
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H radiocyanation of bioactive molecules via sequential iodination/copper-mediated cross-coupling†
Mami
Horikawa
 a,
Stephen T.
Joy
a,
Liam S.
Sharninghausen
a,
Xia
Shao
b,
Anna K.
Mapp
*a,
Peter J. H.
Scott
*b and
Melanie S.
Sanford
*a
a,
Stephen T.
Joy
a,
Liam S.
Sharninghausen
a,
Xia
Shao
b,
Anna K.
Mapp
*a,
Peter J. H.
Scott
*b and
Melanie S.
Sanford
*a
aDepartment of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan 48109, USA. E-mail: amapp@umich.edu; mssanfor@umich.edu
bDepartment of Radiology, University of Michigan, 1301 Catherine, Ann Arbor, Michigan 48109, USA. E-mail: pjhscott@umich.edu
First published on 20th October 2023
Abstract
This report describes a net C–H radiocyanation reaction for the transformation of electron rich (hetero)aromatic substrates into 11CN-labeled products. Electrophilic C(sp2)–H iodination of the (hetero)arene with N-iodosuccinimide is followed by Cu-mediated radiocyanation with K11CN. This sequence is applied to a variety of substrates, including the nucleobases uracil and cytosine, the amino acids tyrosine and tryptophan, and the peptide LYRAGWRAFS, which undergoes selective C–H radiocyanation at the tryptophan (W) residue.
The incorporation of 11C into organic molecules is a valuable approach for generating positron emission tomography (PET) radiotracers.1 [11C]Cyanide is a particularly valuable 11C synthon, as it is readily available from small medical cyclotrons and is an excellent nucleophile for both organic and transition metal-mediated reactions.2,3 We recently reported a Cu-mediated method for the radiocyanation of aryl halides with [11C]cyanide.4 Using CuI in combination with 1,2-dimethylethylene diamine (dmeda) as a ligand5 enables the rapid reaction of aryl halides with [11C]CN− to afford [11CN]aryl nitrile products. This transformation shows extremely high functional group tolerance and thus offers an attractive approach for the radiolabeling of biological molecules such as unprotected peptides. However, a key limitation of the method is that aryl halides are not native functional groups in biology. As such, de novo solid-phase peptide synthesis (SPPS) needs to be employed to access each of the aryl halide-containing precursors required for radiocyanation (Scheme 1A). For instance, the [11C]CN radiolabeling of opioid receptor agonist nociceptin necessitated the synthesis of this 17-amino acid peptide with an iodophenylalanine inserted in place of the native leucine at residue 14. This approach is particularly undesirable in cases where (1) the native peptide or protein is readily available (such that a more direct approach for [11C]cyanation would greatly expedite radiolabeling), (2) the requisite iodo-amino acid building block for SPPS is not readily available (or is unstable), or (3) local SPPS equipment and/or expertise is unavailable (which is the case for most PET radiochemistry facilities).
 | ||
| Scheme 1 (A) Our prior work: de novo solid phase peptide synthesis (SPPS) of iodine-containing peptides for 11CN radiolabeling.5 (B) This work: net C–H radiocyanation of native peptides via electrophilic iodination/Cu-mediated radiocyanation sequence, which opens the door for ultimately labeling full proteins. | ||
A complementary approach would involve directly converting a native peptide (or other bioactive molecule) into a radiocyanated product in a single pot (net C–H radiocyanation; Scheme 1B). We reasoned that this could be accomplished by combining electrophilic C(sp2)–H iodination of a (hetero)arene with subsequent Cu-mediated radiocyanation of the in situ-generated aryl iodide. In this sequence, radiocyanation should occur selectively at the most electron rich C(sp2)–H site of the molecule. Notably, there is literature precedent for the electrophilic iodination of amino acids, peptides, and proteins, supporting the feasibility of the first step of this sequence on such scaffolds.6 In this Edge Article, we demonstrate the viability of this approach for the net C–H radiocyanation of diverse arene and heteroarene substrates, including unprotected peptides. Furthermore, we demonstrate the selective radiolabeling of tryptophan moieties in the presence of other aromatic amino acids.
Our initial studies focused on establishing a one-pot electrophilic iodination/radiocyanation sequence for two substrates: 1,1′-biphenyl (1) and tyrosine (2). These were selected to represent the range of targets of interest, from hydrophobic molecules with modest nucleophilicity (exemplified by 1) to those that are hydrophilic and electron rich and contain other functional groups (exemplified by 2). Key considerations included (i) developing mild conditions for electrophilic iodination that minimize over-functionalization and other side reactions, (ii) identifying an electrophilic iodinating reagent whose by-products do not interfere with the radiocyanation step, and (iii) developing a sequence that is practical and reproducible for both substrates.
We surveyed four reagents that have been employed in the literature for the iodination of electron rich arenes and peptides: N-iodosuccinimide (NIS),7 I(Py)2BF4 (IPy2),8 NaI/chloramine (NaI/Chlor),9 and NaI/Selectfluor® (NaI/Select).10 Initial screens were carried out at 25 °C using dichloromethane/acetonitrile/trifluoroacetic acid as the solvent system, since this mixture effectively dissolves both 1 and 2 and is also straightforward to remove via a nitrogen purge. The yield/selectivity of C–H iodination for each substrate was determined by 1H NMR spectroscopic analysis of the crude reaction mixture (Table 1). For both 1 and 2, NIS and IPy2 afforded similarly high (70–80%) yields of the mono-iodination product. In contrast, significantly lower yields (<1–35%) were obtained with NaI/Chlor and NaI/Select.
|
|
|||
|---|---|---|---|
| Entry | ArH | I+ | Yield ArI |
| a Conditions: 1 equiv. (0.05 mmol) of ArH, 1 equiv. of [I+], 0.2 mL of trifluoroacetic acid (TFA), 2 mL of dichloromethane (DCM), 0.25 mL of acetonitrile (MeCN) at 25 °C for 2 h. b Mass balance was primarily unreacted starting material as determined by 1H NMR spectroscopy. c Mass balance was primarily unreacted starting material and 4% diiodotyrosine as determined by 1H NMR spectroscopy. | |||
| 1 | 1 | NIS | 77% |
| 2 | 1 | IPy2 | 78% |
| 3 | 1 | NaI/chlor | <1%b |
| 4 | 1 | NaI/select | 9%b |
| 5 | 2 | NIS | 75% |
| 6 | 2 | IPy2 | 70% |
| 7 | 2 | NaI/chlor | 35%c |
| 8 | 2 | NaI/select | 16%b |

We next moved forward to the C–H iodination/radiocyanation sequence using NIS and IPy2. Both substrates 1 and 2 were subjected to the iodination conditions in Table 1, and then the volatiles were removed. Cu-mediated radiocyanation was performed on this crude material without further purification, using our previously reported conditions.4 The radiochemical yield (RCY) of each reaction was determined by radio-TLC and/or radio-HPLC. As summarized in Table 2, significant differences in RCY were observed as a function of the choice of I+ reagent. With NIS, 1-11CN was formed in 68 ± 14% RCY (n = 6), while lower RCY (40 ± 4%, n = 3) was obtained with IPy2. Similarly, using 2 as a substrate, the NIS reaction afforded a higher RCY (68 ± 4%, n = 4) than that with IPy2 (54 ± 7%, n = 3). Based on these results, NIS was selected as the iodinating reagent moving forward.11
|
|
|||
|---|---|---|---|
| Entry | ArH | I+ | RCY Ar11CN |
a Iodination conditions: 1 equiv. (0.05 mmol) of ArH, 1 equiv. of [I+], 0.2 mL of trifluoroacetic acid (TFA), 2 mL of dichloromethane (DCM), 0.25 mL of acetonitrile (MeCN) at 25 °C. Radiocyanation conditions: H11CN, 0.5 equiv. of CuI, 1 equiv. of dmeda, 0.25 equiv. of K3PO4, 5.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF/H2O (163 μL).
b 24 h.
c 2 h. RCY = radiochemical yield. :1 DMF/H2O (163 μL).
b 24 h.
c 2 h. RCY = radiochemical yield.
|
|||
| 1 | 1 | NIS | 68 ± 14%b (n = 6) |
| 2 | 1 | IPy2 | 40 ± 4%b (n = 3) |
| 3 | 2 | NIS | 68 ± 4%c (n = 4) |
| 4 | 2 | IPy2 | 54 ± 7%c (n = 3) |

Under these optimal conditions, we evaluated the scope of C–H radiocyanation with a range of arene and heteroarene substrates (Scheme 2). In all cases, selectivity was confirmed by comparison to authentic standards of the 12CN products. Ether and amine-substituted aromatic substrates react under these conditions to form 2-9-11CN in radiochemical yields ranging from 36–86% (Scheme 2A). As anticipated based on the mechanism of electrophilic iodination, these transformations proceed with high selectivity for sites ortho- and/or para- to resonance electron donating substituents. This sequence is effective for simple aromatics (forming 3-5-11CN), as well as arene rings in more highly functionalized bioactive molecules, such as lidocaine (to form 6-11CN), a precursor to the radiotracer PIMBA (to form 7-11CN), nimesulide (to form 8-11CN), and febuxostat (to form 9-11CN).
 | ||
| Scheme 2 Substrate scope for net C(sp2)–H radiocyanation. RCY (radiochemical yield) calculated by radio-HPLC and radio-TLC as detailed in the ESI.† Unless otherwise noted a single regioisomer was detected in both the C–H iodination and radiocyanation steps. i2% of a regioisomeric product was observed. | ||
Electron rich heterocycles are also viable substrates (Scheme 2B). For instance, benzothiophene, pyrazole, pyrazolylpyridine, and aminopyridine derivatives react to form 10-13-11CN in radiochemical yields ranging from 45–79%. The nucleobases uracil and cytosine afford the 11CN-labeled analogues 14-11CN and 15-11CN in 69% and 84% radiochemical yield, respectively. Comparable radiochemical yield (75%) is obtained for the glycosylated base in cytidine (to form 16-11CN). Overall, the examples in Scheme 2 highlight the compatibility of this method with diverse functional groups, including 1° and 2° alcohols, carboxylic acids, amines, amides, and sulfonamides.
Finally, we evaluated three aromatic amino acids, phenylalanine, tyrosine (Tyr, Y), and tryptophan (Trp, W), along with peptides containing one or more of these residues. We sought to establish (1) which individual aromatic amino acids participate in this net C–H radiocyanation sequence and (2) the selectivity of the reaction when multiple aromatic amino acids are present. Consistent with literature reports,12 phenylalanine proved inert towards electrophilic iodination/radiocyanation in all contexts examined (both as a free, unprotected amino acid and in various peptides).
In contrast, tyrosine underwent high yielding iodination/radiocyanation as the unprotected amino acid (yielding 2-11CN in 68 ± 14% RCY, n = 4), as an acyl-methyl protected derivative (to form 17-11CN in 44 ± 5 RCY, n = 4), and in the short peptide Ac-GYG-NH2 (affording 18-11CN in 67 ± 7% RCY, n = 3; Scheme 3A). These results are consistent with literature reports showing selective electrophilic iodination of tyrosine ortho-to the OH substituent as well as the compatibility of phenols with Cu-mediated radiocyanation of aryl halides.4 This radiosynthesis of 18-11CN from tyrosine was automated in a commercial synthesis module, and the product was purified by a preparatory HPLC, affording a 23 ± 9% isolated RCY, 1.2 ± 0.1 Ci μmol−1 molar activity, n = 2; see ESI Section 6.3†).
 | ||
| Scheme 3 (A) Amino acid substrates. (B) Competition study between tyrosine and tryptophan derivatives. | ||
We anticipated challenges with both steps in the net C–H radiocyanation of tryptophan. First, the reaction of tryptophan derivatives with electrophilic iodinating reagents has been reported to form mixtures of products due to the low stability of 2-iodotryptophan as well as competing oxidation and cyclization pathways.14 Second, in our previous studies of Cu-mediated radiocyanation, 2-haloindoles were among the poorest performing substrates.4 These challenges are reflected in our initial results using unprotected tryptophan and the protected derivative Ac-Trp-NH2 as substrates. Under the standard electrophilic iodination/radiocyanation conditions, both afforded very low yields of 11CN-labeled products (none detected and 2%, respectively). We hypothesized that judicious selection of protecting groups (specifically eliminating nucleophilic NH2 groups proximal to the 2-position) might enable the target transformation. Indeed, using Ac-Trp-OMe as a substrate, the radiolabeled product 20-11CN was formed selectively in 10 ± 3% RCY (n = 5).15 The selectivity of C–H radiocyanation for the 2-position was confirmed by comparison with a 12CN-containing authentic sample. While the yield of this transformation is modest, it is sufficient for most radiochemistry applications.
Our final objective was to achieve selective 11CN-radiolabeling of peptides containing multiple aromatic amino acids. To test the selectivity of this C–H iodination/radiocyanation sequence, a competition experiment was conducted between Ac-Trp-OMe and Ac-Tyr-OMe. As shown in Scheme 3B, treatment of a 1:1 mixture of these protected amino acids with 1 equiv. of NIS followed by the standard radiocyanation conditions resulted in selective formation of 20-11CN (in 8 ± 1% RCY, n = 3). 17-11CN was not detected. The observed selectivity is consistent with the higher nucleophilicity of tryptophan versus tyrosine. The modest overall RCC of 20-11CN is likely due to a combination of side reactions during the iodination step, the modest stability of iodotryptophan intermediate, and low efficiency of the radiocyanation at the 2-position of the indole.4,13
Based on the high selectivity of this competition experiment, we pursued the net C–H radiocyanation of the peptide LYRAGWRAFS, which contains three different aromatic amino acids (Scheme 4). Subjecting the peptide to our optimal electrophilic iodination/radiocyanation conditions in an automated radiosynthesis module afforded a single detectable radiocyanated product (as determined by radio-HPLC analysis) in 17 ± 3% RCC. A series of experiments was conducted to establish the site selectivity of radiolabeling, including synthesis of an authentic sample of the 12CN-tyrosine peptide (which does not match the observed product by HPLC) and trypsin digestion of the initial C–H iodination product (which shows that the iodine is incorporated exclusively on the tryptophan fragment). Overall, our data (see ESI,† p. S56–S61 (Section 7) for a complete summary) are fully consistent with selective radiocyanation of the tryptophan residue in this peptide as shown in Scheme 4. Notably, this 11CN-labelled peptide would not be accessible via the original SPPS approach in Fig. 1A,† since the required 2-iodotryptophan building block is not sufficiently stable for isolation and use in peptide synthesis.
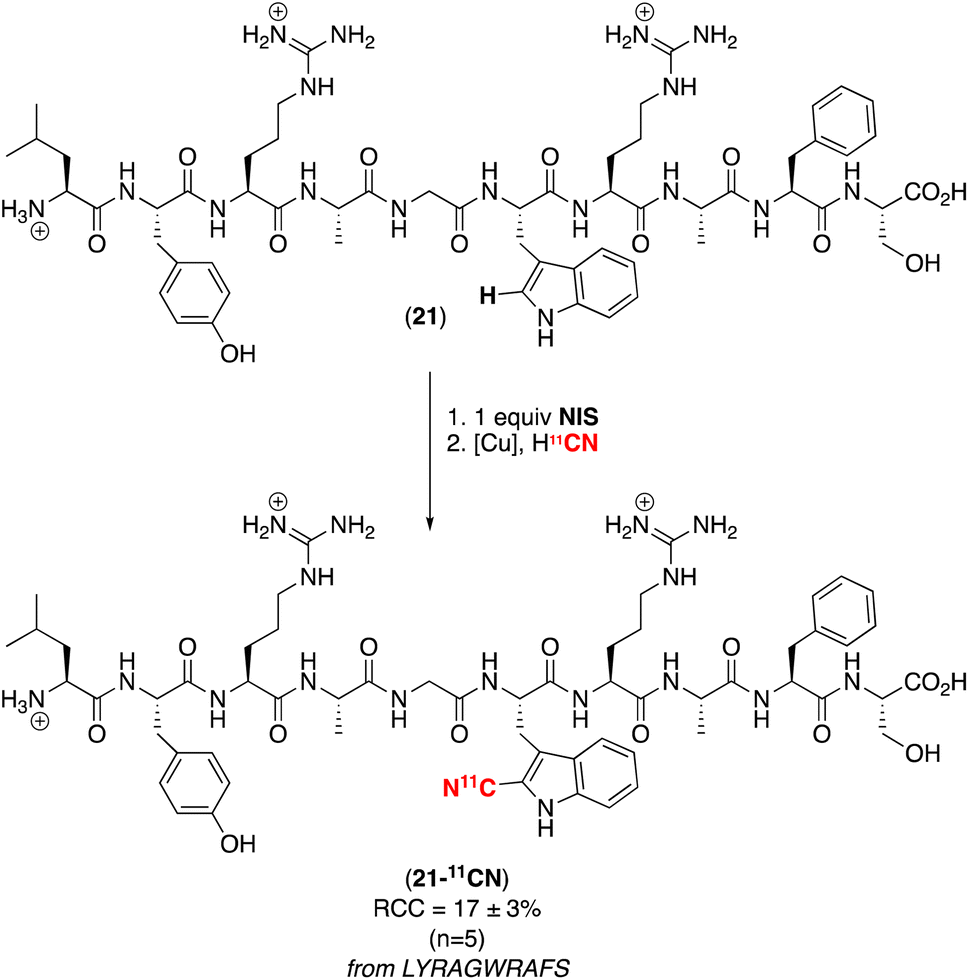 | ||
| Scheme 4 Selective C–H radiocyanation of the tryptophan residue of LYRAGWRAFS. | ||
Conclusions
In conclusion, this report demonstrates a simple two-step net C(sp2)–H radiocyanation reaction. This transformation is operationally simple, proceeds smoothly with peptides, and can be conducted in automated in a commercial radiosynthesis module (substrates 18-11CN and 21-11CN). It provides a straightforward approach to selectively introducing a 11CN moiety on tryptophan residues in a peptide containing multiple other aromatic amino acids without the need to synthesize dedicated labelling precursors. Ongoing work is focused on applying this method to the 11CN-radiolabeling of various bioactive molecules of interest to our PET imaging program, including proteins.Data availability
Further details of experimental methods, additional analysis, and analytical data, as well as supplemental figures including NMR spectra, analytical TLC and HPLC traces, and additional screening and control experiments are included in the ESI.†Author contributions
M. S. S., P. J. H. S. and A. K. M. conceived the idea, provided funding and directed the project. M. H., L. S. S. and S. T. J. synthesized reference standards and labeling precursors. M. H., L. S. S. and X. S. performed radiolabeling studies. M. H., M. S. S. and P. J. H. S. wrote the manuscript. All authors reviewed and approved the final version.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The National Institutes of Health (R01EB021155) is gratefully acknowledged for supporting this work. We thank Dr Eric Webb and Dr Tanpreet Kaur for the preparation of K11CN solutions, Dr Allen Brooks for helpful discussions, and Dr Mónica Rivas for helping with purification of several compounds.Notes and references
- P. Miller, N. Long, R. Vilar and A. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- (a) Y.-P. Zhou, K. J. Makaravage and P. Brugarolas, Nucl. Med. Biol., 2021, 102, 56–86 CrossRef PubMed; (b) Y. Xu and W. Qu, Eur. J. Org Chem., 2021, 33, 4653–4682 CrossRef.
- (a) M. Ponchant, F. Hinnen, S. Demphel and C. Crouzel, Appl. Radiat. Isot., 1997, 48, 755–762 CrossRef CAS; (b) W. Zhao, H. G. Lee, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2017, 139, 7152–7155 CrossRef CAS PubMed.
- L. S. Sharninghausen, S. Preshlock, S. T. Joy, M. Horikawa, X. Shao, W. P. Winton, J. Stauff, T. Kaur, R. A. Koeppe, A. K. Mapp, P. J. H. Scott and M. S. Sanford, J. Am. Chem. Soc., 2022, 144, 7422–7429 CrossRef CAS PubMed.
- (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC; (b) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890–2891 CrossRef CAS PubMed.
- (a) J. Vicente, I. Saura-Llamas and D. Bautista, Organometallics, 2005, 24, 6001–6004 CrossRef CAS; (b) G. Sugiura, H. Kühn, M. Sauter, U. Haberkorn and W. Mier, Molecules, 2014, 19, 2135–2165 CrossRef PubMed; (c) B. Lelesz, G. K. Toth, B. Peitl, C. Hegedus, L. Drimba, R. Sari, Z. Szilvassy and J. Nemeth, J. Radioanal. Nucl. Chem., 2014, 299, 157–164 CrossRef CAS; (d) A. Clerico, G. Iervasi, C. Manfredi, S. Salvadori, M. Marastoni, M. G. Del Chicca, D. Giannessi, S. Del Ry, M. G. Andreassi, L. Sabatino, M. R. Iascone, A. Biagini and L. Donato, Eur. J. Nucl. Med., 1995, 22, 997–1004 CrossRef CAS.
- (a) A. Castanet, F. Colobert and P. Broutin, Tetrahedron Lett., 2002, 43, 5047–5048 CrossRef CAS; (b) P. Bovonsombat, P. Khanthapura, M. M. Krause and J. Leykajarakul, Tetrahedron Lett., 2008, 49, 7008–7011 CrossRef CAS.
- (a) J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc. Chem. Commun., 1992, 14, 1016–1017 RSC; (b) G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 14, 1307–1308 RSC.
- (a) T. Kometani, D. S. Watt and T. Ji, Tetrahedron Lett., 1985, 26, 2043–2046 CrossRef CAS; (b) W. M. Hunter and F. C. Greenwood, Nature, 1962, 194, 495–596 CrossRef CAS PubMed.
- R. Bertrand, M. Wagner, V. Derdau and O. Plettenburg, Bioconjugate Chem., 2016, 27, 2281–2286 CrossRef CAS PubMed.
- We hypothesized that the pyridine released from IPy2 might inhibit the radiocyanation, resulting in lower yields with this oxidant. However, the addition of 2 equiv of pyridine to the radiocyanation of 4-iodobiphenyl led to minimal change in yield compared to the control study (n = 3), suggesting that this is likely not an issue.
- (a) A. Vértes, S. Nagy and Z. Klencsár, Handb. Nucl. Chem., 2003, 1555–1575 Search PubMed; (b) G. Espuña, D. Andreu, J. Barluenga, X. Pérez, A. Planas, G. Arsequell and G. Valencia, Biochemistry, 2006, 45, 5957–5963 CrossRef; (c) G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, J. M. Alvarez-Gutiérrez, A. Ballesteros and J. M. González, Angew. Chem., Int. Ed., 2004, 43, 325–329 CrossRef PubMed.
- (a) P. Sum, D. How, N. Torres, P. J. Petersen, J. Ashcroft, E. I. Graziani, F. E. Koehn and T. S. Mansour, Bioorg. Med. Chem. Lett., 2003, 13, 2805–2808 CrossRef CAS PubMed; (b) M. Schottelius, M. Konrad, T. Osl, A. Poschenrieder and H. Wester, Tetrahedron Lett., 2015, 56, 6602–6605 CrossRef CAS; (c) A. M. Steer, H. L. Bolt, W. D. G. Brittain and S. L. Cobb, Tetrahedron Lett., 2018, 59, 2644–2646 CrossRef CAS.
- (a) G. Mourier, L. Moroder and A. Previero, Z. Naturforsch., B: J. Chem. Sci., 1984, 39, 101–104 CrossRef; (b) A. Coste, M. Toumi, K. Wright, V. Razafimahaleo, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 3841–3844 CrossRef CAS PubMed; (c) M. Toumi, F. Couty, J. Marrot and G. Evano, Org. Lett., 2008, 10, 5027–5030 CrossRef CAS PubMed.
- The reproducibility of reactions forming 20-11CN and 21-11CN was enhanced by the addition of sodium ascorbate (1 equiv.) to the mixture during the radiocyanation step. However, the sodium ascorbate additive did not have a similar favorable impact with the other substrates. With 20-11CN and 21-11CN there is often a significant amount of NIS remaining following the iodination step, and we hypothesize that the role of sodium ascorbate is to quench this so that it does not oxidize the CuI mediator to the unreactive CuII state.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and radioTLC and HPLC tracers. See DOI: https://doi.org/10.1039/d3sc03948j |
| This journal is © The Royal Society of Chemistry 2023 |