
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast C–C and C–N bond formation reactions in water microdroplets facilitated by the spontaneous generation of carbocations†
Ting
Wang
a,
Zheng
Li
a,
Hang
Gao
a,
Jun
Hu
*b,
Hong-Yuan
Chen
a and
Jing-Juan
Xu
 *a
*a
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: xujj@nju.edu.cn
bSchool of Life Sciences and Health Engineering, Jiangnan University, Wuxi 214122, China. E-mail: hujun@jiangnan.edu.cn
First published on 4th October 2023
Abstract
Carbocations are important electrophilic intermediates in organic chemistry, but their formation typically requires harsh conditions such as extremely low pH, elevated temperature, strong oxidants and/or expensive noble-metal catalysts. Herein, we report the spontaneous generation of highly reactive carbocations in water microdroplets by simply spraying a diarylmethanol aqueous solution. The formation of transient carbocations as well as their ultrafast in-droplet transformations through carbocation-involved C–C and C–N bond formation reactions are directly characterized by mass spectrometry. The intriguing formation and stabilization of carbocations are attributed to the super acidity of the positively charged water microdroplets as well as the high electric fields at the water–air interfaces. Without the utilization of external acids as catalysts, we believe that these microdroplet reactions would pose a new and sustainable way for the construction of aryl-substituted compounds.
Introduction
As an important category of carbocations, diarylcarbenium ions are highly reactive electrophiles, which have been widely applied to the synthesis of various kinds of pharmaceuticals, agrochemicals, dyes and functional materials.1–4 For example, they are extensively proposed as the key intermediates in the construction of diarylmethylpiperazine cores, such as cetirizine for anti-allergy,5 lomerizine for migraine,6 almitrine for respiratory diseases,7 and manidipine, which has the effect of lowering blood pressure.8 The industrial production of many dyes (e.g., malachite green and crystal violet9) is also facilitated by the acid-induced formation of diarylcarbenium ions to construct the desired triarylmethane cores. Despite their significance in both industry and academia, diarylcarbenium ions are still not conveniently utilized due to the harsh conditions especially the extremely low pH10 to produce them. Moreover, the electron-deficient nature of diarylcarbenium ions makes them hard to be tamed because of their high reactivities towards many nucleophiles. As a result, their lifetimes in polar solvents (e.g., alcohols)11,12 are fleeting. Measurements of the diarylcarbenium ions generated via laser flash photolysis by transient absorption spectroscopy have revealed that their lifetimes are typically on picosecond levels.12–14 In the bulk solutions, stable diarylcarbenium ions are only found in non-nucleophilic solvents when concentrated acids even super acids are used.10 For example, benzyl alcohol is predominantly protonated in highly concentrated sulfuric acid, facilitating the elimination of water to form the dibenzoyl cation.15 Such extremely acidic media also demonstrated the stabilization of carbocations, in which lifetimes of these cations can be prolongated to microsecond levels and could thus be captured by conventional absorption spectroscopy10 and NMR.16 Nevertheless, concentrated acids, elevated temperatures, extra catalysts or strong oxidants are still needed for producing the diarylcarbenium ions.4,17,18Unlike bulk media, microdroplets are recently found to be able to provide a unique reaction environment, which has allowed the discovery of many unprecedented reaction pathways as well as the acceleration of known reactions by several orders of magnitude (103 to 106).19,20 The elegant studies in microdroplet chemistry during the last few decades have uncovered several aspects of the unique properties of water microdroplets, including the partial solvation,19,21 ordered molecular orientation,22,23 ultrahigh electric fields,24–26 spontaneous redox reactions27 and extreme pH,28,29 which all exhibit remarkable differences with the bulk water. The extreme pH is an intriguing property of a charged microdroplet, which suggests that a positively charged microdroplet alone can be considered as a super acid to promote the acid-catalyzed reactions, and vice versa for a negatively charged microdroplet as a super base.28,30 For example, the use of a strong base, which is a must in the synthesis of benzimidazoles through the nucleophilic addition of o-aryl diamine to carboxylic acids, can be completely avoided when using the microdroplet synthesis.31
Inspired by the recent findings in microdroplet chemistry, we envision the spontaneous formation of diarylcarbenium ions in positively charged water microdroplets because of their super acidity.32,33 By electrospraying their aqueous solutions, positively charged microdroplets containing diarylmethanols are conveniently produced. By further combining with ambient mass spectrometry, our postulation was confirmed by the detection of the expected diarylcarbenium ions, proving the superiority of water microdroplets in generating carbocations as no concentrated acids are required. Moreover, rapid transformations of the in-droplet formed diarylcarbenium ions via the C–C and C–N bond formation reactions were also achieved by adding nucleophiles into the sprayed solutions. It thus demonstrated a new and green approach to the construction of aryl-substituted compounds including useful pharmaceuticals, without the use of concentrated acids, high temperatures, catalysts, strong oxidants and other external conditions.
Results and discussion
The experimental setup used for the spontaneous diarylcarbenium ion production in water microdroplets is shown in Fig. 1a. Ambient mass spectrometry (MS) was also employed to probe these putative cationic species that might have formed in the electrosprayed water droplets. For an initial trial, a submicron-sized ion emitter (with a tip opening of ∼400 nm in diameter) containing the aqueous solution of diarylmethanols (100 μM) was placed in front of the inlet of a mass spectrometer at a distance of 15 mm. A metal (Fe, 0.2 mm in diameter) electrode was also inserted into the ion emitter from its rear end to induce the generation of water microdroplets via electrospray ionization (ESI), which was achieved through the application of a high voltage (∼1.5 kV) onto the metal electrode. It should be noted that the inserted metal electrode was intentionally placed at a distance of at least 4 cm away from the front tip of the ion emitter, to avoid the interferents from electrooxidations of analytes during ESI. The complete experimental setup photograph of MS measurements with the modified ionization source (emitter-MS setup) is shown in Fig. S1.† The mass spectra in Fig. 1c–e show the results obtained from the aqueous solution of 4,4′-dimethoxybenzhydrol (1a), diphenylmethanol (1b) and 4,4′-difluorobenzhydrol (1c), respectively. To our delight, apart from the mass peaks referring to the compound 1a (m/z 267.0991, [1a + Na]+, theoretical m/z 267.0992), the signal of the putative diarylcarbenium ion (m/z 227.1066, [2a]+, theoretical m/z 227.1067) was also observed in Fig. 1c. On the basis of mass difference (Δm = 39.9925, which matched exactly with the mass of [(Na − H) + H2O]), it can thus be concluded that the ion at m/z 227.1066 was derived from protonated 1avia the elimination of water (Fig. 1b). It is necessary to clarify that 2a can be generated without the application of a high voltage. However, the conversion rate is much lower compared with that obtained under a 1.5 kV high voltage, probably due to the diminished acidity in the uncharged microdroplets (Fig. S2†).33 As exhibited in Fig. 1d, a similar result was observed in the case of diphenylmethanol (i.e., 1b), in which the mass peak at m/z 167.0856 was assigned to the diarylcarbenium ion 2b (theoretical m/z 167.0855). We note that a higher percentage of the diarylcarbenium ion is observed with a smaller ion emitter (Fig. S3 and S4†). This can be explained by the higher surface charge-to-volume ratios of smaller microdroplets when a smaller ion emitter was employed.34 | ||
| Fig. 1 Mass spectrometric examination of the spontaneous formation of diarylcarbenium ions in water microdroplets. (a) A schematic illustration of the experimental setup; (b) mechanism for the generation of diarylcarbenium ions in the electrosprayed water microdroplets; (c) mass spectrum of the aqueous solution of 4,4′-dimethoxybenzhydrol (1a, 100 μM); (d) mass spectrum of the aqueous solution of diphenylmethanol (1b, 100 μM) and (e) mass spectrum of the aqueous solution of 4,4′-difluorobenzhydrol (1c, 100 μM). | ||
The relative abundance of the diarylcarbenium ions is also affected by the reaction distance (defined as the distance between the emitter tip and the MS inlet). In our practice, a clear increase in the relative intensity of diarylcarbenium ions was observed when a longer reaction distance was employed (Fig. S5†), which might be attributed to the longer reaction times35 as well as the higher surface charge-to-volume ratio of smaller droplets produced by fission.29 It is also worth noting that the electrophilicity and lifetime of the diarylcarbenium ion can be varied significantly along with the electron-donating abilities of the substituents at para-positions.36 In particular, the lifetime of diarylcarbenium ions with an electron-withdrawing substitution is extremely fleeting and typically on a picosecond level (e.g., the half-life time of 2c is less than 30 ps in methanol water11), making it difficult to be directly observed. Despite no signal that refers to 4,4′-difluorobenzhydrol was observed (probably due to the low electron density of the oxhydryl on 1c to affiliate a proton or a sodium ion), the in-droplet formation of transient diarylcarbenium ion 2c was clearly evidenced by mass peaks at m/z 203.0667 ([2c]+, theoretical m/z 203.0667, Δm = 0 mDa). Besides the evidence from the high-accuracy mass measurements, a detailed examination of these diarylcarbenium ions by tandem mass spectrometry was also carried out. As summarized in Fig. S6,† fragmentations of these ions all matched well with their structures. The above results thus cooperatively supported the spontaneous formation of diarylcarbenium ions in the positively charged water microdroplets.
The effect of solution pH was also investigated. When an alkali aqueous solution of 1a (pH ≥ 9) was sprayed (Fig. S7†), no diarylcarbenium ions were produced. In contrast, a significant increase in the percentage of 2a was observed when lowering the solution pH, which further convinced the critical role of pH in the production of diarylcarbenium ions in the microdroplets.
Encouraged by the solid confirmation of the spontaneous generation of diarylcarbenium ions, we then aimed to explore their in-droplet reactions. We selected electron-rich compounds including 1,3,5-trimethoxybenzene (TMB, 3a) and 3-methylindole (3-MI, 3b) to evaluate their nucleophilic addition to 2a. As shown in Fig. 2a, such C–C bond formations can be mechanistically categorized into the well-known Friedel–Crafts (FC) reactions, which are widely employed in late-stage functionalization of the arenes and heteroarenes.37,38Fig. 2b shows the mass spectrum obtained by electrospraying the aqueous mixture of compounds 1a and 3a. Besides signals of the reactants (m/z 169.0861, [TMB + H]+, theoretical m/z 169.0859, Δm = 0.2 mDa) and the in situ produced diarylcarbenium ion (2a, m/z 227.1066), the mass peak referring to the expected product 4a (m/z 395.1854, theoretical m/z 395.1853, Δm = 0.1 mDa) was also abundantly observed. Further examination of the newly formed product by tandem mass spectrometry was also conducted. Upon collision induced dissociation (CID), the fragmentation patterns of ions located at m/z 395.1854 matched well with that of the respective product (see Fig. S8†). The above experimental results thus clearly demonstrated the efficient in-droplet reaction between the newly produced diarylcarbenium ion and TMB. In another parallel test, the reaction between the diarylcarbenium ion 2a and 3-MI was evaluated. Similarly, product 4b formed through the nucleophilic reaction of 3-MI is confirmed by the observation of a mass peak at m/z 358.1808 (theoretical m/z 358.1802, Δm = 0.6 mDa, Fig. 2c), which is also examined by tandem mass spectrometry (see Fig. S9†). We also note that the reaction times of the microdroplets can be very short (<10 μs),39,40 suggesting that the diarylcarbenium ion-involved reactions are ultrafast and spontaneous in the flying water microdroplets.
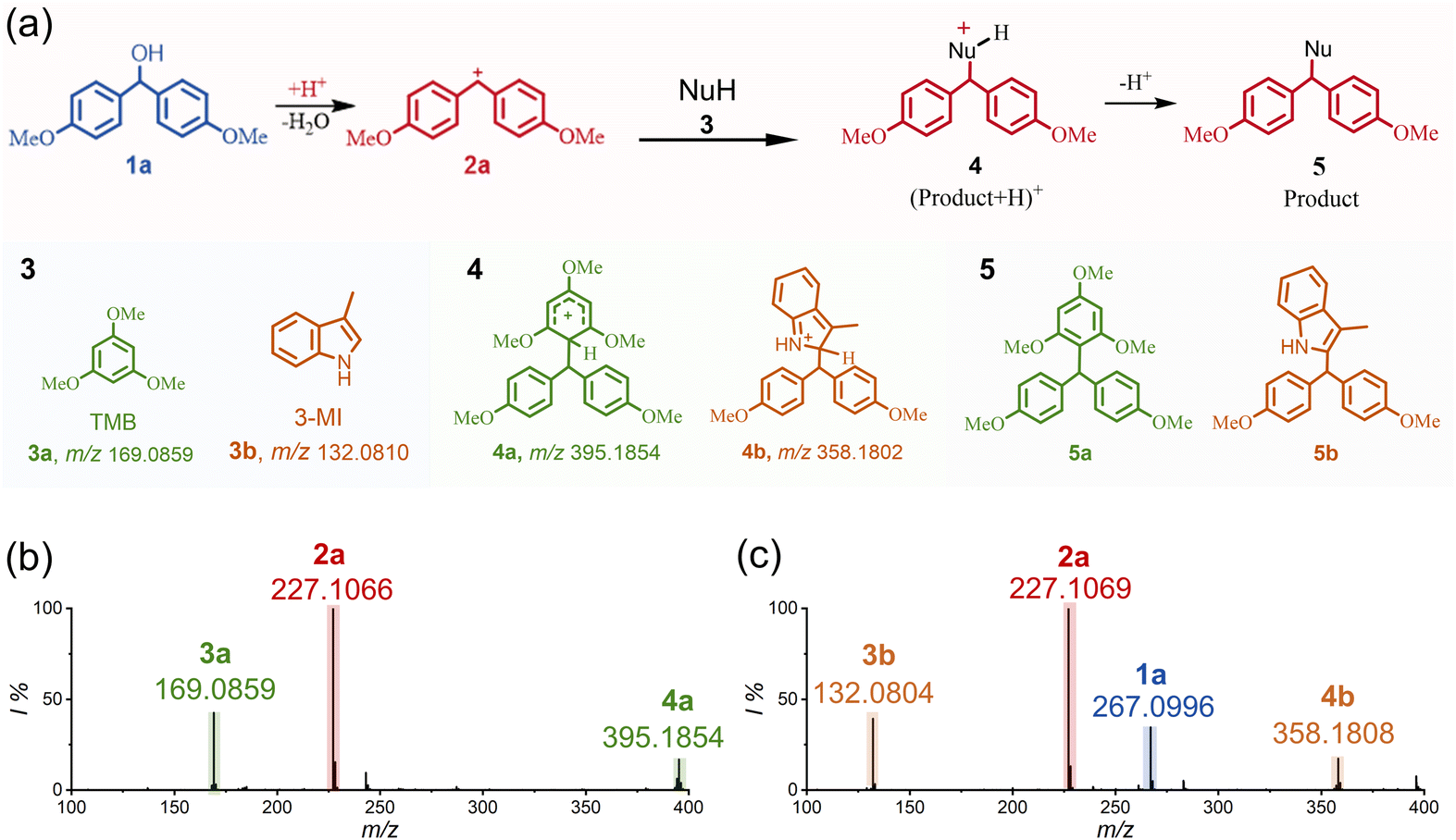 | ||
| Fig. 2 Mass spectrometric analysis of the spontaneous dehydrative C–C coupling in water microdroplets. (a) Proposed reaction mechanism for the spontaneous dehydrative C–C coupling between 1a and 3 in water microdroplets. (b) Mass spectrum showing the spontaneous dehydrative C–C coupling products when spraying the 1a and 3a mixture solution. (c) Mass spectrum showing the spontaneous dehydrative C–C coupling products when spraying the 1a and 3b mixture solution. Uncharged product 5 was yielded by deprotonation of product 4. | ||
Besides the C–C bond formations, we also investigate the feasibility of the diarylcarbenium ions involved in C–N couplings. Nitrogen-containing heterocyclic compounds including pyrazole (pyr, 3c) and piperazine (PZ, 3d) were selected as nucleophiles, which are important structural motifs that are widely found in many biomolecules and pharmacologically active compounds.41,42 As the illustration of the reaction mechanisms in Fig. 3, the trapping of the electron-deficient diarylcarbenium ions by the electron-rich nitrogen atoms is highly selective in constructing C–N bonds.43Fig. 3a, b and S10† summarize the experimental results of the in-droplet reactions between the diarylmethanols (1b and 1c) and the nucleophiles (3c and 3d). On the basis of our mass examination (Fig. 3c, d, S10c and d†), all four combinations succeeded in producing the C–N coupling products (m/z 235.1230, 253.1699, 289.1511 and 271.1041 for products 6a, 6b, 8a and 8b, respectively). Additionally, further examination of these products was carried out by tandem mass spectrometry (see Fig. S11†).
 | ||
| Fig. 3 Mass spectrometric analysis of the spontaneous dehydrative C–N coupling in water microdroplets. (a) Proposed reaction mechanism for the spontaneous dehydrative C–N coupling between 1b and 3c in water microdroplets. (b) Proposed reaction mechanism for the spontaneous dehydrative C–N coupling between 1c and 3d in water microdroplets. (c) Mass spectrum showing the spontaneous dehydrative C–N coupling products when spraying the 1b and 3c mixture solution. (d) Mass spectrum showing the spontaneous dehydrative C–N coupling products when spraying the 1c and 3d mixture solution. The uncharged products 7a and 9a were yielded by deprotonation of products 6 and 8, respectively. | ||
Next, we tried the off-line collection of the products from the microdroplet reaction (Fig. S12†) and then analyzed them using gas chromatography-mass spectrometry (GC-MS) and Proton Nuclear Magnetic Resonance (1H NMR). The experimental results of GC-MS are shown in Fig. S13–S15.† Both C–C reaction products (5a and 5b) and C–N reaction products (7b and 9a) were successfully detected. However, for C–N reaction products (7a and 9b), only a small amount of decomposed products was identified in GC-MS. We infer that these products were not stable under the operating conditions of GC-MS. As an alternative, the reaction yields were determined using 1H NMR. The experimental results of 1H NMR are shown in Fig. S16–S18.† The conversion rates of products 5a, 5b, 7a, 7b, 9a and 9b were calculated to be about 15%, 52%, 1%, 9%, 9%, and 1%, respectively. Although the yield from microdroplet collection remains limited and there is a demand for further research to enable large-scale production, the immense potential of this technique cannot be overlooked. Indeed, it is anticipated to play a significant role in the future evolution of green synthetic chemistry.
Importantly, the aforementioned C–C and C–N reactions are all spontaneous and ultrafast in the electrosprayed water microdroplets, compared to the same bulk reactions that are typically proceeded with the help of elevated temperatures,44 metal catalysts18 or strong oxidants.45 Furthermore, the concentrated acid that is needed to induce the formation of the key reactive intermediates, diarylcarbenium ions, is also discarded. The super acidity is an important but still mysterious aspect of the positively charged water microdroplets. Experimental measurements by a fluorescent method have revealed that a decrease in the apparent pH of sprayed water microdroplets can reach up to 3 units (i.e., pH ≈ 4), compared with the bulk pure water.46,47 However, such acidification seems still far from the conditions of the bulk reactions,48,49 in which highly concentrated acids (pH ≪ 1) are employed. Unlike the common Brønsted acid, a water microdroplet generated in positive ESI contains excess protons and lacks the counter anions. These excess protons are preferred to be distributed at roughly equidistant intervals on its surface to minimize potential energy,50 thus resulting in a highly localized extreme pH on the surface of a water microdroplet. In addition, enrichment of the surface protons as a result of water evaporation during its flying to the MS inlet might also contribute to the super acidity of a water microdroplet.28,30 Apart from the surface confinement and enrichment of protons, partial solvation of analytes might also be an important factor to facilitate the formation and stabilization of the diarylcarbenium ions, in which the regeneration of diarylmethanols via an attack of the diarylcarbenium ion by a water molecule might be alleviated.
In recent studies, a high electric field (>107 V m−1) and molecular orientation at the surface of water microdroplets were recognized as crucial factors in promoting the formation and stabilization of reactive carbocations.33,51–53 The high electric field can promote the formation of a double electric layer near the water–air interface. In this circumstance, the outer layer is rich in protons that can act as a superacid, forming stable diarylcarbenium ions.54,55 Other studies56 opine that aromatic compounds in microdroplets preferentially inhabit the air–water interface, positioning the aromatic ring towards the air side while the –OH group remains embedded in water. Such a high electric field has the potential to break the C–O bond, thus leading to the production of the diarylcarbenium ions at the interface of the microdroplet. Subsequently, these ions then proceed to combine with nucleophiles, culminating in the spontaneous formation of the corresponding C–C and C–N products in the microdroplets.
Conclusions
In conclusion, by taking advantage of the super acidity of positively charged water microdroplets, the spontaneous generation of diarylcarbenium ions was achieved by simply electrospraying the aqueous solutions of diarylmethanols. The in-droplet trapping of the diarylcarbenium ions by various nucleophiles also enabled rapid C–C and C–N bond formation, which avoided the use of concentrated acids, elevated temperatures, external catalysts and strong oxidants compared with the same reactions in bulk media. Taken together, we believe that water microdroplets might offer a new locus to fully exploit the highly reactive carbocations in a simple and green approach. Through the continuous uncovering of the intriguing microdroplet chemistry, we wish applications especially in green organic synthesis to be promoted in the near future.Data availability
All data included in this study are available upon request by contact with the corresponding author.Author contributions
Ting Wang, Jun Hu and Jing-Juan Xu conceived the work and designed the experiments. Ting Wang, Zheng Li, Hang Gao and Jun Hu performed the experiments and analyzed the data. Ting Wang, Jun Hu and Jing-Juan Xu described the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grants no. 22034003 and 22004071), Natural Science Foundation of Jiangsu Province (Grant BK20200668), and Excellent Research Program of Nanjing University (ZYJH004).Notes and references
- S. Minegishi, R. Loos, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2005, 127, 2641–2649 CrossRef CAS PubMed.
- P. Costa, M. Fernandez-Oliva, E. Sanchez-Garcia and W. Sander, J. Am. Chem. Soc., 2014, 136, 15625–15630 CrossRef CAS PubMed.
- P. Singh and R. K. Peddinti, J. Chem. Sci., 2022, 134, 63 CrossRef CAS.
- M. Okajima, K. Soga, T. Nokami, S. Suga and J. i. Yoshida, Org. Lett., 2006, 8, 5005–5007 CrossRef CAS PubMed.
- J. Reiter, P. Trinka, F. L. Bartha, L. Pongó, B. Volk and G. Simig, Org. Process Res. Dev., 2012, 16, 1279–1282 CrossRef CAS.
- C. W. Evans, H. M. Viola, D. Ho, L. C. Hool, S. A. Dunlop, M. Fitzgerald and K. S. Iyer, RSC Adv., 2012, 2, 8587–8590 RSC.
- P. J. Gengo, H. O. Pettit, S. J. O. Neill, K. Wei, R. McNutt, M. J. Bishop and K. J. Chang, J. Pharmacol. Exp. Ther., 2003, 307, 1221–1226 CrossRef CAS PubMed.
- A. Roca-Cusachs and F. Triposkiadis, Drugs, 2005, 65, 11–19 CrossRef CAS PubMed.
- R. Muthyala, A. R. Katritzky and X. F. Lan, Dyes Pigm., 1994, 25, 303–324 CrossRef CAS.
- N. C. Deno, J. J. Jaruzelski and A. Schriesheim, J. Am. Chem. Soc., 1955, 77, 3044–3051 CrossRef CAS.
- J. Peon, D. Polshakov and B. Kohler, J. Am. Chem. Soc., 2002, 124, 6428–6438 CrossRef CAS PubMed.
- B. P. Fingerhut, C. F. Sailer, J. Ammer, E. Riedle and R. de Vivie-Riedle, J. Phys. Chem. A, 2012, 116, 11064–11074 CrossRef CAS PubMed.
- J. Ammer, C. F. Sailer, E. Riedle and H. Mayr, J. Am. Chem. Soc., 2012, 134, 11481–11494 CrossRef CAS PubMed.
- T. A. Nigst, J. Ammer and H. Mayr, J. Phys. Chem. A, 2012, 116, 8494–8499 CrossRef CAS PubMed.
- R. G. Bryant, J. Chem. Educ., 1983, 60, 933–935 CrossRef CAS.
- E. M. Arnett and T. C. Hofelich, J. Am. Chem. Soc., 1983, 105, 2889–2895 CrossRef CAS.
- T. Ohwada, T. Suzuki and K. Shudo, J. Am. Chem. Soc., 1998, 120, 4629–4637 CrossRef CAS.
- H. Hikawa, H. Suzuki and I. Azumaya, J. Org. Chem., 2013, 78, 12128–12135 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- Z. Wei, Y. Li, R. G. Cooks and X. Yan, Annu. Rev. Phys. Chem., 2020, 71, 31–51 CrossRef CAS PubMed.
- S. Vaitheeswaran and D. Thirumalai, J. Am. Chem. Soc., 2006, 128, 13490–13496 CrossRef CAS PubMed.
- I. Nam, J. K. Lee, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 12396–12400 CrossRef CAS PubMed.
- I. Nam, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 36–40 CrossRef CAS PubMed.
- D. Zhang, X. Yuan, C. Gong and X. Zhang, J. Am. Chem. Soc., 2022, 144, 16184–16190 CrossRef CAS PubMed.
- Y. Meng, E. Gnanamani and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 19709–19713 CrossRef CAS PubMed.
- Y. Meng, E. Gnanamani and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 7724–7728 CrossRef CAS PubMed.
- C. Gong, D. Li, X. Li, D. Zhang, D. Xing, L. Zhao, X. Yuan and X. Zhang, J. Am. Chem. Soc., 2022, 144, 3510–3516 CrossRef CAS PubMed.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC.
- S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed.
- K.-H. Huang, Z. Wei and R. G. Cooks, Chem. Sci., 2021, 12, 2242–2250 RSC.
- P. Basuri, L. E. Gonzalez, N. M. Morato, T. Pradeep and R. G. Cooks, Chem. Sci., 2020, 11, 12686–12694 RSC.
- V. Buch, A. Milet, R. Vacha, P. Jungwirth and J. P. Devlin, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 7342–7347 CrossRef CAS PubMed.
- A. Kumar, S. Mondal and S. Banerjee, J. Am. Chem. Soc., 2021, 143, 2459–2463 CrossRef CAS PubMed.
- J. Hu, Q. Y. Guan, J. Wang, X. X. Jiang, Z. Q. Wu, X. H. Xia, J. J. Xu and H. Y. Chen, Anal. Chem., 2017, 89, 1838–1845 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Nat. Commun., 2018, 9, 1562 CrossRef PubMed.
- H. Mayr and A. R. Ofial, Angew. Chem., Int. Ed., 2006, 45, 1844–1854 CrossRef CAS PubMed.
- M. Zeng and S. L. You, Synlett, 2010, 2010, 1289–1301 CrossRef.
- M. Sakhalkar, P. Aduri, S. Lande and S. Chandra, Clean Technol. Environ., 2020, 22, 59–71 CrossRef CAS.
- J. Hu, T. Wang, W. J. Zhang, H. Hao, Q. Yu, H. Gao, N. Zhang, Y. Chen, X. H. Xia, H. Y. Chen and J. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 18494–18498 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, J. Am. Chem. Soc., 2016, 138, 3453–3460 CrossRef CAS PubMed.
- M. Shaquiquzzaman, G. Verma, A. Marella, M. Akhter, W. Akhtar, M. F. Khan, S. Tasneem and M. M. Alam, Eur. J. Med. Chem., 2015, 102, 487–529 CrossRef CAS PubMed.
- S. G. Kucukguzel and S. Senkardes, Eur. J. Med. Chem., 2015, 97, 786–815 CrossRef CAS PubMed.
- M.-J. Luo, G. F. Lv, Y. Li and J. H. Li, Green Chem., 2021, 23, 2044–2048 RSC.
- K. Funabiki, T. Komeda, K. Nishikawa, K. Yamada, Y. Kubota and M. Matsui, Tetrahedron, 2014, 70, 9245–9252 CrossRef CAS.
- P. Bhattacharjee and U. Bora, ACS Omega, 2019, 4, 11770–11776 CrossRef CAS PubMed.
- C. C. Chang, Y. Kazoe, K. Morikawa, K. Mawatari, R. J. Yang and T. Kitamori, Anal. Chem., 2013, 85, 4468–4474 CrossRef CAS PubMed.
- Y. Kazoe, K. Mawatari, Y. Sugii and T. Kitamori, Anal. Chem., 2011, 83, 8152–8157 CrossRef CAS PubMed.
- F. Scholz, D. Himmel, L. Eisele, W. Unkrig and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 1689–1692 CrossRef CAS PubMed.
- A. Martin, A. Arda, J. Désiré, A. Martin-Mingot, N. Probst, P. Sinaÿ, J. Jiménez-Barbero, S. Thibaudeau and Y. Blériot, Nat. Chem., 2016, 8, 186–191 CrossRef CAS PubMed.
- S. Banerjee and S. Mazumdar, Int. J. Anal. Chem., 2012, 2012, 282574 Search PubMed.
- S. Banerjee, Int. J. Mass Spectrom., 2023, 486, 117024 CrossRef CAS.
- A. Kumar, S. Mondal and S. Banerjee, J. Phys. Chem. C, 2023, 127, 6662–6669 CrossRef CAS.
- A. Kumar, S. Mondal, Sandeep, P. Venugopalan, A. Kumar and S. Banerjee, J. Am. Chem. Soc., 2022, 144, 3347–3352 CrossRef CAS PubMed.
- R. G. Cooks, Y. Feng, K. H. Huang, N. M. Morato and L. Qiu, Isr. J. Chem., 2023, 63, e202300034 CrossRef.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CrossRef CAS PubMed.
- A. Nandy, A. Kumar, S. Mondal, D. Koner and S. Banerjee, J. Am. Chem. Soc., 2023, 145, 15674–15679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03870j |
| This journal is © The Royal Society of Chemistry 2023 |