
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometry guided crystallization of anisotropic DNA origami shapes†
Shujing
Huang
,
Min
Ji
,
Yong
Wang
and
Ye
Tian
 *
*
College of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, Jiangsu Key Laboratory of Artificial Functional Materials, Chemistry and Biomedicine Innovation Center, Nanjing University, Nanjing, 210023, China. E-mail: ytian@nju.edu.cn
First published on 3rd October 2023
Abstract
Three-dimensional assembly based on DNA origami structures is an ideal method to precisely fabricate nano-scale materials. Additionally, applying an anisotropic assembly unit facilitates constructing complex materials with extraordinary structure. However, it still remains challenging to crystallize anisotropic DNA nano-structures using simple design, because the assembly of low-symmetry monomers often requires harsh auxiliary conditions and more complicated crystallization processes. In this work, we managed to crystallize the anisotropic elongated octahedral DNA origami frames by non-specific connections, and acquired two kinds of highly ordered superlattices purely by conducting multiple annealing processes and increasing the rigidity of the connection parts. In the case where the connection parts were composed of soft DNA sticky ends, we obtained the theoretically inaccessible simple cubic superlattices by this anisotropic DNA origami shape. Through characterization by small-angle X-ray scattering and scanning electron microscopy, we found that the DNA monomers are arbitrarily arranged due to the stress buffering of the soft DNA SEs, while in the stiffer case, simple tetragonal superlattices with translational arrangement of most anisotropic DNA origami shapes were synthesized as expected. This work deepened the understanding of geometry-guided crystallization of DNA origami shapes and provided a new path for constructing three-dimensional functional devices with simple design.
Introduction
Compared with inorganic colloidal nanoparticles, DNA nano-structures synthesized by DNA origami technology often have more accurate sizes and well-defined shapes.1–6 In addition, programmable modification of functional sites on DNA origami nano-structures endows them with excellent ability for loading guest objects and further assembly.7–11 Therefore, DNA origami technology has fully shown its unique advantages in the design, manufacture, and application of nanomaterials.12–17 Moreover, DNA origami nano-shapes have been demonstrated to crystallize into distinct kinds of three-dimensional (3D) superlattices.18–22 As generally acknowledged, crystallization is a thermodynamics-dominant process in pursuit of the most stable state, in which the hybridization of connection sites is maximized.23–27 For the DNA origami monomers with high symmetry, the assembly results are always identical regardless of which sites the monomer attachment occurs in. Hence non-specific connection is sufficient to crystallize the monomers into an ordered assembly, and guarantee the high assembly yields. However, in the crystallization process of the DNA origami monomers with low symmetry, the design of sticky ends (SEs), used for connection, is crucial to achieve the thermodynamic optimum state of DNA origami assembly.28–30 According to the symmetry of DNA origami monomers, it is necessary to adopt appropriate SE pairs to simultaneously guarantee the maximization of SE hybridization and the correct binding modes between DNA building blocks. Therefore, for the DNA monomers with lower symmetry (or cocrystallization of multi-units of DNA origami nano-shapes), the types of SEs should be increased accordingly to satisfy the specific arrangement of the building blocks.21,27–30 However, the cumbersome design requirements in crystallizing low-symmetry monomers usually bring excessive connection restrictions and increase difficulty in the crystallization process.31,32 Hence, it is important to explore a simple method for low-symmetry DNA building blocks to assemble into well-defined 3D lattices. In this work, by reasonably applying reannealing technology, we managed to crystallize the anisotropic DNA origami monomers—the elongated octahedral (E-octa) DNA origami frames (DOFs) with D4h shape symmetry by endowing the functional sites with identical DNA SE pairs, though this is considered to potentially cause the aperiodic connections brought by the relatively low symmetry of the E-octa DOFs. Surprisingly, by simply and reasonably increasing the cycles of the annealing process, the E-octa DOFs could assemble into an unexpected 3D superlattice with a highly ordered simple cubic (SC) structure. This lattice type is theoretically difficult to realize under a rational arrangement of E-octa DOFs. In addition, to improve the influence of the shape of the anisotropic monomers in the crystallization process, the rigidity of the soft DNA SEs is increased and the crystal structure could shift from SC structure to simple tetragonal (ST) structure under the same annealing conditions, promoting the achievement of the shape-mediated ordered crystals. In this work, the annealing process is carefully explored and demonstrated to be a crucial step for fabricating ordered 3D assemblies composed of anisotropic DNA origami monomers. This approach offers an opportunity to crystallize anisotropic DNA origami nano-structures with easy design of the functional DNA SEs, and provides more paths for assembling large-scaled and highly ordered DNA lattices with unique symmetries and complex structures.Results and discussion
The anisotropic E-octa DOF is composed of twelve six-helix bundles. The length of the four bundles in the middle plane is ∼28.56 nm, while the length of the other eight bundles is ∼35.70 nm (Fig. 1a and S1†). The traditional connection design of anisotropic DOFs during the crystallization process required strict and specific manipulation of the DNA SEs stretched out from the vertices, on the basis of satisfying the rule of maximizing the DNA hybridizations and guaranteeing the correct binding modes of the DNA origami monomers. Here, we only used one type of complementary DNA SE pair to explore the geometry effect of anisotropic DOFs during the crystallization. The complementary SEs utilized here were called SE-A and SE-B (Fig. 1b, depicted as cones and cylinders), and the corresponding assembly units were called E-octa-A and E-octa-B, respectively (Fig. 1b). In addition, 10 nm gold nanoparticles (AuNPs) were placed in the body center of each E-octa DOF to facilitate the structural characterization and further analysis by small-angle X-ray scattering (SAXS).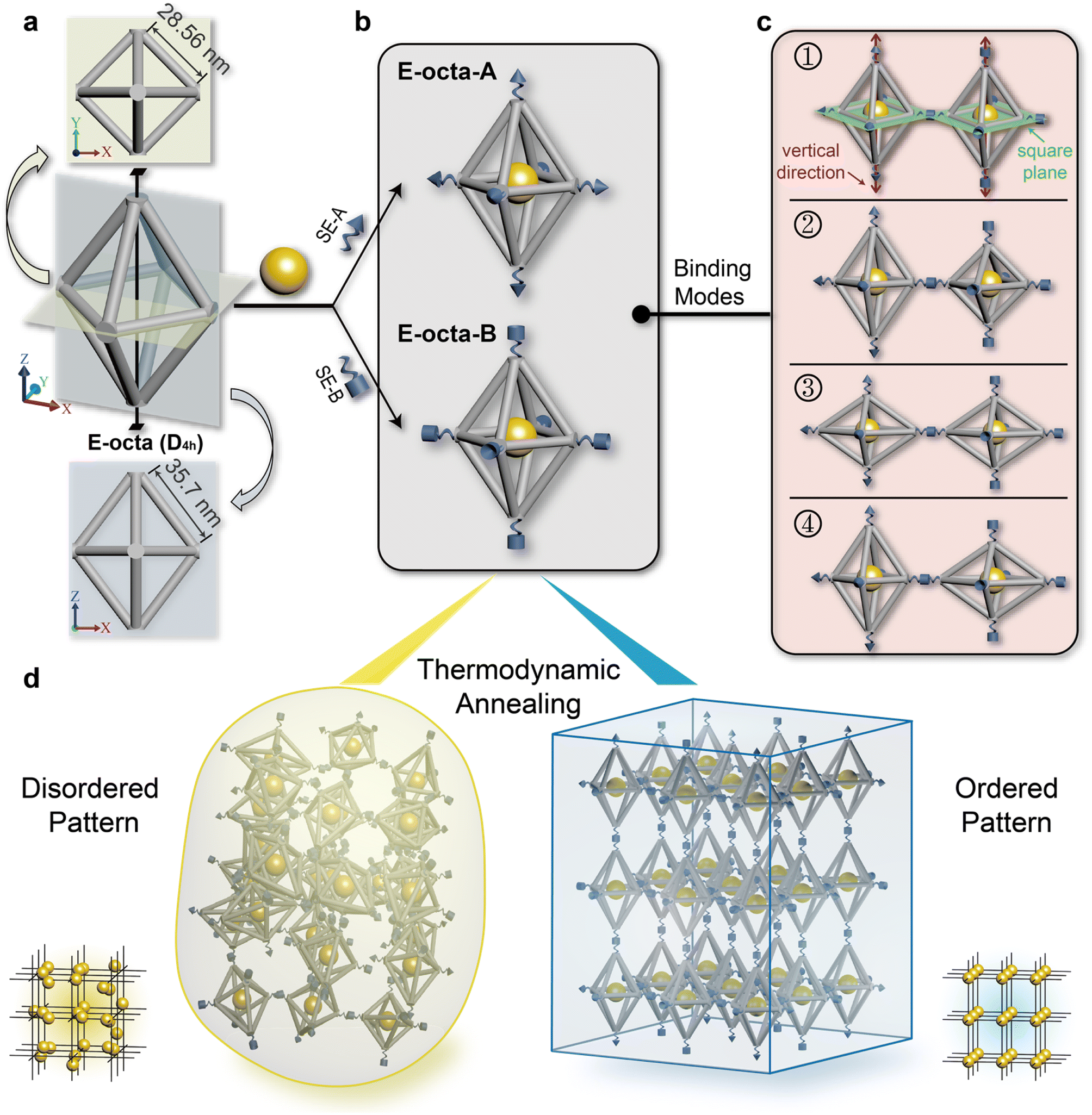 | ||
| Fig. 1 Schematic illustration of assembling E-octa DOFs. (a) The shape of E-octa DOF has D4h symmetry (middle) with a four-fold symmetry axis through the top and bottom vertices. The corresponding top view and side view are shown beside. The edge lengths in the middle plane are ∼28.56 nm while the others are ∼35.7 nm. (b) AuNPs and SEs are mounted onto the E-octa DOFs to form the building blocks, which are called E-octa-A and E-octa-B, respectively. The AuNP is positioned at the body center of the E-octa DOF, while SEs are installed at the six vertices. (c) Four possible binding modes between interconnected E-octa DOFs during assembly. Binding modes 1 and 2 are between vertices in the square plane; binding mode 3 is between vertices along the vertical direction; binding mode 4 is between vertices in the square plane and along the vertical direction. Square plane and vertical direction are marked in mode 1. (d) Possible 3D assemblies after thermodynamic annealing, including the disordered pattern and ordered pattern. Corresponding arrangement of AuNPs are shown beside each assembly. | ||
Unlike the previous reports adopting strict connection design in the crystallization of E-octa DOFs,29 the nonspecific connection utilized here would result in an arbitrary binding mode between nearby building blocks. As shown in Fig. 1c, four possible binding modes may simultaneously occur, which are formed between vertices in the middle square plane (numbers 1 and 2 shown in Fig. 1c), or along the vertical direction (number 3), or vertices both in the square plane and along the vertical direction (number 4). Theoretically, it is hard to synthesize a type of superlattice that simultaneously contains all four kinds of binding modes discussed above and maintains a highly ordered arrangement of the assembly units. Consequently, disordered or amorphous assemblies may be generated (Fig. 1d, left). However, owing to the assembly composed of indiscriminate binding modes, it will inevitably lead to a low efficiency of DNA hybridization for SEs, which would result in an unstable state with high energy. In theory, the degree of the crystal order is inversely correlated with the energy of the system. Hence, a strategy for further reducing the system energy may help fabricate the ordered DNA origami superlattices, considering the potentially unexpected binding modes caused by the anisotropic geometry of the DNA building blocks (Fig. 1d, right).
We commonly adopt an annealing process from high to low temperature to fabricate the DNA origami superlattices, as shown in Fig. 2a (50 °C to 20 °C with a rate of 0.2 °C h−1). Equal amounts of E-octa-A and E-octa-B were mixed, and then annealed. The obtained assembly deposited at the bottom of the tube was further characterized by SAXS to explore the inner arrangement of the DOFs. The two-dimensional (2D) ring and one-dimensional (1D) S(q) curve both exhibited a possible amorphous structure, with approximately three broad peaks (Fig. 2c, purple curve with sample name “Eocta-S1”). Note that the signals indicated in the SAXS results came from the AuNPs located at the center of the E-octa DOFs, which can indirectly reveal the arrangement of the E-octa DOFs.
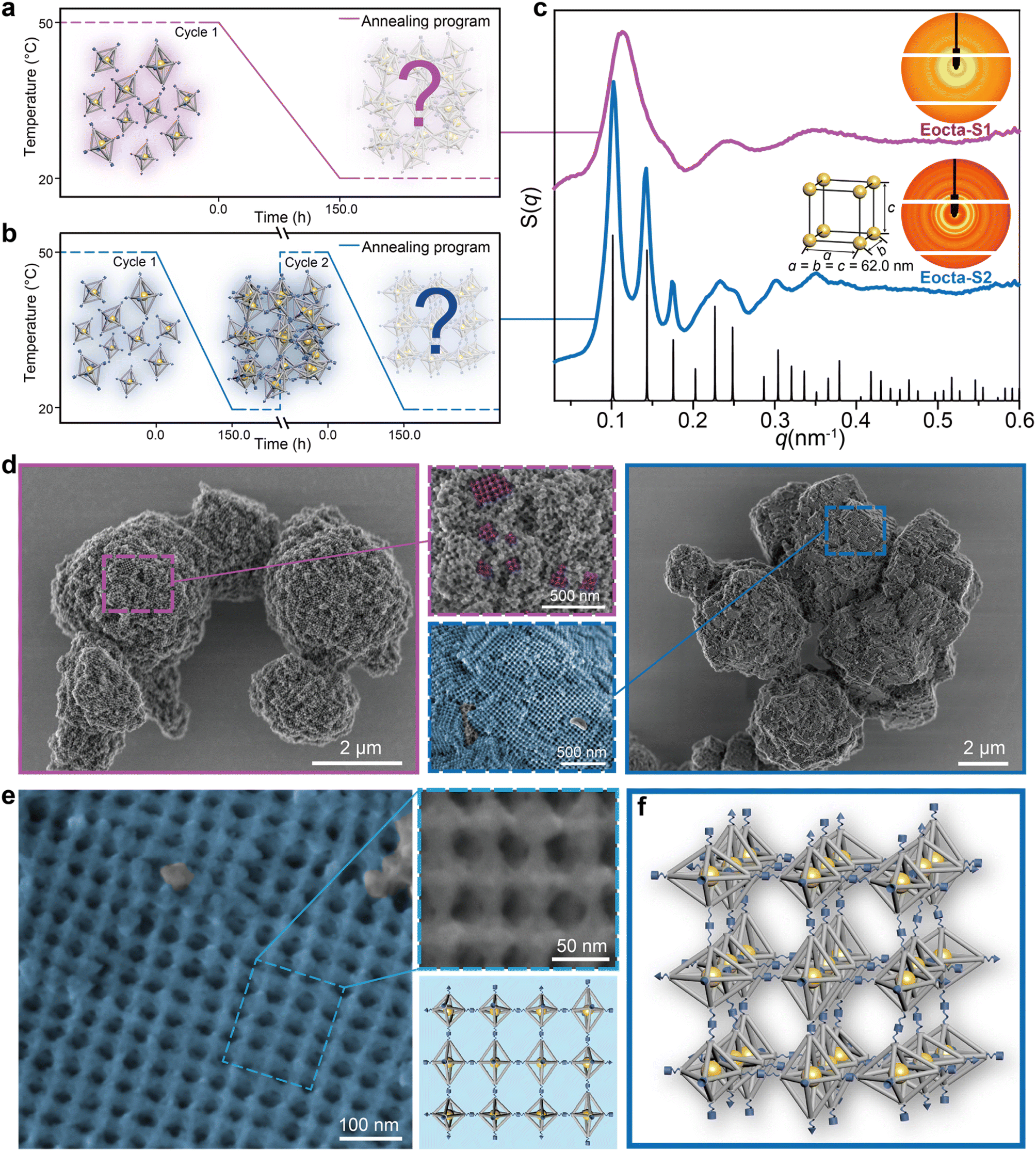 | ||
| Fig. 2 Assembling E-octa DOFs by different annealing cycles. (a) Assembling E-octa DOFs by one annealing cycle, and the corresponding assembly is called Eocta-S1. The annealing procedure is from 50 °C to 20 °C with the rate of 0.2 °C h−1. (b) Assembling E-octa DOFs by two annealing cycles, and the corresponding assembly is called Eocta-S2. The annealing procedures of the two cycles are both from 50 °C to 20 °C with the rate of 0.2 °C h−1. (c) Experimental SAXS results of Eocta-S1 (purple) and Eocta-S2 (blue), and standard scattering peaks of SC with parameter of 62.0 nm (black). Corresponding 2D patterns and AuNP unit cell are shown as inset. (d) Representative low-magnification SEM images of Eocta-S1 (left, framed in the purple box) and Eocta-S2 (right, framed in the blue box) after being coated with a layer of silica. Close-up views (middle) of the regions labelled by dotted boxes, in which grid patterns are painted in false colors (purple for Eocta-S1, blue for Eocta-S2). Scale bars, 2 μm (left and right) and 500 nm (middle). (e) Representative high-magnification SEM image of Eocta-S2 with grid pattern painted in blue (left), close-up view of the region framed in the blue dotted box (top-right) and corresponding model (bottom-right). Scale bars, 100 nm and 50 nm. (f) Proposed model of Eocta-S2. | ||
Annealing time is always an important parameter when growing the artificial crystals. Increasing the annealing cycle discussed above from one to two imposed a positive effect on the crystal order (Fig. 2b). Different from annealing in the discrete monomer state (the first annealing cycle), reannealing is based on the already formed 3D structures. In the second annealing cycle, the SEs in the assembly formed during the first annealing will de-hybridize when the temperature is up to 50 °C. However, the assembly units will not be completely dispersed into the solution and remain largely condensed at the bottom of the tube (Fig. S2†). This will facilitate the rearrangement of the E-octa DOFs to the equilibrium state due to the reduced free energy in the following annealing process.24 Hence, this strategy is beneficial to transforming the amorphous assembly into a thermodynamic steady state, resulting in the formation of an ordered superlattice. The newly formed assembly is termed as “Eocta-S2” and was characterized with more than seven recognizable 1D peaks when tested by SAXS (the blue curve as shown in Fig. 2c). However, increasing the annealing cycles to three did not obviously improve the order of samples (Fig. S3†). Surprisingly, by comparing the experimental scattering curve with the standard peaks (black), the obtained DNA origami assembly was proved to be a simple cubic (SC) nanoparticle superlattice (a = b = c = 62.0 nm). Notably, the SC structure is theoretically difficult to be obtained via rational crystallization of E-octa DOFs. Hence, a further characterization of the deposited aggregates is required to visually observe the arrangement of the DOFs in detail.
In order to retain the inherent structural details and morphologies of the DNA origami superlattices during the scanning electron microscopy (SEM) characterization, the superlattices were coated with a thin layer of silica.33,34 At low magnification, the overall appearance of Eocta-S1 and Eocta-S2 showed distinct differences. Eocta-S2 showed more regions with right-angled domains and flat surfaces (Fig. 2d, framed in the blue box on the right), while for Eocta-S1, the DNA origami monomers preferred to aggregate together randomly (Fig. 2d, framed in the purple box on the left). Furthermore, two typical regions of the samples were selected and magnified to carefully compare the differences in detail between the two superlattices (purple dotted box for Eocta-S1 and blue dotted box for Eocta-S2). For better observation, the grid pattern, which is considered to occur when the monomers are arranged in order, is painted in corresponding false color (purple for Eocta-S1 and blue for Eocta-S2). It could be found that most surface regions of Eocta-S2 had a grid pattern, and the domain size of the grid pattern in Eocta-S2 was notably larger than the same region in Eocta-S1 (see more examples in Fig. S4 and S5†). Compared with Eocta-S1, Eocta-S2 could organize into ordered structures with larger domain sizes, which verifies the positive effect of the reannealing process on crystal order.
Furthermore, a large grid pattern area in Eocta-S2 was selected to confirm the actual arrangement of E-octa DOFs. A representative grid pattern region was magnified (Fig. 2e, framed in the blue dotted box) to clearly observe the details of the binding modes between adjacent E-octa DOFs (see more in Fig. S6†). As illustrated in the right part of Fig. 2e, the magnified region captured twelve monomers with the corresponding model shown below. Each connection site in this region was found to be completely hybridized. Therefore, it can be speculated that most of the DNA SEs in Eocta-S2 have been hybridized after the second annealing cycle, and the system has almost reached the stable state. This may also account for the phenomenon that the ordering of the lattice did not improve significantly after three cycles of annealing (Fig. S3†). Besides, the arbitrary arrangement of multiple binding modes (numbers 1, 2, and 4 as shown in Fig. 1c) could be observed in this representative region, which revealed an irregular arrangement of the DNA origami building blocks. Moreover, the parameter of the unit cell of Eocta-S2 calculated from SAXS results (∼62.0 nm) is close to the theoretical average lattice parameters (∼64.3 nm) of the expected ST structure assembled by E-octa DOFs (see detailed calculation in Fig. S7†), which also proved that the DNA origami monomers were randomly arranged in a 3D manner. Theoretically, though it is unreasonable for the rigid DOFs to assemble in a random way, the maximization of SE hybridizations after reannealing contributes to generating a thermodynamically stable state. Furthermore, it is hypothesized that the softness of DNA SEs allows for deforming to some extent, which underlies the maximum hybridization of connection sites to counteract the stress generated from the disordered arrangement of anisotropic E-octa DOFs. Accordingly, the proposed model of the Eocta-S2 is shown in Fig. 2f.
To avoid the stress buffering caused by the soft DNA SEs during the assembly, the stiffness of single-stranded DNA SEs was strengthened through transforming them into double strand-dominated structures. In this work, DNA SEs are designed as 30-nucleotide (nt) long including a poly-T non-complementary part (22T) and an 8 nt complementary part. Hence, sequences of poly-A were introduced during the assembly to bind with the poly-T part of DNA SEs to increase the rigidity of the connection parts (Fig. 3a). This strategy perfected the definition of the rigid shape of the DNA origami monomers, which promoted the ordered arrangement of E-octa DOFs. On demand of achieving a stable state, the E-octa systems with rigid connection tended to maximize the SE hybridizations under the little deformation of monomers. Thus, a newly formed assembly was obtained after undergoing two cycles of annealing, which was termed as “Eocta-R2”. Similar to the system with soft DNA SEs (Eocta-S2), the new synthesized assemblies were then characterized by SAXS and SEM as well. As shown in the S(q) curve (green curve, Fig. 3b and S8†) extracted from the 2D rings (inset in Fig. 3b), more than twenty sharp scattering peaks indicated that Eocta-R2 was highly ordered. Besides, these peaks can match well with the standard ST peaks (black curve shown in Fig. 3b), with the parameter of the unit cell: a = b = 61.4 nm, c = 76.6 nm. Compared with Eocta-S2, the type of the superlattice has been transformed from SC into ST, which is more consistent with the expectation and previously reported results.29 Furthermore, Eocta-R2 exhibited more clear cuboid structures when scanned under SEM after silica coating (Fig. 3c and S9†). The corresponding crystal domains were ∼1 μm2 in size, and the surface grid patterns were flatter and larger than Eocta-S1 and Eocta-S2. To confirm the arrangement of the DOFs in Eocta-R2, a 4 × 4 region selected from a large grid pattern area was magnified for further observation, and the corresponding model is shown on the right (Fig. 3e). It can be found that the DOFs in the first line framed in the red box are arranged in parallel exposing the middle square planes while the other DOFs are also in a parallel arrangement but expose rhombic planes. This indicates that Eocta-R2 presents as the polycrystalline structure instead of the single crystal. In addition, a small number of disordered structures can also be found in the Eocta-R2 (Fig. S10†). In summary, enhancing the rigidity of the connection sites of E-octa DOFs can deplete the stress buffering brought by soft DNA SEs, and lead to a geometry-guided crystallization of the anisotropic DNA origami building blocks (Fig. 3f).
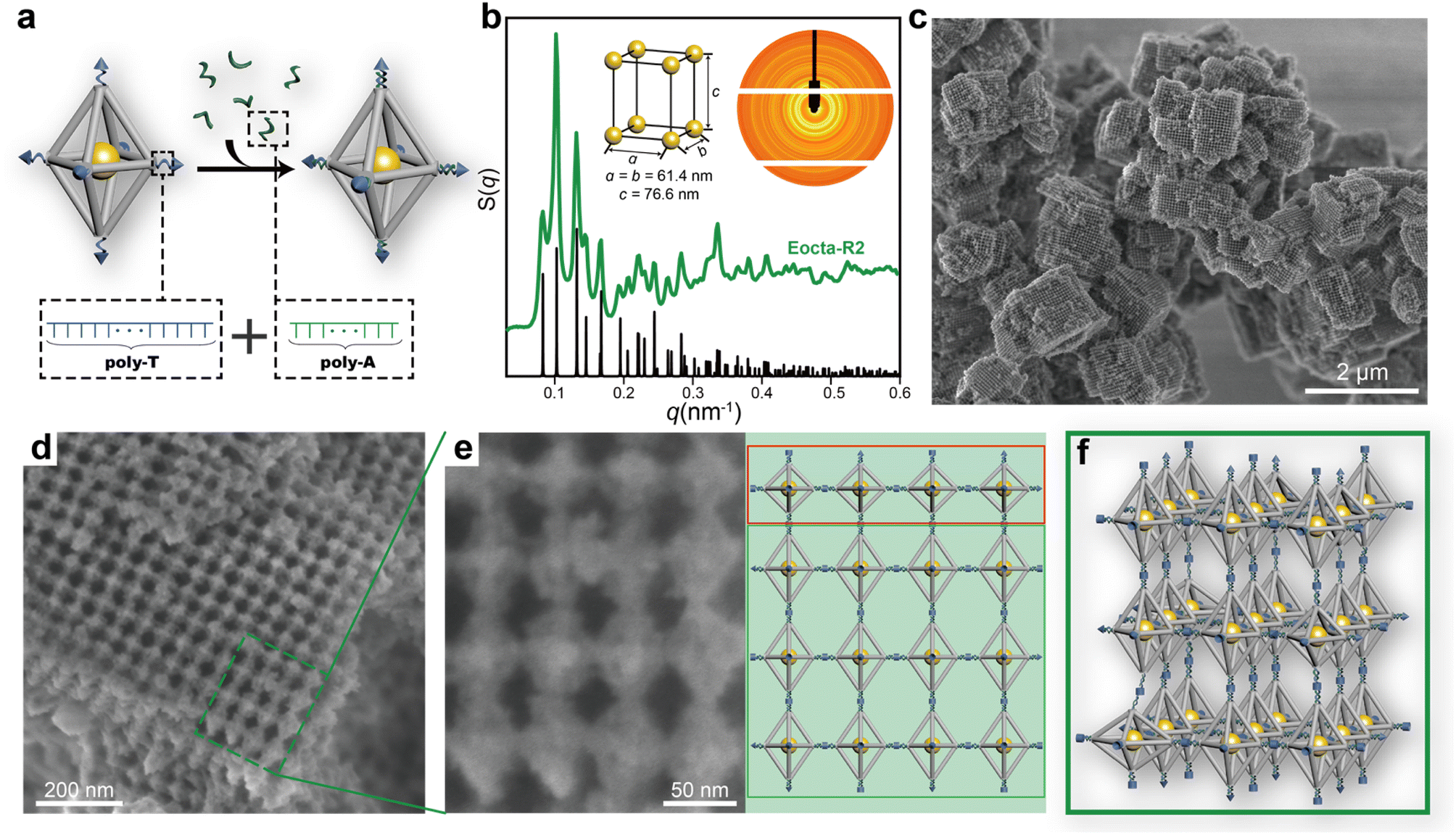 | ||
| Fig. 3 Crystallizing E-octa DOFs by adding additional poly-A sequences. (a) Added poly-A sequences during the assembly process can hybridize with the poly-T portion of SEs which is not involved in the connection between DOFs. (b) Experimental SAXS curve of nanoparticle superlattice Eocta-R2 (green) and corresponding standard ST peaks (black). Corresponding 2D pattern and AuNP unit cell with the ST structure (a = b = 61.4 nm, c = 76.6 nm) are shown as inset. (c) Representative low-magnification SEM image of Eocta-R2. Scale bar, 2 μm. (d) Representative high-magnification SEM image of Eocta-R2. Scale bar, 200 nm. (e) The close-up view of the region framed in the green dotted box (left) and corresponding model with the two types of regions framed in the red box and green box, respectively (right). Scale bar, 50 nm. (f) Proposed model of Eocta-R2. | ||
To quantitatively analyze the order degree of the superlattices mentioned above (Eocta-S1, Eocta-S2 and Eocta-R2), the superlattices presented in the SEM images were classified as four categories, Type I to IV (Fig. 4a). From Type I to IV, the size ranges of the regions with grid pattern are 0–0.2 μm, 0.2–0.5 μm, 0.5–1 μm, and >1 μm, respectively. Normally, a larger size of grid pattern often indicates a higher order of the superlattice. For clarity, each type of region is colored with distinct false color: orange for Type I, yellow for Type II, green for Type III, and blue for Type IV. For Eocta-S1, the DOF monomers in most regions are randomly arranged and only a few regions exhibit the grid pattern. Hence, most regions in Eocta-S1 are amorphous and belong to Type I (Fig. S11†). In comparation, the sample of Eocta-S2 shows some cuboid shapes, which belong to Type III and Type IV, while the amorphous regions still account for a large proportion (Fig. S12†). For the sample of Eocta-R2, most regions have block morphology and larger size, which leads to the predominance of Type IV regions (Fig. S13†). Moreover, the region types of these three superlattices are quantitatively analyzed and the total statistical areas are 325.5 μm2, 292.8 μm2, and 302.5 μm2, respectively. As shown in Fig. 4b, the proportion of Type I sharply decreases from Eocta-S1 to Eocta-S2 to Eocta-R2, while the proportion of Type II shows the opposite trend. In addition, the regions of Types III and IV can only be observed in Eocta-S2 and Eocta-R2, and the proportion of Type IV region statistics from the latter sample is more than three times that of the former one. These results demonstrate that both of the two strategies, increasing the cycles of the annealing process and strengthening the stiffness of monomer shapes, can enhance the orderliness of E-octa DOF/AuNP superlattices, which is consistent with the results measured by SAXS.
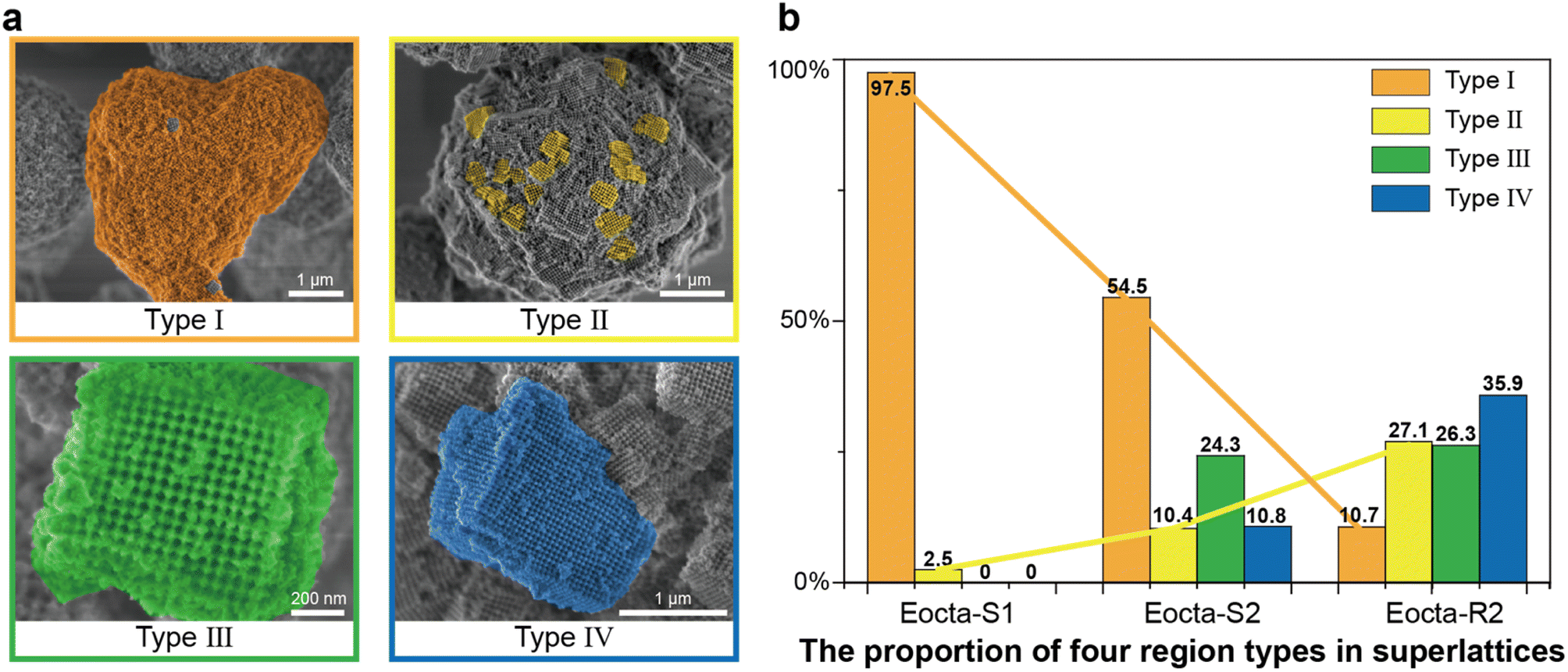 | ||
| Fig. 4 The proportion of four typical domains in each superlattice. (a) Four region types in superlattices are colored in orange (Type I), yellow (Type II), green (Type III) and blue (Type IV), respectively. From Type I to IV region, the size ranges of the grid pattern regions are 0–0.2 μm, 0.2–0.5 μm, 0.5–1 μm, and larger than 1 μm, respectively. Scale bars, 1 μm (Type I, II and IV) and 200 nm (Type III). (b) The proportion statistics of the four region types in superlattices Eocta-S1 (Type I, 97.5%; Type II, 2.5%; Type III, 0%; Type IV, 0%), Eocta-S2 (Type I, 54.5%; Type II, 10.4%; Type III, 24.3%; Type IV, 10.8%), and Eocta-R2 (Type I, 10.7%; Type II, 27.1%; Type III, 26.3%; Type IV, 35.9%). From Eocta-S1 to Eocta-S2 to Eocta-R2, the proportions of Type I gradually decrease while the proportions of Type II gradually increase. | ||
Conclusion
In summary, we have realized the geometry-guided crystallization of anisotropic DOFs with non-specific connections by strategies including increasing the cycles of the annealing process and the stiffness of DNA SEs. During the thermodynamic governed crystallization process, the energy of the system can be reduced effectively by multiple annealing processes, so as to improve the orderliness of assembly. In this work, the reannealing technology has been proved to be able to occupy an important position in the crystallization of anisotropic nanoparticles. In addition, the DNA SEs can play a crucial role in determining the types of the formed crystals. The shape characteristics of the anisotropic monomers are weakened because of the deformability of soft DNA SEs, resulting in random arrangement of E-octa DOFs and achieving SC superlattices. By strengthening the stiffness of the SEs, the monomer shape becomes the prominent factor in assembly, which prompts DOFs to form shape-mediated 3D arrangements rather than random ones. This strategy stresses the significance of annealing in crystallization and provides an access to simplifying the connection design for crystallizing anisotropic monomers, lowering the threshold for crystallization of anisotropic nanoparticles. These results break through the traditional cognition of assembling anisotropic DNA origami monomers, and deepen the understanding of their geometry-mediated crystallization. We believe that these achievements can stimulate advances in the assembly methodology of nanomaterials and the manufacture of functional devices with simpler design and cheaper cost.Data availability
All detailed experimental and characterization data associated with this work are available in the ESI.†Author contributions
Y. T. conceived and directed the project. S. H. performed the experiments. S. H., M. J. and Y. W. contributed to data analysis. S. H., M. J. and Y. T. wrote the manuscript. All authors participated in results, discussion and manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 92056114, Grant No. 22372077 and Grant No. 21971109), the Excellent Youth Fund of Jiangsu Province (Grant No. BK20220124) and the funding from State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2301). We thank the staff members at the BL19U2 beamline of the National Facility for Protein Science in Shanghai (NFPS), Shanghai Advanced Research Institute, Chinese Academy of Sciences, for providing technical support and assistance in data collection and analysis. We thank the staff from the BL16B1 beamline at the Shanghai Synchrotron Radiation Facility for assistance during data collection.References
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed
.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hoegberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed
- Y. Ke, N. V. Voigt, K. V. Gothelf and W. M. Shih, J. Am. Chem. Soc., 2012, 134, 1770–1774 CrossRef CAS PubMed
- F. Zhang, S. Jiang, S. Wu, Y. Li, C. Mao, Y. Liu and H. Yan, Nat. Nanotechnol., 2015, 10, 779–784 CrossRef CAS PubMed
- C. Chen, X. Wei, M. F. Parsons, J. Guo, J. L. Banal, Y. Zhao, M. N. Scott, G. S. Schlau-Cohen, R. Hernandez and M. Bathe, Nat. Commun., 2022, 13, 4935 CrossRef CAS PubMed
- X. Shen, C. Song, J. Wang, D. Shi, Z. Wang, N. Liu and B. Ding, J. Am. Chem. Soc., 2012, 134, 146–149 CrossRef CAS PubMed
- Y. Li, Z. Liu, G. Yu, W. Jiang and C. Mao, J. Am. Chem. Soc., 2015, 137, 4320–4323 CrossRef CAS PubMed
- R. Iinuma, Y. Ke, R. Jungmann, T. Schlichthaerle, J. B. Woehrstein and P. Yin, Science, 2014, 344, 65–69 CrossRef CAS PubMed
- T. A. Meyer, C. Zhang, G. Bao and Y. Ke, Nano Lett., 2020, 20, 2799–2805 CrossRef CAS PubMed
- Y. Li, J. Pei, X. Lu, Y. Jiao, F. Liu, X. Wu, J. Liu and B. Ding, J. Am. Chem. Soc., 2021, 143, 19893–19900 CrossRef CAS PubMed
- X. Lan, X. Lu, C. Shen, Y. Ke, W. Ni and Q. Wang, J. Am. Chem. Soc., 2015, 137, 457–462 CrossRef CAS PubMed
- Q. Jiang, Q. Liu, Y. Shi, Z.-G. Wang, P. Zhan, J. Liu, C. Liu, H. Wang, X. Shi, L. Zhang, J. Sun, B. Ding and M. Liu, Nano Lett., 2017, 17, 7125–7130 CrossRef CAS PubMed
- W. Fang, S. Jia, J. Chao, L. Wang, X. Duan, H. Liu, Q. Li, X. Zuo, L. Wang, L. Wang, N. Liu and C. Fan, Sci. Adv., 2019, 5, eaau4506 CrossRef PubMed
- H. Pei, R. Sha, X. Wang, M. Zheng, C. Fan, J. W. Canary and N. C. Seeman, J. Am. Chem. Soc., 2019, 141, 11923–11928 CrossRef CAS PubMed
- L. Xin, C. Zhou, X. Duan and N. Liu, Nat. Commun., 2019, 10, 5394 CrossRef PubMed
- P. Wang, J.-H. Huh, H. Park, D. Yang, Y. Zhang, Y. Zhang, J. Lee, S. Lee and Y. Ke, Nano Lett., 2020, 20, 8926–8932 CrossRef CAS PubMed
- W. Liu, M. Tagawa, H. L. Xin, T. Wang, H. Emamy, H. Li, K. G. Yager, F. W. Starr, A. V. Tkachenko and O. Gang, Science, 2016, 351, 582–586 CrossRef CAS PubMed
- Y. Tian, Y. Zhang, T. Wang, H. L. Xin, H. Li and O. Gang, Nat. Mater., 2016, 15, 654–661 CrossRef CAS PubMed
- T. Zhang, C. Hartl, K. Frank, A. Heuer-Jungemann, S. Fischer, P. C. Nickels, B. Nickel and T. Liedl, Adv. Mater., 2018, 30, 1800273 CrossRef PubMed
- Y. Tian, J. R. Lhermitte, L. Bai, T. Vo, H. L. Xin, H. Li, R. Li, M. Fukuto, K. G. Yager, J. S. Kahn, Y. Xiong, B. Minevich, S. K. Kumar and O. Gang, Nat. Mater., 2020, 19, 789–796 CrossRef CAS PubMed
- S.-T. Wang, B. Minevich, J. Liu, H. Zhang, D. Nykypanchuk, J. Byrnes, W. Liu, L. Bershadsky, Q. Liu, T. Wang, G. Ren and O. Gang, Nat. Commun., 2021, 12, 3702 CrossRef CAS PubMed
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed
- E. Auyeung, T. I. N. G. Li, A. J. Senesi, A. L. Schmucker, B. C. Pals, M. O. de la Cruz and C. A. Mirkin, Nature, 2014, 505, 73–77 CrossRef PubMed
- D. J. Lewis, L. Z. Zornberg, D. J. D. Carter and R. J. Macfarlane, Nat. Mater., 2020, 19, 719–724 CrossRef CAS PubMed
- Y. Kim, R. J. Macfarlane, M. R. Jones and C. A. Mirkin, Science, 2016, 351, 579–582 CrossRef CAS PubMed
- M. Ji, J. L. Liu, L. Z. Dai, L. Wang and Y. Tian, J. Am. Chem. Soc., 2020, 142, 21336–21343 CrossRef CAS PubMed
- Y. Wang, L. Dai, Z. Ding, M. Ji, J. Liu, H. Xing, X. Liu, Y. Ke, C. Fan, P. Wang and Y. Tian, Nat. Commun., 2021, 12, 3011 CrossRef PubMed
- M. Ji, Z. Zhou, W. Cao, N. Ma, W. Xu and Y. Tian, Sci. Adv., 2022, 8, eadc9755 CrossRef CAS PubMed
- C. Zhang, M. Zheng, Y. P. Ohayon, S. Vecchioni, R. Sha, N. C. Seeman, N. Jonoska and C. Mao, J. Am. Chem. Soc., 2022, 144, 8741–8754 CrossRef CAS PubMed
- T. Zhang and B. Wei, J. Am. Chem. Soc., 2022, 144, 18479–18484 CrossRef CAS PubMed
- E. Auyeung, R. J. Macfarlane, C. H. J. Choi, J. I. Cutler and C. A. Mirkin, Adv. Mater., 2012, 24, 5181–5186 CrossRef CAS PubMed
- X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Nature, 2018, 559, 593–598 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02722h |
| This journal is © The Royal Society of Chemistry 2023 |