
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinitrogen cleavage by a dinuclear uranium(III) complex†
Nadir
Jori
 a,
Megan
Keener
a,
Thayalan
Rajeshkumar
b,
Rosario
Scopelliti
c,
Laurent
Maron
*b and
Marinella
Mazzanti
*a
a,
Megan
Keener
a,
Thayalan
Rajeshkumar
b,
Rosario
Scopelliti
c,
Laurent
Maron
*b and
Marinella
Mazzanti
*a
aInsititut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, 31077 Toulouse Cedex 4, France
cX-Ray Diffraction and Surface Analytics Platform, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 6th November 2023
Abstract
Understanding the role of multimetallic cooperativity and of alkali ion-binding in the second coordination sphere is important for the design of complexes that can promote dinitrogen (N2) cleavage and functionalization. Herein, we compare the reaction products and mechanism of N2 reduction of the previously reported K2-bound dinuclear uranium(III) complex, [K2{[UIII(OSi(OtBu)3)3]2(μ-O)}], B, with those of the analogous dinuclear uranium(III) complexes, [K(2.2.2-cryptand)][K{UIII(OSi(OtBu)3)3}2(μ-O)], 1, and [K(2.2.2-cryptand)]2[{UIII(OSi(OtBu)3)3}2(μ-O)], 2, where one or two K+ ions have been removed from the second coordination sphere by addition of 2.2.2-cryptand. In this study, we found that the complete removal of the K+ ions from the inner coordination sphere leads to an enhanced reducing ability, as confirmed by cyclic voltammetry studies, of the resulting complex 2, and yields two new species upon N2 addition, namely the U(III)/U(IV) complex, [K(2.2.2-cryptand)][{UIII(OSi(OtBu)3)3}(μ-O){UIV(OSi(OtBu)3)3}], 3, and the N2 cleavage product, the bis-nitride, terminal-oxo complex, [K(2.2.2-cryptand)]2[{UV(OSi(OtBu)3)3}(μ-N)2{UVI(OSi(OtBu)3)2(κ-O)}], 4. We propose that the formation of these two products involves a tetranuclear uranium–N2 intermediate that can only form in the absence of coordinated alkali ions, resulting in a six-electron transfer and cleavage of N2, demonstrating the possibility of a three-electron transfer from U(III) to N2. These results give an insight into the relationship between alkali ion binding modes, multimetallic cooperativity and reactivity, and demonstrate how these parameters can be tuned to cleave and functionalize N2.
Introduction
Multimetallic cooperativity plays an important role both in the biological and industrial reduction and cleavage of dinitrogen (N2) to ammonia (NH3), but the involved mechanisms remain ambiguous.1,2 Although the binding and reduction of N2 by molecular metal complexes have been intensively studied, in most cases only a few compounds have been reported that can cleave N2 without the assistance of supporting ligands, or the addition of external reducing agents.2–4The first example of N2 cleavage by a metal complex was reported more than 20 years ago when Cummins and coworkers showed that the Mo(III) complex, [MoIII(N(tBu)Ar)3] (Ar = 3,5-C6H3(CH3)2), cleaves N2 to yield a Mo(VI) nitride via an end-on bridging intermediate.5,6 An analogous reactivity was reported recently for a Mo(III) siloxide complex, [MoIII(OSi(OtBu)3)3].7 Remarkably to date, these complexes remain the only examples of N2 cleavage involving mononuclear metal complexes in the absence of reducing agents. A low-valent heteromultimetallic NbIIIx/Nay complex was also reported that can cleave N2, but the inner sphere cation was proposed to be key in the binding and polarization of N2.8,9
Alkali metals are often employed as external reducing agents in combination with metal complexes, and their key role in the binding, activation, and cleavage of N2 has been demonstrated by many recent studies.10–19 In particular, it was proposed that alkali ions are key in promoting the assembly of multimetallic iron complexes that effect the cleavage of N2, but the hypothesis was difficult to verify due to the absence of key intermediates.14 More recently, Holland and coworkers showed that removing the potassium (K+) cation from the K-bridged, dinuclear FeNNFe complex, led to a high degree of activation of the N2 in the resulting mononuclear Fe(N2)2 complex.20
Although the number of studies reporting binding and reduction of N2 by uranium complexes has significantly increased in recent years,3,18,19,21–35 only a handful of examples of N2 cleavage to nitrides by uranium have been identified, in which all required the use of an external reducing agent,18,19,24,33,36 or assistance from a non-innocent ligand,34 although very small amounts of a N2 cleavage side product were identified in one case in the reaction of a putative U(III) with N2.35 We previously reported the reactivity of N2 with a series of dinuclear U(III) oxo-bridged complexes supported by (tris-tert-butoxy)siloxide ligands which contained different alkali metal ions, [M2{[UIII(OSi(OtBu)3)3]2(μ-O)}] (M = K, Rb, Cs) B–D (Scheme 1).19,30 We demonstrated that N2 binding is less favored with larger cations by steric factors, but that in all the N2 complexes, a four-electron reduction of N2 occurs. Full cleavage of N2 could only be effected by further reduction of the bound N24− moiety with an external reducing agent, leading to dinuclear and tetranuclear uranium nitride complexes.18,19
 | ||
| Scheme 1 Previous works on dinitrogen (N2) reduction by multimetallic U(III)–alkali metal ion complexes bearing tris(tert-butoxy)siloxide ligands.19,30 | ||
However, the role of the alkali metal ions in the N2 cleavage by these multimetallic systems remained ambiguous, and in particular, we were interested in understanding if the presence of the alkali metal ions is indeed essential for N2 cleavage, or if U(III) ions can cooperatively cleave N2.
Herein, we demonstrate that removal of the alkali metal ion from the second coordination sphere has an unexpected outcome on the reactivity of the diuranium(III) complexes with N2. Removal of one alkali metal ion forming complex, [K(2.2.2-cryptand)][K{UIII(OSi(OtBu)3)3}2(μ-O)], 1, and further reactivity with N2, resulted in the formation a highly activated (N2)4− species. In contrast, both the reaction of the full alkali metal ion-sequestered complex, [K(2.2.2-cryptand)]2[{UIII(OSi(OtBu)3)3}2(μ-O)], 2, with N2, and removal of the K+ cation from the isolated (N2)4− complex, E, resulted in the immediate cleavage of N2. Overall, this study provides the first example of direct stoichiometric N2 cleavage by an isolated uranium(III) compound without the assistance of the supporting ligand or external alkali metal reducing agents.
Results and discussion
Synthesis of anionic diuranium(III) complexes
To investigate how the presence of K+ cations in the second coordination sphere may affect the reaction of dinuclear U(III) complexes with N2, we first pursued the synthesis of K+ cation-sequestered complexes.The addition of 1 equiv. of 2.2.2-cryptand to a d8-THF solution of B at −80 °C resulted in the consumption of B, with the appearance of a new resonance at −0.27 ppm, and signals corresponding to [K(2.2.2-cryptand)]+, as evidenced by 1H NMR spectroscopy (Fig. S1b†). Single crystals suitable for XRD analysis of [K(2.2.2-cryptand)][K{UIII(OSi(OtBu)3)3}2(μ-O)], complex 1, were obtained from layering a concentrated THF solution with n-hexanes at −40 °C in 72% yield (Scheme 2).
 | ||
| Scheme 2 Sequestration of the K+ cations from complex B. | ||
Similarly, addition of 1 equiv. of 2.2.2-cryptand to 1 in d8-THF at −80 °C, led to the disappearance of 1, with the appearance of a single new resonance at 1.03 ppm and signals corresponding to [K(2.2.2-cryptand)]+, as observed by 1H NMR spectroscopy (Fig. S1c†). Dark red single crystals suitable for X-ray diffraction studies of [K(2.2.2-cryptand)]2[{UIII(OSi(OtBu)3)3}2(μ-O)], complex 2, were isolated by layering a concentrated THF solution with n-hexanes at −40 °C in 76% yield (Scheme 2). It is important to note that the complete removal of the second K+ cation proved more difficult, and the isolation of analytically pure 2 in high yields required an excess (5 equiv.) of 2.2.2-cryptand.
Complexes 1 and 2 are insoluble in toluene and showed low thermal stability in THF solutions at 25 °C, resulting in complete decomposition after 6 hours. Removal of one or two K+ cations in complexes 1 and 2 results in a decreased stability when compared to the previously reported K+ cation-bound complex, B, which is stable in THF at 25 °C for 12 hours.
The solid-state molecular structure of complex 1 (left, Fig. 1) shows an anionic complex with two U(III) ions bridged by an oxo ligand. The inner-sphere K+ cation (2.851(15) Å) is coordinated to the bridging oxo moiety and by four oxygen atoms of three siloxide ligands, at a shorter distance than that reported for B (2.913(4) Å), suggesting a stronger K1–Ooxo interaction.30 The presence of a stronger K–Ooxo bonding is consistent with the fact that an excess of 2.2.2-cryptand is required to fully remove the K+ cations to yield complex 2. In 2, the second K+ cation is coordinated by 2.2.2-cryptand as a [K(2.2.2-cryptand)]+ counterion. The values of the UIII–O–UIII bond distances (U1–O105: 2.123(13) and U2–O105: 2.155(13) Å) in 1 are slightly longer than those found in the UIV–O–UIV complex A (2.085(1), 2.137 (1) Å),30 and compare well with those found in the UIII–O–UIII complexes, B–D (2.100(5)–2.178(3) Å).19,30 The U1–O105–U2 core is slightly bent (172.7(8)°), and is consistent with the previously reported K+ cation-bound UIII–O–UIII complex, B (167.4(2)°).30
 | ||
| Fig. 1 Solid-state molecular structures of the anion of (left) 1 and (right) 2 with 50% probability ellipsoids. Color code: uranium (green), potassium (light blue), oxygen (red), carbon (grey), silicon (orange). Hydrogen atoms and methyl groups on the –OSi(OtBu)3 ligands were omitted for clarity. | ||
The solid-state molecular structure of 2 (right, Fig. 1) shows an anionic complex with two U(III) ions bridged by an oxo ligand, with two outer-sphere [K(2.2.2-cryptand)]+ counterions. The values of the UIII–O–UIII bond distances (U1–O1 and U2–O1: 2.1061(3) Å) are identical, and longer than those found for the UIV–O–UIV complex A (2.085(1), 2.1376(13) Å), but shorter than those observed in 1 (2.123(13) Å, 2.155(13) Å), while comparing well with the previously reported UIII–O–UIII complexes, B–D (2.100(5)–2.178(3) Å). Additionally, the UIII–O–UIII bond angle in 2 is linear (180°; Table 1).
| Complex | U–U | U1–Ooxo | U2–Ooxo | U1–O–U2 | M1–Ooxo | M2–Ooxo |
|---|---|---|---|---|---|---|
| a Values from ref. 19 and 30. | ||||||
| A | 4.2128(9) | 2.0852(13) | 2.1376(13) | 172.19(8) | — | — |
| B | 4.2619(10) | 2.178(3) | 2.120(3) | 167(4) | 2.913(4) | 3.392(4) |
| 1 | 4.2697(12) | 2.123(12) | 2.155(13) | 172.7(8) | 2.851(15) | — |
| 2 | 4.2123(8) | 2.1061(3) | 2.1061(3) | 180 | — | — |
| D | 4.247(1) | 2.137(7) | 2.126(7) | 177.9(4) | 3.336(8) | 3.434(8) |
Electrochemical studies
To determine and compare the reducing ability of complexes, 2 and 1, with those of the previously reported complexes B–D, cyclic voltammetry studies were carried out under argon with [Bu4N][BPh4] (0.1 M in THF) as the supporting electrolyte (Fig. 2).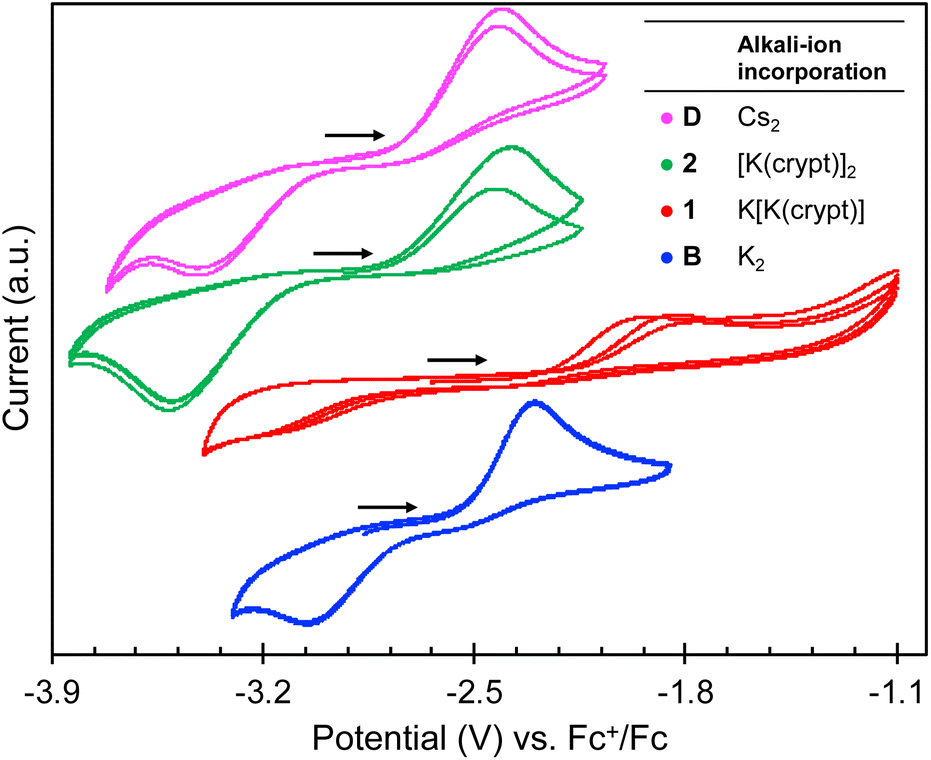 | ||
| Fig. 2 [−3.9 V; −1.1 V] region of cyclic voltammogram of 3.0 mM THF solutions of complexes D (pink),192 (green), 1 (red), and B (blue)19 recorded in 0.1 M [NBu4][BPh4] under Ar (scan rate = 100 mV s−1; referenced against [Fe(C5H5)2]+/0). | ||
As previously observed for complexes B–D (ref. 19) distinctive irreversible oxidation events at Epa = −1.89 V and −2.25 V are observed in the cyclic voltammograms of 1 and 2, respectively, which are assigned to the U(III)/U(IV) couple (Fig. 2). The corresponding reduction events at Epc = −3.23 V and −3.48 V, respectively, are only observed after initial oxidation, in which these redox events can be attributed to the U(IV)/U(III) couple. The reduction potentials for 1 and 2 are more negative compared to the previously reported complex B (Table 2), with Epc values comparable to that observed for D. This suggests that partial and full sequestration of the K+ cations by use of 2.2.2-cryptand results in a higher reducing ability for both 1 and 2, and is comparable to what was previously observed when the K+ cation is replaced by a weaker Lewis acid, such as Cs+.
The higher oxidation potential observed for 1 (Epa = −1.89 V) compared to complexes, 2 (Epa = −2.25 V) and B (Epa = −2.30 V), could be attributed to the stronger K–Ooxo interaction, which renders the removal of the K+ cation from the inner coordination sphere more difficult, and therefore the overall process more irreversible.
Dinitrogen binding and cleavage
Considering the high reducing ability of complexes 1 and 2, we set out to investigate how removal of one or two K+ cations from the inner coordination sphere of the complex could affect the reactivity with N2, when compared to the previously reported B–D complexes.First, exposing a dark red solution of 2 in THF to N2 at −40 °C, resulted in an immediate color change to dark orange. Analysis by 1H NMR spectroscopy of the reaction mixture at −40 °C revealed the consumption of 2, and the formation of a 1H NMR silent species, with the appearance of the signal corresponding to [K(2.2.2-cryptand)][OSi(OtBu)3] (formation of 0.5 equiv. determined by quantitative 1H NMR spectroscopy) (Fig. S11†). Instead, analysis of the same reaction mixture at −80 °C by 1H NMR spectroscopy revealed refined resonances at δ = 35.48, 6.61, and −14.81 ppm (Fig. S12†), corresponding to the U(III)/U(IV) anion, [{UIII(OSi(OtBu)3)3}(μ-O){UIV(OSi(OtBu)3)3}]−. The assignment of the putative, K-sequestered U(III)/U(IV) species, [K(2.2.2-cryptand)][{UIII(OSi(OtBu)3)3}(μ-O){UIV(OSi(OtBu)3)3}], 3, was confirmed by the independent synthesis of the U(III)/U(IV) complex, [K{UIII(OSi(OtBu)3)3}(μ-O){UIV(OSi(OtBu)3)3}], 3-K, and further addition of 2.2.2-cryptand (Scheme S1†). Notably, the 1H NMR spectrum of complex 3-K in the presence of 1 equiv. 2.2.2-cryptand in d8-THF at −80 °C, displayed identical resonances (Fig. S10†) as observed in the reaction mixture obtained after addition of N2 to 2.
From the reaction mixture obtained after exposing 2 to N2, golden crystals suitable for XRD analysis of the bridging bis-nitride, terminal-oxo complex, [K(2.2.2-cryptand)]2[{UV(OSi(OtBu)3)3}(μ-N)2{UVI(OSi(OtBu)3)2(κ-O)}], 4, were isolated from a mixture of n-hexanes and THF (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at −40 °C in 67% yield (yield is provided considering the reaction stoichiometry in Scheme 3). The 1H NMR spectrum of 4 in d8-THF is silent at −40 °C and −80 °C, and the complex is insoluble in non-polar solvents.
:1) at −40 °C in 67% yield (yield is provided considering the reaction stoichiometry in Scheme 3). The 1H NMR spectrum of 4 in d8-THF is silent at −40 °C and −80 °C, and the complex is insoluble in non-polar solvents.
 | ||
| Scheme 3 (a) Dinitrogen cleavage by 2 and upon addition of 2.2.2-cryptand to E. (b) Dinitrogen activation by 1. | ||
X-Band EPR studies of the in situ reaction mixture obtained after addition of N2 to 2 (20 mM frozen THF/Et2O solution), showed two independent sets of signals at 6 K, in which the first is very intense and was fit to an axial set of g-values (g1 = 2.35; g2 = 2.05; g3 = 2.05), whereas the second signal is at higher field (g = 1.13 and g = 1.09), and much less intense (Fig. S49†). The first signal was attributed to the U(III)/U(IV) complex, 3, whereas the second signal could be assigned to the U(V)/U(VI) bis-nitride, terminal-oxo complex, 4. These assignments were confirmed by independent measurement of the EPR spectra for the putative U(III)/U(IV) complex, 3, and of isolated 4, both in frozen THF:Et2O solutions (Fig. S52–S58†) and are consistent with g-values reported for U(III) complexes37,38 and U(V) terminal oxo complexes.39–41
The solid-state molecular structure of 4 (Fig. 3) shows the presence of a dinuclear complex, in which there are two molecules per asymmetric unit, where the two uranium ions are bridged by two nitrides, and are overall bound by five –OSi(OtBu)3 ligands, indicating the loss of one ligand.
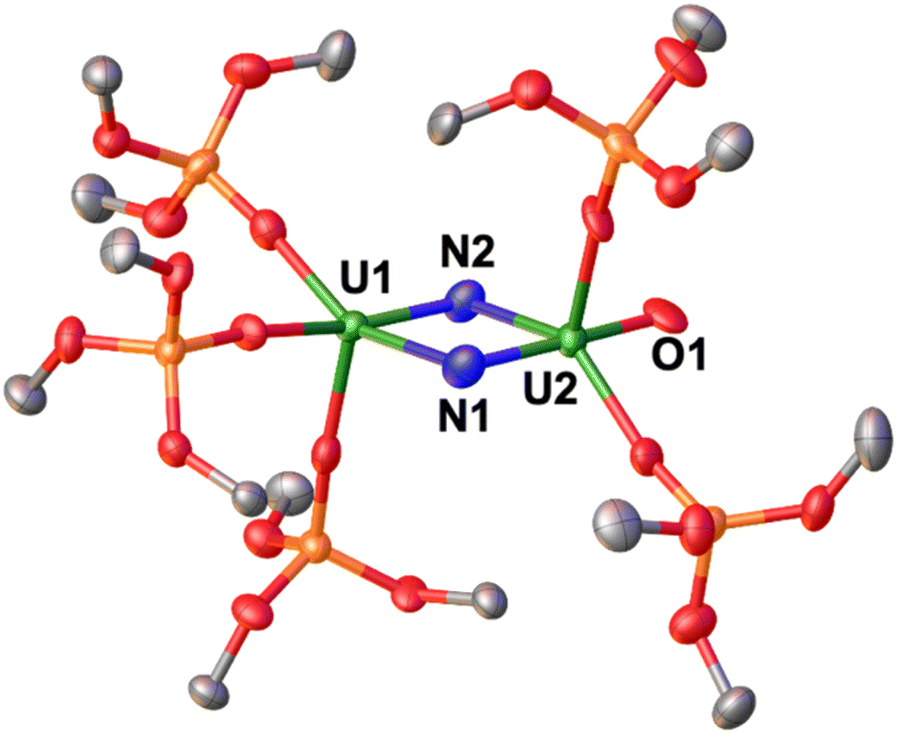 | ||
| Fig. 3 Solid-state molecular structure of the anionic moiety [{UV(OSi(OtBu)3)3}(μ-N)2{UVI(OSi(OtBu)3)2(κ-O)}]2−, 4, (50% probability ellipsoids). Color code: uranium (green), oxygen (red), carbon (grey), nitrogen (blue), and silicon (orange). Hydrogen atoms, solvent molecules and methyl groups the –OSi(OtBu)3 were omitted for clarity. | ||
The overall charge of complex 4 is consistent with the presence of U(V)/U(VI) centers. The two U ions are pentacoordinated and display a distorted square pyramidal geometry, and are bridged by two nitride ligands with a short U⋯U distance of 3.3672(5) Å. The U1 ion is coordinated by three siloxide ligands and the two nitrides, while U2 is coordinated by two siloxide ligands, two nitrides, and a terminal oxo moiety. The U2N2O core is planar, with a N1–N2 separation of 2.543(11) Å, ruling out the presence of a bond between the two nitrogen atoms. The bridging U–N bond distances are asymmetric, featuring a combination of short (U1–N2: 1.950(7) Å, U2–N1: 1.892(8) Å) and elongated (U1–N1: 2.315(8) Å, U2–N2: 2.251(7) Å) bond distances. This is consistent with the presence of U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N multiple bonds and singly bound U–N, respectively, as observed in previously reported U(VI)/U(VI) and U(VI)/U(V) bis-nitride bridged complexes.35,42 The bond valence sum analysis and the computational data (vide infra), suggest that U1 and U2 are formally +5 and +6, respectively; however, a delocalized valence cannot be fully ruled out. Overall, the solid-state molecular structure of 4 displays a unique nitride-substituted analogue of the uranyl(VI) ion, which is only the third example of such OUN motif. The distances observed in the trans oxo-nitrido moiety [OUN] found in 4 (U2–O1: 1.845(6) Å; U2–N1 = 1.892(8) Å) are significantly longer than those found in the trans sodium-capped oxo-nitrido U(VI) complex reported by Hayton and coworkers (U–Ooxo = 1.797(7) Å; U–Nnitride = 1.818(9) Å),43 and are comparable to those found in the analogous cesium-capped [OUN] motif in the previously reported complex, [Cs3{U(OSi(OtBu)3)3}(μ-N)2{U(OSi(OtBu)3)2(κ-O)}][CsOSi(OtBu)3], (U–Ooxo: 1.856(4) Å; U2–N2nitride = 1.85(1) Å).19
N multiple bonds and singly bound U–N, respectively, as observed in previously reported U(VI)/U(VI) and U(VI)/U(V) bis-nitride bridged complexes.35,42 The bond valence sum analysis and the computational data (vide infra), suggest that U1 and U2 are formally +5 and +6, respectively; however, a delocalized valence cannot be fully ruled out. Overall, the solid-state molecular structure of 4 displays a unique nitride-substituted analogue of the uranyl(VI) ion, which is only the third example of such OUN motif. The distances observed in the trans oxo-nitrido moiety [OUN] found in 4 (U2–O1: 1.845(6) Å; U2–N1 = 1.892(8) Å) are significantly longer than those found in the trans sodium-capped oxo-nitrido U(VI) complex reported by Hayton and coworkers (U–Ooxo = 1.797(7) Å; U–Nnitride = 1.818(9) Å),43 and are comparable to those found in the analogous cesium-capped [OUN] motif in the previously reported complex, [Cs3{U(OSi(OtBu)3)3}(μ-N)2{U(OSi(OtBu)3)2(κ-O)}][CsOSi(OtBu)3], (U–Ooxo: 1.856(4) Å; U2–N2nitride = 1.85(1) Å).19
The molecular structure of 4 is similar to that of the bis-nitride, terminal-oxo U(V)/U(V) complex, [Cs3{U(OSi(OtBu)3)3}(μ-N)2{U(OSi(OtBu)3)2(κ-O)}][CsOSi(OtBu)3]. However, it should be noted that in order to promote full cleavage of N2 from D, the addition of 2 equiv. of an alkali metal reducing agent (CsC8) under N2 atmosphere were required, despite the similar redox potentials measured for 2 and D. In contrast, the diuranium(III) complex, 2, is able to effect the direct, stoichiometric cleavage of N2 without further addition of reducing agent or assistance from supporting ligand, which is unprecedented in f elements chemistry. Additionally, it is remarkable that one uranium ion is able to transfer three electrons to N2, yielding a U(VI) ion, as these multielectron transformations still remain rare for uranium.
The formation of the U(VI)/U(V) bridging bis-nitride, terminal-oxo complex, 4, requires the binding and reduction of N2 by two molecules of complex 2, most likely through a proposed tetranuclear intermediate, which is supported by the isolation of 4 and the formation of 3 as observed by 1H NMR spectroscopy (Scheme 3a).
In our previous works, the reaction of the alkali metal-bound diuranium(III) complexes, B–D were shown to form the diuranium(V)–N24−E–G complexes (Scheme 1), in which each had different binding constants; however, no direct N2 cleavage was observed. Considering that the measured reduction potentials for 1 and 2 by cyclic voltammetry studies were greater than for B, but very similar to that of D, the observed differences in reactivity can be interpreted in terms of unfavorable steric interactions for the alkali ion-bound complexes, which most likely prevent the formation of the tetranuclear intermediate. To further verify this hypothesis, we next studied how removal of the K+ cation from complex E could affect the bound hydrazido (N24−) moiety.
The addition of 2 equiv. of 2.2.2-cryptand to complex E in d8-THF at −40 °C, resulted in an immediate color change from dark brown to dark orange. Analysis by 1H NMR spectroscopy indicated the formation of [K(2.2.2-cryptand)][OSi(OtBu)3] and 3 (Fig. S15†), similar to the reaction mixture of 2 and N2. Golden crystals of 4 were isolated in 50% yield (per 1.0 equiv. of E) by leaving a concentrated hexane:toluene (10:1) solution at −40 °C over the course of two days.
Overall, these results suggest that removal of the coordinated K+ cations from complex E, results in an important structural change which promotes further reactivity of the bound N2. The cleavage of N2, and the formation of the putative UIII–O–UIV complex 3, requires two dimers to fully reduce one molecule of N2. We propose that due to steric factors, removal of the K+ cations results in a weaker binding of N2, where an equilibrium between the U(III)/U(III) complex, 2, and the U(V)/U(V)–N24− complex, exists (vide infra), as previously observed for D. However, the removal of the bound alkali metal cations allows two dimeric complexes to interact and form the proposed tetranuclear intermediate, which can then effect the six-electron transfer and subsequent cleavage of one molecule of N2, yielding the U(VI)/U(V) bis-nitride, terminal-oxo complex 4.
We next investigated the reaction of 1 with N2 to assess if the tetranuclear intermediate may be accessible when one potassium is still bound in the second coordination sphere.
Exposing a dark red solution of 1 in THF at −40 °C to N2, resulted in an immediate color change to dark brown-orange. Analysis of the reaction mixture by 1H NMR spectroscopy in d8-THF at −80 °C, showed the complete consumption of 1 and the formation of the putative U(III)/U(IV) complex, 3, suggesting a similar reaction pathway (Fig. S20†). However, the formation of [K(2.2.2-cryptand)][OSi(OtBu)3], which had been observed during N2 cleavage to form complex 4, is not observed in this reaction mixture (Scheme 3a and b).
Indeed, the X-band EPR spectrum of the reaction mixture for 1 and N2 in a THF:Et2O (1:1) frozen glass solution at 6 K shows the presence of the U(III)/U(IV) complex, 3. However, there were no additional signals suggestive of a U(V) species, indicating that 4 is most likely not formed in this reaction (Fig. S59†).
Attempts to isolate the N-containing species from this reaction mixture proved unsuccessful. However, through an alternative route, single crystals of a U(V)/U(IV) hydrazido (N24) complex, [K(2.2.2-cryptand)][K2{UV(OSi(OtBu)3)3}{UIV(OSi(OtBu)3)3}(μ-O)(μ-N2)], 5 (Fig. S39†), could be isolated upon addition of 1 equiv. of 2.2.2-cryptand to E in toluene at −40 °C. It is important to note that complex 5 could only be obtained once, as attempts to isolate analytically pure material were unsuccessful due to the low solubility of the reactants and products in toluene. Isolation of the U(IV)/U(V) N24− complex, 5, provides further support for the proposed tetranuclear intermediate (Scheme 3b).
Overall, the reaction of 1 with N2 also involves four uranium centers; however, N2 cleavage is not observed due the presence of K+ cations in the inner coordination sphere, decreasing the reducing ability of the U ions, preventing the transfer of one additional electron to reduce N2. In contrast, the reaction of complexes B–D with N2 only involves two uranium centers due to steric hindrance.
Reactivity of the N-containing complexes
To further investigate the N-containing species formed upon addition of N2 to complexes 2 and 1, as well as the addition of 2.2.2-cryptand to E, we next pursued their reactivity with acids (H+) and CO.The addition of excess HCl(Et2O) to the residue of the reaction mixture obtained from 2 and N2, yielded NH4Cl in 72% yield (per 1 equiv. of 2). To further determine whether the NH4Cl was unambiguously derived from N2, the reactivity with labelled 15N2 was investigated, leading to the formation of isotopically enriched 15NH4Cl as evidenced by 1H NMR spectroscopy (Fig. S24†). Similarly, addition of excess HCl(Et2O) to the residue of the reaction mixture obtained from E and 2.1 equiv. of 2.2.2-cryptand in THF, also yielded NH4Cl in 74% yield (per 1 equiv. of E). Overall, these results are consistent with the formation of the bis-nitride, terminal-oxo complex, 4, as the primary N-containing species (Scheme 4). Finally, the addition of excess HCl(Et2O) to isolated 4 resulted in the formation of NH4Cl in 92% yield (1.84 equiv., 100% conversion corresponding to 2 equiv. of NH4Cl), consistent with the presence of the two nitride ligands (Scheme 4).
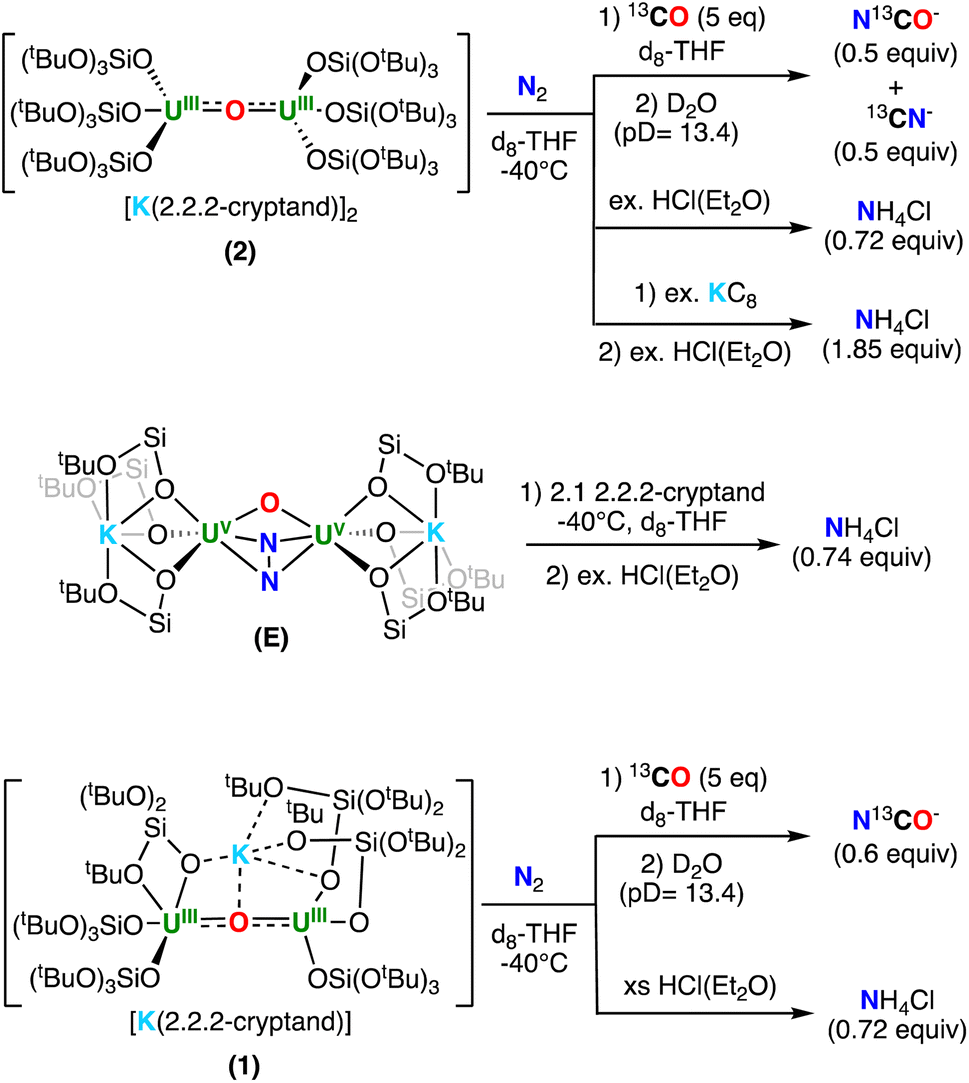 | ||
| Scheme 4 Reactivity of (top) 2, (middle) E and 2.2.2-cryptand, and (bottom) 1, after addition of N2 with H+ and 13CO. | ||
We also investigated the reactivity with 13CO. The addition of 5 equiv. of 13CO to the reaction mixture obtained after addition of N2 to 2, led to an immediate color change from dark orange to brown. The products of the reaction could not be isolated; however, quenching the reaction mixture with basic (pD = 13.4) D2O, revealed the presence of 0.5 equiv. of N13CO− and 0.5 equiv. of 13CN− (per 1 equiv. of 2), as evidenced by quantitative 13C NMR spectroscopy (Fig. S29†). The amount of NCO−/CN− is consistent with the formation of 0.5 equiv. of the bis-nitride, terminal-oxo complex, 4 (top, Scheme 4). The formation of NCO−/CN− has been previously observed in the reactivity of uranium nitride complexes with CO.18,19,42,44–46 In particular, the previously reported dinuclear U(V) bis-nitride complex, [K2{UV(OSi(OtBu)3)3}2(μ-N)2], showed similar reactivity with CO yielding a 1:1 ratio of N13CO−:13CN−.
Alternatively, the addition of 5 equiv. of 13CO to the reaction mixture obtained after addition of N2 to 1, led to a series of color changes over 48 hours. The products of the reaction could not be isolated, but quenching the reaction mixture with basic (pD = 13.4) D2O, revealed the presence of 0.5 equiv. of N13CO− (per 1 equiv. of 1) by quantitative 13C NMR spectroscopy (Fig. S32† and Scheme 4). This reactivity is most consistent with the formation of 0.5 equiv. of the N24− complex, 5, as the formation of NCO− with concomitant release of N2 was previously observed for the reaction of CO and the U(V)/U(V) hydrazido–amide complex, [K2{UV(OSi(OtBu)3)3}2(μ-NH)(μ-N2)].29 Interestingly, addition of excess HCl(Et2O) to the residue obtained from the reaction mixture of 1 with N2 yielded NH4Cl in 74% yield (per 1 equiv. of 1). Notably, the formation of NH4Cl was not previously observed upon addition of HCl to the U(V)/U(V) N24− complexes, E–G,19,30 suggesting that the N24− in the U(IV)/U(V) complex, 5, is more activated.
Computational studies
To gain insight into the N2 reduction pathways promoted by complexes, 1 and 2, DFT calculations (B3PW91) were performed, including solvent and dispersion forces. Formation of the isolated U(V)/U(VI) bis-nitride, terminal-oxo complex, 4, from the reaction of N2 and 2, was shown to be thermodynamically favorable by −34.0 kcal mol−1 (−26.5 kcal mol−1 in Gibbs free energy) (Scheme 5). Unfortunately, the calculations of the proposed tetranuclear intermediate were intractable; however, the calculations demonstrate that complete reduction of N2 may involve two intermediate species, namely a U(IV)/U(IV) N22− (A-N2) and a U(V)/U(V) N24− complex (B-N2), in which both intermediate species involve a step-wise, two-electron reduction to N2.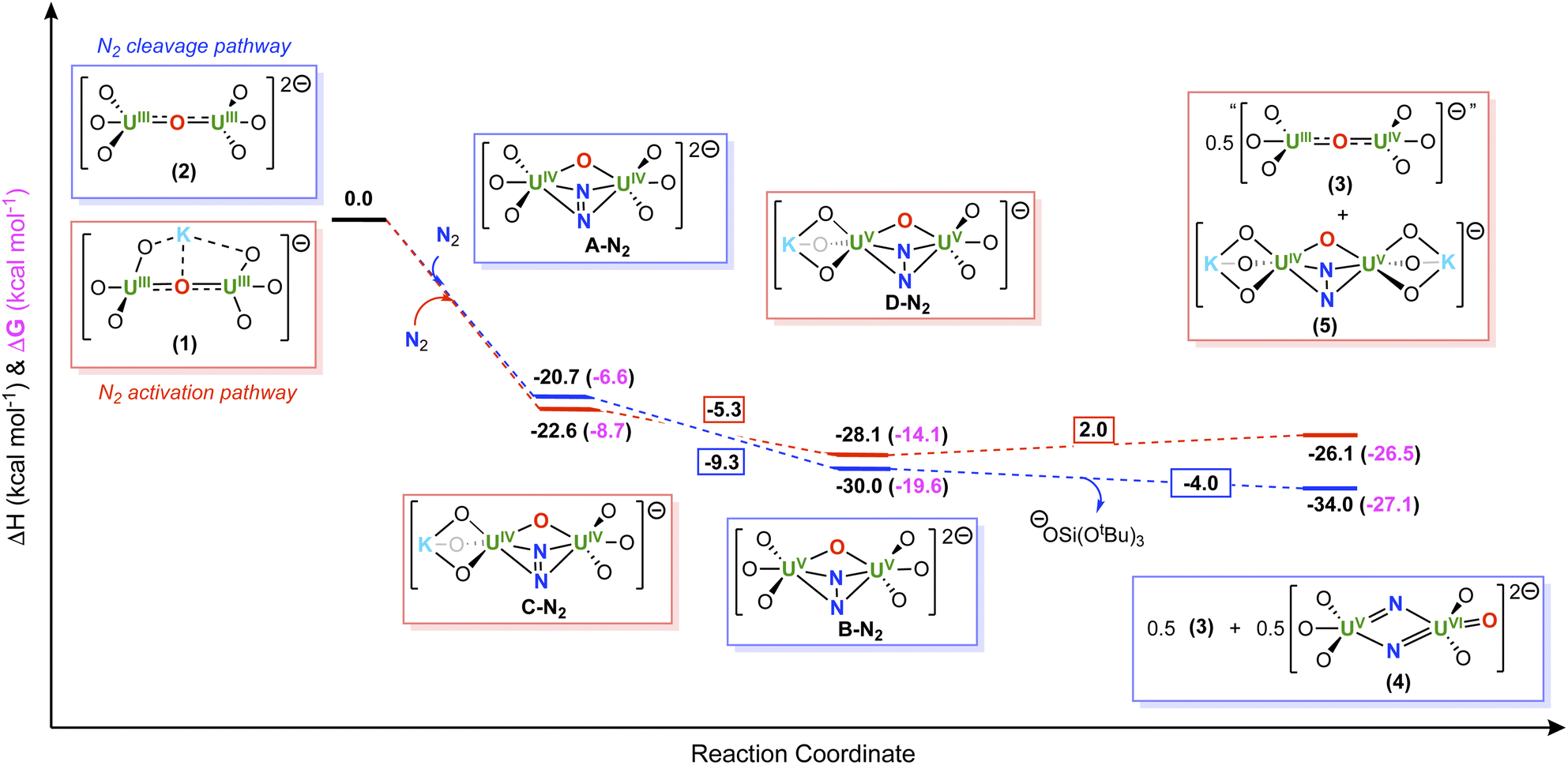 | ||
| Scheme 5 Computed enthalpy and Gibbs free energy (ΔH (black) and ΔG (pink) in kcal mol−1) profiles for the formation of (blue) 4 and (red) 5. | ||
Interestingly, the first two-electron reduction of N2, forming intermediate A-N2, is computed to be exothermic by −20.7 kcal mol−1, (−6.6 kcal mol−1 in Gibbs free energy), as well as the second two-electron reduction to yield the N24− intermediate, B-N2, is exothermic by −9.3 kcal mol−1 (−7.5 kcal mol−1 in Gibbs Free energy). The final N–N bond cleavage step, which involves the proposed tetranuclear uranium intermediate, is also exothermic by −4.0 kcal mol−1 (−12.4 kcal mol−1 in Gibbs free energy).
Next, the oxidation states of the complexes, 2 and 4, as well as the intermediates, A-N2 and B-N2, were verified by computing the different spin states.
For complex 2, the septet (s = 3), quintet (s = 2), and triplet (s = 1) spin states were considered. As expected for a U(III)/U(III) system, the septet (s = 3; six unpaired electrons) was found to be the ground state, with the other spin states higher than 14.7 kcal mol−1 in energy. The unpaired spin localization clearly shows that the unpaired electrons are fully localized at the uranium centers (Fig. 4).
 | ||
| Fig. 4 Unpaired spin density plot for (a) 2 and (b) 4. | ||
The spin states for the N2-bound intermediate species, namely, A-N2 and B-N2, were also computed considering the quintet (s = 2), triplet (s = 1), and singlet (s = 1) ground states. Interestingly, the quintet (s = 2) is 3.1 kcal mol−1 higher in energy than the triplet (s = 1), while the singlet (s = 0) is higher than 44.0 kcal mol−1 in energy. Scrutinizing the unpaired spin density on the U centers for the two lower spin states (quintet and triplet), clearly shows that the quintet (s = 2) corresponds to the U(IV)/U(IV) N22− (A-N2), whereas the triplet (s = 1) corresponds to the U(V)/U(V) N24− (B-N2) intermediate. Interestingly, in the quintet (s = 2) spin-state, the SOMO-2 and the SOMO-3 (SOMO = singly occupied molecular orbital), indicates occupation of two degenerate N–N π* ligand orbitals, suggesting that the N2 has undergone a two-electron reduction to a N22− moiety, yielding formal U(IV)/U(IV) 5f2 centers. Therefore, some unpaired spin density is observed on the N2 moiety with a ferromagnetic coupling with the unpaired spins at the uranium.
For the bis-nitride, terminal-oxo complex, 4, a doublet (s = 1/2) spin state was calculated, which is consistent with a mixed-valent 5f1 U(V)/U(VI) complex. The unpaired spin density is located on only one uranium, suggesting that this is most likely the U(V) 5f1 ion, whereas the second uranium ion does not display any unpaired spin density, suggesting this is a U(VI) 5f0 ion (Fig. 4), which is in line with the observed bond valence sum analysis. Interestingly, the two bridging nitride moieties are negatively charged (−0.8, −0.9), indicating a nucleophilic character.
Similar analysis was performed for the reduction of N2 by complex 1, in which calculations for the proposed tetranuclear intermediate were also intractable. In this system, the two step-wise, two-electron reductions of N2, forming the intermediate, U(IV)/U(IV) N22− (C-N2) and U(V)/U(V) N24− (D-N2) species, were found to be thermodynamically favorable by −22.6 and −5.3 kcal mol−1 (−8.7 and −10.9 kcal mol−1 in Gibbs free energy, respectively) respectively. Whereas, subsequent formation of the bis-nitride, terminal-oxo complex, 4, was endothermically unfavorable by 34.6 kcal mol−1. Therefore, the overall six-electron reduction and cleavage of N2 is endothermic by 6.7 kcal mol−1. In contrast, the formation of the U(IV)/U(V) N24− complex, 5, is slightly endothermic by 2.0 kcal mol−1 (exergonic by 7.5 kcal mol−1) leading to an overall thermodynamically favorable reaction pathway (−26.1 kcal mol−1 in enthalpy, −27.1 kcal mol−1 in Gibbs free energy). Overall, these results are consistent with the experimental findings, suggesting that generation of the U(IV)/U(V) N24− species, 5, is more favorable when partial K+ cation sequestration has occurred, whereas N2 cleavage to the U(V)/U(VI) bis-nitride, terminal-oxo complex, 4, is more favorable when all K+ cations have been removed from the inner coordination sphere.
Conclusions
Herein, we have compared the mechanism and resulting products of N2 reduction by previously reported K2-bound dinuclear uranium(III) complexes with those of the analogous uranium(III) dinuclear complexes, 1 and 2, where one or two K+ ions have been removed from the inner coordination sphere by addition of 2.2.2-cryptand. The complete sequestration of the K+ cations resulted in an enhanced reducing ability of complex 2, leading to the formation of two products upon N2 addition, namely, the U(III)/U(IV) complex, 3, and the U(V)/U(VI) bis-nitride complex, 4. The formation of these two products requires four uranium centers to be involved in N2 cleavage, and is proposed to occur via a tetranuclear N2 intermediate that can only form in the absence of coordinated alkali metal ions, resulting in a six-electron N2 cleavage. Removal of only one K+ cation and subsequent reactivity with N2 led to the formation of the U(III)/U(IV) complex, 3, and to the formation of a U(V)/U(IV) N24− complex, 5, where the N24− is more activated than in the analogous U(V)/U(V) N24−, E–G complexes, as indicated by its high reactivity with electrophiles, such as acid (H+) and CO. Most notably, we had previously shown that N2 evolution occurred upon addition of a strong acid (HCl) to the complexes E and F, whereas in this study, we found HCl addition to complex 5 resulted in NH4Cl formation. Additionally, computational studies indicate that N2 cleavage by 2, with concomitant formation of the bis-nitride complex, 4, is thermodynamically favored. Both the U(V)/U(VI) bis-nitride 4, and the U(V)/U(IV) N24−, 5, complexes react with CO in ambient conditions leading to CN− and NCO− or solely NCO−, respectively. Overall, these results provide an important insight into the relationship between alkali ion-binding modes and the multimetallic cooperativity and reactivity within a unique uranium system that cleaves and functionalizes N2, demonstrating the possibility of a three-electron transfer from U(III) to N2.Data availability
The data that support the findings of this study are openly available in the Zenodo repository at https://doi.org/10.5281/zenodo.10044157.Author contributions
N. J. designed and carried most experiments and analyzed the data; M. K. identified the conditions for the isolation of the key complex 4. M. M. designed and supervised the project; T. R. and L. M. carried out the computational study; R. S. measured and analyzed the X-Ray data; N. J., M. K., and M. M. wrote the manuscript with contributions of all authors, and all authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 212723 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Farzaneh Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses. We thank Dr A. Sienkiewicz for EPR data collections. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.References
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS.
- F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem.–Eur. J., 2021, 27, 3892–3928 CrossRef CAS PubMed.
- D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed.
- S. J. K. Forrest, B. Schluschass, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef CAS PubMed.
- C. E. Laplaza, M. J. A. Johnson, J. C. Peters, A. L. Odom, E. Kim, C. C. Cummins, G. N. George and I. J. Pickering, J. Am. Chem. Soc., 1996, 118, 8623–8638 CrossRef CAS.
- M. Pucino, F. Allouche, C. P. Gordon, M. Worle, V. Mougel and C. Coperet, Chem. Sci., 2019, 10, 6362–6367 RSC.
- A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1998, 120, 437–438 CrossRef CAS.
- A. Caselli, E. Solari, R. Scopelliti, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, J. Am. Chem. Soc., 2000, 122, 3652–3670 CrossRef CAS.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed.
- K. P. Chiang, S. M. Bellows, W. W. Brennessel and P. L. Holland, Chem. Sci., 2014, 5, 267–274 RSC.
- K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807–16816 CrossRef CAS PubMed.
- Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. Garcia-Serres and L. J. Murray, Angew Chem. Int. Ed. Engl., 2015, 54, 1499–1503 CrossRef CAS PubMed.
- S. F. McWilliams and P. L. Holland, Acc. Chem. Res., 2015, 48, 2059–2065 CrossRef CAS PubMed.
- G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- L. R. Doyle, A. J. Wooles, L. C. Jenkins, F. Tuna, E. J. L. McInnes and S. T. Liddle, Angew Chem. Int. Ed. Engl., 2018, 57, 6314–6318 CrossRef CAS PubMed.
- L. R. Doyle, A. J. Wooles and S. T. Liddle, Angew Chem. Int. Ed. Engl., 2019, 58, 6674–6677 CrossRef CAS PubMed.
- N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2021, 143, 11225–11234 CrossRef CAS PubMed.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. Zivkovic, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232–9242 RSC.
- S. F. McWilliams, E. Bill, G. Lukat-Rodgers, K. R. Rodgers, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2018, 140, 8586–8598 CrossRef CAS PubMed.
- A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS.
- P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS.
- G. F. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef PubMed.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew Chem. Int. Ed. Engl., 2002, 41, 3433–3436 CrossRef CAS.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS.
- M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, N. Kaltsoyannis and S. T. Liddle, Nat. Chem., 2019, 11, 806–811 CrossRef CAS PubMed.
- P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS PubMed.
- X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- P. L. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Q. Zhu, Angew Chem. Int. Ed. Engl., 2021, 60, 473–479 CrossRef CAS PubMed.
- M. Keener, F. Fadaei-Tirani, R. Scopelliti, I. Zivkovic and M. Mazzanti, Chem. Sci., 2022, 13, 8025–8035 RSC.
- X. Q. Xin, I. Douair, Y. Zhao, S. O. Wang, L. Maron and C. Q. Zhu, Natl. Sci. Rev., 2023, 10, nwac144 CrossRef PubMed.
- I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC.
- J. Jung, S. T. Loffler, J. Langmann, F. W. Heinemann, E. Bill, G. Bistoni, W. Scherer, M. Atanasov, K. Meyer and F. Neese, J. Am. Chem. Soc., 2020, 142, 1864–1870 CrossRef CAS.
- D. Gourier, D. Caurant, J. C. Berthet, C. Boisson and M. Ephritikhine, Inorg. Chem., 1997, 36, 5931–5936 CrossRef CAS PubMed.
- W. W. Lukens, N. M. Edelstein, N. Magnani, T. W. Hayton, S. Fortier and L. A. Seaman, J. Am. Chem. Soc., 2013, 135, 10742–10754 CrossRef CAS PubMed.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS.
- L. Barluzzi, F. C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew Chem. Int. Ed. Engl., 2014, 53, 10412–10415 CrossRef CAS.
- M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew Chem. Int. Ed. Engl., 2016, 55, 12290–12294 CrossRef CAS PubMed.
- L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, XRD, electrochemistry, EPR, IR, UV/vis/near IR, data, computational details. CCDC 277300–2277304. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc05253b |
| This journal is © The Royal Society of Chemistry 2023 |