
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvoking C2+ production from electrochemical CO2 reduction by the steric confinement effect of ordered porous Cu2O†
Longlong
Fan
a,
Qinghong
Geng
a,
Lian
Ma
a,
Chengming
Wang
b,
Jun-Xuan
Li
a,
Wei
Zhu
a,
Ruiwen
Shao
c,
Wei
Li
 d,
Xiao
Feng
a,
Yusuke
Yamauchi
ef,
Cuiling
Li
*ab and
Lei
Jiang
bgh
d,
Xiao
Feng
a,
Yusuke
Yamauchi
ef,
Cuiling
Li
*ab and
Lei
Jiang
bgh
aKey Laboratory of Cluster Science, Ministry of Education, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: licuiling@mail.ipc.ac.cn
bCAS Key Laboratory of Bio-Inspired Materials and Interface Science, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cBeijing Advanced Innovation Center for Intelligent Robots and Systems and Institute of Engineering Medicine, Beijing Institute of Technology, Beijing 100081, China
dDepartment of Chemistry, Laboratory of Advanced Materials, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, iChEM and State Key Laboratory of Molecular Engineering of Polymers, Fudan University, Shanghai 200433, China
eSchool of Chemical Engineering and Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane 4072, Australia
fJST-ERATO Yamauchi Materials Space-Tectonics Project and Department of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8603, Japan
gKey Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry, Beihang University, Beijing 100191, China
hSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 101407, China
First published on 11th November 2023
Abstract
Selective conversion of carbon dioxide (CO2) to multi-carbon products (CO2-to-C2+) at high current densities is in essential demand for the practical application of the resultant valuable products, yet it remains challenging to conduct due to the lack of efficient electrocatalysts. Herein, three-dimensional ordered porous cuprous oxide cuboctahedra (3DOP Cu2O–CO) were designed and synthesized by a molecular fence-assisted hard templating approach. Capitalizing on the merits of interconnected and uniformly distributed pore channels, 3DOP Cu2O–CO exhibited outstanding electrochemical CO2-to-C2+ conversion, achieving faradaic efficiency and partial current density for C2+ products of up to 81.7% and −0.89 A cm−2, respectively, with an optimal formation rate of 2.92 mmol h−1 cm−2 under an applied current density of −1.2 A cm−2. In situ spectroscopy and simulation results demonstrated that the ordered pores of 3DOP Cu2O–CO can effectively confine and accumulate sufficient *CO adsorption during electrochemical CO2 reduction, which facilitates efficient dimerization for the formation of C2+ products. Furthermore, the 3DOP structure induces a higher local pH value, which not only enhances the C–C coupling reaction, but also suppresses competing H2 evolution.
Introduction
The electrochemical conversion of carbon dioxide (CO2) to value-added multi-carbon (C2+) products with higher energy density and market value provides a promising avenue to mitigate or solve the energy and environmental crisis that currently exists due to the excessive emission of CO2 wastes.1–3 However, the one-carbon (C1) nature and chemical stability of CO2 molecules impose a great obstacle for the imperative carbon–carbon (C–C) bond-coupling reaction, which limits the efficiency of CO2-to-C2+ conversion.4–7 It has been demonstrated that adequate coverage of adsorbed CO intermediates (*CO) on catalyst surfaces is essential for dimerization and protonation to form C2+ products.4,8–10 Thus far, catalyst design strategies that can promote *CO coverage and/or suppress CO escape are promising for enabling deep CO2 reduction to valuable C2+ products with high selectivity and efficiency.Among all the strategies, catalysts with concavities have demonstrated extraordinary confinement of reaction intermediates.4,11,12 For instance, Cu2O cavities enabled a C2+ faradaic efficiency (FE) of 75.2 ± 2.7% by spatial confinement for carbon intermediates,4 and a maximum C2+ FE of 77.0 ± 0.3% was realized by optimizing the confinement effect of Cu2O hollow multishell structures.11 Unfortunately, the confinement effect reported in these studies is insufficient for high C2+ selectivity to occur at ampere-level current densities, thus retarding their practical applications. Additionally, understanding of the structure–performance relationship is lacking, which hinders the production of elaborate designs for electrocatalysts with greater efficiency. To address these issues, ordered porous Cu2O has been proposed to advance the production and selectivity of C2+ products by imparting a well-organized spatial confinement effect with its unique electrocatalytic CO2 performance.
Three-dimensional ordered porous (3DOP) materials, characterized by their well-defined pore arrangements, tunable pore sizes, and smooth mass transfer of the 3D interconnected pore channels, have demonstrated remarkable catalytic performance.13–19 Employing hard templates, including mesoporous materials (e.g., mesoporous silica and carbon)13,19–22 and colloidal crystals (e.g., polystyrene and silica spheres),20,23–29 has been the most extensively explored method for fabricating 3DOP materials. Despite significant efforts in this area, it remains a substantial challenge to achieve precise control over the nanoarchitectures within the templates to create 3DOP materials, especially for those beyond the well-studied silica-, carbon- and metal–organic framework (MOF)-based materials.30–33
The main challenge for the nanospace-confined synthesis of 3DOP metal oxides lies in how to introduce metal precursors into the confined spaces and suppress the migration of metal species out of the confined spaces during the reaction processes. The notorious migration of metal species leads to fluctuations in the pore ordering and the overall morphologies, and even results in nonporous products.13,31,34,35 Therefore, irregular porous metal oxides with particle sizes ranging from tens of nanometers to several micrometers have been created by different researchers.13,31 It is crucial to develop a comprehensive understanding and precise control over the growth mechanism of metal oxides inside confined template spaces. By achieving simultaneous regulation of overall morphologies and structural ordering, we can anticipate significant advancements and superior applications of porous metal oxides.
To actualize this concept into practice, we proposed the synthesis of 3DOP Cu2O crystals using a hard-templating strategy. The synthesis of Cu2O is a multistep reaction involving different reagents and, therefore, conferring its confined growth inside hard templates requires several cycles of infiltration reaction procedures.36,37 The processes that fulfill the voids of hard templates with reagents and prevent their leaching during the subsequent wet chemical reaction processes are crucial for preparing ordered porous Cu2O crystals with satisfactory repeatability.
In this work, we developed a molecular fence-assisted hard-templating strategy, in which Cu2+ ions are converted to [Cu(NH3)n]2+ complexes for the synthesis of 3DOP Cu2O crystals (Scheme 1). The loose and less soluble [Cu(NH3)n]2+ (Fig. S1†) allowed reagents to infiltrate into the inner region of the well-patterned polystyrene (PS) monolith by capillary effect. Once the aqueous reagents entered the voids of the PS monoliths, the pre-adsorbed Cu2+ ions were dissolved and reacted to form Cu(OH)2, thereby inhibiting the migration of Cu2+ ions to the outside of the template. Taking advantage of the capsulating effect of the molecular fence and the shaping effect of the assembled PS template, 3DOP Cu2O cuboctahedra (abbreviated as 3DOP Cu2O–CO) were successfully synthesized. The spatial confinement effect of 3DOP Cu2O–CO was clarified in the electrocatalyzing CO2 reduction reaction (CO2RR), with FEs for C2+ products up to 73.4% and 81.7% that were attained due to the 3DOP Cu2O–CO in an H-type reactor and flow cell, respectively. More impressively, 3DOP Cu2O–CO provides a maximum partial current density of C2+ products up to −0.89 A cm−2 at an applied current density of −1.2 A cm−2 in a flow cell.
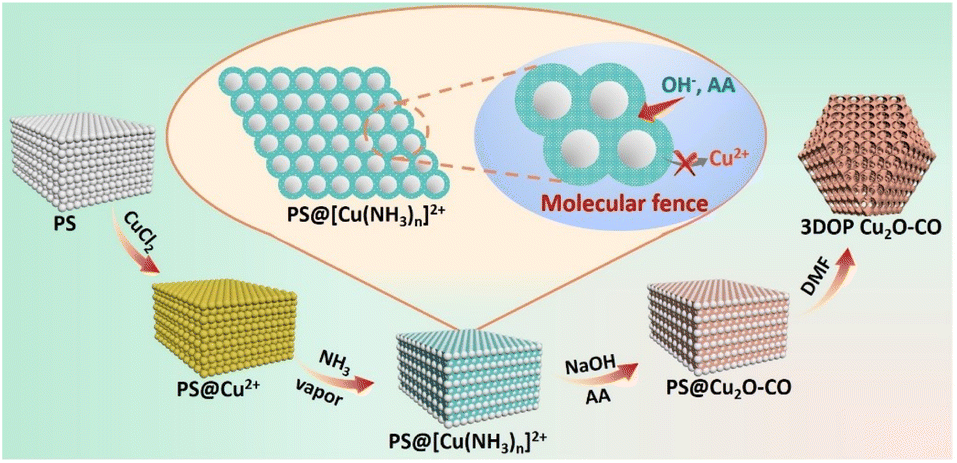 | ||
| Scheme 1 Schematic diagram illustrating the procedure for synthesis of 3DOP Cu2O–CO using a molecular fence-assisted hard templating strategy. | ||
Results and discussion
Synthesis and characterization of 3DOP Cu2O–CO
The molecular fence strategy for synthesizing 3DOP Cu2O–CO is schematically illustrated in Scheme 1. As the essence of this approach, ammonia vapor was utilized to stabilize Cu2+ and prevent its dissolution from the template. In the synthetic procedure, the 3D-ordered PS template pre-adsorbed with Cu2+ (denoted as PS@Cu2+) was treated in an airtight container saturated with ammonia vapor. During this process, the color of the PS cake changed from light yellow to bright cyan (Fig. S2†), indicating that Cu2+ was converted to other forms. The cyan color began to form at the outmost layer and then gradually expanded from the exterior to the interior of the PS@Cu2+ monoliths, depending on the treatment time (Fig. S3†). To reveal the exact change, the products formed at this stage were collected and characterized by powder X-ray diffraction (XRD) and Fourier transform infrared (FTIR) spectrometry.The XRD results revealed that after ammonia treatment, the cyan-colored complex is a mixture of [Cu(NH3)2]2+ and [Cu(NH3)4]2+ complexes (Fig. S4†). For simplicity, we denote the copper–ammonia complex formed after ammonia treatment as [Cu(NH3)n]2+. Distinctly different from the FTIR profile of the CuCl2 precursor, several new peaks appeared in the FTIR spectra for the sample after ammonia vapor treatment. Typically, the sharp maximum peaks at approximately 3350 cm−1 and 3130 cm−1 were ascribed to the N–H stretching vibrations of NH3. Additionally, the antisymmetric bending fundamental mode of NH4+ occurred as an intense absorption at approximately 1406 cm−1, and the NH3 wagging of NH3 in [Cu(NH3)n]2+ appeared at approximately 1254 cm−1 (Fig. S5†).38,39 Based on the above results and the influence of synthetic conditions on the complexation of Cu, the [Cu(NH3)n]2+ compound was formed under exposure to ammonia vapor. In the successive wet-chemical reactions, the preformed [Cu(NH3)n]2+ compound ensured the infiltration of reagents to the PS voids yet inhibited the dissolution of the adsorbed Cu2+ into the solutions (Fig. S6†), thus dictating that the reactions occur in the confined space of the templates.
After completion of the reaction and removal of the PS template by dimethylformamide (DMF), the products obtained under typical conditions were collected. The XRD diffraction patterns were consistent with the standard patterns of Cu2O nanocrystals (JCPDS no. 05-0667) (Fig. S7†), demonstrating the formation of Cu2O in the synthesis. No other diffraction peaks were observed, excluding the formation of undesired impurities. The results showed that the [Cu(NH3)n]2+ compound formed during the ammonia treatment only serves as a unidirectional encapsulating agent without influencing the formation of Cu2O.40
Low-magnification scanning electron microscopy (SEM) images showed that cuboctahedra with uniform and well-patterned pores were obtained (Fig. 1a). Although the crystal size slightly deviated, there are sharp edges on the cuboctahedra, and a dominant edge length of 1.5 μm was confirmed by statistical analysis (Fig. S8a†). As is known, a cuboctahedron possesses eight triangular facets and six square facets. To more closely examine the pore organization inside the polyhedron, individual particles projected in the directions of the [110] and [100] zone axes were selected and characterized by SEM, transition electron microscopy (TEM), and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1e–g and i–k). The corresponding schematic models are also displayed for comparison (Fig. 1d and h).
 | ||
| Fig. 1 Morphological characterization of 3DOP Cu2O–CO. (a and b) Representative low- and high-magnification SEM images, respectively. (c) Atomic resolution aberration-corrected HAADF-STEM image of the concave surface of the 3DOP Cu2O–CO. False color is applied to enhance the contrast. (d–k) Structural characterization of an individual 3DOP Cu2O–CO in the (d–g) [100] and (h–k) [110] zone axes: (d and h) schematic illustration, (e and i) SEM, (f and j) TEM, and (g and k) HAADF-STEM images. (l) HAADF-STEM image and corresponding elemental maps of Cu (yellow), O (red), and their overlap (orange). | ||
All the results indicate that ordered macropores (approximately 215 nm) interconnected by mesopore channels with an average length of 50 nm were evenly arranged inside the Cu2O cuboctahedra (Fig. 1b, d–k, S8b and S9†). Thus, the resultant porous Cu2O cuboctahedra were characterized by the close-packed order of monodisperse PS templates without any decrease in ordering (Fig. S10†). Thus, the aforementioned results indicated that the 3DOP Cu2O cuboctahedra were successfully synthesized. The selected-area electron diffraction (SAED) pattern of one individual 3DOP Cu2O–CO crystal demonstrated its poly-crystalline nature by showing diffraction rings (Fig. S11†). This was consistent with the small grain size (23.6 nm) of 3DOP Cu2O–CO, as calculated by the Scherrer equation based on the most intense (111) diffraction peak (Table S1†).
The surface structure around the nanopores of the 3DOP Cu2O–CO was further investigated using aberration-corrected atomic-resolution HAADF-STEM (Fig. S12†). The lattice fringes with distances of 0.21 nm and 0.25 nm were attributed to the (200) and (111) planes of Cu2O (Fig. S12a†). The magnified HAADF-STEM image illustrated in Fig. 1c shows that the concave surface possesses a large number of atomic defect sites, which usually enable the adsorption of intermediates in electrocatalysis.41,42 The HAADF-STEM and corresponding elemental mapping images (Fig. 1l) show the uniform distribution of Cu and O over the entire particle, and the Cu/O atomic ratio was determined to be 2/1 (Fig. S13†). All these results consistently demonstrate the formation of high-quality ordered porous Cu2O cuboctahedra without impurities.
To the best of our knowledge, this study presents the first synthesis of 3DOP Cu2O cuboctahedra. Elucidating the mechanism behind the simultaneous regulation of porous structure and morphology holds paramount significance in the development of a diverse range of innovative porous materials. Considering the synthetic conditions in the present study, the surfactant (polyvinylpyrrolidone (PVP)) and assembled PS template may be the two factors responsible for morphology control. Therefore, we conducted control experiments to reveal the reasons for the formation of 3DOP Cu2O–CO. Although PVP is effective in regulating the geometry of Cu2O in wet-chemical synthesis,36,37 irregularly shaped products were obtained under the same synthetic procedure to synthesize 3DOP Cu2O–CO using PVP as an additive but without the PS template (Fig. S14†).
When the well-assembled PS@[Cu(NH3)n]2+ monolith was ground to very small pieces for the subsequent synthesis, the ordered porous structure and cuboctahedral morphology remained in the obtained products (Fig. S15†). All of these results indicate that the 3D-ordered assembly of the PS template, and not the PVP surfactant, shaped the morphology of the porous Cu2O. Therefore, an inspection of the PS assembly was carried out by SEM, which revealed that PS spheres assumed a face-centered cubic (fcc) structure to form a closely packed PS template (Fig. S16†). In a basic fcc unit, each PS sphere is surrounded by 12 other spheres (Fig. S17a†) that are located at the vertexes of a cuboctahedron (Fig. S17b†). Consequently, the space around each sphere in the assembled template is divided into 14 parts, with different orientations by the 12 adjacent spheres.34 When Cu2O crystals grow inside the closely packed PS template, the crystals can move in only 14 directions (6 in the [100] direction and 8 in the [111] direction) under the confinement effect imposed by the adjacent 12 spheres. The consecutive growth of Cu2O by the same limitations of each PS sphere template will exclusively result in the formation of cuboctahedral morphology (Fig. 2a).
 | ||
| Fig. 2 Effect of the assembled PS template on crystal morphology. Schematic illustration of the crystal growth manner and the resultant products using (a) a closely packed and (b) a disordered PS template. (c) HAADF-STEM image and corresponding elemental maps of (d) Cu and (e) O of one individual porous Cu2O sphere. | ||
To validate the nanospace-confined growth model, control experiments were carried out by changing the assembly ordering of the PS template. It was found that only the closely packed PS template produced porous cuboctahedra, which were gradually transformed to porous spheres after increasing the chaos of the assembled PS template (Fig. S18†). The HAADF-STEM image consistently revealed that the formed pores were randomly distributed inside the spheres (Fig. 2c–e). Except for the above-mentioned shaping and pore-directing effect of the assembled PS template, fulfilling the template voids by reagents is critical for producing high-quality 3DOP Cu2O–CO because the severe and distinct volume shrinkage occurring during the conversion of copper precursors to Cu2O can easily deform the porous structures and aggravate the irregular morphologies.13,31 In a typical synthesis without ammonia treatment of the PS@Cu2+, the pre-adsorbed Cu precursors will be dissolved and migrate out of the PS template, resulting in irregular products containing porous and nonporous particles (Fig. S19†). Therefore, the success of 3DOP Cu2O–CO production in the present study relies on the shape and templating effect of the 3DOP PS template and the subtle utilization of a molecular fence, which guarantees the fulfillment of Cu2+.
Electrochemical CO2RR performance in an H-type cell
As a proof-of-concept, the confined enhancement of the 3DOP Cu2O–CO for the electrochemical CO2 reduction reaction (CO2RR) was conducted using a three-electrode H-type cell in CO2-saturated 0.1 M KHCO3 electrolyte. For comparison, nonporous Cu2O cuboctahedra (abbreviated as Cu2O–CO) (Fig. S20†) were synthesized and examined under the same conditions. The linear sweep voltammetry (LSV) plots obtained in CO2-saturated electrolyte catalyzed by the two different catalysts are displayed in Fig. 3a. The 3DOP Cu2O–CO exhibited a higher current response over the entire potential range, indicating that CO2RR is kinetically favored on the 3DOP Cu2O–CO in comparison to the nonporous catalyst.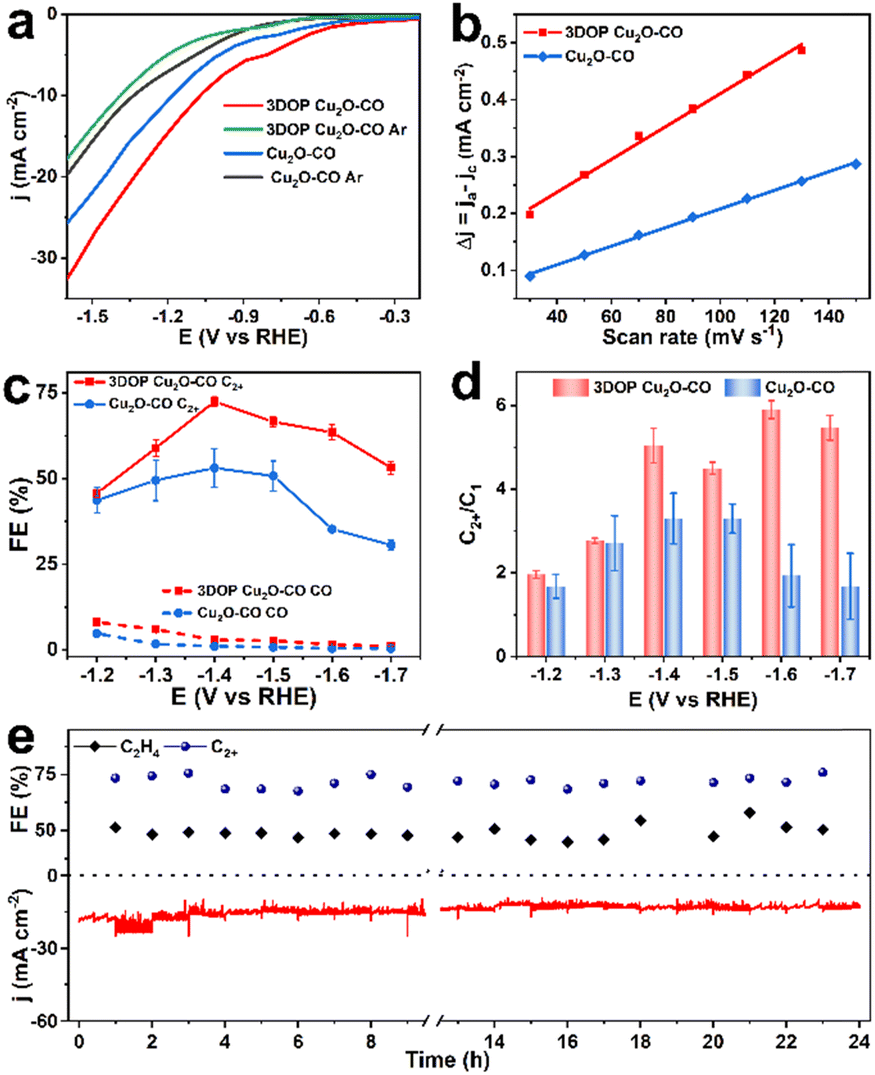 | ||
| Fig. 3 Electrochemical CO2RR performance measured in an H-type cell. (a) Current–voltage curves obtained in 0.1 M KHCO3 electrolyte saturated with CO2 or Ar. (b) Half of the capacitive current density at 0.28–0.4 V vs. RHE as a function of the scan rate for 3DOP Cu2O–CO and Cu2O–CO. (c) C2H4 and C2+ FEs and (d) C2+-to-C1 product selectivity at different applied potentials for different catalysts. (e) Stability test over a span of 24 h for CO2RR catalyzed by 3DOP Cu2O–CO in 0.1 M KHCO3 at −1.4 V vs. RHE. | ||
To investigate the number of active sites and their accessibility, the electrochemically active surface areas (ECSA) of different catalysts were then estimated by the electrochemical double-layer capacitance (Cdl) method after electrode activation treatment. The 3DOP Cu2O–CO produced a larger Cdl value of 2.9 mF cm−2 as compared to that of Cu2O–CO (1.6 mF cm−2) (Fig. 3b and S21†), indicating the high exposure and accessibility of the active sites in 3DOP Cu2O–CO. The sequence of the ECSA value was in agreement with that of the Brunauer–Emmett–Teller (BET) surface area calculated from N2 sorption isotherms (Fig. S22†), in which 3DOP Cu2O–CO possessed a higher surface area of 66.0 m2 g−1 in comparison to Cu2O–CO (5.0 m2 g−1). More importantly, the capacity of 3DOP Cu2O–CO to adsorb CO2 molecules (4.96 m3 g−1) was much larger than that of Cu2O–CO (2.79 m3 g−1), suggesting the promising advantages of using 3DOP Cu2O–CO for CO2 electrolysis (Fig. S23†).
The gaseous and liquid products derived from electrocatalyzing CO2RR at different potentials were quantitatively analyzed by online gas chromatography and 1H nuclear magnetic resonance (1H-NMR) spectroscopy, respectively. Based on the number of carbon atoms in the products, there were three categories of products: C2+ products (C2H4, C2H5OH, CH3COOH, and n-C3H7OH), C1 products (CO, CH4, and HCOOH), and H2 (Fig. S24†). 3DOP Cu2O–CO showed superior selectivity for C2+ products in comparison to that of the nonporous Cu2O–CO sample (Fig. 3c, S24, and Table S2†). 3DOP Cu2O–CO provided a maximum C2+ FE of 73.4% at −1.4 V versus a reversible hydrogen electrode (vs. RHE), exceeding that of Cu2O–CO (56.2%) (Fig. 3c and Table S2†). The 3DOP Cu2O sample promoted the conversion of CO2 to C2+ products other than the C1 products, with relatively high C2+-to-C1 ratios at all the examined potentials, suggesting that the ordered porous structures evoke additional C–C coupling during the CO2RR (Fig. 3d).
Even when compared with the reported results, the 3DOP Cu2O–CO provided top-level FE for the valuable C2+ products (Table S3†). To assess the catalytic stability and selectivity of the catalysts, the electrochemical CO2RR was carried out under steady-state conditions at −1.4 V vs. RHE. It is worth noting that 3DOP Cu2O–CO possessed robust long-term stability in current response and product selectivity for C2H4 and C2+ during a 24 hour CO2RR electrolysis test (Fig. 3e). Structural degradation and composition regression are the two key factors that decrease the catalytic activity in the CO2RR catalyzed by Cu2O.43,44 By imparting porosity to Cu2O crystals to generate interconnected ordered porous structures, superior structural integrity prevented the 3DOP Cu2O–CO from undergoing such degradation (Fig. S25 and S26†). All the above results demonstrate that the as-prepared 3DOP Cu2O–CO promotes the conversion of CO2 into C2+ products with a high catalytic durability.
Mechanism investigation
To explore the origins of the superior C2+ selectivity of 3DOP Cu2O–CO, finite-element method (FEM) simulations were implemented to track the key reaction species of CO2RR in the confined space of 3DOP architectures. In the simulations, CO2 molecules diffused into the nanopores of the 3DOP architecture, where the CO2 molecules were adsorbed onto the pore walls and reduced to C1 intermediates (Fig. 4a and S27†). The local concentration of C1 intermediates will accumulate and then dimerize to C2 species by increasing likelihood of C–C coupling (Fig. 4b). All the results validate the concentration of C1 species by nanopores via the steric confinement effect, and significantly enhanced accumulation was achieved in the internal pores of the 3DOP architecture, indicating the coherent effect of the ordered porous structure in confining C1 intermediates for C–C coupling reactions. However, in the case of nonporous Cu2O–CO, the CO2 molecules can only be adsorbed on the external surfaces, and the formed C1 intermediates are easily released as C1 products (CO, CH4, and HCOOH) (Fig. S28†).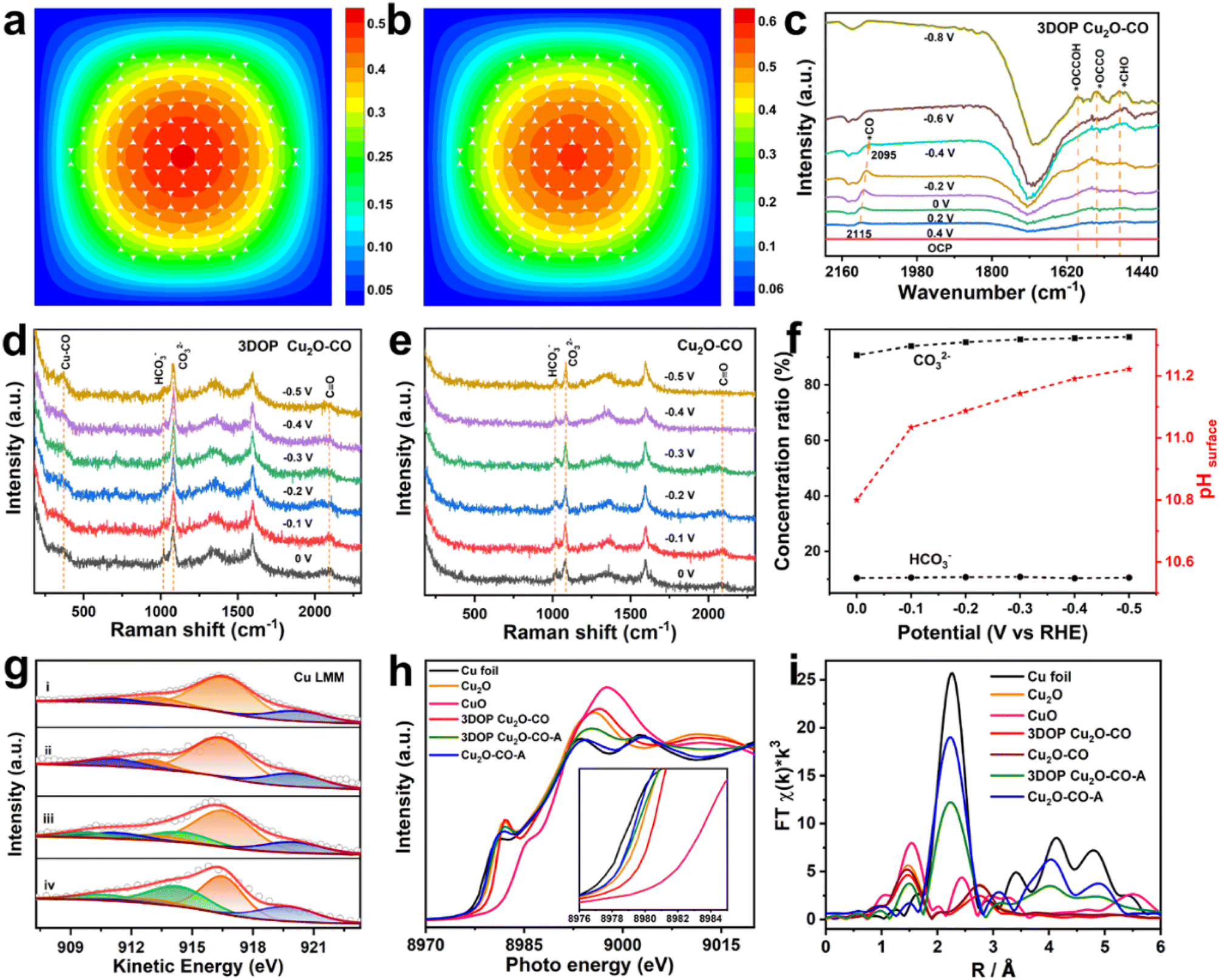 | ||
| Fig. 4 FEM simulations and in situ spectroscopy characterizations. The FEM simulated distributions of (a) C1 and (b) C2 over 3DOP Cu2O–CO. (c) In situ ATR-SEIRAS spectra of 3DOP Cu2O–CO in CO2-saturated 0.1 M KHCO3 aqueous solution. In situ Raman spectra of (d) 3DOP Cu2O–CO and (e) Cu2O–CO in CO2-saturated 0.1 M KHCO3 aqueous solution. (f) Ratios of [HCO3−]/[Ctotal] and [CO32−]/[Ctotal] and corresponding calculated pHsurface. (g) Cu LMM, (h) Cu K-edge XANES and (i) Fourier transform (FT) Cu K-edge EXAFS data for the indicated samples. The compositions of the fitted plots in panel (g) are Cu2O (orange plot), CuO (blue plot and satellite peak), and Cu (green plot). Representative example of catalyst XANES data fitting with a linear combination of reference spectra. 3DOP Cu2O–CO-A and Cu2O–CO-A represent the corresponding samples after electrolyzing CO2 for 5 h. | ||
To provide mechanistic insights into the enhanced C2+ production, we performed in situ attenuated total reflectance-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) under CO2RR conditions to identify the key intermediates and their evolution as the applied potentials were varied. As displayed in the ATR-SEIRAS spectra of 3DOP Cu2O–CO, the vibrational band centered at approximately 2100 cm−1, which corresponds to the atop configuration of *CO adsorption, appeared at 0.2 V vs. RHE and gradually increased to the maximum at −0.2 V vs. RHE (Fig. 4c). As the applied potential bias increased further, the peaks assigned to *CO adsorption disappeared, and new peaks ascribed to *CHO (1490 cm−1), *OCCOH (1593 cm−1), and *OCCO (1548 cm−1),45 which are the key reaction intermediates for forming C2 products, appeared in the ATR-SEIRAS spectra (Fig. 4c).
The *CO is widely used to evaluate the *CO coverage on a catalyst,9,46 and, therefore, it can be concluded that the *CO first accumulates on 3DOP Cu2O–CO, and then is consumed by the C–C coupling reaction to form multi-carbon products. As two-factor authentication, the peaks corresponding to the Cu–CO stretching (369 cm−1) and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching (2090 cm−1) of adsorbed *CO were also observed in the in situ Raman spectra for 3DOP Cu2O–CO (Fig. 4d).47 In great contrast to 3DOP Cu2O–CO, no obvious adsorption peaks related to the above-mentioned intermediates were detected for the nonporous Cu2O–CO sample (Fig. 4e and S29†). All the above results supply evidence that the 3DOP nanoarchitectures can entrap CO intermediates to form an adequate amount of *CO ready for C–C coupling, thus evoking the formation of C2+ products.
O stretching (2090 cm−1) of adsorbed *CO were also observed in the in situ Raman spectra for 3DOP Cu2O–CO (Fig. 4d).47 In great contrast to 3DOP Cu2O–CO, no obvious adsorption peaks related to the above-mentioned intermediates were detected for the nonporous Cu2O–CO sample (Fig. 4e and S29†). All the above results supply evidence that the 3DOP nanoarchitectures can entrap CO intermediates to form an adequate amount of *CO ready for C–C coupling, thus evoking the formation of C2+ products.
It was demonstrated that the product selectivity of the electrochemical CO2RR was pH sensitive. A great pH gradient always occurs at the electrode surfaces with large roughness factors,48 and, therefore, monitoring the local pH near the electrode surface is critical to elucidating the correlation between the local pH environment and the catalytic activity and selectivity. In the CO2/bicarbonate system, the in situ Raman measurement, which displays the adsorbed HCO3− (1015 cm−1) and CO32− (1078 cm−1) signals, is an effective method for providing the local pH values.45,49 The local pH value of 3DOP Cu2O–CO increased from 10.8 to 11.2 when the potential bias changed from 0 to −0.5 V vs. RHE, whereas the pH value remained lower than 10.8 for Cu2O–CO (Fig. 4f and Table S4†), suggesting the steric confinement of the OH− of the ordered porous structures. The high local alkalinity triggered by the 3DOP nanoarchitecture not only suppresses the competing reaction of the hydrogen evolution reaction (HER), but also greatly benefits the C–C coupling reaction of *CO in the formation of C2+ products.48
The chemical states of Cu2O during the electrocatalysis of CO2 were investigated using X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS). The deconvolution of the Cu 2p spectra and the related analysis of the component fitting for different samples (Fig. S30 and Table S5†) were carried out according to previous studies.50 The Cu 2p and Auger Cu LMM XPS spectra revealed that the surface compositions of both samples transformed from Cu2O to a Cu2O and Cu mixture, indicating that Cu2O is partially reduced to Cu after electrolyzing CO2 (Fig. 4g, S30, and Table S6†).51 The existence of CuO for all the samples may be associated with the exposure of the samples to air before injection into the vacuum chamber of the XPS apparatus.
The pre-edge features of Cu K-edge X-ray absorption near-edge spectra (XANES) further verified the composition regression of both Cu2O samples during electrolysis of CO2 by showing a less intense profile shifting to a lower energy for the spectrum of a metallic Cu foil (Fig. 4h). The Cu local structure was probed by extended X-ray absorption fine structure spectroscopy (EXAFS), and showed the typical Cu–Cu bond (2.3 Å) for Cu foil, and Cu–Cu (2.8 Å) and Cu–O (1.5 Å) bonds for the Cu2O reference sample (Fig. 4h). Transformation of the Cu2O-related peaks to the Cu–Cu bond in Cu foil was clearly recognized for both samples after electrocatalysis of the CO2RR, and additional Cu–O bonds were retained in the 3DOP Cu2O–CO sample, suggesting that less conversion occurs in 3DOP Cu2O–CO (Fig. 4i).
To quantitatively analyze the coordination number (CN) of the Cu–O and Cu–Cu bonds in each sample, an EXAFS curve-fitting analysis was carried out for the samples before and after electrolysis of CO2. The smaller CN value for the 3DOP Cu2O–CO sample suggests a larger number of unsaturated sites (Fig. S31 and Table S7†), which was in agreement with the abundant atomic defects recognized on the concave surface of 3DOP Cu2O–CO and the grain boundaries in the Cu2O skeletons (Fig. 1c and S12†). Such unsaturated atomic sites have been proven to be favorable for the adsorption of reaction intermediates.8,52 The abundant adsorption of *CO, *OCCOH, and *OCCO intermediates is capable of preventing the reduction of Cu2O during electrocatalysis of the CO2RR and results in a stable catalytic performance with little decay in product selectivity.4,43
Electrochemical CO2RR performance for practical applications
High C2+ selectivity at industrial current density is critical to realize the potential utilization of valuable products, and remains challenging. Targeted to CO2-to-C2+ conversion at ampere-level current density, the electrochemical CO2RR of the 3DOP Cu2O–CO was then performed in a flow cell reactor, which enabled gaseous CO2 to diffuse to the catalyst layer directly from the gas-diffusion layer.53–55 Catalyst ink consisting of 3DOP Cu2O–CO or Cu2O–CO was directly spray-coated on hydrophobic carbon paper to form a gas-diffusion electrode (GDE), which was then constructed as the cathode in the flow cell reactor (Fig. 5a). | ||
| Fig. 5 Electrochemical CO2RR performance measured in a flow cell reactor. (a) Components of the flow cell reactor and the diffusion of 3DOP Cu2O–CO. (b) FEs for C2+ and H2 products over 3DOP Cu2O–CO and Cu2O–CO samples. (c) C2+ partial current densities and (d) C2+ formation rate under different applied current densities over 3DOP Cu2O–CO and Cu2O–CO samples. (e) Comparison of FEs and partial current densities for C2+ products over 3DOP Cu2O–CO with the reported state-of-the-art electrocatalysts. (f) Long-term stability of 3DOP Cu2O–CO at a current density of −0.6 A cm−2. All measurements were performed in a flow cell reactor using 1.0 M KOH as the electrolyte. | ||
The flow cell measurement revealed that 3DOP Cu2O–CO exhibited high C2+ selectivity and largely depressed H2 and CO production under the entire range of applied current densities (Fig. 5b and S32†). At a current density of −1.0 A cm−2, the 3DOP Cu2O–CO sample achieved the highest FE of 81.7% for C2+ products. However, the FE for C2+ sharply decreased from 41.2% to 6.7% over nonporous Cu2O–CO when the applied current density decreased from −0.2 to −1.0 A cm−2 (Fig. 5b). More impressively, the partial current density of C2+ products over 3DOP Cu2O–CO continued to increase, and reached −0.89 A cm−2 at an applied current density of −1.2 A cm−2, outperforming that of Cu2O–CO (no higher than −0.12 A cm−2) (Fig. 5c). Additionally, the C2+ formation rate consistently increased to 2.92 mmol h−1 cm−2 under a current density of −1.2 A cm−2 (Fig. 5d).
A careful comparison of the CO2-to-C2+ electrochemical catalytic performance revealed that 3DOP Cu2O–CO exhibited superiority in the FE and partial current density of C2+ products as compared to the state-of-the-art samples (Fig. 5e and Table S3†). The durability of 3DOP Cu2O–CO in the electrochemical CO2RR was measured at a current density of −0.6 A cm−2. The FE for C2+ products remained higher than 68.5%, with no obvious decay for the recorded potential during a time period of 19 h (Fig. 5f). The 3DOP Cu2O–CO catalyst exhibited high and stable CO2-to-C2+ conversion at ampere-level current density.
Conclusions
Ordered porous Cu2O was designed to selectively convert CO2 to value-added C2+ products by imparting spatial confinement engineering to an effective Cu2O catalyst. A molecular fence-assisted hard templating strategy was used to successfully synthesize 3DOP Cu2O–CO in a confined space for the first time. The resultant 3DOP Cu2O–CO exhibited a three-dimensional interconnected porous structure obtained from the well-patterned templates. The integrity of the 3DOP construction not only prevented structural deformation, but also entrapped additional CO intermediates to form an adequate amount of *CO, which was ready for C–C coupling to form C2+ products under CO2 electrolysis.Therefore, the as-designed 3DOP Cu2O–CO exhibited excellent performance for CO2 electrolysis, with FEs for C2+ up to 73.4% in the H-type reactor, 81.7% in the flow cell reactor, and satisfactory retention of product selectivity. Importantly, 3DOP Cu2O–CO achieved CO2-to-C2+ conversion with a partial current density of −0.89 A cm−2 at a formation rate of 2.92 mmol h−1 cm−2 under an applied current density of −1.2 A cm−2. This work introduces novel insights into the design of highly active and selective electrocatalysts for CO2RR, with great promise for practical applications.
Data availability
All relevant data are presented in the main text and ESI.†Author contributions
L. F. designed and performed the experiments and analyzed the data. Q. G. drew the scheme and assisted with the electrochemical measurements in the flow cell reactor. R. S. performed the TEM characterization. L. M., C. W. and J. L. carried out the product analysis of CO2 electrolysis. W. Z. performed synthetic experiments. W. L. created sliced samples using the FIB technique. X. F. and Y. Y. revised the manuscript. C. L. wrote the manuscript. C. L. and L. J. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Analysis and Testing Center in Beijing Institute of Technology for technical support. This research is supported by the National Natural Science Foundation (21988102) and National Key Research and Development Program of China (No. 2022YFA1503000).Notes and references
- P. Wei, D. Gao, T. Liu, H. Li, J. Sang, C. Wang, R. Cai, G. Wang and X. Bao, Nat. Nanotechnol., 2022, 18, 299 CrossRef.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789 CrossRef CAS.
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400 CrossRef CAS.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Adv. Mater., 2020, 32, e1908398 CrossRef.
- J. D. Yi, R. Xie, Z. L. Xie, G. L. Chai, T. F. Liu, R. P. Chen, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS.
- Y. Lum and J. W. Ager, Nat. Catal., 2018, 2, 86 CrossRef.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606 CrossRef CAS.
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063 CrossRef CAS.
- J. Y. Kim, G. Kim, H. Won, I. Gereige, W. B. Jung and H. T. Jung, Adv. Mater., 2022, 34, e2106028 CrossRef.
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202113498 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946 CrossRef CAS.
- B. T. Yonemoto, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2014, 136, 8895 CrossRef CAS.
- J. Fang, L. Zhang, J. Li, L. Lu, C. Ma, S. Cheng, Z. Li, Q. Xiong and H. You, Nat. Commun., 2018, 9, 521 CrossRef.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834 CrossRef CAS.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688 CrossRef CAS.
- H. Liang, L. Liu, N. Wang, W. Zhang, C.-T. Hung, X. Zhang, Z. Zhang, L. Duan, D. Chao, F. Wang, Y. Xia, W. Li and D. Zhao, Adv. Mater., 2022, 34, 2202873 CrossRef CAS.
- K. Lan, L. Liu, J. Yu, Y. Ma, J.-Y. Zhang, Z. Lv, S. Yin, Q. Wei and D. Zhao, JACS Au, 2023, 3, 1141 CrossRef CAS.
- Y. Ren, Z. Ma, L. Qian, S. Dai, H. He and P. G. Bruce, Catal. Lett., 2009, 131, 146 CrossRef CAS.
- C. Zhu, D. Du, A. Eychmuller and Y. Lin, Chem. Rev., 2015, 115, 8896 CrossRef CAS.
- H. Li, H. Lin, S. Xie, W. Dai, M. Qiao, Y. Lu and H. Li, Chem. Mater., 2008, 20, 3936 CrossRef CAS.
- R. Ryoo, S. H. Joo and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- X. Zheng, J. Han, Y. Fu, Y. Deng, Y. Liu, Y. Yang, T. Wang and L. Zhang, Nano Energy, 2018, 48, 93 CrossRef CAS.
- J. Wang, X. Yang, C. Yang, Y. Dai, S. Chen, X. Sun, C. Huang, Y. Wu, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 40100 CrossRef CAS.
- N. Wu, F. T. Li, W. Zhu, J. Cui, C. A. Tao, C. Lin, P. M. Hannam and G. Li, Angew. Chem., Int. Ed., 2011, 50, 12518 CrossRef.
- H. Hong, J. Liu, H. Huang, C. Atangana Etogo, X. Yang, B. Guan and L. Zhang, J. Am. Chem. Soc., 2019, 141, 14764 CrossRef CAS.
- H. J. Han, A. V. Shneidman, D. Y. Kim, N. J. Nicolas, J. E. S. van der Hoeven, M. Aizenberg and J. Aizenberg, Angew. Chem., Int. Ed., 2022, 61, e202111048 CrossRef.
- E. J. Crossland, N. Noel, V. Sivaram, T. Leijtens, J. A. Alexander-Webber and H. J. Snaith, Nature, 2013, 495, 215 CrossRef CAS.
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque, Y. Li and B. Chen, Science, 2018, 359, 206 CrossRef CAS.
- H. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 14526 CrossRef CAS.
- X. Sun, Y. Shi, P. Zhang, C. Zheng, X. Zheng, F. Zhang, Y. Zhang, N. Guan, D. Zhao and G. D. Stucky, J. Am. Chem. Soc., 2011, 133, 14542 CrossRef CAS.
- S. Lee, M. Choun, Y. Ye, J. Lee, Y. Mun, E. Kang, J. Hwang, Y.-H. Lee, C.-H. Shin, S.-H. Moon, S.-K. Kim, E. Lee and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 9230 CrossRef CAS.
- J. Hwang, C. Jo, K. Hur, J. Lim, S. Kim and J. Lee, J. Am. Chem. Soc., 2014, 136, 16066 CrossRef CAS.
- Q. Li, Z. Dai, J. Wu, W. Liu, T. Di, R. Jiang, X. Zheng, W. Wang, X. Ji, P. Li, Z. Xu, X. Qu, Z. Xu and J. Zhou, Adv. Energy Mater., 2020, 10, 1903750 CrossRef CAS.
- H. Y. Hsueh, H. Y. Chen, Y. C. Huang, Y. C. Ling, S. Gwo and R. M. Ho, Adv. Mater., 2013, 25, 1780 CrossRef CAS.
- D.-F. Zhang, H. Zhang, L. Guo, K. Zheng, X.-D. Han and Z. Zhang, J. Mater. Chem., 2009, 19, 5220 RSC.
- M. J. Siegfried, J. Am. Chem. Soc., 2006, 128, 10356 CrossRef CAS.
- B. Pommier and P. Gélin, Phys. Chem. Chem. Phys., 1999, 1, 1665 RSC.
- T. López, E. Ortiz-Islas, P. Guevara and E. Gómez, NSTI Nanotech, 2012, 3, 230 Search PubMed.
- C. Lu, L. Qi, J. Yang, D. Zhang, N. Wu and J. Ma, J. Phys. Chem. B, 2004, 108, 17825 CrossRef CAS.
- D. Du, Q. Geng, L. Ma, S. Ren, J.-X. Li, W. Dong, Q. Hua, L. Fan, R. Shao, X. Wang, C. Li and Y. Yamauchi, Chem. Sci., 2022, 13, 3819 RSC.
- C. Li, B. Jiang, N. Miyamoto, J. H. Kim, V. Malgras and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 11558 CrossRef CAS.
- J. Kibsgaard, Y. Gorlin, Z. Chen and T. F. Jaramillo, J. Am. Chem. Soc., 2012, 134, 7758 CrossRef CAS.
- T. Moller, F. Scholten, T. N. Thanh, I. Sinev, J. Timoshenko, X. Wang, Z. Jovanov, M. Gliech, B. Roldan Cuenya, A. S. Varela and P. Strasser, Angew. Chem., Int. Ed., 2020, 59, 17974 CrossRef.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936 CrossRef CAS.
- Y. Katayama, F. Nattino, L. Giordano, J. Hwang, R. R. Rao, O. Andreussi, N. Marzari and S.-H. Yang, J. Phys. Chem. C, 2019, 123, 5951 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and R. Cuenya, ACS Catal., 2021, 11, 7694 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610 CrossRef CAS.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101 CrossRef CAS.
- Y. Wu, C. Chen, X. Yan, S. Liu, M. Chu, H. Wu, J. Ma and B. Han, Green Chem., 2020, 22, 6340 RSC.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478 CrossRef CAS.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783 CrossRef CAS.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527 CrossRef CAS.
- D. Gao, P. Wei, H. Li, L. Lin, G. Wang and X. Bao, Acta Phys.-Chim. Sin., 2020, 37, 2009021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04840c |
| This journal is © The Royal Society of Chemistry 2023 |