
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general approach to stereospecific Pd-catalyzed cross-coupling reactions of benzylic stereocenters†
Meruyert
Binayeva
ab,
Xinghua
Ma
ab,
Pejman
Ghaemimohammadi
ab and
Mark R.
Biscoe
 *ab
*ab
aDepartment of Chemistry and Biochemistry, The City College of New York (CCNY), New York, NY 10031, USA. E-mail: mbiscoe@ccny.cuny.edu
bThe Graduate Center of the City University of New York (CUNY), 365 Fifth Avenue, New York, NY 10016, USA
First published on 24th November 2023
Abstract
We have developed a general process for the formation of enantioenriched benzylic stereocenters via stereospecific Pd-catalyzed cross-coupling reactions of enantioenriched benzylic tricyclohexyltin nucleophiles. This process proceeds with excellent stereospecificity for a remarkably broad scope of electrophilic coupling partners including aryl and heteroaryl halides and triflates, acid chlorides, thioesters, chloroformates, and carbamoyl chlorides. Thus, enantioenriched 1,1-diarylalkanes as well as formal products of asymmetric enolate arylation are readily accessed using this approach. We additionally provide the first demonstration of a Sn-selective cross-coupling reaction using a vicinal alkylborylstannane nucleophile. In these reactions, the presence of cyclohexyl spectator ligands on tin is essential to ensure selective transfer of the secondary benzylic unit from tin to palladium.
Aryl-substituted stereocenters are ubiquitous structural components of biologically active molecules.1 Of potential aryl-containing stereocenters, 1,1-diaryl stereocenters and stereocenters formed from α-arylated carbonyl groups are particularly common structural motifs found in compounds emerging from the drug-discovery process (Scheme 1).1–3 In most instances, a single enantiomer of a racemic mixture constitutes the therapeutically active species, while the opposite stereoisomer may be inactive, reduce the efficacy of the active species, or result in deleterious side effects.4 Therefore, synthetic strategies to rapidly and reliably introduce or manipulate aryl-substituted stereocenters with precise control of absolute stereochemistry are of significant medicinal importance.
 | ||
| Scheme 1 Examples of benzylic stereocenters in small-molecule drugs. | ||
Stereospecific transition metal-catalyzed cross-coupling reactions employing configurationally stable enantioenriched nucleophiles (1) have emerged as a powerful alternative approach to asymmetric synthesis (Scheme 2).5,6 Though this approach requires the preparation of an enantioenriched main group organometallic nucleophile, stereochemical information can often be reliably transferred via a stereoretentive or stereoinvertive transmetallation event in ensuing cross-coupling processes.5e Previous studies of Pd-catalyzed stereospecific Stille reactions suggest that the stereoretentive course of transmetallation is strongly preferred for enantioenriched alkyltin nucleophiles when Cu(I) is employed as a co-transmetallating agent, and is often independent of the electronic and steric properties of the nucleophilic and electrophilic coupling partners.7 Thus, stereospecific Stille cross-coupling reactions have tremendous potential as a general synthetic strategy to predictably modify three-dimensional molecular structure.
 | ||
| Scheme 2 Stereospecific transition metal-catalyzed cross-coupling reactions using enantioenriched main group organometallic nucleophiles. | ||
Because organostannanes used in cross-coupling reactions possess four organic units, each of which is theoretically capable of undergoing transmetallation to Pd, it is essential that selective transfer of a single unit be achieved during the cross-coupling process. The selective transfer of a C(sp) or C(sp2) group from an organostannane is commonly achieved by incorporation of three alkyl units (e.g., Me or n-Bu) onto tin, which serve as spectator ligands due to the slow rate of alkyl transfer from tin (Scheme 3).8 Slow alkyl transfer from tin therefore complicates efforts to effect the selective transfer of a single alkyl unit from a tetraalkylstannane. We have demonstrated that incorporation of a carbastannatrane backbone9 into the alkylstannane enables the selective transfer of an otherwise unactivated secondary alkyl group in Pd-catalyzed cross-coupling reactions (Scheme 4a) when CuCl is employed as a co-transmetallating agent and JackiePhos10 (2) as the supporting phosphine ligand.7d,g,h However, in the absence of a carbastannatrane activating group, significant electronic differentiation of single alkyl unit of the tetraalkylstannane has been necessary to effect selective alkyl transfer. Hoppe11 and Falck7a have separately demonstrated the ability of highly activated secondary alkyl groups to undergo selective and stereospecific transfer to Pd in the presence of n-butyl ligands. In these instances, electronic activation (i.e., inclusion of an α-heteroatom or α-C(sp2) substituent) in combination with a remote coordinating group was required to promote selective alkyl transfer (Scheme 4b and c). More recently, to effect selective transmetallation of less activated secondary alkyl units, we replaced the n-butyl spectator ligands of the enantioenriched tetraalkylstannane with cyclohexyl spectator ligands.7i,12 This modification enabled the selective and stereospecific transfer of nitrogen-containing stereocenters (Scheme 4d) to Pd, and was thereafter extended to use with racemic α-oxygenated secondary alkylstannanes not bearing a remote directing/activating oxygen-containing group.13 These results suggested that selective and stereospecific transfer of marginally activated secondary alkyl groups might be broadly achievable for other enantioenriched alkyltricyclohexylstannane nucleophiles. Herein, we describe the use of enantioenriched benzylic tricyclohexylstannanes in stereospecific Pd-catalyzed reactions with aryl and acyl electrophiles (Scheme 4e). In this system, incorporation of cyclohexyl ligands into the organostannane nucleophile is essential to achieve selective transfer of the benzylic unit to Pd. In line with previous stereospecific Stille couplings, this process proceeds with high stereospecificity while tolerating an extraordinarily broad scope of electrophilic coupling partners, including aryl and heteroaryl halides and triflates, acid chlorides, thioesters, chloroformates, and carbamoyl chlorides. Thus, in addition to products from stereospecific arylation reactions (i.e., 1,1-diaryl stereocenters),14 formal products of asymmetric enolate arylation reactions can also be readily obtained using this approach.15–19 This process will therefore serve as a general synthetic strategy for the stereocontrolled formation of new benzylic stereocenters.
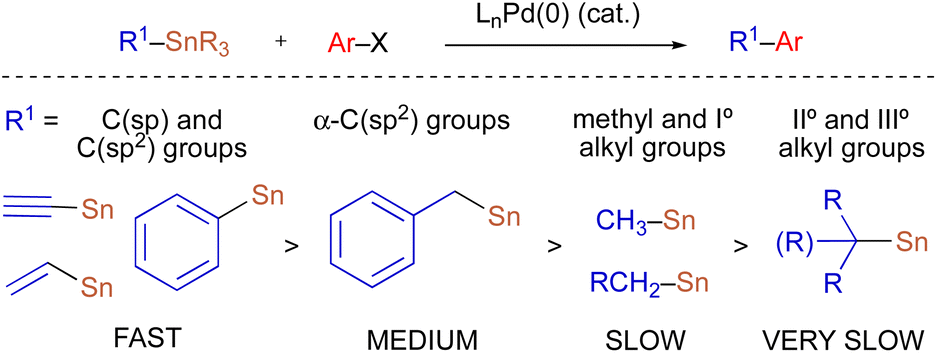 | ||
| Scheme 3 Relative rates of group transfer in Pd-catalyzed cross-coupling reactions of tetraorganostannanes. | ||
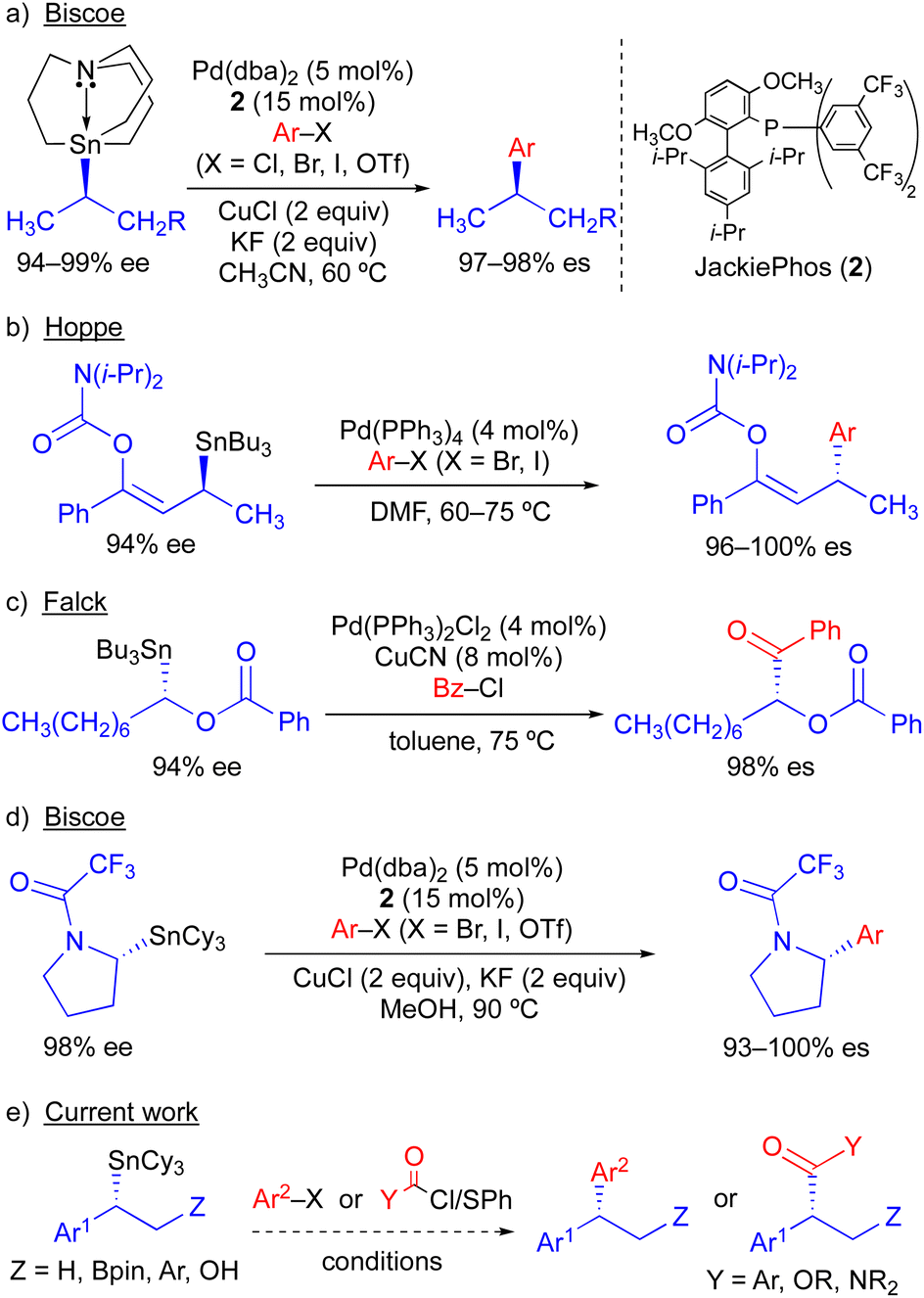 | ||
| Scheme 4 Stereospecific Pd-catalyzed Stille cross-coupling reactions using enantioenriched secondary alkylstannanes. | ||
We have previously found that the use of JackiePhos (2) as a supporting phosphine ligand facilitates transmetallation of alkyl groups from organostannanes in Pd-catalyzed Stille cross-coupling reactions when CuCl is included as a co-transmetallating agent (see Scheme 4a and d).7d,g–i Thus, 2 was a logical choice of supporting phosphine ligand for use in exploratory Pd-catalyzed cross-coupling studies using enantioenriched benzylic organostannanes. We initiated our investigations using secondary benzylic tributylstannane 3a in cross-coupling reactions with 4-bromoacetophenone (Table 1). Use of 3a as the nucleophilic coupling partner resulted in low yields of coupling product 4a and/or significant n-butyl transfer (5a) for all solvents examined. In contrast, when tricyclohexylstannane analogue 3b was employed, significantly improved yields of 4a were obtained alongside only nominal evidence (less than 2% by gas chromatography) of competitive cyclohexyl coupling (5b). We found that reactions conducted at 110 °C provided optimal reaction conversion across the range of conditions examined. These results indicate that although a secondary benzylic stereocenter is insufficiently activated to outcompete transfer of an unactivated primary alkyl group from tin, selective transfer can be achieved in the presence of an unactivated secondary alkyl unit. To investigate potential stereochemical transfer in these reactions, enantioenriched 3b was prepared using a stereoselective sulfoxide-directed metalation strategy previously reported by Ruano and Padwa.20 When enantioenriched 3b was employed under the optimized cross-coupling reaction conditions, exceptional stereofidelity was observed (Table 1, entries 9 and 10). Thus, a selective and enantiospecific Pd-catalyzed coupling was achieved using an enantioenriched secondary benzylic tricyclohexylstannane nucleophile.
|
|
||||
|---|---|---|---|---|
| Entry | R | Solvent | 4a yielda (%) | 5 yielda (%) |
| a Calibrated GC or 1H NMR yields. b Using (S)-3b (96% ee); % es = [% ee of product]/[% ee of starting material]. | ||||
| 1 | n-Bu (3a) | CH3CN | 29 | 45 (5a) |
| 2 | n-Bu | Toluene | 11 | 8 |
| 3 | n-Bu | CH3OH | 18 | 14 |
| 4 | n-Bu | 1,4-Dioxane | 20 | 9 |
| 5 | n-Bu | t-BuOH | 26 | 19 |
| 6 | Cy (3b) | CH3CN | 69 | <2 (5b) |
| 7 | Cy | Toluene | 57 | <2 |
| 8 | Cy | CH3OH | 32 | <2 |
| 9 | Cy | 1,4-Dioxane | 72 (99% es)b | <2 |
| 10 | Cy | t-BuOH | 82 (97% es)b | <2 |
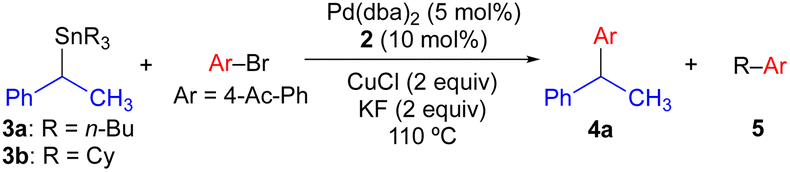
Using the optimized conditions of Table 1, we investigated the scope of electrophilic coupling partners tolerated in stereospecific cross-coupling reaction with enantioenriched organostannane 3b (Table 2). Electron-rich, electron-neutral, and electron-deficient aryl electrophiles all undergo efficient Pd-catalyzed cross-coupling reactions, generating 1,1-diaryl stereocenters (4) with high enantiospecificity. The use of heteroaryl electrophiles is also acceptable, as is the use of an aryl electrophile bearing an ortho substituent. Thus, enantiospecificity is largely independent of the steric and electronic properties of the electrophilic aryl coupling partner, which is consistent with our past observations for stereospecific Pd-catalyzed Stille reactions. Additionally, acyl electrophiles can be successfully employed using the same reaction conditions (6a–6e), generating formal products of asymmetric enolate arylation reactions. Asymmetric mono-arylation reactions that form tertiary α-keto stereocenters constitute a major synthetic challenge due to the potential for diarylation as well as the propensity of the arylated α-keto stereocenter to racemize under basic conditions.16 Our ability to access products 6a–6e indicates that formal products of the asymmetric enolate arylation of ketones, esters, and amides can be broadly achieved using a stereospecific benzylic coupling strategy, thus circumventing the problems commonly associated with asymmetric enolate arylation. In cases where an enantioenriched acyl electrophile is employed, high reagent-controlled diastereoselectivity is achieved (6d, 6e). For all cross-coupling reactions in Table 2, no evidence of cyclohexyl transfer was observed.

As a more general approach to the preparation of enantioenriched benzylic stannanes, we adapted the asymmetric copper-catalyzed borylstannation of styrene developed by Liao using chiral sulfinylphosphine ligand 7.21 Using t-amyl alcohol as solvent and increasing the CuCl loading, we found that tricyclohexyltin could be incorporated into this process affording α-aryl-β-borylstannane 8 in high enantiomeric excess (Scheme 5). Styrene derivatives with aryl groups bearing an electron-donating group, electron-withdrawing group, and ortho-substituent could each be employed in this process. However, heteroaryl styrene derivatives appear to be incompatible with this transformation. Conceptually, enantioenriched 8 constitutes a powerful building block for use in stereospecific cross-coupling reactions, which could be employed in tin-selective or boron-selective cross-coupling processes. Using a known process for Pd-catalyzed Suzuki reactions of primary alkylboronate esters with 8a,21 we achieved a boron-selective cross-coupling reaction with no measurable erosion of enantioenrichment. Alternatively, when we employed our standard conditions from Table 2 with 8a, we successfully achieved a tin-selective cross-coupling reaction with efficient transfer of stereochemistry. Though Sn-selective cross-coupling reactions of vicinal borylstannyl alkenes have been demonstrated,22 to the best of our knowledge, there is no prior report of a Sn-selective cross-coupling reaction of a C(sp3)–Sn bond in the presence of a C(sp3)–B bond.23
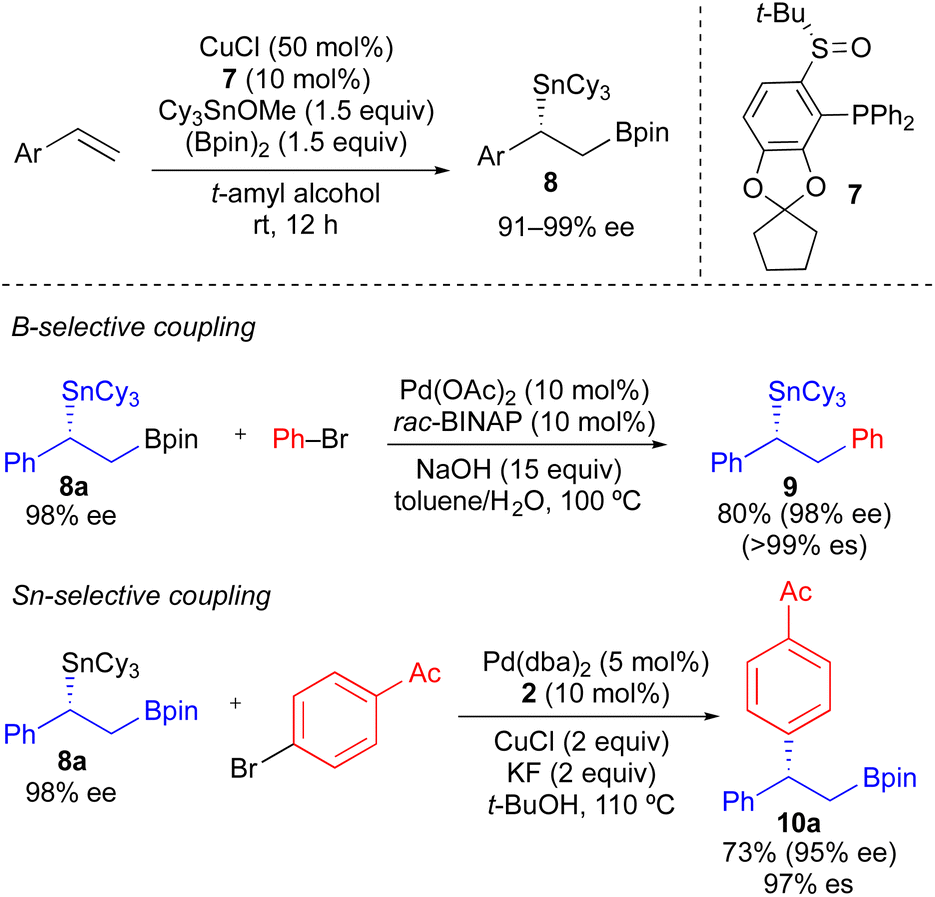 | ||
| Scheme 5 Formation of enantioenriched α-aryl-β-borylstannanes via asymmetric Cu-catalyzed borylstannation of styrenes, and their use in B-selective and Sn-selective cross-coupling reactions. | ||
Our initial studies using borylstannane 8a (and aryl analogues thereof) focused on stereospecific Sn-selective couplings. Such a process leaves the primary Bpin group on the cross-coupling product, which could serve as a versatile functional handle for further structural modification.24 Using the Sn-selective stereospecific cross-coupling conditions, highly enantioenriched alkylboron products containing a 1,1-diaryl stereocenter could be readily prepared and isolated (Table 3, 10a, 10e). As we found that oxidation of the primary Bpin group facilitated chiral-phase HPLC analysis, we incorporated an optional oxidative workup into our general cross-coupling procedure and isolated the hydroxyl derivatives in the majority of our examples. Again, high enantiospecificities could be achieved in cross-coupling reactions using α-aryl-β-borylstannane nucleophiles regardless of the electronic or steric properties (electron-rich, electron-deficient, ortho-substituted) of the electrophilic coupling partner. Additionally, we found that α-aryl-β-borylstannane nucleophiles (8) derived from styrene precursors with sterically and electronically differentiated aryl rings could be efficiently employed in these stereospecific benzylation reactions. Identical reaction conditions to those of Table 2 were employed for cross-coupling reaction of 8, with no reoptimization of individual reactions. Thus, we have established a single set of reaction conditions that can be universally employed in these stereospecific benzylation reactions. Though these standard conditions do result in slightly diminished yields for products of Table 3 when compared to products of Table 2, we note that yields can be improved ca. 10–30% by increasing the loading of organostannane from 1.2 equiv. to 2.0 equiv. Finally, enantioenriched benzylic stannanes (11) prepared via boron-selective cross-coupling or oxidation reactions were employed in stereospecific Pd-catalyzed arylation and acylation reactions (Table 4). Again, high stereofidelity was observed in all of these reactions. It is particularly noteworthy that this cross-coupling reaction cleanly tolerates the presence of a free β-hydroxyl group (12b) on the enantioenriched organostannane.
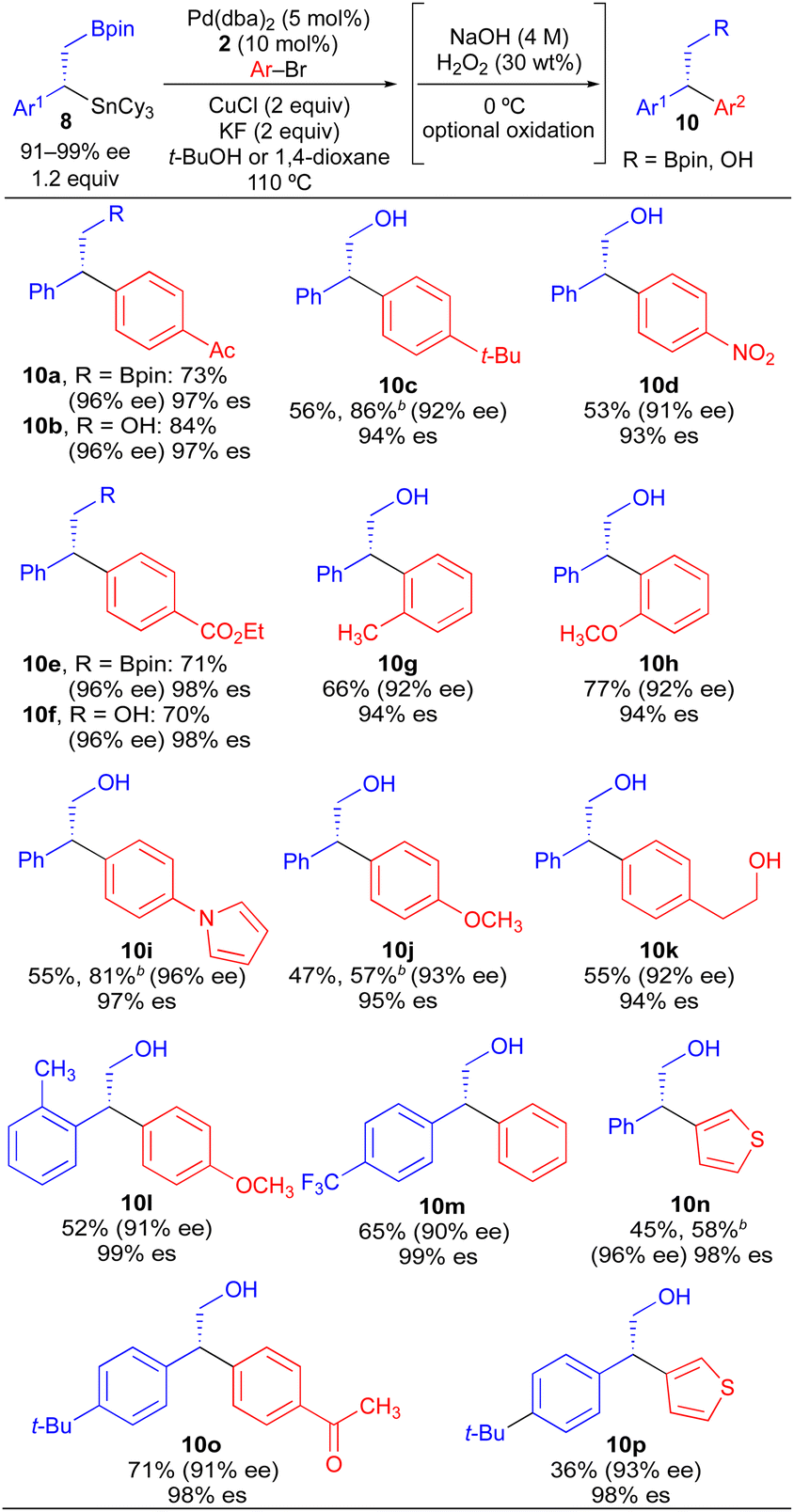
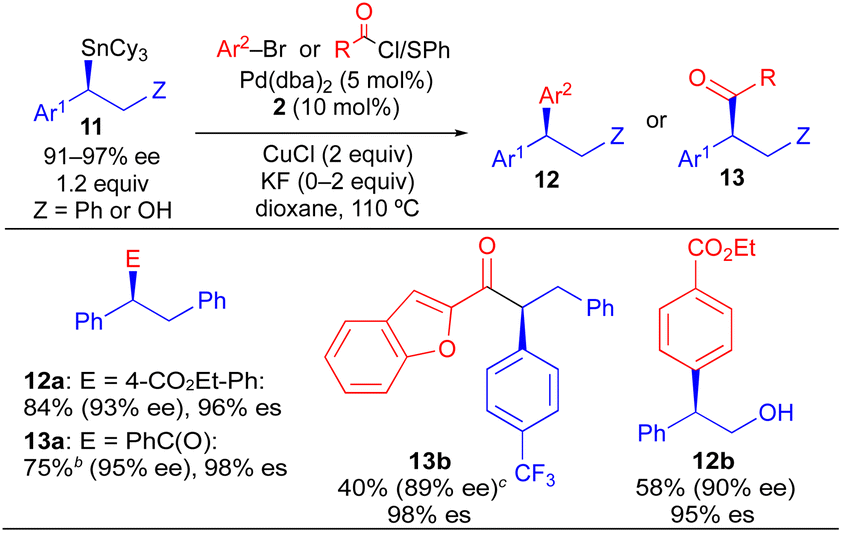
This stereospecific benzylation process, in combination with our previous studies using enantioenriched secondary alkylstannane nucleophiles, suggests that the stereochemical course of alkyltin transmetallation is largely independent of the electronic and steric properties of the nucleophilic and electrophilic coupling partners when Pd/Cu are employed. The uniformity of reaction conditions (i.e., Pd/Cu/JackiePhos) across diverse classes of alkylstannane nucleophiles (benzylic, nitrogen-containing,7i oxygen-containing,13 and unactivated stereocenters7d) is a particularly significant feature of this chemistry, which will facilitate its future application to diversity-oriented synthesis. In contrast, the dominant stereochemical course of alkylboron transmetallation in analogous stereospecific Suzuki cross-coupling processes is dictated by subtle electronic and steric perturbations of the coupling partners.5e Thus, it is uncommon for the reaction conditions for a stereospecific Suzuki transformation using a specific class of alkylboron nucleophile to be successfully extended to couplings that employ alternative, electronically differentiated alkylboron nucleophiles. In addition to facilitating selective alkyl transfer to copper and palladium, use of RSnCy3 compounds in place of RSnBu3 compounds offers important practical benefits. ClSnCy3 is crystalline and odorless, and exhibits significantly lower toxicity than commonly used ClSnBu3.25 RSnCy3 derivatives also tend to be highly crystalline, in contrast to RSnBu3 derivatives which tend to be oils. Thus, the crystallinity and solubility profile of tricyclohexyltin byproducts should enable tin to be readily purged from waste streams via solvent swaps and crystallization. As concerns regarding organotin toxicity and the removal of tin byproducts can heavily influence the decision to incorporate the Stille reaction into a synthetic sequence when SnBu3 (or SnMe3) nucleophiles are required, we expect that the use of SnCy3-based nucleophiles will extend beyond cross-coupling reactions of activated secondary alkyl groups and may find application in traditional C(sp2)–C(sp2) Stille cross-coupling reactions where aryl and vinyl SnBu3 derivatives are commonly employed.
Conclusions
In summary, we have developed a general strategy for the formation of enantioenriched benzylic stereocenters via stereospecific Pd-catalyzed cross-coupling reactions of enantioenriched benzylic tricyclohexyltin nucleophiles. The use of cyclohexyl spectator ligands on the benzylic stannanes is vital to ensure selective stereospecific transfer of the secondary benzylic unit. In addition to aryl bromide and triflate electrophiles, chloroformates, carbamoyl chlorides, acid chlorides, and thioesters can be efficiently employed in these stereospecific cross-coupling reactions. Thus, enantioenriched 1,1-diarylalkanes as well as formal products of asymmetric enolate arylation are readily accessed using this approach. For all electrophiles and nucleophiles employed, excellent enantiospecificity or diasterospecificity was observed, indicating that the stereochemical transfer is widely independent of the electronic and steric properties of both the electrophilic and nucleophilic coupling partners employed. This work further illustrates the broad synthetic advantages achievable by incorporating cyclohexyl spectator ligands into organostannanes used in Stille cross-coupling reactions. Based on these advantages, we feel that applications of organotricyclohexylstannanes (RSnCy3) may transcend C(sp2)–C(sp3) cross-coupling reactions and find utility in C(sp2)–C(sp2) cross-coupling reactions where RSnBu3 or RSnMe3 nucleophiles are more commonly employed but less desirable.Data availability
All data have been included in the ESI.†Author contributions
Xinghua Ma initiated this project and developed the standard reaction conditions. Xinghua Ma, Meruyert Binayeva, and Pejman Ghaemimohammadi all contributed to cross-coupling reactions of Table 2. Meruyert Binayeva conducted all experiments shown in other schemes and tables.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the City College of New York, the National Science Foundation (CHE-1955472: early exploratory work on factors influencing selective alkyl transfer from tin), and the National Institutes of Health (R01GM131079: diversification of benzylic stereocenters through transition metal catalysis) for support of this work.Notes and references
- D. Ameen and T. J. Snape, Med. Chem. Commun., 2013, 4, 893 RSC.
- T. Jia, P. Cao and J. Liao, Chem. Sci., 2018, 9, 546 RSC.
- Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955 CrossRef CAS.
- M. C. Nunez, M. E. Garcia-Rubino, A. Conejo-Garcia, O. Cruz-Lopez, M. Kimatrai, M. A. Gallo, A. Espinosa and J. M. Campos, Curr. Med. Chem., 2009, 16, 2064 CrossRef CAS PubMed.
- Reviews articles on stereospecific cross-coupling reactions: (a) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082 CrossRef CAS PubMed; (b) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105 RSC; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed; (d) E. L. Lucas and E. R. Jarvo, Nat. Rev. Chem., 2017, 1, 65 CrossRef; (e) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584 CrossRef CAS PubMed; (f) J. Xu, O. P. Bercher, M. R. Talley and M. P. Watson, ACS Catal., 2021, 11, 1604 CrossRef CAS PubMed.
- For stereospecific cross-coupling approaches to benzylic stereocenters using enantioenriched benzylic electrophiles, see: (a) A. Lopez-Perez, J. Adrio and J. C. Carretero, Org. Lett., 2009, 11, 5514 CrossRef CAS PubMed; (b) B. L. H. Taylor, E. C. Swift, J. D. Waetzig and E. R. Jarvo, J. Am. Chem. Soc., 2011, 133, 389 CrossRef CAS PubMed; (c) B. L. H. Taylor, M. R. Harris and E. R. Jarvo, Angew. Chem., Int. Ed., 2012, 51, 7790 CrossRef CAS PubMed; (d) P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280 CrossRef CAS PubMed; (e) M. R. Harris, L. E. Hanna, M. A. Greene, C. E. Moore and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 3303 CrossRef CAS PubMed; (f) Q. Zhou, H. D. Srinivas, S. Dasgupta and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 3307 CrossRef CAS PubMed; (g) I. M. Yonova, A. G. Johnson, C. A. Osborne, C. E. Moore, N. S. Morrisette and E. R. Jarvo, Angew. Chem., Int. Ed., 2014, 53, 2422 CrossRef CAS PubMed; (h) M. O. Konev, L. E. Hanna and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 6730 CrossRef CAS PubMed; (i) Q. Zhou, H. D. Srinivas, S. Zhang and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 11989 CrossRef CAS PubMed; (j) Q. Zhou, K. M. Cobb, T. Tan and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 12057 CrossRef CAS PubMed; (k) C. Li, Y. Zhang, Q. Sun, T. Gu, H. Peng and W. Tang, J. Am. Chem. Soc., 2016, 138, 10774 CrossRef CAS PubMed; (l) M. Brambilla and M. Tredwell, Angew. Chem., Int. Ed., 2017, 56, 11981 CrossRef CAS PubMed; (m) S.-Q. Zhang, B. L. H. Taylor, C.-L. Ji, Y. Gao, M. R. Harris, L. E. Hanna, E. R. Jarvo, K. N. Houk and X. Hong, J. Am. Chem. Soc., 2017, 139, 12994 CrossRef CAS PubMed; (n) J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577 CrossRef CAS PubMed; (o) J. Xu, O. P. Bercher and M. P. Watson, J. Am. Chem. Soc., 2021, 143, 8608 CrossRef CAS PubMed.
- (a) J. Ye, R. K. Bhatt and J. R. Falck, J. Am. Chem. Soc., 1994, 116, 1 CrossRef CAS; (b) K. W. Kells and J. M. Chong, J. Am. Chem. Soc., 2004, 126, 15666 CrossRef CAS PubMed; (c) P. M. Krizkova and F. Hammerschmidt, Eur. J. Org Chem., 2013, 5143 CrossRef PubMed; (d) L. Li, C.-Y. Wang, R. Huang and M. R. Biscoe, Nat. Chem., 2013, 5, 607 CrossRef CAS PubMed; (e) N. Theddu and E. Vedejs, J. Org. Chem., 2013, 78, 5061 CrossRef CAS PubMed; (f) F. Zhu, M. J. Rouke, T. Yang, J. Rodriguez and M. A. Walczak, J. Am. Chem. Soc., 2016, 138, 12049 CrossRef CAS PubMed; (g) C.-Y. Wang, G. Ralph, J. Derosa and M. R. Biscoe, Angew. Chem., Int. Ed., 2017, 56, 856 CrossRef CAS PubMed; (h) G. Ralph and M. R. Biscoe, Organometallics, 2019, 38, 3912 CrossRef CAS PubMed; (i) X. Ma, H. Zhao, M. Binayeva, G. Ralph, M. Diane, S. Zhao, C.-Y. Wang and M. R. Biscoe, Chem, 2020, 6, 781 CrossRef CAS PubMed.
- J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 6129 CrossRef CAS.
- For pioneering carbastannatrane studies, see: (a) K. Jurkschat, A. Tzschach, J. Meunier-Piret and M. Van Meerssche, J. Organomet. Chem., 1985, 290, 285 CrossRef CAS; (b) K. Jurkschat, A. Tzschach and J. Meunier-Piret, J. Organomet. Chem., 1986, 315, 45 CrossRef CAS; (c) E. Vedejs, A. R. Haight and W. O. Moss, J. Am. Chem. Soc., 1992, 114, 6556 CrossRef CAS; (d) M. S. Jensen, C. Yang, Y. Hsiao, N. Rivera, K. M. Wells, J. Y. L. Chung, N. Yasuda, D. L. Hughes and P. J. Reider, Org. Lett., 2000, 2, 1081 CrossRef CAS PubMed; (e) E. Fillion and N. J. Taylor, J. Am. Chem. Soc., 2003, 125, 12700 CrossRef CAS PubMed.
- J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720 CrossRef CAS PubMed.
- R. Kalkofen and D. Hoppe, Synlett, 2006, 1959 CAS.
- (a) R. J. Linderman and J. M. Siedlecki, J. Org. Chem., 1996, 61, 6492 CrossRef CAS PubMed; (b) M. J. Movassaghi, D. S. Siegel and S. Han, Chem. Sci., 2010, 1, 561 RSC.
- H. Zhao, A. T. Jose, A. Asany, S. M. Khan and M. R. Biscoe, Org. Lett., 2022, 24, 8714 CrossRef CAS PubMed.
- For asymmetric dual Cu/Pd-catalyzed approaches using styrenes, see: (a) K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228 CrossRef CAS PubMed; (b) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760 CrossRef CAS PubMed; (c) S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372 CrossRef CAS PubMed; (d) K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 851 CrossRef CAS PubMed; (e) B. Chen, P. Cao, X. Yin, Y. Liao, L. Jiang, J. Ye, M. Wang and J. Liao, ACS Catal., 2017, 7, 2425 CrossRef CAS; (f) J. Lee, S. Radomkit, S. Torker, J. del Pozo and A. H. Hoveyda, Nat. Chem., 2018, 10, 99 CrossRef CAS PubMed; (g) A. M. Bergmann, S. K. Dorn, K. B. Smith, K. M. Logan and M. K. Brown, Angew. Chem., Int. Ed., 2019, 58, 1719 CrossRef CAS PubMed; (h) S. Feng, Y. Dong and S. L. Buchwald, Angew. Chem., Int. Ed., 2022, 61, e202206692 CrossRef CAS PubMed.
- For stereospecific Pd-catalyzed Suzuki reaction using enantioenriched benzylic organoboranes, see: (a) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024 CrossRef CAS PubMed; (b) S. C. Matthew, B. W. Glasspoole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828 CrossRef CAS PubMed; (c) B. Roh, A. O. Farah, B. Kim, T. Feoktistova, F. Moeller, K. D. Kim, P. H.-Y. Cheong and H. G. Lee, J. Am. Chem. Soc., 2023, 145, 7075 CrossRef CAS PubMed.
- Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J. S. Yu, ACS Catal., 2020, 10, 955 CrossRef CAS.
- For approaches to tertiary α-keto stereocenters using asymmetric mono-arylation reaction, see: (a) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS PubMed; (b) A. Bigot, A. E. Williamson and M. J. Gaunt, J. Am. Chem. Soc., 2011, 133, 13778 CrossRef CAS PubMed; (c) J. S. Harvey, S. P. Simonovich, C. R. Jamison and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 13782 CrossRef CAS PubMed; (d) Z. Huang, Z. Liu and J. Zhou, J. Am. Chem. Soc., 2011, 133, 15882 CrossRef CAS PubMed; (e) K. Kobayashi, Y. Yamamoto and N. Miyaura, Organometallics, 2011, 30, 6323 CrossRef CAS; (f) Z. Huang, L. H. Lim, Z. Chen, Y. Li, F. Zhou, H. Su and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 4906 CrossRef CAS PubMed; (g) Z. Huang, Z. Chen, L. H. Lim, G. C. P. Quang, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 5807 CrossRef CAS PubMed; (h) J. Yang and J. Zhou, Org. Chem. Front., 2014, 1, 365 RSC.
- For stereoconvergent metal-catalyzed approaches to enolate arylation, see: (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442 CrossRef CAS PubMed; (b) A. H. Cherney and S. E. Reisman, Tetrahedron, 2014, 70, 3259 CrossRef CAS; (c) R. Oost, A. Misale and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 4587 CrossRef CAS PubMed.
- For an umpolung approach using α-keto electrophiles in stereoconvergent metal-catalyzed arylation reactions, see: (a) X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS PubMed; (b) P. M. Lundin, J. Esquivas and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 154 CrossRef CAS PubMed; (c) S. Lou and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 1264 CrossRef CAS PubMed; (d) P. M. Lundin and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11027 CrossRef CAS PubMed; (e) J. Mao, F. Liu, M. Wang, L. Wu, B. Zheng, S. Liu, J. Zhong, Q. Bian and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 17662 CrossRef CAS PubMed; (f) M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS PubMed; (g) B. Li, T. Li, M. A. Aliyu, Z. H. Li and W. Tang, Angew. Chem., Int. Ed., 2019, 58, 11355 CrossRef CAS PubMed; (h) T. Iwamoto, C. Okuzono, L. Adak, M. Jin and M. Nakamura, Chem. Commun., 2019, 55, 1128 RSC.
- J. L. G. Ruano, J. Aleman and A. Padwa, Org. Lett., 2004, 6, 1757 CrossRef CAS PubMed.
- T. Jia, P. Cao, D. Wang, Y. Lou and J. Liao, Chem.–Eur. J., 2015, 21, 4918 CrossRef CAS PubMed.
- (a) R. R. Singidi and T. V. RajanBabu, Org. Lett., 2010, 12, 2622 CrossRef CAS PubMed; (b) Y. Takemoto, H. Yoshida and K. Takaki, Chem.–Eur. J., 2012, 18, 14841 CrossRef CAS PubMed; (c) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2015, 51, 6291 Search PubMed; (d) K. Suzuki, N. Sugihara, Y. Nishimoto and M. Tasuda, Angew. Chem., Int. Ed., 2022, 61, e202201883 CrossRef CAS PubMed.
- A boron-selective cross-coupling reaction of the SnBu3 analogue of 8 has been demonstrated by Liao. See ref. 21.
- (a) H. Wang, C. Jing, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 16859 CrossRef CAS PubMed; (b) Z. Wang, N. Wierich, J. Zhang, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2023, 145, 8770 CrossRef CAS PubMed; (c) C. D. Roy and H. C. Brown, J. Organomet. Chem., 2007, 692, 784 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2290981. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04519f |
| This journal is © The Royal Society of Chemistry 2023 |