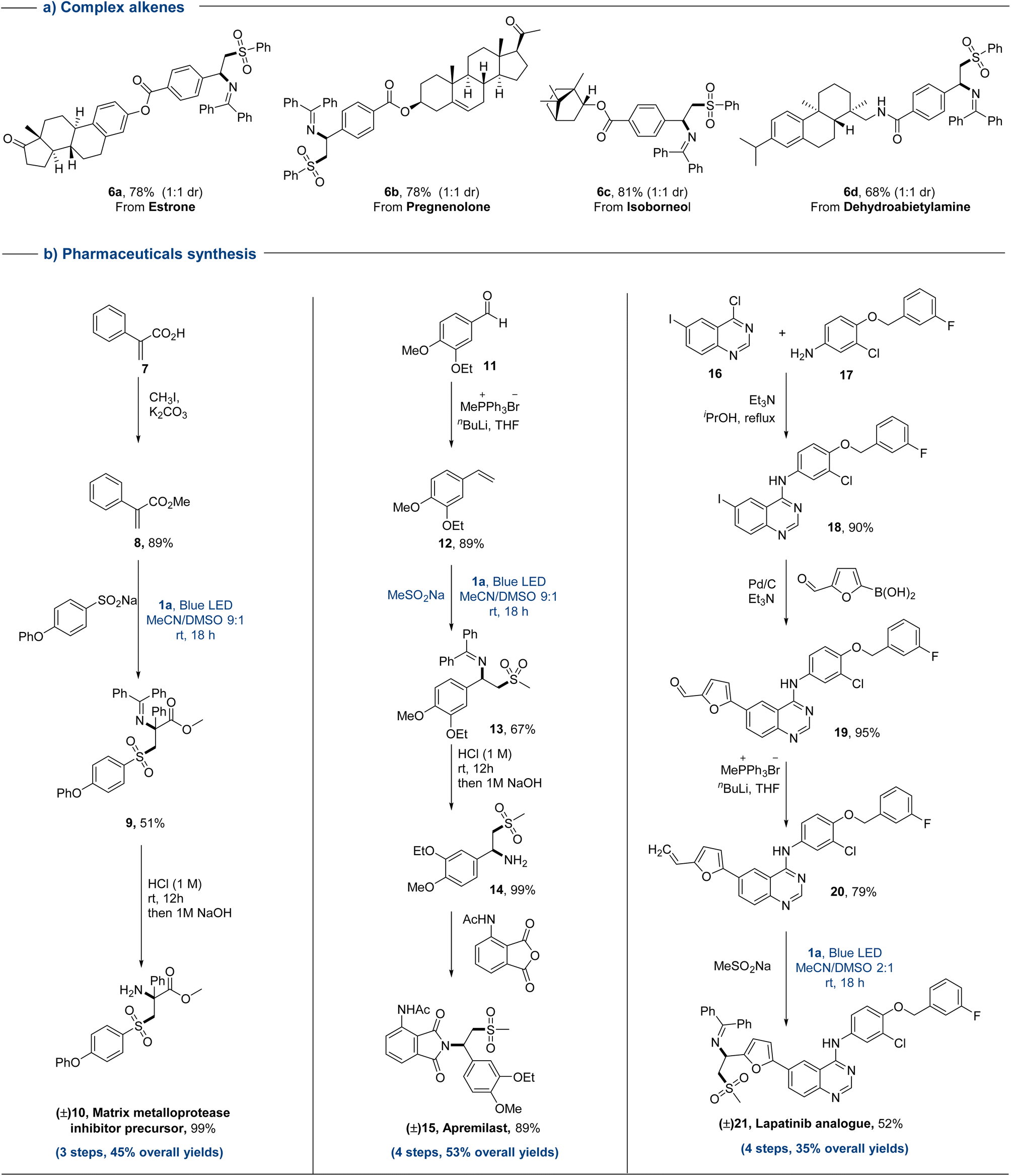 | ||
| Fig. 3 Substrate scope of complex alkenes and synthesis of pharmaceuticals. | ||
Generally, de novo synthesis is used to synthesize drugs containing an amino-sulfone functionality. For changing key functional groups in drugs such as sulfonyl groups, it is often necessary to replace the corresponding substrate and re-synthesize from scratch. This greatly slows down the synthesis efficiency of drug molecules. Consequently, achieving modular and streamlined synthesis of drug molecules is critical for the rapid expansion of drug libraries. This sulfonylimination strategy was demonstrated efficaciously, which allowing different drugs to be synthesized in a concise manner (10, 15, and 21) with decent overall yields (35–53%, Fig. 3b). Only 3–4 steps were required to create these high functionalized molecules from simple starting materials. For drugs 15 and 21, the reaction conditions for the introduction of amino and sulfone groups in traditional synthesis are relatively harsh (strong acid and strong base).2e,i Meanwhile, these groups can't be introduced at the same time. In the synthesis of these three drugs, the construction of key alkenes, the functionalization of alkenes, and subsequent simple modifications could bypass the one-to-one correspondence between drugs and starting substrates, and could efficiently synthesize drugs in a modular and gentle manner. In addition, this sulfonylimination strategy provided a way to quickly enrich the drug library, by only simply changing different oximes and sulfinates in a desired step.
To gain insight into the sulfonylimination mechanism, we performed a series of UV-vis spectroscopic measurements. An obvious increase in absorption intensity (400–500 nm) was observed by the addition of colorless 3a into 1a, accompanied by a certain degree of yellow color deepening. To further investigate the interaction between 1a and 3a, a series of experiments were performed. The Job's plot measurement16 revealed a 1:1 stoichiometry of the corresponding EDA complex (Fig. S2 in the ESI†). In addition, NMR titration further revealed the presence of weak interaction between 1a an 3a (Fig. S3 in the ESI†). And the association constant KEDA between them was calculated to be 3.65 M−1 using the Benesi–Hildebrand method17 (Fig. S4 in the ESI†). This was proposed to result from the formation of an EDA complex (Fig. 4a). Next, free radical inhibition and capture experiments were performed. The addition of radical scavenger TEMPO (2,2,6,6-tetramethylpiperidinoxy) to the reaction mixture completely suppressed the reactivity. And in the presence of BHT, sulfonyletherification of olefin (7a:7b = 10:1) rather than the desired product 4a was formed mainly (Fig. 4b). Radical clock experiments with different cyclopropanes led to the corresponding ring-opening products 23 and 25 (Fig. 4c). And the products contained only sulfone functional groups, indicating that the order of addition of sulfone groups was preferential. These results hinted towards the radical nature of the reaction. Finally, different competitive experiments were carried out separately (Fig. 4d). An evaluation of the reactivities of sulfinate 3a and 3a′ gave the corresponding products 4a and 4a′ with 49% and 50% yield, respectively. This indicated that the overall rate of the generation of corresponding free radicals is probably comparable between aryl and alkyl sulfinates via EDA activation. When different alkenes reacted simultaneously with sulfinate 3a, more electron-rich product 4c was more inclined to be formed over 4f. This perhaps resulted from the stabilization of carbon radicals by increasing electron density.
 | ||
| Fig. 4 Mechanistic studies and the proposed mechanism. | ||
Based on the aforementioned mechanistic studies and further DFT calculations, a plausible reaction mechanism is shown in Fig. 4e. First, the EDA complex 1A is formed via Coulomb interaction and π–π stacking between oxime 1a and sulfinate 3a. The existence of complex 1A is further supported by time-dependent density functional theory (TDDFT),9c,18 which indicates that the excitation results in a single electron transfer (SET) from 3a to 1a. Then under the irradiation of visible light, the complex 1A in the ground state is excited to give the first singlet excited state 1A*. The SET promotes the homolytic N–O bond cleavage of 1a. There are two possible mechanisms for N–O cleavage. It may occur in the first singlet excited state, directly giving persistent iminyl radical B (ref. 19) and ion pair intermediate C. Alternatively, the singlet 1A* undergoes intersystem crossing (ISC),20 reaching the triplet 3A, followed by N–O bond cleavage with a barrier of 4.2 kcal mol−1 (TS1). Although it is hard to determine which mechanism is actually involved, it is certain that iminyl radical B is generated under blue light irradiation. Subsequently, the resulting intermediate C undergoes intramolecular single electron transfer to deliver transient sulfonyl radical E. Through transition state TS2 (ΔG‡2 = 8.4 kcal mol−1), the active radical E attacks alkene 2a to produce C-centered radical F. In contrast, kinetically unfavorable radical additions between iminyl radical B, the sulphinic radical and alkene 2a need to overcome a higher free energy barrier, 17.2 kcal mol−1 (viaTS2N) and 24.4 kcal mol−1 (viaTS2O), respectively. Finally, this C-centered radical F and the long-lived iminyl radical B participate in a radical–radical cross-coupling process to yield the desired β-imino sulfone product 4a. Considering the large free energy difference between TS2 and TS2N, the formation of isomer 4aN can be kinetically suppressed, which theoretically explains the excellent regioselectivity of this transformation.
In order to test the generality of this EDA strategy, different donors were evaluated for the alkene difunctionalizations (Fig. 5a). Trifluoromethylimination of olefin could be achieved by using sodium trifluoromethanesulfonate as the donor (27a). As for potassium ethyl xanthate, the corresponding product could also be effectively obtained (27b). Organophosphorus compounds have a wide range of applications in the fields of organic synthesis, materials, coordination chemistry, medicine and agrochemicals due to their unique properties.21 Specifically, β-aminophosphine derivatives have been widely used to build a diversity of ligands or organocatalysts.2g,j,21f,g Therefore, the ability to rapidly construct this building block is crucial for expanding the diversity of organophosphorus libraries. With diarylphosphine oxides as donors, the corresponding β-iminophosphines could be obtained in moderate yields (27c–27e).
 | ||
| Fig. 5 Construction of β-amino compounds. Conditions: (a) 1 (0.12 mmol), 2′ (0.10 mmol), 26 (0.18 mmol), MeCN/DMSO (9:1,1.0 mL), rt, 18 h; (b) 1 (0.12 mmol), 2′ (0.10 mmol), 26 (0.18 mmol), Cs2CO3 (0.10 mmol), MeCN/DMSO (9:1, 1.0 mL), rt, 18 h; (c) 1 (0.12 mmol), 2′ (0.10 mmol), 3 (0.18 mmol), Cs2CO3 (0.10 mmol), MeCN (1.0 mL), rt, 18 h; (d) 1 (0.12 mmol), 2′ (0.10 mmol), 26 (0.18 mmol), Cs2CO3 (0.15 mmol), MeCN/DMSO (9:1, 1.0 mL), rt, 18 h. Isolated yields are given in all cases. | ||
Vinyl sulfones are excellent Michael acceptors and they have widespread applications in biological research as covalent protease inhibitors.22 On the basis of the understanding of the reaction mechanism, this photo-induced intermolecular charge transfer strategy is further demonstrated in the construction of functionalized vinyl sulfones. After a quick reaction optimization (Tables S7–S10 in the ESI†), α-halogenated olefins bearing aromatic rings and an ester group all reacted smoothly (Fig. 5b), delivering the corresponding β-imino vinyl sulfones with acceptable yields (28a–28d). Interestingly, alpha carboxyl substituted olefin could be used to produce the desired product by decarboxylation (28a, X = CO2H). For different substituted sulfinates, the reaction system could also be well compatible (28e–28j). Moreover, this protocol worked in the late-stage functionalization of complex alkenes through direct vinylsulfonylimination (28k). Meanwhile, the β-imino vinylphosphines could be obtained by using α-bromostyrene as the capture reagent (28l and 28m).
Conclusions
In conclusion, a general strategy for imino functionalizations of alkenes has been developed by rationally utilizing the components involved in the formation of EDA. This strategy is photocatalyst-free, and features mild reaction conditions, excellent regioselectivity and broad functional group tolerance. With sulfinates as the donors, different substituted alkenes, even sterically encumbered tetra-substituted olefins could smoothly participate in the reaction. And it can rapidly construct structurally diverse β-imino sulfones, which provides a direct route for accessing this privileged motif. A series of experimental and computational studies suggest that an EDA complex between the electron-rich sulfinate and the electron-poor oxime was in situ formed, followed by the generation of a long-lived iminyl radical and a transient sulfonyl radical via intermolecular charge transfer. The large reactivity difference of these two radical species explains that the reaction has outstanding regional selectivity. Based on the understanding of the mechanism, different donors can also be compatible, and the corresponding β-aminovinyl compounds can also be obtained. This transition metal and catalyst free protocol provides a simple and tunable method to the corresponding β-amino compounds.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Q.-A. C. provided resources and supervision. Q.-A. C. and X.-X. Z. conceptualized the work. X.-X. Z. performed the investigations and the writing of original draft. X.-X. Z., H. Z., Y.-K. M., Y. L., Y.-Y. L., D.-W. J., and B.-S. W. analyzed the data. All authors contributed to the final version of the manuscript (writing, review and editing).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Natural Science Foundation of China (22271277 and 21971234) is acknowledged.Notes and references
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed; (b) M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- (a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed; (b) D. P. Becker, G. DeCrescenzo, J. Freskos, D. P. Getman, S. L. Hockerman, M. Li, P. Mehta, G. E. Munie and C. Swearingen, Bioorg. Med. Chem. Lett., 2001, 11, 2723–2725 CrossRef CAS PubMed; (c) C. K. Wada, J. H. Holms, M. L. Curtin, Y. Dai, A. S. Florjancic, R. B. Garland, Y. Guo, H. R. Heyman, J. R. Stacey, D. H. Steinman, D. H. Albert, J. J. Bouska, I. N. Elmore, C. L. Goodfellow, P. A. Marcotte, P. Tapang, D. W. Morgan, M. R. Michaelides and S. K. Davidsen, J. Med. Chem., 2002, 45, 219–232 CrossRef CAS PubMed; (d) V. Aranapakam, G. T. Grosu, J. M. Davis, B. Hu, J. Ellingboe, J. L. Baker, J. S. Skotnicki, A. Zask, J. F. DiJoseph, A. Sung, M. A. Sharr, L. M. Killar, T. Walter, G. Jin and R. Cowling, J. Med. Chem., 2003, 46, 2361–2375 CrossRef CAS PubMed; (e) H. W. Man, P. Schafer, L. M. Wong, R. T. Patterson, L. G. Corral, H. Raymon, K. Blease, J. Leisten, M. A. Shirley, Y. Tang, D. M. Babusis, R. Chen, D. Stirling and G. W. Muller, J. Med. Chem., 2009, 52, 1522–1524 CrossRef CAS PubMed; (f) T. A. Rano and G. H. Kuo, Org. Lett., 2009, 11, 2812–2815 CrossRef CAS PubMed; (g) V. A. Stepanova and I. P. Smoliakova, Curr. Org. Chem., 2012, 16, 2893–2920 CrossRef CAS; (h) M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819–833 RSC; (i) A. Lyu, L. Fang and S. Gou, Eur. J. Med. Chem., 2014, 87, 631–642 CrossRef CAS PubMed; (j) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657–1677 RSC.
- (a) J. S. Lin, X. Y. Dong, T. T. Li, N. C. Jiang, B. Tan and X. Y. Liu, J. Am. Chem. Soc., 2016, 138, 9357–9360 CrossRef CAS PubMed; (b) J. A. Li, P. Z. Zhang, K. Liu, A. Shoberu, J. P. Zou and W. Zhang, Org. Lett., 2017, 19, 4704–4706 CrossRef CAS PubMed; (c) Y. Xiong, Y. Sun and G. Zhang, Org. Lett., 2018, 20, 6250–6254 CrossRef CAS PubMed; (d) C. Chen, Y. Bao, J. Zhao and B. Zhu, Chem. Commun., 2019, 55, 14697–14700 RSC; (e) Z. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457–1463 CrossRef CAS; (f) W. H. Rao, L. L. Jiang, X. M. Liu, M. J. Chen, F. Y. Chen, X. Jiang, J. X. Zhao, G. D. Zou, Y. Q. Zhou and L. Tang, Org. Lett., 2019, 21, 2890–2893 CrossRef CAS PubMed; (g) L. J. Wang, J. M. Chen, W. Dong, C. Y. Hou, M. Pang, W. B. Jin, F. G. Dong, Z. D. Xu and W. Li, J. Org. Chem., 2019, 84, 2330–2338 CrossRef CAS PubMed; (h) W. Zhang and X. Yang, J. Fluorine Chem., 2021, 248, 109823 CrossRef CAS.
- (a) R. Mao, Z. Yuan, Y. Li and J. Wu, Chem. - Eur. J., 2017, 23, 8176–8179 CrossRef CAS PubMed; (b) J. Zhang, F. Zhang, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 3891–3894 RSC; (c) Y. Wu, Y. Zhang, M. Jiang, X. Dong, H. B. Jalani, G. Li and H. Lu, Chem. Commun., 2019, 55, 6405–6408 RSC; (d) S.-Q. Guo, H.-Q. Yang, A.-L. Wang, Y.-Z. Jiang, G.-Q. Xu, Y.-C. Luo, Z.-X. Chen and P.-F. Xu, Green Chem., 2021, 23, 9571–9576 RSC.
- (a) J. Davies, S. G. Booth, S. Essafi, R. A. Dryfe and D. Leonori, Angew. Chem., Int. Ed., 2015, 54, 14017–14021 CrossRef CAS PubMed; (b) J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed.
- (a) T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 3172–3177 CrossRef CAS PubMed; (b) S. Q. Lai, B. Y. Wei, J. W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997–22003 CrossRef CAS PubMed; (c) J. Li, Y. Yuan, X. Bao, T. Sang, J. Yang and C. Huo, Org. Lett., 2021, 23, 3712–3717 CrossRef CAS PubMed; (d) T. Patra, M. Das, C. G. Daniliuc and F. Glorius, Nat. Catal., 2021, 4, 54–61 CrossRef CAS; (e) D. S. Lee, V. K. Soni and E. J. Cho, Acc. Chem. Res., 2022, 55, 2526–2541 CrossRef CAS PubMed; (f) J. Majhi, R. K. Dhungana, Á. Rentería-Gómez, M. Sharique, L. Li, W. Dong, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 15871–15878 CrossRef CAS PubMed; (g) G. Tan, M. Das, H. Keum, P. Bellotti, C. Daniliuc and F. Glorius, Nat. Chem., 2022, 14, 1174–1184 CrossRef CAS PubMed; (h) G. Tan, F. Paulus, A. Renteria-Gomez, R. F. Lalisse, C. G. Daniliuc, O. Gutierrez and F. Glorius, J. Am. Chem. Soc., 2022, 144, 21664–21673 CrossRef CAS PubMed; (i) G. Y. Tan, M. Das, R. Kleinmans, F. Katzenburg, C. Daniliuc and F. Glorius, Nat. Catal., 2022, 5, 1120–1130 CrossRef CAS; (j) Y. Zheng, Z. J. Wang, Z. P. Ye, K. Tang, Z. Z. Xie, J. A. Xiao, H. Y. Xiang, K. Chen, X. Q. Chen and H. Yang, Angew. Chem., Int. Ed., 2022, e202212292 CAS; (k) J. E. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Humpel, D. Rana, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS PubMed; (l) X. L. Luo, S. S. Li, Y. S. Jiang, F. Liu, S. H. Li and P. J. Xia, Org. Lett., 2023, 25, 1742–1747 CrossRef CAS PubMed; (m) C. P. Yuan, Y. Zheng, Z. Z. Xie, K. Y. Deng, H. B. Chen, H. Y. Xiang, K. Chen and H. Yang, Org. Lett., 2023, 25, 1782–1786 CrossRef CAS PubMed; (n) Y. Zheng, Z. Liao, Z. Xie, H. Chen, K. Chen, H. Xiang and H. Yang, Org. Lett., 2023, 25, 2129–2133 CrossRef CAS PubMed.
- R. Foster, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS.
- (a) S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed; (b) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (c) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- (a) M. Nappi, G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 4921–4925 CrossRef CAS PubMed; (b) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adan and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485–1489 CrossRef CAS PubMed; (c) B. Liu, C. H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed; (d) Q. Guo, M. Wang, H. Liu, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 4747–4751 CrossRef CAS PubMed; (e) J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227–15231 CrossRef CAS PubMed; (f) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed; (g) H. E. Ho, A. Pagano, J. A. Rossi-Ashton, J. R. Donald, R. G. Epton, J. C. Churchill, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Chem. Sci., 2019, 11, 1353–1360 RSC; (h) K. Liang, N. Li, Y. Zhang, T. Li and C. Xia, Chem. Sci., 2019, 10, 3049–3053 RSC; (i) S. Xie, D. Li, H. Huang, F. Zhang and Y. Chen, J. Am. Chem. Soc., 2019, 141, 16237–16242 CrossRef CAS PubMed; (j) X. D. Su, B. B. Zhang, Q. Liu, J. T. Cheng, Z. X. Wang and X. Y. Chen, Org. Lett., 2021, 23, 8262–8266 CrossRef CAS PubMed; (k) M. Sharique, J. Majhi, R. K. Dhungana, L. M. Kammer, M. Krumb, A. Lipp, E. Romero and G. A. Molander, Chem. Sci., 2022, 13, 5701–5706 RSC.
- (a) Y. Cheng and S. Yu, Org. Lett., 2016, 18, 2962–2965 CrossRef CAS PubMed; (b) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638–4641 CrossRef CAS PubMed; (c) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619–12623 CrossRef CAS PubMed; (d) M. C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (e) J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697–5701 CrossRef CAS PubMed; (f) E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed; (g) M. Kim, E. You, S. Park and S. Hong, Chem. Sci., 2021, 12, 6629–6637 RSC; (h) A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. M. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 43–52 CrossRef CAS PubMed; (i) W. Zhou, S. Wu and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 8914–8919 CrossRef CAS PubMed.
- (a) S. Engl and O. Reiser, Org. Lett., 2021, 23, 5581–5586 CrossRef CAS PubMed; (b) Y. X. Chen, Z. J. Wang, J. A. Xiao, K. Chen, H. Y. Xiang and H. Yang, Org. Lett., 2021, 23, 6558–6562 CrossRef CAS.
- (a) Y. C. Hu, D. W. Ji, C. Y. Zhao, H. Zheng and Q. A. Chen, Angew. Chem., Int. Ed., 2019, 58, 5438–5442 CrossRef CAS PubMed; (b) C. S. Kuai, D. W. Ji, C. Y. Zhao, H. Liu, Y. C. Hu and Q. A. Chen, Angew. Chem., Int. Ed., 2020, 59, 19115–19120 CrossRef CAS PubMed; (c) S. Y. Guo, F. Yang, T. T. Song, Y. Q. Guan, X. T. Min, D. W. Ji, Y. C. Hu and Q. A. Chen, Nat. Commun., 2021, 12, 6538 CrossRef CAS PubMed; (d) W. S. Jiang, D. W. Ji, W. S. Zhang, G. Zhang, X. T. Min, Y. C. Hu, X. L. Jiang and Q. A. Chen, Angew. Chem., Int. Ed., 2021, 60, 8321–8328 CrossRef CAS PubMed; (e) G. Zhang, C.-Y. Zhao, X.-T. Min, Y. Li, X.-X. Zhang, H. Liu, D.-W. Ji, Y.-C. Hu and Q.-A. Chen, Nat. Catal., 2022, 5, 708–715 CrossRef CAS; (f) C. Y. Zhao, Y. Y. Liu, X. X. Zhang, G. C. He, H. Liu, D. W. Ji, Y. C. Hu and Q. A. Chen, Angew. Chem., Int. Ed., 2022, 61, e202207202 CrossRef CAS PubMed.
- (a) T. Mikami and K. Narasaka, Chem. Lett., 2000, 29, 338–339 CrossRef; (b) M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2008, 81, 539–547 CrossRef CAS.
- (a) R. T. McBurney and J. C. Walton, J. Am. Chem. Soc., 2013, 135, 7349–7354 CrossRef CAS PubMed; (b) J. C. Walton, Acc. Chem. Res., 2014, 47, 1406–1416 CrossRef CAS PubMed.
- (a) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem. - Eur. J., 2018, 24, 12154–12163 CrossRef CAS PubMed; (b) K. A. Rykaczewski, E. R. Wearing, D. E. Blackmun and C. S. Schindler, Nat. Syn., 2022, 1, 24–36 CrossRef.
- Y. Lu, Q. Liu, Z. X. Wang and X. Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202116071 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- (a) M. J. James, F. Strieth-Kalthoff, F. Sandfort, F. J. R. Klauck, F. Wagener and F. Glorius, Chem. - Eur. J., 2019, 25, 8240–8244 CrossRef CAS PubMed; (b) A. Lipp, S. O. Badir, R. Dykstra, O. Gutierrez and G. A. Molander, Adv. Synth. Catal., 2021, 363, 3507–3520 CrossRef CAS PubMed.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed; (b) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- Y. Lu, C. Z. Fang, Q. Liu, B. L. Li, Z. X. Wang and X. Y. Chen, Org. Lett., 2021, 23, 5425–5429 CrossRef CAS PubMed.
- (a) A. M. Jiménez and M. J. Navas, Crit. Rev. Anal. Chem., 1997, 27, 291–305 CrossRef; (b) L. Weber, Coord. Chem. Rev., 2005, 249, 741–763 CrossRef CAS; (c) T. Baumgartner and R. Reau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (d) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC; (e) M. Marinozzi, F. Pertusati and M. Serpi, Chem. Rev., 2016, 116, 13991–14055 CrossRef CAS PubMed; (f) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (g) H. Ni, W. L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed.
- D. C. Meadows and J. Gervay-Hague, Int. J. Med. Res. Rev., 2006, 26, 793–814 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2223172, 2224155, 2224158 and 2248860. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03667g |
| This journal is © The Royal Society of Chemistry 2023 |