
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold-catalyzed alkenylation and arylation of phosphorothioates†
Urvashi
,
Sampoorna
Mishra
and
Nitin T.
Patil
 *
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhauri, Bhopal – 462 066, India. E-mail: npatil@iiserb.ac.in
First published on 30th October 2023
Abstract
Reported herein is the ligand-enabled gold-catalyzed alkenylation and arylation of phosphorothioates using alkenyl and aryl iodides. Mechanistic studies revealed a crucial role of the in situ generated Ag–sulfur complex, which undergoes a facile transmetalation with the Au(III) intermediate, thereby leading to the successful realization of the present reaction. Moreover, for the first time, the alkenylation of phosphoroselenoates under gold redox catalysis has been presented.
Phosphorothioates, a class of compounds with a unique chemical structure, have garnered considerable attention because of their remarkable biological activities and wide-ranging applications in pharmaceutical and agrochemical industries.1 For instance, medicines like echothiophate, employed in the treatment of glaucoma,2 and amifostine, used for cancer chemotherapy,3 belong to the family of phosphorothioates. The role of phosphorothioates as potent synthetic intermediates further enhances their significance.4 Especially, S-aryl phosphorothioates are well-known for their anticholinesterase, antiproliferative, and antibacterial properties, along with their established role as pesticides (Scheme 1a).5 Classical methods for synthesizing phosphorothioates are centered around the P–S bond formation through the nucleophilic substitution reactions of (RO)2P(O)X or R-SX.6 However, this approach is associated with several drawbacks viz the use of toxic and moisture sensitive reagents, harsh reaction conditions and low functional group tolerability. As far as transition-metal catalysis is concerned, a new approach which employs copper-catalyzed reductive coupling of aryl sulfonyl chlorides/aryl sulfonyl hydrazides with H-phosphonates was developed (Scheme 1b, Path I).7 Nonetheless, the requirement of toxic reagents and harsh reaction conditions such as high temperature and high catalyst loading limits the applicability of this method. Subsequently, the transition metal-catalyzed cross-dehydrogenative coupling of aryl thiols with H-phosphonates has also been reported for obtaining S-aryl phosphorothioates (Scheme 1b, Path II).8
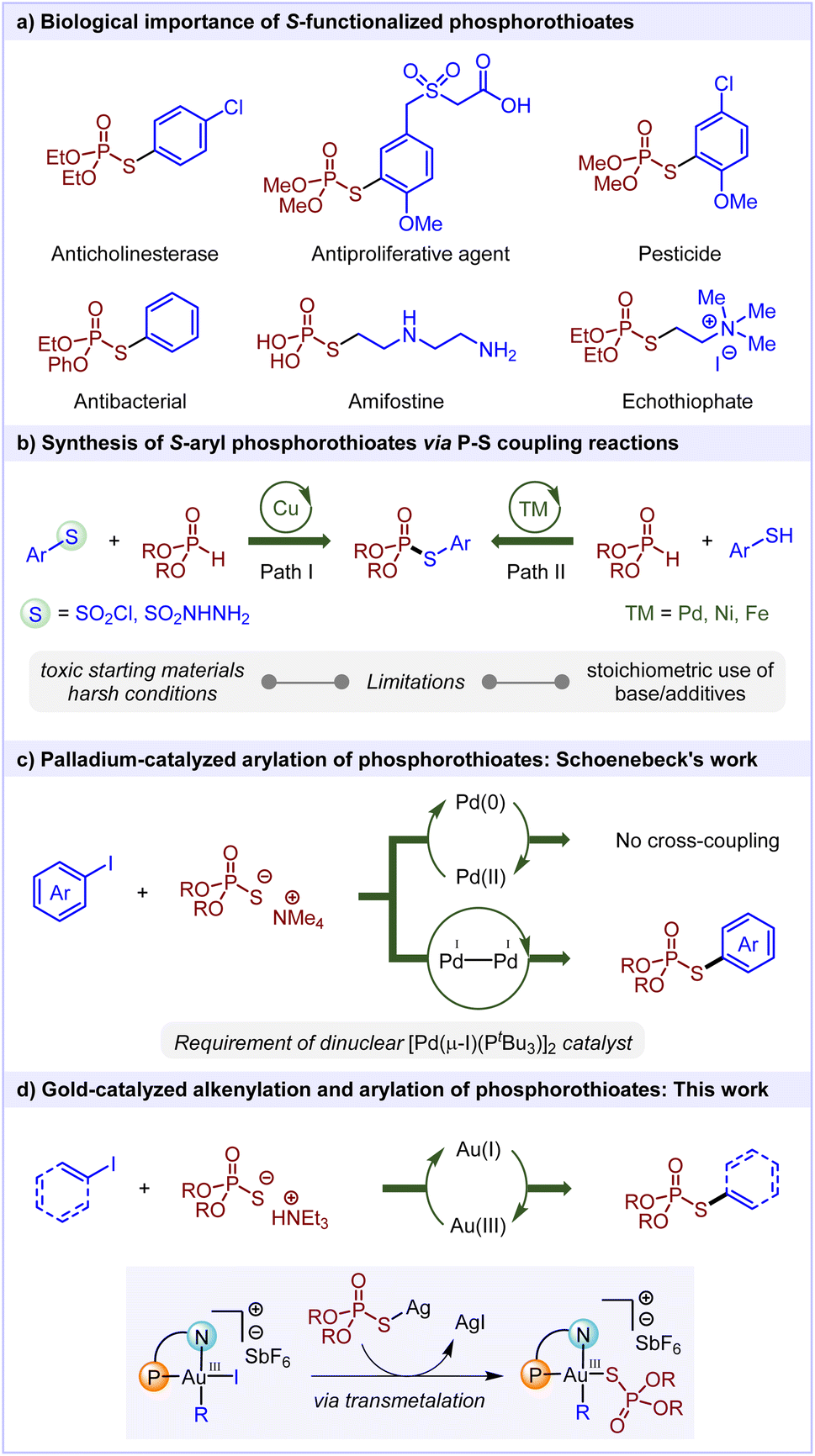 | ||
| Scheme 1 Transition metal-catalyzed synthesis of S-alkenyl/aryl phosphorothioates: known and present work. | ||
One of the most appealing strategies to access S-functionalized phosphorothioates would be transition metal-catalyzed C(sp2)-S cross-coupling reactions. Interestingly, while significant strides have been made in the domain of transition metal-catalyzed C–S cross-coupling reactions,9 the direct cross-coupling between phosphorothioate salts and organohalides has remained underexplored. In 2019, Schoenebeck and co-workers showed that the cross-coupling of phosphorothioates with aryl iodides could not be accomplished under conventional Pd(0)/Pd(II) redox catalysis (Scheme 1c).10 This failure can be attributed to the unfavourable ligand exchange at the Pd(II) center and high barrier for reductive elimination, as supported by computational studies. Rather, the employment of dinuclear Pd(I) catalysis allowed an efficient access to S-aryl phosphorothioates through a distinct mechanistic paradigm. Clearly, there is a need for development of a new transition metal-catalyzed redox paradigm for achieving the C(sp2)-S functionalization of phosphorothioates with organohalides. Notably, there have been no prior reports on alkenylation of phosphorothioates under transition metal catalysis, emphasizing the need for further development in this field.
In recent years, ligand-enabled redox gold catalysis has emerged as a promising technique to achieve various cross-coupling reactions.11 As compared to Pd(0) complexes, the oxidative addition of Au(I) into C(sp2)–I bonds poses a considerable challenge due to the high redox potential of the Au(I)/Au(III) couple.12 Accordingly, the reductive elimination step being the microscopic reverse of oxidative addition is supposed to be facile from the Au(III) intermediate. With this background, we envisioned that utilizing Au(I)/Au(III) redox catalysis could resolve the challenging reductive elimination of C(sp2)-SPO(OR)2, presenting a direct method for obtaining S-alkenyl/aryl phosphorothioates. As far as gold redox catalysis is concerned, a significant hurdle in C–S bond formation arises from the highly thiophilic nature of gold complexes, which may make the Au(I) species catalytically inactive.13 An additional challenge would be the transmetalation or ligand exchange processes at the Au(III) center due to the strong nature of the Au–I bond.14 Banking on the role of silver salts as a halide scavenger in ligand-enabled redox gold catalysis,11 we hypothesized that the presence of silver salts might promote the formation of an Ag–SPO(OR)2 complex (Scheme 1d). The resulting silver complex could undergo transmetalation with the Au(III) intermediate, subsequently triggering reductive elimination to afford S-alkenyl/aryl phosphorothioates. Herein, for the first time, we report the gold-catalyzed cross-coupling reactions of phosphorothioates with alkenyl iodides and aryl iodides to access S-alkenyl and S-aryl phosphorothioates.15
We began our reaction development with the use of (2-iodovinyl)benzene 1a (1.0 equiv.) (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 97:3) and O,O-diphenyl phosphorothioate 2a (1.0 equiv.) in the presence of 1 mol% MeDalPhosAuCl as the catalyst along with AgOTf (1.1 equiv.) as the halide scavenger in DCM at 40 °C for 3 h. Pleasingly, the desired product 3a was obtained in 28% yield (E:Z = 97:3) (Table 1, entry 1). In an effort to enhance the yield of 3a, we turned our attention towards the screening of silver salts. To our delight, the use of AgBF4 and AgSbF6 led to an increase in yield (61% and 66%, respectively) (entry 4–5); meanwhile, other silver salts like AgNTf2 and AgOTs, bearing strongly coordinating counterions, were found to be almost ineffective for this transformation (entry 6–7). This observation suggests that silver salts with non-coordinating counterions are more efficient for this transformation. Subsequently, various solvents like o-DCB, DCE, MeOH, CHCl3 and 1,4-dioxane were screened; amongst them, the use of DCE provided an excellent yield of 98% (entry 9).16 Furthermore, while screening other (P, N)-ligated gold complexes, it was found that MorDalPhosAuCl could also catalyse the reaction giving a 56% yield of the desired product (entry 11).16
:Z = 97:3) and O,O-diphenyl phosphorothioate 2a (1.0 equiv.) in the presence of 1 mol% MeDalPhosAuCl as the catalyst along with AgOTf (1.1 equiv.) as the halide scavenger in DCM at 40 °C for 3 h. Pleasingly, the desired product 3a was obtained in 28% yield (E:Z = 97:3) (Table 1, entry 1). In an effort to enhance the yield of 3a, we turned our attention towards the screening of silver salts. To our delight, the use of AgBF4 and AgSbF6 led to an increase in yield (61% and 66%, respectively) (entry 4–5); meanwhile, other silver salts like AgNTf2 and AgOTs, bearing strongly coordinating counterions, were found to be almost ineffective for this transformation (entry 6–7). This observation suggests that silver salts with non-coordinating counterions are more efficient for this transformation. Subsequently, various solvents like o-DCB, DCE, MeOH, CHCl3 and 1,4-dioxane were screened; amongst them, the use of DCE provided an excellent yield of 98% (entry 9).16 Furthermore, while screening other (P, N)-ligated gold complexes, it was found that MorDalPhosAuCl could also catalyse the reaction giving a 56% yield of the desired product (entry 11).16
|
|
||
|---|---|---|
| Entry | Deviation from “standard conditions” | Yield 3ab (%) |
| a Reaction conditions: 0.10 mmol 1a, 0.10 mmol 2a, 1 mol% MeDalPhosAuCl, 1.1 equiv. AgOTf, DCM (0.1 M), 40 °C, and 3 h. b Isolated yields. c No reaction. | ||
| 1 | None | 28 |
| 2c | Without MeDalPhosAuCl | — |
| 3c | Without AgOTf | — |
| 4 | 1.1 equiv. AgBF4 | 61 |
| 5 | 1.1 equiv. AgSbF6 | 66 |
| 6 | 1.1 equiv. AgNTf2 | <5 |
| 7 | 1.1 equiv. AgOTs | 18 |
| 8 | AgSbF6 and o-DCB | 83 |
| 9 | AgSbF 6 and DCE | 98 |
| 10c | AgSbF6 and MeOH | — |
| 11 | 1 mol% MorDalPhosAuCl, AgSbF6 and DCE | 56 |

With the optimized reaction conditions in hand, we turned our attention to explore the scope of alkenyl iodides 1 by treating them with O,O-diphenyl phosphorothioate 2a (Scheme 2). To our delight, several styrenyl-based alkenyl iodides having electron-donating (–OMe and –Me) and electron-withdrawing substituents (–CO2Me and –CF3) at the para position worked well in the reaction to afford S-alkenyl phosphorothioates (3b–3i) in moderate to good yields (31–89%). Also, meta and ortho substituted styrenyl iodides reacted to deliver the products (3j–3m) in 48–84% yields. Notably, the styrenyl iodides bearing iodo, bromo and chloro substituents react in a chemoselective fashion to deliver the products (3d, 3f, 3g, 3k and 3m) in 48–87% yields. Next, naphthyl-based alkenyl iodides (1n and 1o) and disubstituted styrenyl iodides (1p and 1q) coupled efficiently to give desired products (3n–3q) in 54–93% yields. Also, octenyl-derived alkenyl iodide 1r and ethyl-3-iodoacrylate 1s provided the corresponding products (3r and 3s) with high yields (75% and 91%, respectively). Certainly, the (Z)-isomer of 1s delivers the (Z)-isomer of product 3s selectively. Delightfully, estrone-derived alkenyl iodide 1t also worked well to afford the product 3t in 78% yield. Furthermore, we sought to investigate the scope of phosphorothioates 2. Both O,O-diphenyl phosphorothioate 2a and O,O-dialkyl phosphorothioates (2u–2w) were found to react well with (2-iodovinyl)benzene 1a to afford the desired products (3a, 3u–3w) in moderate to excellent yields (62–98%).
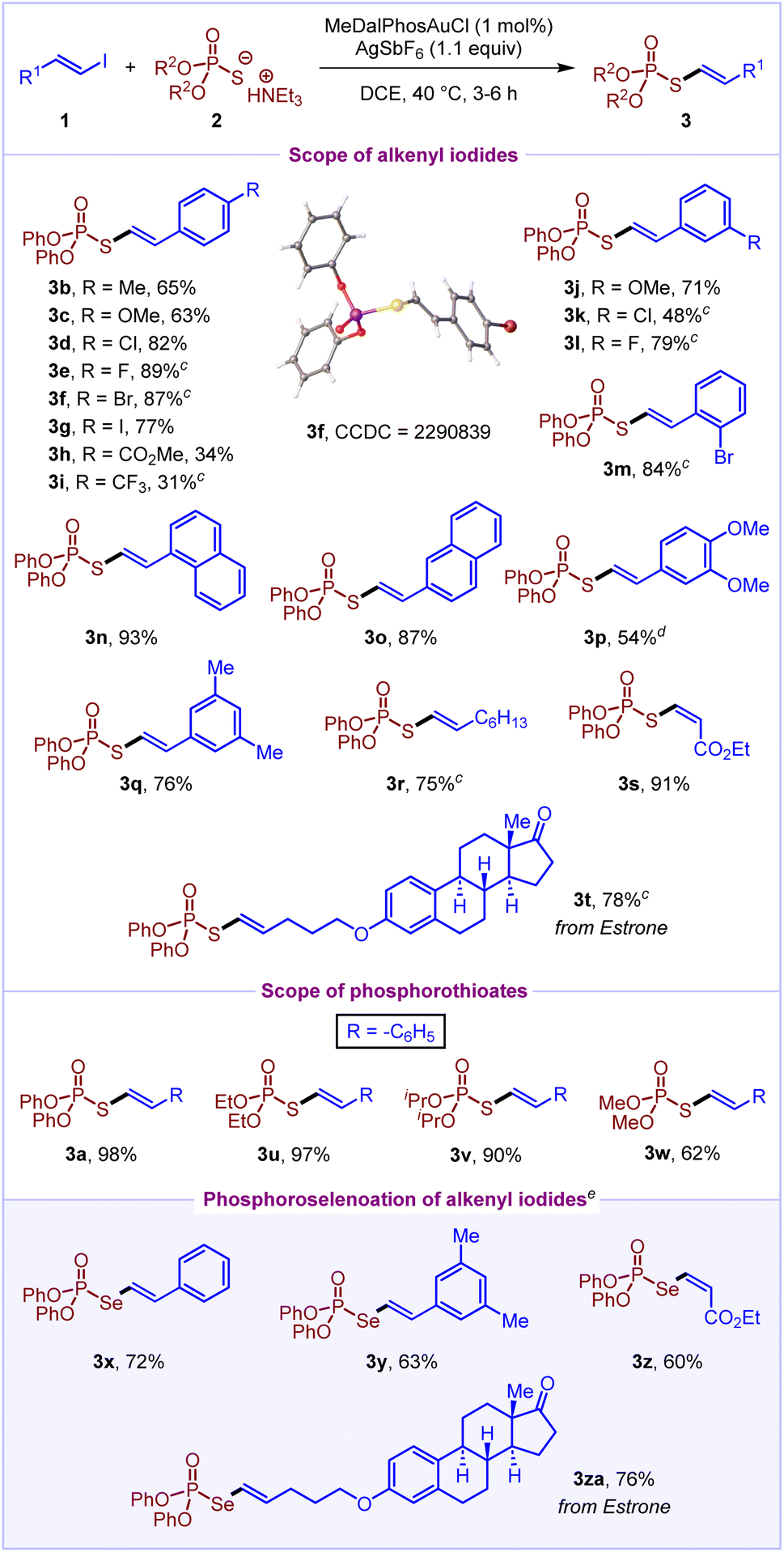 | ||
| Scheme 2 Scope of gold-catalyzed alkenylation of phosphorothioatesa,b. aReaction conditions: 0.20 mmol 1, 0.20 mmol 2, 1 mol% MeDalPhosAuCl, 1.1 equiv. AgSbF6, DCE (0.1 M), 40 °C, and 3–6 h. bIsolated yields. cReaction was performed with 2.5 mol% MeDalPhosAuCl at 70 °C for 2 h. dReaction was performed at 30 °C. eReaction was performed with 10 mol% MeDalPhosAuCl at 80 °C for 12 h. | ||
With the scope of S-alkenylation of phosphorothioates established, we wondered whether the reaction scope could be broadened by using phosphoroselenoates as potential coupling partners. Remarkably, the Se-alkenylation of phosphoroselenoates was achieved, providing an efficient access to Se-alkenyl phosphoroselenoates. Pleasingly, both styrenyl and alkyl derived alkenyl iodides worked well to deliver the products 3x–3za in 60–76% yields. In addition, the reaction of alkenyl iodide 1a with phosphoric acid was performed under the optimised reaction conditions both in the presence and absence of a base; however, it did not result in the formation of the desired C–O cross-coupling product.
In order to establish the generality of this reactivity, we further envisaged the development of arylation of phosphorothioates under Au(I)/Au(III) catalysis. To our delight, aryl iodides were efficiently cross-coupled with phosphorothioates to deliver the desired S-aryl phosphorothioates (Scheme 3). At first, the scope of aryl iodides was investigated by utilising O,O-diphenyl phosphorothioate 2a as the model substrate. Delightfully, various aryl iodides with electron-donating (–OMe, –Me, –NHTs, and –NMeMs) and electron-withdrawing (–CN, –COMe, –CO2Me, –NO2, and –CF3) substituents at the ortho/meta/para positions reacted well to deliver the S-aryl phosphorothioates (5b–5p) in 61–96% yields. Notably, aryl iodides bearing halo groups (–Cl and –Br) and pseudohalides (–OTs) were well tolerated in the reaction to afford the products (5f, 5g and 5n) in 62–85% yields. Disubstituted aryl iodides (4q and 4r), 1-iodonaphthalene 4s and 9H-fluorene based aryl iodide 4u were also well compatible (71–97%) under the optimized reaction conditions. Pleasingly, heteroaromatic scaffolds such as 2-iodothiophene and carbazole based iodoarenes also reacted well to deliver the products 5t and 5v in 68% and 99% yields, respectively. Complex natural product (menthol, dehydroabietylamine, and estrone) derived aryl iodides were successfully cross-coupled to deliver the corresponding products (5w–5y) in moderate to excellent yields (42–91%). Furthermore, the scope of phosphorothioates was evaluated and it was found that O,O-dialkyl phosphorothioates react well to afford products 5z, 5za and 5zb in moderate to good yields. However, axially chiral phosphorothioate 2zc did not react to deliver the desired cross-coupled product, likely due to its incompatibility with the silver salt in the reaction mixture.
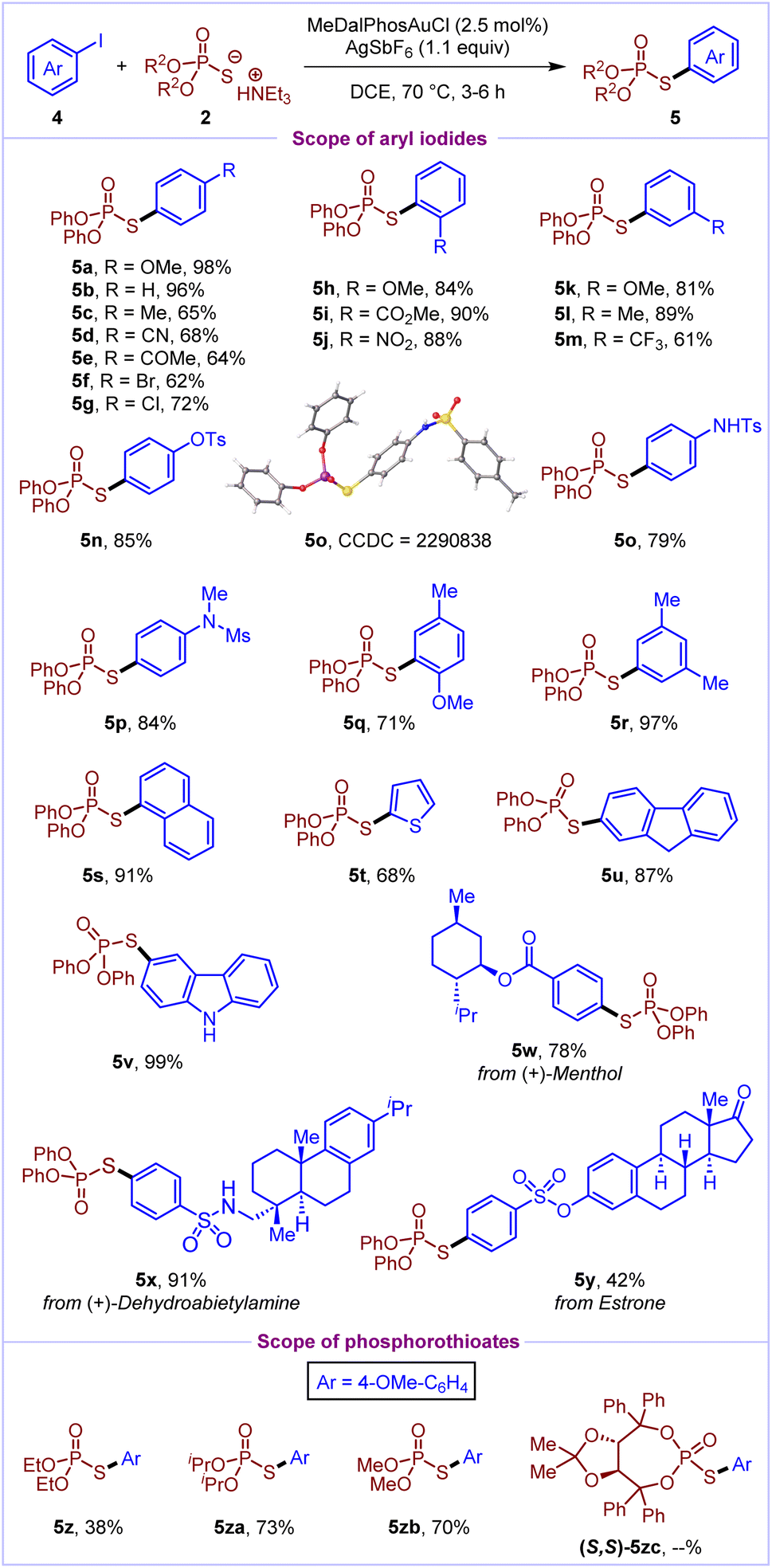 | ||
| Scheme 3 Scope of gold-catalyzed arylation of phosphorothioatesa,b. aReaction conditions: 0.20 mmol 4, 0.20 mmol 2, 2.5 mol% MeDalPhosAuCl, 1.1 equiv. AgSbF6, DCE (0.1 M), 70 °C, and 3–6 h. bIsolated yields. | ||
Since sulfur compounds are well-known to exhibit strong coordination with gold,13 the regeneration of active catalyst is the key for achieving C–S cross-coupling reactivity. In order to gain insights into the reaction mechanism, some control experiments were performed. Initially, to investigate the role of silver salts, AgSPO(OPh)2 complex A was formed by treating AgSbF6 and O,O-diphenyl phosphorothioate 2a in a 1:1 ratio.16 The 31P NMR spectra for this compound show a single peak at 34.5 ppm. Upon addition of 1 equivalent of MeDalPhosAuCl to this mixture, the formation of Au(I)SPO(OPh)2 complex B was observed (peaks at 58.7 ppm and 34.7 ppm), which suggests that the AgSPO(OPh)2 complex A is capable of undergoing ligand exchange with MeDalPhosAuCl. Next, we intended to check the catalytic activity of this Au(I)SPO(OPh)2 complex B. In a stoichiometric reaction of 4-iodoanisole 4a with pre-formed complex B, no oxidative addition of the Au(I) complex with the iodoarene was observed (Scheme 4A). The subsequent addition of 1 equivalent of AgSbF6 led to the generation of the desired product 5a (16.4 ppm). This indicates that silver plays a role in the reactivation of the complex B to form active cationic Au(I) species, necessary for the realization of catalytic reactivity. Furthermore, the formation of Au(III) intermediates D and E during the reaction was confirmed by 31P NMR and mass spectrometric analysis (Scheme 4B).
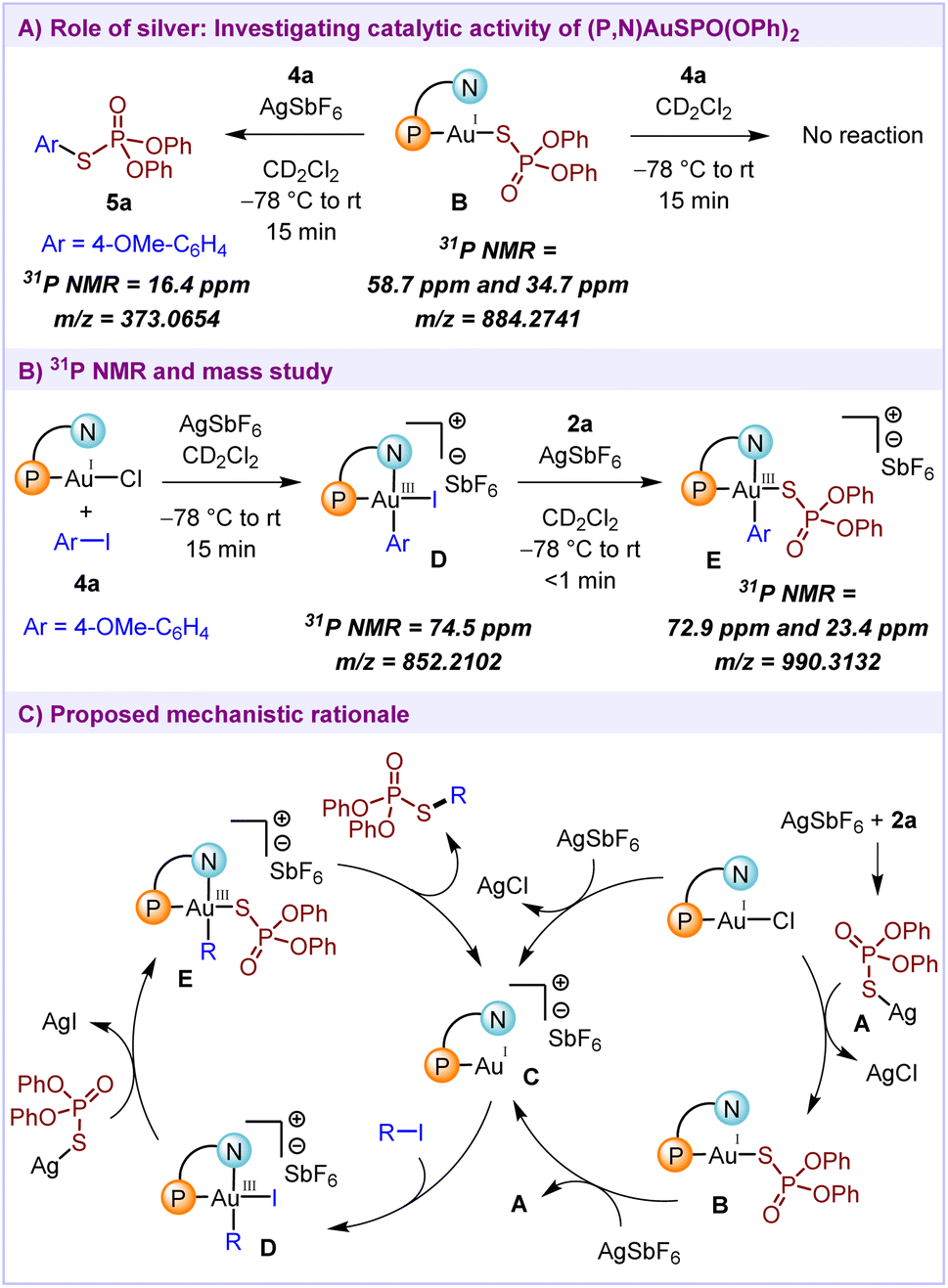 | ||
| Scheme 4 Mechanistic investigations and plausible mechanism. | ||
Based on the control experiments and 31P NMR studies, the mechanistic cycle for the cross-coupling of phosphorothioates with organohalides is proposed (Scheme 4C). Initially, the reaction of MeDalPhosAuCl with AgSbF6 would generate the active gold catalyst C. Alternatively, first AgSPO(OPh)2 complex A could react with MeDalPhosAuCl to generate the Au(I)SPO(OPh)2 complex B which acts as a catalyst resting state. In the presence of a silver salt, this complex B would undergo a ligand exchange to generate the active cationic Au(I) complex C. Next, in the presence of alkenyl/aryl iodides 1/4, the cationic Au(I) species C would undergo oxidative addition to generate the Au(III) intermediate D (74.5 ppm). A subsequent transmetalation between the AgSPO(OPh)2 complex A and Au(III) intermediate D would lead to the generation of Au(III) intermediate E (72.9 ppm and 23.4 ppm, doublet with J = 10 Hz). Further, a facile reductive elimination from intermediate E would afford the desired product 3/5 along with the regeneration of the active catalyst.
Conclusions
In summary, we have developed the first gold-catalyzed alkenylation and arylation of phosphorothioates with organohalides via a C(sp2)–S cross-coupling reaction under Au(I)/Au(III) redox catalysis. This methodology offers direct access to the biologically relevant S-alkenyl and S-aryl phosphorothioates at a catalyst loading as low as 1 mol%. Moreover, the gold-catalyzed alkenylation of phosphoroselenoates has also been accomplished to afford S-alkenyl phosphoroselenoates. Mechanistic studies revealed a crucial role of the in situ generated Ag–sulfur complex, which undergoes a facile transmetalation with the Au(III) intermediate, thereby leading to the successful realization of the desired cross-coupling reactivity. It is anticipated that harnessing the potential of this transmetalation strategy in gold catalysis could allow access to cross-coupling reactions that have remained elusive under conventional redox transition metal catalysis.Data availability
The ESI† includes experimental procedures and characterization data supporting this article.Author contributions
Urvashi and N. T. P. conceptualized the project. Urvashi and S. M. performed and analysed the experiments. Urvashi and N. T. P. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support by SERB, New Delhi (CRG/2022/000195 and SCP/2022/000063) is gratefully acknowledged. We also acknowledge the financial assistance from BRNS (58/14/30/2022-BRNS/37101). Urvashi thanks the Ministry of Education, Government of India for the award of the Prime Minister's Research Fellowship (PMRF).References
- Books and Reviews: (a) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley Interscience, New York, 2000 Search PubMed; (b) P. J. Murphy, Organophosphorus Reagents, Oxford University Press, Oxford, UK, 2004 CrossRef; (c) N. S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257–1269 CrossRef CAS PubMed . Selected Reports:; (d) S. Cogoi, V. Rapozzi, F. Quadrifoglio and L. Xodo, Biochemistry, 2001, 40, 1135–1143 CrossRef CAS PubMed; (e) H. Yan, X. Wang, R. KuoLee and W. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 5631–5634 CrossRef CAS; (f) S.-W. Rhee, R. P. Iyer, J. E. Coughlin, S. Padmanabhan and J. P. Malerich, J. Labelled Compd. Radiopharm., 2012, 55, 197–200 CrossRef CAS; (g) T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J.-B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902–914 CrossRef CAS PubMed; (h) R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278–282 CrossRef CAS.
- W. R. Morton, S. M. Drance and M. Fairclough, Am. J. Ophthalmol., 1969, 68, 1003–1010 CrossRef CAS PubMed.
- (a) D. Antonadou, M. Pepelassi, M. Synodinou, M. Puglisi and N. Throuvalas, Int. J. Radiat. Oncol., Biol., Phys., 2002, 52, 739–747 CrossRef CAS; (b) D. J. Grdina, Y. Kataoka and J. S. Murley, Drug Metab. Drug Interact., 2000, 16, 237–279 CrossRef CAS.
- Selected Reports: (a) M. Fukuoka, S. Shuto, N. Minakawa, Y. Ueno and A. Matsuda, J. Org. Chem., 2000, 65, 5238–5248 CrossRef CAS PubMed; (b) A. M. Lauer, F. Mahmud and J. Wu, J. Am. Chem. Soc., 2011, 133, 9119–9123 CrossRef CAS PubMed; (c) N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245–249 CrossRef CAS; (d) A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138–5141 CrossRef CAS PubMed; (e) F. J. Robertson and J. Wu, J. Am. Chem. Soc., 2012, 134, 2775–2780 CrossRef CAS; (f) Y. Qiu, J. C. Worch, D. N. Chirdon, A. Kaur, A. B. Maurer, S. Amsterdam, C. R. Collins, T. Pintauer, D. Yaron, S. Bernhard and K. J. T. Noonan, Chem. - Eur. J., 2014, 20, 7746–7751 CrossRef CAS.
- (a) L. L. Murdock and T. L. Hopkins, J. Agric. Food Chem., 1968, 16, 954–958 CrossRef CAS; (b) A.-H. Lee and R. L. Metcalf, Pestic. Biochem. Physiol., 1973, 2, 408–417 CrossRef CAS; (c) A.-H. Lee, R. L. Metcalf and G. M. Booth, Ann. Entomol. Soc. Am., 1973, 66, 333–343 CrossRef CAS; (d) H. Kazuo, S. Katsuki, H. Mitsuo, N. Masaru, T. Kenji and Y. Masaaki, Jpn. Tokkyo Koho., JP48018461B 19730606, 1973; (e) S. Tadao, K. Hiroshi, S. Tadashi and T. Akira, Jpn. Kokai Tokkyo Koho., JP49069836A 19740705, 1974; (f) E. P. Reddy, M. V. R. Reddy and S. C. Bell, PCT Int. Appl., WO2005089269A2 20050929, 2005.
- Books: (a) M. Gulea, Progress in the Chemistry of Phosphorothioates, in Advances in Organic Synthesis, ed. A.Rahman, vol. 12, 2018 Search PubMed; (b) Selected Reports: R. G. Harvey, H. I. Jacobson and E. V. Jensen, J. Am. Chem. Soc., 1963, 85, 1623–1626 CrossRef CAS; (c) B. Kaboudin, Tetrahedron Lett., 2002, 43, 8713–8714 CrossRef CAS; (d) Y.-X. Gao, G. Tang, Y. Cao and Y.-F. Zhao, Synthesis, 2009, 1081–1086 Search PubMed; (e) Y.-J. Ouyang, Y.-Y. Li, N.-B. Li and X.-H. Xu, Chin. Chem. Lett., 2013, 24, 1103–1105 CrossRef CAS; (f) Y.-C. Liu and C.-F. Lee, Green Chem., 2014, 16, 357–364 RSC; (g) X. Bi, J. Li, F. Meng, H. Wang and J. Xiao, Tetrahedron, 2016, 72, 706–711 CrossRef CAS; (h) Y. Moon, Y. Moon, H. Choi and S. Hong, Green Chem., 2017, 19, 1005–1013 RSC; (i) S. A. Rather, M. Y. Bhat, F. Hussain and Q. N. Ahmed, J. Org. Chem., 2021, 86, 13644–13663 CrossRef CAS PubMed.
- (a) J. Bai, X. Cui, H. Wang and Y. Wu, Chem. Commun., 2014, 50, 8860–8863 RSC; (b) G. Kumaraswamy and R. Raju, Adv. Synth. Catal., 2014, 356, 2591–2598 CrossRef CAS; (c) X. Zhang, D. Wang, D. An, B. Han, X. Song, L. Li, G. Zhang and L. Wang, J. Org. Chem., 2018, 83, 1532–1537 CrossRef CAS PubMed.
- (a) Y. Zhu, T. Chen, S. Li, S. Shimada and L.-B. Han, J. Am. Chem. Soc., 2016, 138, 5825–5828 CrossRef CAS PubMed; (b) H. Huang, J. Ash and J. Y. Kang, Org. Biomol. Chem., 2018, 16, 4236–4242 RSC; (c) J.-W. Xue, M. Zeng, S. Zhang, Z. Chen and G. Yin, J. Org. Chem., 2019, 84, 4179–4190 CrossRef CAS PubMed.
- Reviews: (a) C.-F. Lee, Y.-C. Liu and S. S. Badsara, Chem.–Asian J., 2014, 9, 706–722 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed.
- X.-Y. Chen, M. Pu, H.-G. Cheng, T. Sperger and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 11395–11399 CrossRef CAS PubMed.
- Reviews: (a) B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707–720 CrossRef CAS PubMed; (b) V. W. Bhoyare, A. G. Tathe, A. Das, C. C. Chintawar and N. T. Patil, Chem. Soc. Rev., 2021, 50, 10422–10450 RSC . Selected Reports:; (c) A. Zeineddine, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef; (d) J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183–7192 RSC; (e) M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101–8105 CrossRef CAS PubMed; (f) J. Rodriguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94–97 RSC; (g) J. Rodriguez, D. Vesseur, A. Tabey, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, ACS Catal., 2022, 12, 993–1003 CrossRef CAS; (h) S. R. Mudshinge, Y. Yang, B. Xu, G. B. Hammond and Z. Lu, Angew. Chem., Int. Ed., 2022, 61, e202115687 CrossRef CAS PubMed; (i) A. G. Tathe and N. T. Patil, Org. Lett., 2022, 24, 4459–4463 CrossRef CAS PubMed; (j) P. Font, H. Valdés, G. Guisado-Barrios and X. Ribas, Chem. Sci., 2022, 13, 9351–9360 RSC; (k) W. Li, Y. Chen, Y. Chen, S. Xia, W. Chang, C. Zhu, K. N. Houk, Y. Liang and J. Xie, J. Am. Chem. Soc., 2023, 145, 14865–14873 CrossRef CAS PubMed; (l) A. Das and N. T. Patil, ACS Catal., 2023, 13, 3847–3853 CrossRef CAS; (m) G. Chen and B. Xu, ACS Catal., 2023, 13, 1823–1829 CrossRef CAS; (n) V. W. Bhoyare, E. D. Sosa Carrizo, C. C. Chintawar, V. Gandon and N. T. Patil, J. Am. Chem. Soc., 2023, 145, 8810–8816 CrossRef CAS PubMed; (o) G. Chen and B. Xu, Org. Lett., 2023, 25, 6334–6339 CrossRef CAS PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- (a) Z. Lu, G. B. Hammond and B. Xu, Acc. Chem. Res., 2019, 52, 1275–1288 CrossRef CAS PubMed; (b) Z. Lu, J. Han, O. E. Okoromoba, N. Shimizu, H. Amii, C. F. Tormena, G. B. Hammond and B. Xu, Org. Lett., 2017, 19, 5848–5851 CrossRef CAS PubMed.
- During the final stage of preparation of this manuscript, similar work on gold-catalysed C(sp2)–S cross-coupling appeared on ChemRxiv from Gagosz and co-workers, Muratov, K.; Zaripov, E.; Berezovski M. V., Gagosz. F. DFT-guided Development of New Hemilabile (P^N) Ligands for gold-(i/iii) Redox Catalysis: Application to the Thiotosylation of Aryl Iodides (DOI: https://doi.org/10.26434/chemrxiv-2023-s9w9p). There are considerable differences between our work and Gagosz’s work. Hence, the C(sp2)–S cross-coupling of organohalides with phosphorothioates to achieve S-alkenylation/arylation of phosphorothioates presented herein is complementary to the work demonstrated by Gagosz and co-workers.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2290838 and 2290839. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04888h |
| This journal is © The Royal Society of Chemistry 2023 |