
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence18F-Labeled brain-penetrant EGFR tyrosine kinase inhibitors for PET imaging of glioblastoma†
Maruthi Kumar
Narayanam
ab,
Jonathan E.
Tsang
a,
Shili
Xu
ab,
David A.
Nathanson
a and
Jennifer M.
Murphy
 *ab
*ab
aDepartment of Molecular and Medical Pharmacology, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA
bCrump Institute for Molecular Imaging, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA. E-mail: jmmurphy@mednet.ucla.edu
First published on 9th November 2023
Abstract
Significant evidence suggests that the failure of clinically tested epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (e.g. erlotinib, lapatinib, gefitinib) in glioblastoma (GBM) patients is primarily attributed to insufficient brain penetration, resulting in inadequate exposure to the targeted cells. Molecular imaging tools can facilitate GBM drug development by visualizing drug biodistribution and confirming target expression and localization. To assess brain exposure via PET molecular imaging, we synthesized fluorine-18 isotopologues of two brain-penetrant EGFR tyrosine kinase inhibitors developed specifically for GBM. Adapting our recently reported radiofluorination of N-arylsydnones, we constructed an ortho-disubstituted [18F]fluoroarene as the key intermediate. The radiotracers were produced on an automated synthesis module in 7–8% activity yield with high molar activity. In vivo PET imaging revealed rapid brain uptake in rodents and tumor accumulation in an EGFR-driven orthotopic GBM xenograft model.
Introduction
The epidermal growth factor receptor (EGFR) plays an important role in the development and progression of a variety of malignant tumors including glioblastoma (GBM), one of the most lethal malignancies with a median survival of approximately 16 months.1 Novel anti-tumor therapies that target the ATP-binding site of the tyrosine kinase domain of EGFR have become popular and have resulted in the development of several tyrosine kinase inhibitors (TKIs), with some demonstrating significant clinical efficacy in EGFR mutant lung cancer.2 By contrast, in GBM, first- and second-generation EGFR TKIs have failed to improve outcomes for these patients.3 Their limited efficacy is largely attributed to the presence of a physical barrier – the blood–brain barrier (BBB) – which precludes the delivery of more than 98% of all therapeutics.4 To achieve therapeutic efficacy in GBM, BBB permeability is considered an essential property to maximize patient benefit and clinical outcome.Positron emission tomography (PET) molecular imaging provides a unique opportunity to non-invasively study disease and has contributed a key role in accelerating drug development, particularly in the fields of neurology and oncology.5 The availability of a radiolabeled isotopologue of the small molecule drug candidate in the early stage of CNS drug development can provide information on whole body biodistribution, pharmacokinetics and target engagement.5b,6 In the context of GBM, where brain penetration of drugs is critical, molecular imaging enables non-invasive measurements of differences between brain and plasma pharmacokinetics, indispensable data to establish the optimal therapeutic dose.7 To date, several small molecule TKIs have been radiolabeled and evaluated as PET tracers in an effort to noninvasively assess the in vivo pharmacological properties of the drug.8
Radiotracers based on clinically approved TKIs have been described, two of which have been labeled with fluorine-18: 18F-Gefitinib and 18F-Afatinib (Fig. 1).8e Gefitinib, a first-generation EGFR TKI approved for the treatment of advanced NSCLC, was radiolabeled with fluorine-18 in three steps, in a decay-corrected radiochemical yield (RCY) of 17%.9 Preclinical in vivo studies with the radiotracer revealed limited brain penetration and a lack of uptake in EGFR-expressing tumor xenografts, which is consistent with human clinical experience with gefitinib treatment.2b,8a,8b,10 Of note, a one-step synthesis of 18F-Gefitinib was recently reported via Cu-mediated fluorination of the corresponding aryl pinacol borane with a 22% radiochemical conversion based on radio-TLC and radio-HPLC analysis of the crude material.11 Afatinib, a second-generation EGFR TKI, is an irreversible inhibitor that contains a Michael acceptor for covalent conjugation to a cysteine residue in the ATP binding domain of EGFR (Fig. 1).8e Synthesis of 18F-Afatinib utilizes a similar approach to that of 18F-Gefitinib with a slight difference in the third step which employs a BOP-mediated condensation reaction with the 4-quinazolinone core.12 Construction of 18F-Afatinib was achieved in 17% RCY (decay-corrected) and in vivo evaluation in preclinical studies demonstrated similarly low brain uptake.12,13 Of note, a feasibility study of 18F-Afatinib in NSCLC patients was recently reported.8d
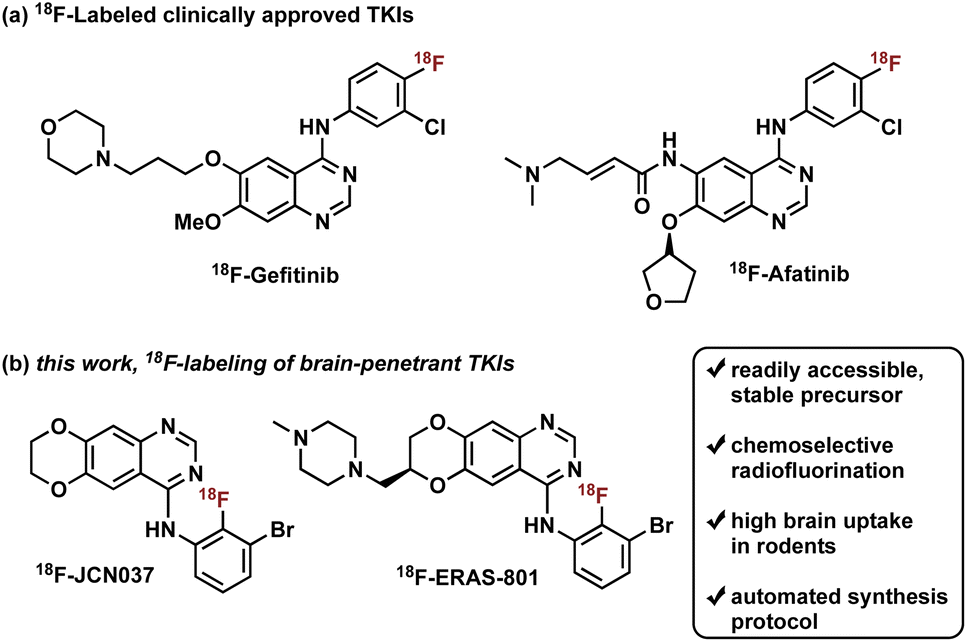 | ||
| Fig. 1 (a) 18F-Labeled tracers based on clinically approved tyrosine kinase inhibitors. (b) This work, synthesis and in vivo imaging of 18F-JCN037 and 18F-ERAS-801, potent brain-penetrant tyrosine kinase inhibitors. | ||
To address the pharmacological problem of current TKIs for the treatment of EGFR-driven GBMs, potent EGFR TKIs designed specifically to penetrate the BBB, JCN037 and its clinical derivative ERAS-801, have been developed that are both highly selective for extracellular domain mutant (e.g., EGFRvIII) and amplified WT EGFR, alterations in EGFR that are specific to GBM.14 These novel TKIs exhibit the 4-anilinoquinazoline core of clinically approved gefitinib and afatinib but incorporate a ring fusion of the 6,7-dialkoxy groups as well as a meta-bromine and ortho-fluorine on the aniline ring (Fig. 1). The structural modifications of JCN037 and ERAS-801, relative to TKIs erlotinib and lapatinb, resulted in considerably improved potency and BBB penetration. Superior efficacy was demonstrated in a GBM orthotopic patient-derived xenograft model (GliomaPDOX) where JCN037 was shown to significantly extend the survival of EGFR-altered, tumor-bearing mice.14a Notably, ERAS-801 which contains an N-methylpiperazine moiety is currently being investigated in phase I clinical trials for treatment of patients with GBM (NCT05222802).
We rationalized that a PET tracer for these TKIs could enable noninvasive measurements of drug biodistribution and accumulation and, importantly, inform on BBB penetration, providing key data largely responsible for the predictive measure of therapeutic efficacy and success. In this work, we report the synthesis and in vivo biodistribution of fluorine-18 isotopologues of JCN037 and ERAS-801, namely 18F-JCN037 and 18F-ERAS-801 (Fig. 1). To avoid differences in their in vivo behavior, the radiotracer and the parent compound have exactly the same chemical structure and, as a result, identical biodistribution, affinity, lipophilicity, polar surface area, etc. Additionally, in vivo imaging was conducted in EGFR-altered GliomaPDOX to evaluate target expression and accumulation of 18F-ERAS-801 in EGFR-driven tumors. We anticipate this novel radiotracer could provide unique insights into the intratumoral drug distribution in the brain for EGFR-driven GBM tumors and facilitate clinical development.
Results and discussion
Radiosynthetic strategy
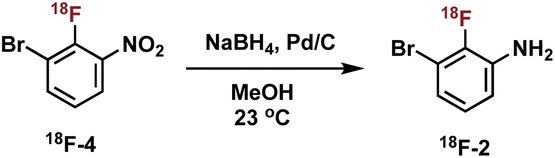
Construction of these radiotracers was envisioned to arise from the conventional SNAr between aniline 18F-2 and the 4-chloroquinazoline pharmacophore 3 to forge the key carbon–nitrogen bond (Scheme 1). The 4-chloroquinazoline core for 18F-JCN037 and 18F-ERAS-801 could be synthesized following literature protocol; thus, to achieve our goal, the critical synthetic step would be generation of radiolabeled aniline 18F-2.14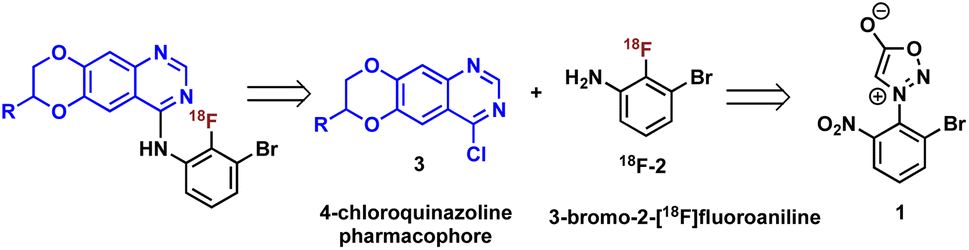 | ||
| Scheme 1 Retrosynthetic analysis. | ||
A particularly noteworthy characteristic of 18F-2 is that it exhibits a sterically hindered ortho-disubstituted [18F]fluoroaryl moiety which is a considerable synthetic challenge. Radiolabeling methodologies that tolerate bulky substituents ortho to the reactive site are limited with few examples of ortho-disubstituted [18F]fluoroarenes in the literature.15 We have previously reported the nucleophilic radiofluorination of N-arylsydnones which facilitates large ortho-substitution and readily affords [18F]fluoroarenes in high yields.16 Utilizing this methodology, we proposed to generate 18F-2via radiofluorination of the corresponding sydnone 1 followed by Pd-catalyzed reduction (Scheme 1).
First, we began our synthetic strategy with preparation of 2-bromo-6-nitrophenyl sydnone precursor 1 as shown in Scheme 2. Formation of the N-aryl glycine 5 was accomplished in 79% yield via SNAr with bromo-2-fluoro-3-nitrobenzene 4. The N-aryl glycine was converted to the corresponding nitrosamine with tbutyl nitrate followed by cyclodehydration to form the sydnone 1 in 72% yield from 5.
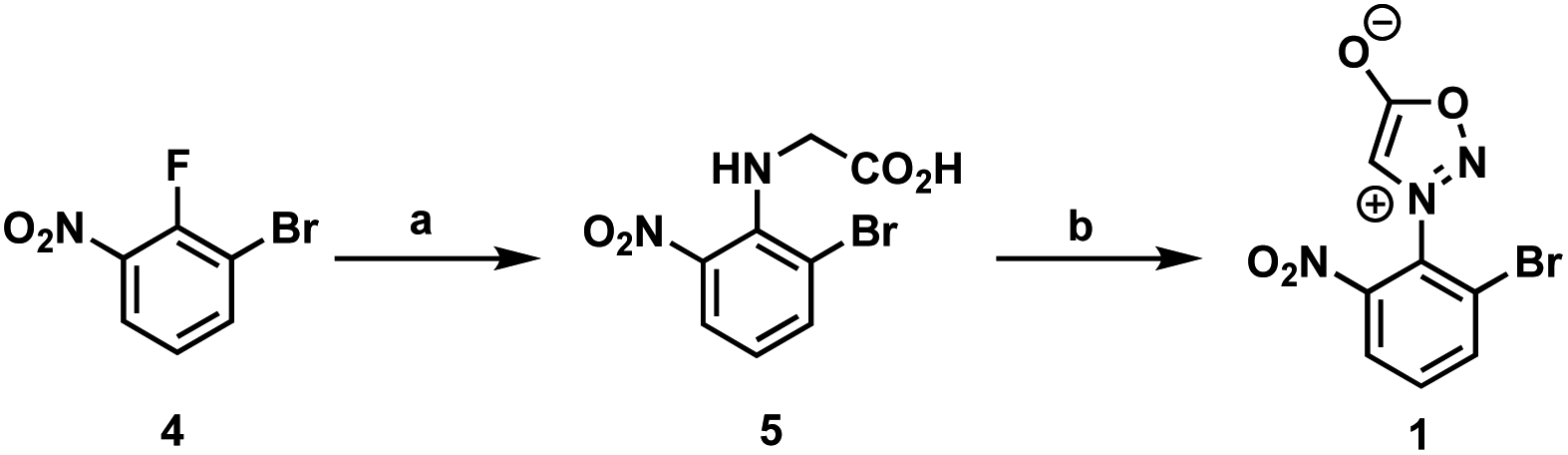 | ||
Scheme 2 Synthesis of 2-bromo-6-nitrophenyl sydnone 1. Reagents and conditions: (a) glycine, K2CO3, 1,4-dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:3), 16 h, 50 °C, 79%; (b) tert-butyl nitrite, THF, 3 h, 23 °C; trifluoroacetic anhydride, 4 h, 23–40 °C, 72%. :water (1:3), 16 h, 50 °C, 79%; (b) tert-butyl nitrite, THF, 3 h, 23 °C; trifluoroacetic anhydride, 4 h, 23–40 °C, 72%. | ||
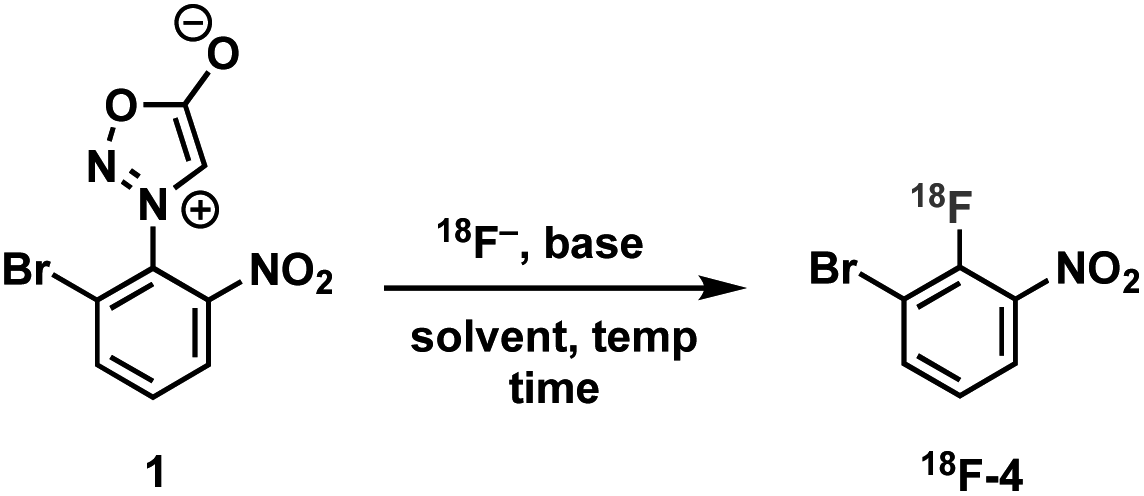
We initially evaluated various conditions for radiofluorination of precursor 1 directly on an automated radiosynthesis module (Table S1†). Given the strong activating nature of the ortho-nitro substituent towards SNAr, conversion of precursor 1 to 18F-4 was nearly quantitative after 10 min at 70 °C (Table 1). Investigation of lower temperatures revealed that the radiofluorination proceeded efficiently at 30 °C in 8 min to cleanly afford the desired nitrobenzene 18F-4 in 97% RCY.
|
|
|||||
|---|---|---|---|---|---|
| Entryb | Base | Solvent | Time (min) | Temp. (°C) | RCYc (%) |
| a Conditions: sydnone precursor 1 (∼1.5 mg), base (K222 (12 mg)/K2CO3 (2 mg) or Et4NHCO3 (4.2 mg)), [18F]fluoride, solvent (400 μL). b Conducted on the ELIXYS automated radiosynthesis module. c RCY was determined by radio-TLC analysis of the crude product 18F-4. | |||||
| 1 | K222/K2CO3 | DMSO | 10 | 70 | 96 |
| 2 | Et4NHCO3 | DMSO | 10 | 70 | 96 |
| 3 | K 222 /K 2 CO 3 | DMSO | 8 | 30 | 97 |
| 4 | Et4NHCO3 | DMSO | 8 | 30 | 70 |
| 5 | K222/K2CO3 | CH3CN | 8 | 30 | 65 |
We next sought to prepare aniline 18F-2. Formation of 18F-2 proceeded in 48% RCY upon the reduction of 18F-4 in methanol with NaBH4 in the presence of Pd/C at ambient temperature for 5 min (Table 2, entry 1). We first focused on the amounts of Pd/C and NaBH4, keeping a consistent ratio of around 2- to 3-fold excess reducing agent over transition metal (Table 2, entries 1–4). Lowering the mass amount of NaBH4 provided cleaner HPLC traces with less side product formation. Significantly lowering the mass of palladium was unproductive and completely suppressed the reduction (see Table 2, entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Pd/C (mg) | NaBH4 (mg) | Time (s) | RCYb (%) |
| a Conditions: 18F-4 (∼185 MBq), methanol (1.2–1.5 mL), 23 °C. b RCY was determined by radio-HPLC analysis of the crude product 18F-2. | ||||
| 1 | 5.0 | 15.0 | 300 | 48 |
| 2 | 5.0 | 13.0 | 300 | 58 |
| 3 | 4.6 | 10.8 | 300 | 53 |
| 4 | 1.4 | 5.0 | 300 | 1 |
| 5 | 5.2 | 3.5 | 150 | 70 |
| 6 | 5.2 | 3.0 | 160 | 60 |
| 7 | 5.5 | 4.1 | 160 | 74 |
| 8 | 5.5 | 4.0 | 140 | 61 |
| 9 | 5.2 | 4.6 | 150 | 85 |
| 10 | 4.8 | 4.6 | 150 | 80 |
Altering the mass ratios between the reducing agent and transition metal to where the palladium was in slight excess proved to further minimize undesirable side product formation and provide cleaner product composition in half the reaction time (see Table S3†). In 2.5 min, conversion to 18F-2 was achieved in 60–70% RCY with ∼3 mg NaBH4 and 5.2 mg Pd/C (Table 2, entries 5 and 6). Further refinement continued to improve the conversion and generate 18F-2 in 85% RCY within 2.5 min (Table 2, entry 9).
With aniline 18F-2 in hand, we focused on the substitution reaction with 4-chloroquinazoline 3a to produce 18F-JCN037 and rigorous screening was conducted. Initially, we used crude 18F-2, without further purification, and found that the addition of 3a into the reaction vial in two equal portions, with the second portion being added after 15 min, revealed greater consumption of 18F-2 and increased product formation (Table S6†). Therefore, a two-part addition of 3a was continued throughout the optimization process. Product formation was observed in 41% RCY upon treatment of crude 18F-2 with 1.3 mg of 3a in IPA at 95 °C for 20 min (Table 3, entry 1). Screening solvents revealed IPA to be superior and increasing the mass amount of 4-chloroquinazoline 3a yielded slightly higher RCYs (Table 3, entries 2–5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3a (mg) | Temp. (°C) | Solvent | Time (min) | RCYb (%) |
| a Conditions: cartridge-purified 18F-2, solvent (0.8–1.2 mL), manual addition of 3a to the reaction vial of the synthesis module, 3a was split into two equal batches and added portion-wise. b RCY was determined by radio-HPLC analysis of the crude product 18F-JCN037. c Crude 18F-2. d Automated addition of 3avia the synthesis module. | |||||
| 1c | 1.3 | 95 | IPA | 20 | 41 |
| 2c | 1.3 | 95 | DMF | 25 | 30 |
| 3c | 3.3 | 95 | IPA | 20 | 45 |
| 4c | 3.6 | 95 | MeOH | 25 | 35 |
| 5c | 5.5 | 95 | IPA | 25 | 49 |
| 6 | 3.5 | 95 | IPA | 30 | 58 |
| 7 | 5.0 | 95 | IPA | 30 | 76 |
| 8 | 4.5d | 95 | MeOH | 30 | 65 |
| 9 | 5.5d | 95 | CH3CN | 30 | 75 |
| 10 | 5.0 | 100 | CH 3 CN | 30 | 78 |
| 11 | 5.0d | 100 | CH3CN | 25 | 62 |
| 12 | 5.0d | 105 | CH3CN | 30 | 68 |
| 13 | 5.0d | 110 | CH3CN | 30 | 65 |
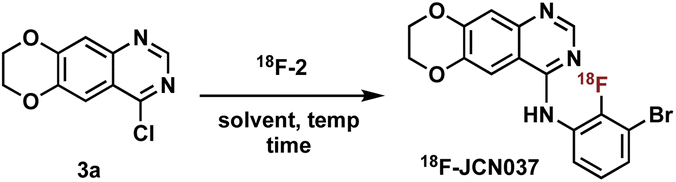
Intermediate HPLC purification of 18F-2 was briefly investigated, generating 18F-JCN037 in 95% RCY albeit adding excessive time constraints to the overall process (Table S7†). Alternatively, to avoid the lengthy HPLC purification, C18 Sep-Pak cartridge purification of 18F-2 was pursued. In IPA, 18F-JCN037 was generated in 76% RCY upon treatment of cartridge-purified 18F-2 with 5.0 mg of 3a in IPA at 95 °C for 30 min (Table 3, entry 7). When this step was translated to an automated radiosynthesis module, the moderate solubility of 4-chloroquinazoline 3a in IPA created an unsatisfactory heterogeneous reagent mixture. Alternatively, solubility of 3a in acetonitrile enabled the consistent and complete addition of the 4-chloroquinazoline to the reaction vial in an automated process. With acetonitrile as the solvent, treatment of 18F-2 with 5.0 mg of 3a at 100 °C for 30 min generated 18F-JCN037 in 78% RCY (Table 3, entry 10).
Having optimized the synthetic protocol, we streamlined the process to ensure the production of 18F-JCN037 on the ELIXYS radiosynthesis module, with minimal manual manipulation. The radiofluorination and substitution steps as well as the intermediate cartridge purifications were fully automated. The reduction step was conducted under inert atmosphere; as such, intermediate 18F-4 was pushed into a sealed vial equipped with an argon balloon, situated on a stir plate next to the radiochemical synthesis module (Fig. S8†). Following the reduction, the vial was manually unscrewed and placed back into the ELIXYS for the completion of the automated synthesis. HPLC purification of 18F-JCN037 was fully automated and the tracer was obtained in >99% radiochemical purity and 24 ± 5% isolated RCY, based on starting [18F]fluoride activity (decay corrected, n = 4). The activity yield was 7.0 ± 0.7% (non-decay corrected, n = 4) and the molar activity was 40.7 ± 5.6 GBq μmol−1 (1.1 ± 0.1 Ci μmol−1), (n = 3) (Fig. 2A).
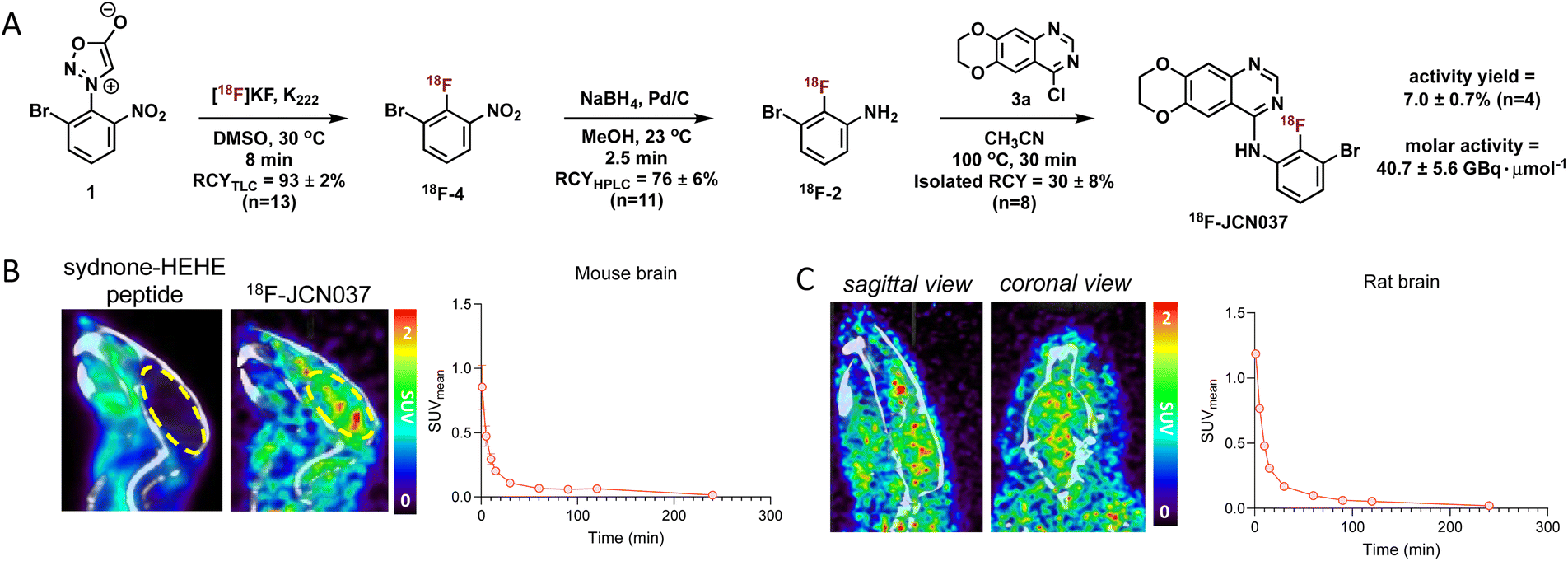 | ||
| Fig. 2 (A) Radiosynthesis of 18F-JCN037. (B) Representative co-registered PET/CT images of sydnone-HEHE-peptide and brain-penetrant 18F-JCN037 biodistribution in a mouse at 1 min post-injection of the tracer and the time–activity curve of 18F-JCN037 in the mouse brain. (C) Representative co-registered PET/CT images of 18F-JCN037 biodistribution in a rat brain at 5 min post-injection of the tracer and the associated time–activity curve. | ||
Molecular imaging of 18F-JCN037
To evaluate the biodistribution of JCN037 in vivo, preclinical imaging studies with 18F-JCN037 were conducted. Micropositron emission tomography-computed tomography (microPET/CT) imaging studies were performed in naïve C57BL6 mice and in a Sprague Dawley (SD) rat at 0–2 h (dynamic scan) and 4 h (static scan) post-injection (p.i.) of the tracer (Fig. 2). Rapid accumulation of 18F-JCN037 was observed in the mouse brain, with a SUVmean value of 0.85 at 1 min p.i., followed by a rapid decrease in the probe concentration in the brain (Fig. S12†). This data is consistent with the previously reported short half-life of JCN037 due to rapid clearance.14a Our laboratory recently reported work involving a cyclic peptide-based radiotracer, sydnone-HEHE-peptide, which fails to cross the BBB.17 Focusing on brain distribution, we compared the PET/CT images between mice that were administered either sydnone-HEHE-peptide or 18F-JCN037 (Fig. 2B). The stark contrast between the in vivo images further highlights the rapid brain uptake of 18F-JCN037. Finally, rapid accumulation of 18F-JCN037 occurred in the rat brain, with a SUVmean value of 1.19 at 1 min p.i., followed by rapid decrease in the brain (Fig. 2C).Preparation of 18F-ERAS-801
Encouraged by the imaging results of 18F-JCN037 we next pursued the synthesis of 18F-ERAS-801. In an effort to secure a fully automated radiosynthesis of 18F-ERAS-801, the sodium borohydride reduction step to afford aniline 18F-2 was revised and reoptimized. Nitroarene 18F-4 was converted to the corresponding aniline via iron reduction in the presence of acetic acid.18 Optimization of the iron reduction was completed and the process was fully automated on the ELIXYS radiosynthesis module to afford 18F-2 in 83 ± 3 RCY (Tables S13 and S14†).Synthesis of 4-chloroquinazoline core 3b was achieved following published protocols14b and the previously optimized conditions for the SNAr between aniline 18F-2 and 3a were applied to the system using 3b as the appropriate 4-chloroquinazoline (Fig. 3A). Fine tuning of the reaction conditions revealed that 18F-ERAS-801 was generated in acetonitrile at 105 °C in 4 min in 86% RCY, based on radio-HPLC analysis of the crude product (Table S16†). Unlike 3a, the addition of 3b was performed in one-pot and, after HPLC-purification, afforded 18F-ERAS-801 in 31 ± 6 isolated RCY (n = 9) (Table S17†).
 | ||
| Fig. 3 (A) Radiosynthesis of 18F-ERAS-801. (B) and (C) Representative co-registered PET/CT images of brain-penetrant 18F-ERAS-801 biodistribution in (B) a mouse and (C) a rat at indicated timepoints post-injection of the tracer, with corresponding time–activity curves of 18F-ERAS-801 uptake in the brains of each animal. | ||
Production of 18F-ERAS-801via the three-step process was fully automated on the ELIXYS radiosynthesis module (Fig. S13†) and was achieved in >99% radiochemical purity and 22 ± 4% isolated RCY, based on starting [18F]fluoride activity (decay corrected, n = 9). The activity yield of 18F-ERAS-801 was 8 ± 2% (non-decay corrected, n = 9) and the molar activity was 41.8 ± 14.1 GBq μmol−1 (1.1 ± 0.4 Ci μmol−1), (n = 4) (Fig. 3A). Inductively coupled plasma mass spectrometry (ICP-MS) analysis was performed to detect iron in the formulated 18F-ERAS-801 samples and the residual iron content was determined to be <10 ppb (n = 2 samples), which is well below the acceptable limit for in-human injection.19 Additional quality control (QC) testing was performed on the formulated dose which met or exceeded the specified criteria for clinical applications (Table S21†).
Molecular imaging of 18F-ERAS-801
With an automated synthesis of 18F-ERAS-801 in hand, microPET/CT imaging studies were conducted in rodents to evaluate overall brain penetration and tissue accumulation of the radiotracer (Fig. 3). Rapid uptake of 18F-ERAS-801 was observed in the mouse brain, with a SUVmean value of 0.71 at 1 min p.i. followed by significantly improved retention and a slower clearance than what was observed for 18F-JCN037 (Fig. 3B). Remarkably, the in vivo biodistribution of 18F-ERAS-801 in a SD rat revealed a slow initial uptake of the radiotracer in the rat brain, with a SUVmean value of 0.33 at 2 h p.i., followed by a long retention time of activity in the brain at 4 h p.i. (Fig. 3C).Encouraged by the significantly improved pharmacokinetic properties and brain retention of 18F-ERAS-801, microPET/CT imaging and biodistribution studies were conducted in mice with GliomaPDOX tumors implanted in the brain to assess targeting and sensitivity (Fig. 4). In parallel, non-tumor bearing NSG mice were evaluated as a control group. To better visualize the brain anatomy and the implanted glioblastoma, MRI was performed in addition to PET and CT due to its superior soft tissue contrast (Fig. 4A). The PET signal is significantly higher in the whole-brain of the tumor-bearing mice compared to the whole-brain of the healthy control mice (Fig. 4B). After 1 hour tracer uptake, 18F-ERAS-801 displayed 1.7-fold higher PET signal in tumor tissue than healthy brain tissue, with relatively high retention in the tumor over the next three hours (Fig. 4C). Notably, at the 4-hour time point, 18F-ERAS-801 accumulation was nearly 3-fold higher than tracer signal in the healthy tissue of the corresponding brain, confirming target specificity (Fig. 4C).
 | ||
| Fig. 4 (A) Representative co-registered PET/MRI/CT images of the control brain and the glioblastoma-bearing brain in mice, 4 hours post-injection of 18F-ERAS-801. Dashed areas, glioblastoma. (B) Time-activity curves of 18F-ERAS-801 in the healthy control mouse brain and in the glioblastoma-bearing mouse brain. (C) Tumor-to-brain ratio of 18F-ERAS-801 uptake over 4 hours post-injection of the tracer. *p < 0.05. | ||
The greater uptake of 18F-ERAS-801 in the brain suggests that ERAS-801 may overcome the poor BBB penetrating properties of other EGFR TKIs that have been tested and failed in GBM. It remains to be determined if these improved properties of ERAS-801 will translate into a clinical benefit for GBM patients. However, the improved outcomes for EGFR-mutant NSCLC patients with brain metastasis treated with the CNS penetrant EGFR TKI Tagrisso20 suggests that higher brain exposures of an EGFR TKI can be effective and non-toxic in the brain.
Conclusions
In summary, we have developed two PET tracers, 18F-JCN037 and 18F-ERAS-801, that are fluorine-18 isotopologues of JCN037 and its clinical derivative ERAS-801, recently developed TKIs against EGFR-driven GBM. PET imaging studies of these tracers enabled noninvasive, whole-body pharmacological measurements including brain accumulation and retention, valuable data for CNS drug development. The synthesis of 18F-ERAS-801 was fully automated on a commercial radiosynthesis module in 8 ± 2% activity yield. PET imaging studies of 18F-ERAS-801 confirmed BBB penetration and, importantly, provided insights into the intratumoral drug distribution in the brain for EGFR-driven GBM tumors.PET tracers that can validate tumor engagement during pre-clinical drug development, visualize and quantify EGFR expression in vivo, differentiate between EGFR mutational statuses, and potentially predict patients' response to TKI treatment are of great interest. PET imaging was recently reported to enable precise quantification of EGFR mutation status in NSCLC patients and improve overall patient management and care.8c The beneficial role of 18F-ERAS-801 to provide this type of predictive information for the clinical assessment of ERAS-801 treatment remains to be seen and additional studies are ongoing to validate this potential value.
Ethical statement
All mice were kept under defined pathogen-free conditions at the AAALAC-approved animal facility of the Division of Laboratory Animals at UCLA. All studies were in accordance with UCLA Office of Animal Resource Oversight (OARO) protocol guidelines and with UCLA Animal Research Committee protocol guidelines.Data availability
Experimental procedures, optimization, characterization data, NMR spectra, in vivo imaging methods and analyses are available in the ESI.†Author contributions
M. K. N. carried out the radiochemistry experiments and optimization with guidance from J. M. M. Imaging experiments were designed by S. X. and conducted by J. E. T. and S. X. with input from D. A. N. The manuscript was written by M. K. N. and J. M. M. and edited by all authors.Conflicts of interest
M. K. N., J. E. T., D. A. N., and J. M. M. are inventors on a patent application covering chemical matter in this publication that is assigned to The Regents of the University of California. D. A. N. is a co-founder, shareholder and consultant for Katmai Pharmaceuticals, which has licensed IP related to this work.Acknowledgements
Financial support was provided in part by the Crump Institute for Molecular Imaging and the Jonsson Comprehensive Cancer Center (JCCC) Career Development Seed Grant. The GNEXT microPET/CTscanner (Sofie Biosciences) was funded by an NIH Shared Instrumentation for Animal Research (SIFAR) Grant (1S10OD026917-01A1). The microPET/CT studies were supported in part by the JCCC Support Grant (2P30CA016042-44). We thank Jeffrey Collins for providing [18F]fluoride for these studies and Mikayla Tamboline for performing the microPET/CT imaging studies.Notes and references
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- (a) K. Kobayashi and K. Hagiwara, Targeted Oncology, 2013, 8, 27–33 CrossRef PubMed; (b) P. Ballard, J. W. T. Yates, Z. Yang, D.-W. Kim, J. C.-H. Yang, M. Cantarini, K. Pickup, A. Jordan, M. Hickey, M. Grist, M. Box, P. Johnström, K. Varnäs, J. Malmquist, K. S. Thress, P. A. Jänne and D. Cross, Clin. Cancer Res., 2016, 22, 5130–5140 CrossRef CAS PubMed.
- (a) A. B. Lassman, M. R. Rossi, J. R. Razier, L. E. Abrey, F. S. Lieberman, C. N. Grefe, K. Lamborn, W. Pao, A. H. Shih, J. G. Kuhn, R. Wilson, N. J. Nowak, J. K. Cowell, L. M. DeAngelis, P. Wen, M. R. Gilbert, S. Chang, W. A. Yung, M. Prados and E. C. Holland, Clin. Cancer Res., 2005, 11, 7841–7850 CrossRef CAS PubMed; (b) I. Vivanco, H. I. Robins, D. Rohle, C. Campos, C. Grommes, P. L. Nghiemphu, S. Kubek, B. Oldrini, M. G. Chheda, N. Yannuzzi, H. Tao, S. Zhu, A. Iwanami, D. Kuga, J. Dang, A. Pedraza, C. W. Brennan, A. Heguy, L. M. Liau, F. Lieberman, W. K. A. Yung, M. R. Gilbert, D. A. Reardon, J. Drappatz, P. Y. Wen, K. R. Lamborn, S. M. Chang, M. D. Prados, H. A. Fine, S. Horvath, N. Wu, A. B. Lassman, L. M. DeAngelis, W. H. Yong, J. G. Kuhn, P. S. Mischel, M. P. Mehta, T. F. Cloughesy and I. K. Mellinghoff, Cancer Discovery, 2012, 2, 458–471 CrossRef CAS PubMed.
- W. M. Pardridge and J. Cereb, Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- (a) R. Uppoor, P. Mummaneni, E. Cooper, H. Pien, A. Sorensen, J. Collins, M. Mehta and S. Yasuda, Clin. Pharmacol. Ther., 2008, 84, 69–74 CrossRef CAS PubMed; (b) P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175–186 CrossRef CAS PubMed.
- L. Cunha, K. Szigeti, D. Mathé and L. F. Metello, Drug Discovery Today, 2014, 19, 936–948 CrossRef CAS PubMed.
- O. Gefvert, M. Bergström, B. Långström, T. Lundberg, L. Lindström and R. Yates, Psychopharmacology, 1998, 135, 119–126 CrossRef CAS PubMed.
- (a) P. Slobbe, A. J. Poot, A. D. Windhorst and G. A. M. S. van Dongen, Drug Discovery Today, 2012, 17, 1175–1187 CrossRef CAS PubMed; (b) V. Bernard-Gauthier, J. J. Bailey, S. Berke and R. Schirrmacher, Molecules, 2015, 20, 22000–22027 CrossRef CAS PubMed; (c) X. Sun, Z. Xiao, G. Chen, Z. Han, Y. Liu, C. Zhang, Y. Sun, Y. Song, K. Wang, F. Fang, X. Wang, Y. Lin, L. Xu, L. Shao, J. Li, Z. Cheng, S. S. Gambhir and B. Shen, Sci. Transl. Med., 2018, 10, eaan8840 CrossRef PubMed; (d) E. A. van de Stadt, M. Yaqub, A. A. Lammertsma, A. J. Poot, P. R. Schober, R. C. Schuit, E. F. Smit, I. Bahce and N. H. Hendrikse, EJNMMI Res., 2020, 10, 97 CrossRef CAS PubMed; (e) A. Högnäsbacka, A. J. Poot, D. J. Vugts, G. A. M. S. van Dongen and A. D. Windhorst, Pharmaceuticals, 2022, 15, 450 CrossRef PubMed; (f) E. Van De Stadt, M. Yaqub, A. A. Jahangir, H. Hendrikse and I. Bahce, Front. Oncol., 2022, 12, 900450 CrossRef CAS PubMed.
- (a) T. Läppchen, M. L. H. Vlaming, E. Custers, J. Lub, C. F. Sio, J. DeGroot and O. C. Steinbach, Appl. Radiat. Isot., 2012, 70, 205–209 CrossRef PubMed; (b) Y. Seimbille, M. E. Phelps, J. Czernin and D. H. S. Silverman, J. Labelled Compd. Radiopharm., 2005, 48, 829–843 CrossRef CAS.
- H. Su, Y. Seimbille, G. Z. Ferl, C. Bodenstein, B. Fueger, K. J. Kim, Y.-T. Hsu, S. M. Dubinett, M. E. Phelps, J. Czernin and W. A. Weber, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 1089–1099 CrossRef CAS PubMed.
- N. J. Taylor, E. Emer, S. Preshlock, M. Schedler, M. Tredwell, S. Verhoog, J. Mercier, C. Genicot and V. Gouverneur, J. Am. Chem. Soc., 2017, 139, 8267–8276 CrossRef CAS PubMed.
- P. Slobbe, A. D. Windhorst, M. S.-v. Walsum, R. C. Schuit, E. F. Smit, H. G. Niessen, F. Solca, G. Stehle, G. A. M. S. van Dongen and A. J. Poot, Nucl. Med. Biol., 2014, 41, 749–757 CrossRef CAS PubMed.
- (a) P. Slobbe, A. D. Windhorst, M. Stigter-van Walsum, E. F. Smit, H. G. Niessen, F. Solca, G. Stehle, G. A. M. S. van Dongen and A. J. Poot, EJNMMI Res., 2015, 5, 14 CrossRef PubMed; (b) N. Colclough, K. Chen, P. Johnström, N. Strittmatter, Y. Yan, G. L. Wrigley, M. Schou, R. Goodwin, K. Varnäs, S. J. Adua, M. Zhao, D. X. Nguyen, G. Maglennon, P. Barton, J. Atkinson, L. Zhang, A. Janefeldt, J. Wilson, A. Smith, A. Takano, R. Arakawa, M. Kondrashov, J. Malmquist, E. Revunov, A. Vazquez-Romero, M. M. Moein, A. D. Windhorst, N. A. Karp, M. R. V. Finlay, R. A. Ward, J. W. T. Yates, P. D. Smith, L. Farde, Z. Cheng and D. A. E. Cross, Clin. Cancer Res., 2021, 27, 189–201 CrossRef CAS PubMed.
- (a) J. E. Tsang, L. M. Urner, G. Kim, K. Chow, L. Baufeld, K. Faull, T. F. Cloughesy, P. M. Clark, M. E. Jung and D. A. Nathanson, ACS Med. Chem. Lett., 2020, 11, 1799–1809 CrossRef CAS PubMed; (b) D. A. Nathanson, M. E. Jung, J. E. Tsang, L. M. Urner, P. M. Clark, T. F. Cloughesy and G. Kim, US Pat., US011377451B011377452, 2022 Search PubMed.
- (a) B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CrossRef CAS PubMed; (b) A. V. Mossine, A. F. Brooks, K. J. Makaravage, J. M. Miller, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Org. Lett., 2015, 17, 5780–5783 CrossRef CAS PubMed; (c) L. S. Sharninghausen, A. F. Brooks, W. P. Winton, K. J. Makaravage, P. J. H. Scott and M. S. Sanford, J. Am. Chem. Soc., 2020, 142, 7362–7367 CrossRef CAS PubMed.
- M. K. Narayanam, G. Ma, P. A. Champagne, K. N. Houk and J. M. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13006–13010 CrossRef CAS PubMed.
- M. K. Narayanam, B. T. Lai, J. M. Loredo, J. A. Wilson, A. M. Eliasen, N. A. LaBerge, M. Nason, A. L. Cantu, B. K. Luton, S. Xu, H. D. Agnew and J. M. Murphy, Bioconjugate Chem., 2021, 32, 2073–2082 CrossRef CAS PubMed.
- D. C. Whritenour, S. J. Brenek and N. J. Tom, Org. Process Res. Dev., 2001, 5, 539–541 CrossRef CAS.
- Guidelines for Elemental Impurities, https://www.ema.europa.eu/en/ich-q3d-elemental-impurities-scientific-guideline, accessed 12-April-2023.
- T. S. Mok, Y.-L. Wu, M.-J. Ahn, M. C. Garassino, H. R. Kim, S. S. Ramalingam, F. A. Shepherd, Y. He, H. Akamatsu, W. S. M. E. Theelen, C. K. Lee, M. Sebastian, A. Templeton, H. Mann, M. Marotti, S. Ghiorghiu and V. A. Papadimitrakopoulou, N. Engl. J. Med., 2017, 376, 629–640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04424f |
| This journal is © The Royal Society of Chemistry 2023 |