DOI:
10.1039/D3SC05305A
(Edge Article)
Chem. Sci., 2023,
14, 13879-13884
Facile synthesis of 1,2-aminoalcohols via α-C–H aminoalkylation of alcohols by photoinduced hydrogen-atom transfer catalysis†
Received
7th October 2023
, Accepted 9th November 2023
First published on 16th November 2023
Abstract
1,2-Aminoalcohols are common motifs found in a wide range of natural products and pharmaceutical compounds. Here we report a photocatalytic method for the direct conversion of readily available aliphatic alcohols into synthetically valuable 1,2-aminoalcohols. A dual catalytic system consisting of an acridinium photoredox catalyst and a cationic hydrogen-atom transfer (HAT) catalyst based on 1,4-diazabicyclo[2.2.2]octane (DABCO) enables an efficient and site-selective HAT from the α-C–H bonds of unprotected primary and secondary alcohols. The subsequent radical addition to a newly designed chiral N-sulfinyl α-iminoester afforded various 1,2-aminoalcohols, including enantiomerically enriched ones, under mild photochemical conditions with high atom and step economy.
1 Introduction
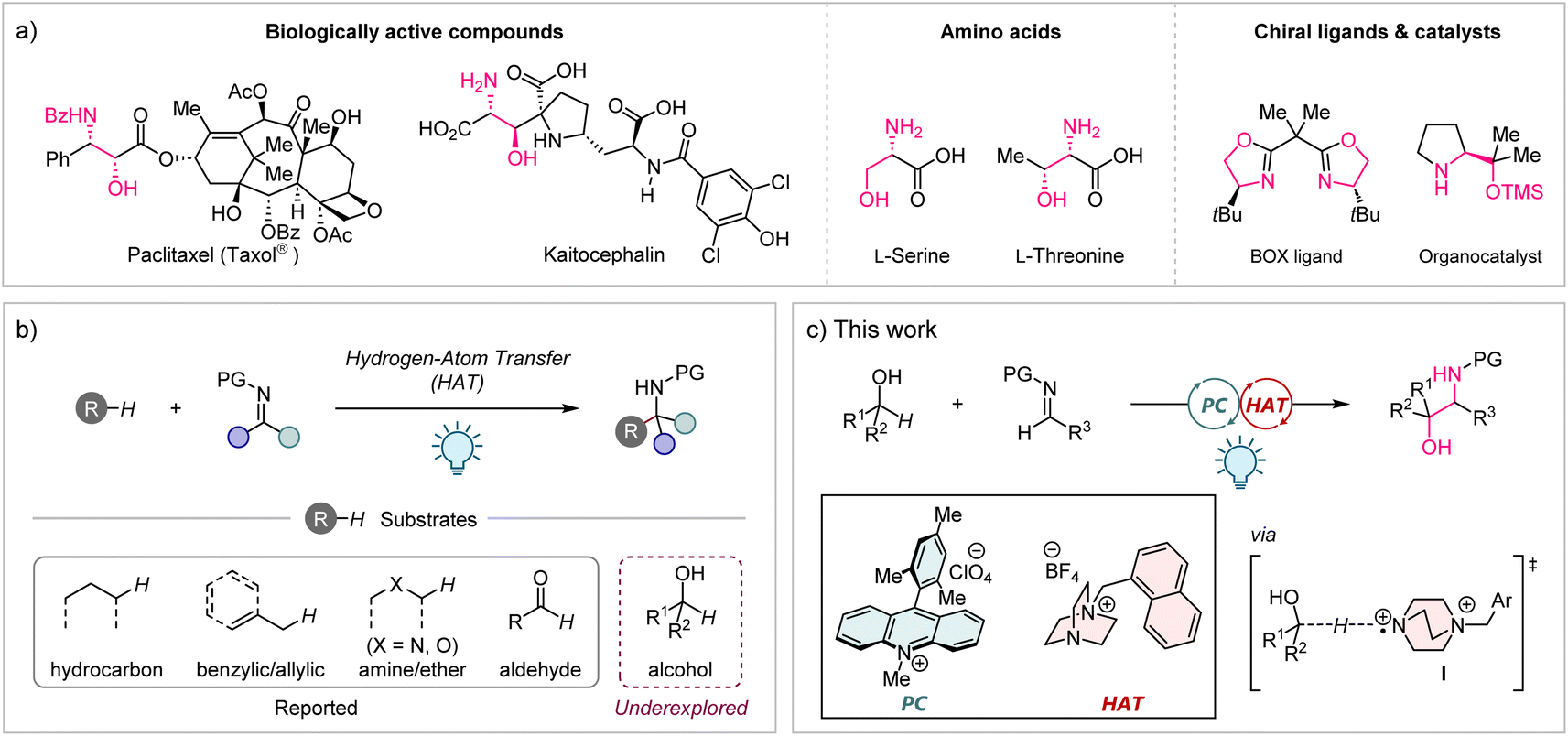
1,2-Aminoalcohols are a ubiquitous structural motif found in a diverse array of natural products, agrochemicals, and pharmaceuticals (Fig. 1a).1 They also play a pivotal role in organic synthesis as privileged ligands and catalysts.2 Therefore, the development of efficient methods for the preparation of these motifs, particularly in a stereoselective manner, is undoubtedly an important area of chemical research. While various approaches, including the oxyamination of alkenes,3a–c ring-opening of epoxides,3d reductive cross-coupling of carbonyls with imines,3e and others,3f–m have been reported to date, most of them suffer from intrinsic drawbacks. These include the necessity for noble metals and/or toxic chemicals, as well as multi-step synthesis for the preparation of specific substrates. Given the abundance and wide availability of aliphatic alcohols as feedstock chemicals, the direct conversion of primary or secondary alcohols into synthetically valuable 1,2-aminoalcohols would be an ideal approach. However, such methods are currently limited to a few reports where primary alcohols are the only applicable substrates.4
 |
| | Fig. 1 (a) 1,2-Aminoalcohol as a ubiquitous structural motif. (b) C–H Aminoalkylation via HAT process. (c) Synthesis of 1,2-aminoalcohols from alcohols and imines by PC/HAT dual catalysis. | |
Hydrogen-atom transfer (HAT) is a powerful strategy to functionalize C(sp3)–H bonds via the generation of carbon-centred radicals.5 The recent development of photoinduced HAT catalytic systems, most of which are enabled by the combination with a photoredox catalyst (PC), has offered a unique opportunity to manipulate the C(sp3)–H bonds of a wide range of substrates under mild conditions in high atom and step economy.6 Imine derivatives are attractive acceptors for the carbon-centred radicals generated by the HAT process, providing structurally diverse amine derivatives. Indeed, photochemical methods for C–H aminoalkylation of a variety of substrates with different HAT catalysts have been reported in recent years (Fig. 1b).7 Surprisingly, however, the use of alcohols as substrates for radical addition to imines via the HAT process has not been included in these reports, although site-selective HAT from the α-C–H bond of aliphatic alcohols has been achieved in several other C–H functionalization reactions.8 Herein, we addressed this underexplored area by using a cationic HAT catalyst based on 1,4-diazabicyclo[2.2.2]octane (DABCO), which was recently developed by our group (Fig. 1c).9 We envisioned that the highly electrophilic dicationic aminium radical I would be suitable for selective HAT from a strong but hydridic α-C–H bond of an alcohol with the aid of a favourable polar effect.10 Meanwhile, imine derivatives were investigated in detail to identify an appropriate structure as an acceptor for the carbon-centred radical generated in situ from the alcohol substrate.
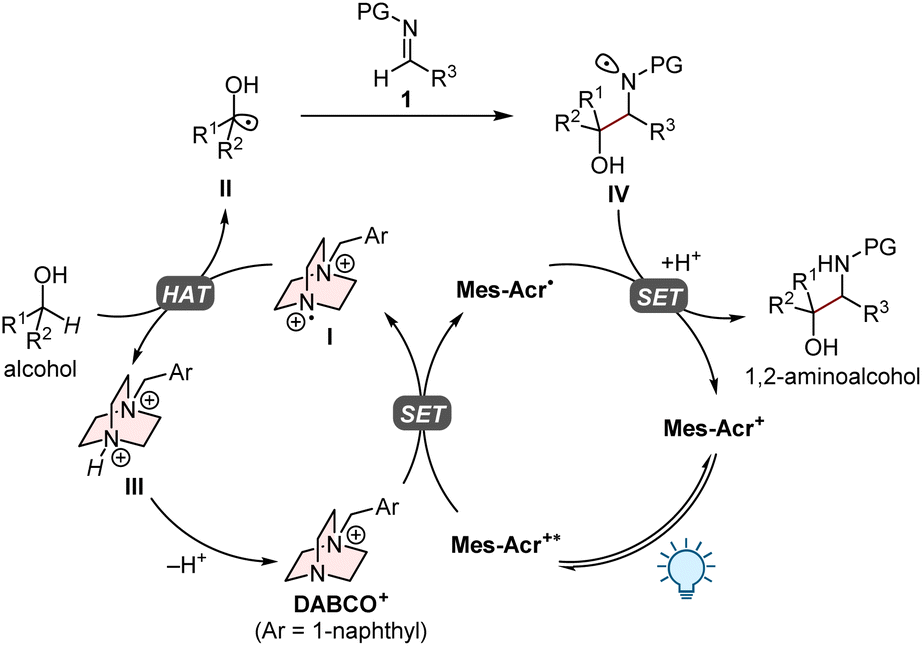
Based on our previous research,9 the proposed mechanism for the present PC/HAT dual catalytic system is outlined in Fig. 2. Upon irradiation with visible light, the organophotoredox catalyst 9-mesityl-10-methylacridinium perchlorate (Mes-Acr+) transitions to a long-lived excited state (Mes-Acr+*) with a high oxidation potential (Ered[Mes-Acr+*/Mes-Acr˙] of 2.06 V vs. saturated calomel electrode (SCE) in MeCN),11 which oxidizes the HAT catalyst (DABCO+) via single-electron transfer (SET) to generate the dicationic aminium radical I. The subsequent HAT process between the alcohol and I furnishes the corresponding α-hydroxy carbon-centred radical II, which undergoes the radical addition to imine 1 to give the nitrogen-centred radical IV. The catalytic cycle would be closed by the single-electron reduction of IV by acridine radical Mes-Acr˙ and the subsequent proton transfer, affording the desired 1,2-aminoalcohol.
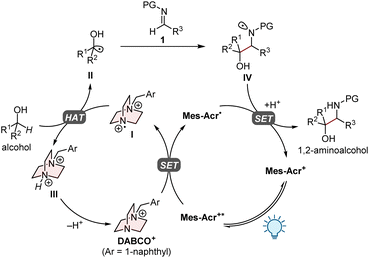 |
| | Fig. 2 Proposed catalytic cycle. | |
2 Results and discussion
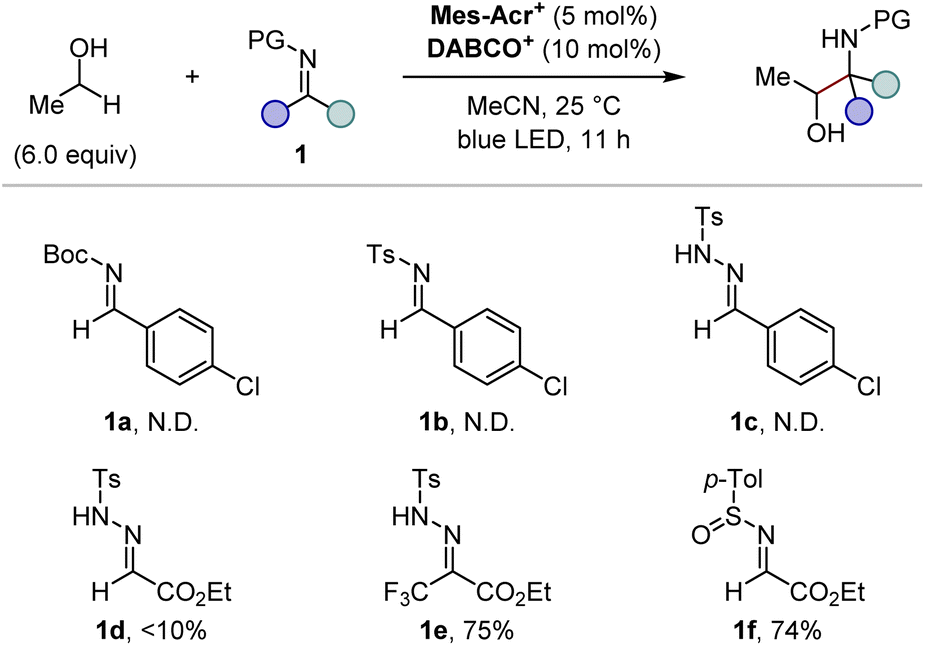
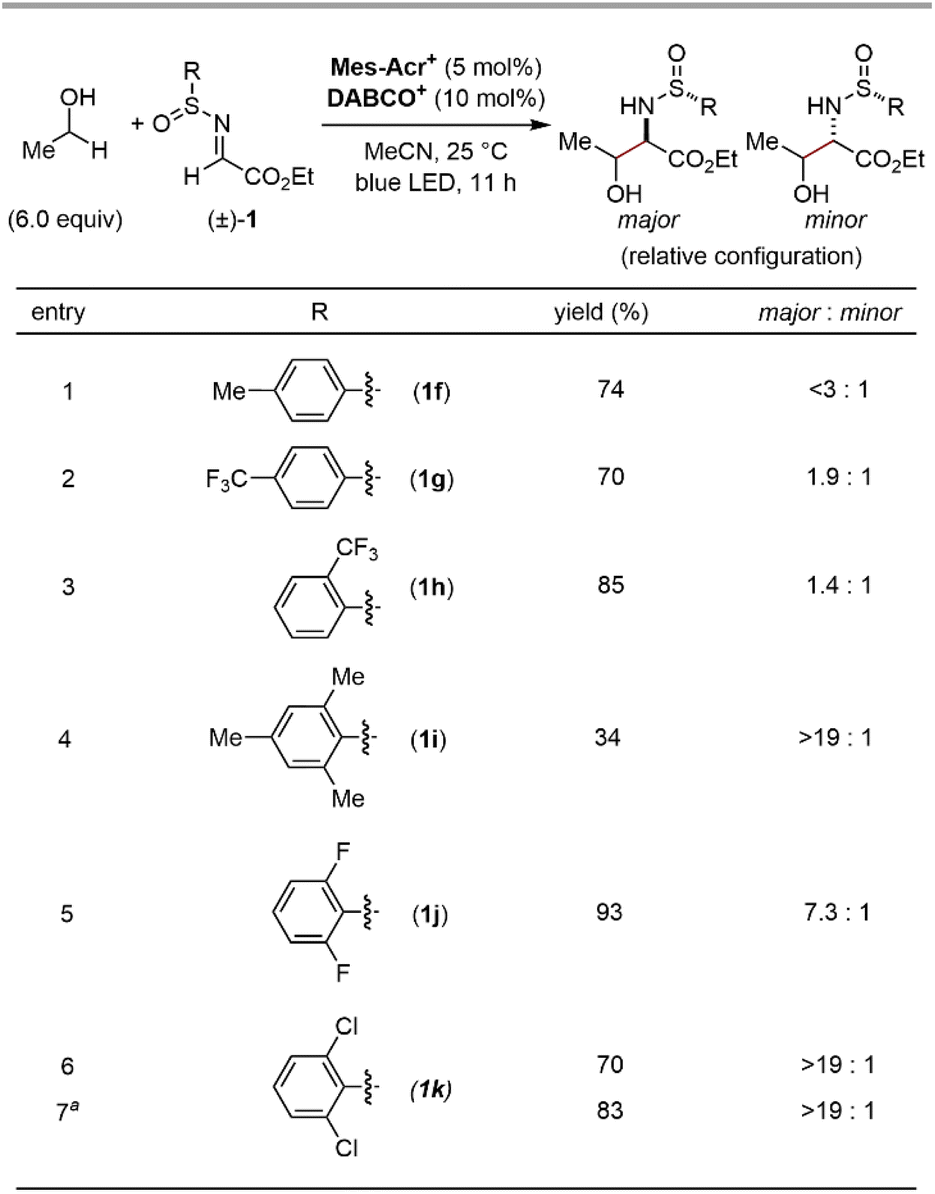
We commenced our investigation by screening imine derivatives as acceptors for the α-hydroxy carbon-centred radical generated in situ from ethanol (Fig. 3). Imine derivatives from p-chlorobenzaldehyde, including N-Boc imine 1a, N-tosyl imine 1b, and N-tosylhydrazone 1c, provided none of the corresponding products. A more electrophilic N-tosylhydrazone derived from ethyl glyoxylate (1d) gave a detectable amount of the expected 1,2-aminoalcohol product, and the further introduction of an electron-withdrawing trifluoromethyl group at the sp2 carbon of the hydrazone moiety (1e) significantly increased the yield. These trends indicate that the highly electrophilic character at the imine moiety is crucial to achieve sufficient reactivity. Given the good balance between electrophilicity and chemical stability, applicability to asymmetric synthesis, and ease of deprotection, we focused on N-sulfinyl α-iminoesters as the imine partners. Gratifyingly, the reaction of ethyl (E)-N-(p-toluenesulfinyl)iminoacetate (1f) with ethanol gave the 1,2-aminoalcohol product in a good yield. We then moved on further investigations of the substituent of the sulfinyl moiety (Fig. 4). Since Ellman's tert-butyl-substituted N-sulfinyl imine is known to be incompatible with radical-mediated conditions, we focused on aryl-substituted N-sulfinyl derivatives.12 Although para- or ortho-monosubstituted phenyl groups (1f–h) gave good yields of 1,2-aminoalcohol products, the stereochemistry of the α-carbon relative to the chiral sulfur centre in the sulfinyl moiety was not well controlled (<3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, Fig. 4, entries 1–3). On the other hand, the sterically more hindered mesityl group (1i) showed high diastereoselectivity at the α-carbon, albeit with a lower yield (Fig. 4, entry 4). Thus, we expected that the introduction of electron-withdrawing groups at the two ortho-positions of the phenyl ring would facilitate the transfer of chirality from the sulfur centre to the α-carbon while maintaining the high electrophilicity at the imine carbon, thereby delivering both high reactivity and diastereoselectivity. Indeed, the ortho-difluorophenyl group (1j) efficiently produced the product with an increased diastereoselectivity (Fig. 4, entry 5). Finally, the slightly bulkier ortho-dichlorophenyl group (1k) was found to be the optimal substituent of the sulfinyl moiety, affording the desired 1,2-aminoalcohol in a good yield with a high diastereoselectivity at the α-carbon (Fig. 4, entry 6). Although the syn/anti selectivity of the 1,2-aminoalcohol moiety was not controlled even after the optimization of reaction conditions (<1.3:1 in all cases), it was found that both the equivalents of ethanol and the reaction time could be reduced (Fig. 4, entry 7). Notably, other HAT catalysts, including those used in the reported conditions for radical addition to imine derivatives,7 did not provide the desired radical adduct from ethanol and (±)-1k (See the ESI for details†). These results demonstrate an advantage of our photocatalytic system where the exceptionally electrophilic aminium radical I is responsible for an efficient HAT process from the α-C–H bond of the alcohol.
:1 dr, Fig. 4, entries 1–3). On the other hand, the sterically more hindered mesityl group (1i) showed high diastereoselectivity at the α-carbon, albeit with a lower yield (Fig. 4, entry 4). Thus, we expected that the introduction of electron-withdrawing groups at the two ortho-positions of the phenyl ring would facilitate the transfer of chirality from the sulfur centre to the α-carbon while maintaining the high electrophilicity at the imine carbon, thereby delivering both high reactivity and diastereoselectivity. Indeed, the ortho-difluorophenyl group (1j) efficiently produced the product with an increased diastereoselectivity (Fig. 4, entry 5). Finally, the slightly bulkier ortho-dichlorophenyl group (1k) was found to be the optimal substituent of the sulfinyl moiety, affording the desired 1,2-aminoalcohol in a good yield with a high diastereoselectivity at the α-carbon (Fig. 4, entry 6). Although the syn/anti selectivity of the 1,2-aminoalcohol moiety was not controlled even after the optimization of reaction conditions (<1.3:1 in all cases), it was found that both the equivalents of ethanol and the reaction time could be reduced (Fig. 4, entry 7). Notably, other HAT catalysts, including those used in the reported conditions for radical addition to imine derivatives,7 did not provide the desired radical adduct from ethanol and (±)-1k (See the ESI for details†). These results demonstrate an advantage of our photocatalytic system where the exceptionally electrophilic aminium radical I is responsible for an efficient HAT process from the α-C–H bond of the alcohol.
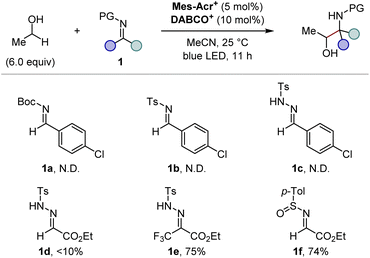 |
| | Fig. 3 Screening of imine derivatives. Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. Syn/anti selectivity of the 1,2-aminoalcohol moiety was <1.6:1 in all cases. N.D. = Not Detected. | |
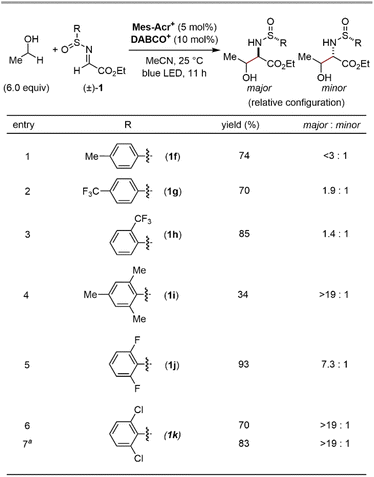 |
| | Fig. 4 Screening of N-sulfinyl α-iminoesters. Yields and ratios of isomers were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. Syn/anti selectivity of the 1,2-aminoalcohol moiety was <1.3:1 in all cases. aThe reaction was run for 6 h using 3 equiv. of EtOH. | |
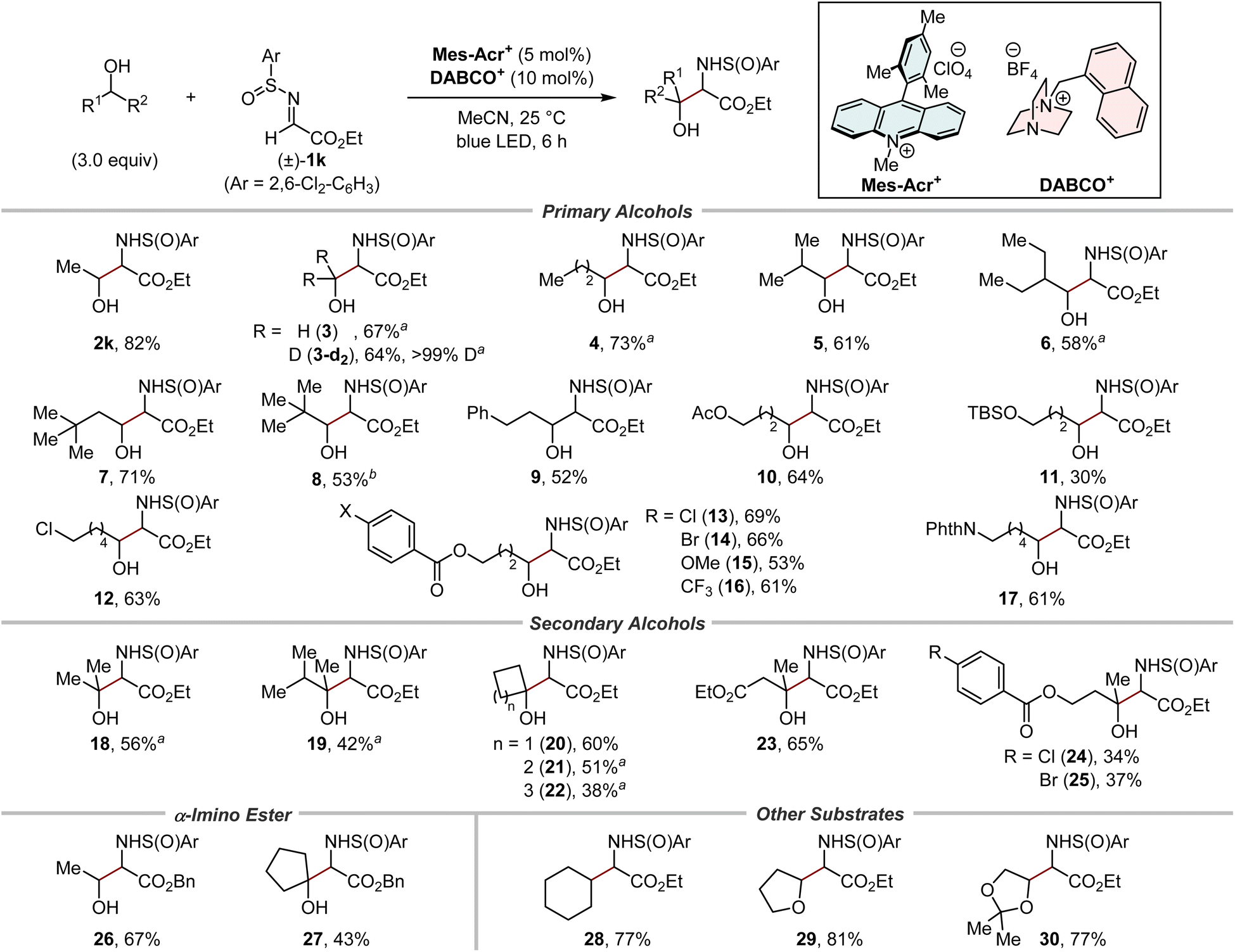
With the optimal N-sulfinyl α-iminoester in hand, the scope of primary and secondary alcohols was evaluated (Fig. 5). The relative stereochemistry between the α-carbon and the sulfur centre in the 1,2-aminoalcohol products was fully controlled in all cases (>19:1 dr). A series of (deuterated) methanol and primary alcohols with varying degrees of steric bulk were good substrates for hydrogen-atom abstraction by the cationic DABCO-based HAT catalyst, affording the corresponding products in moderate to high yields (2k and 3–9). Alcohols bearing several functional groups, such as acetoxy (10), siloxy (11), chloro (12), para-substituted benzoyloxy (13–16), and phthaloyl groups (17), were well tolerated. Of note, these reactions occurred selectively at the α-C–H bond to the hydroxy group. Moreover, despite the increased steric hindrance, both acyclic (18, 19, and 23–25) and cyclic (20–22) secondary alcohols successfully afforded bulky 1,2-aminoalcohols. It was also possible to switch the ester group in the N-sulfinyl α-iminoester from ethyl to benzyl (26 and 27), suggesting the utility of our method for the synthesis of amino acids by the subsequent removal of the benzyl protecting groups. In addition to alcohols, the reactivity of other substrates was briefly examined. Cyclohexane, a representative of hydrocarbons containing stronger C–H bonds than the α-C–H bond of alcohols, afforded the α-monoalkylated amino acid derivative 28 in a good yield. The etheric substrates THF and 2,2-dimethyl-1,3-dioxolane were also suitable substrates, furnishing the corresponding products 29 and 30, respectively, in high yields.
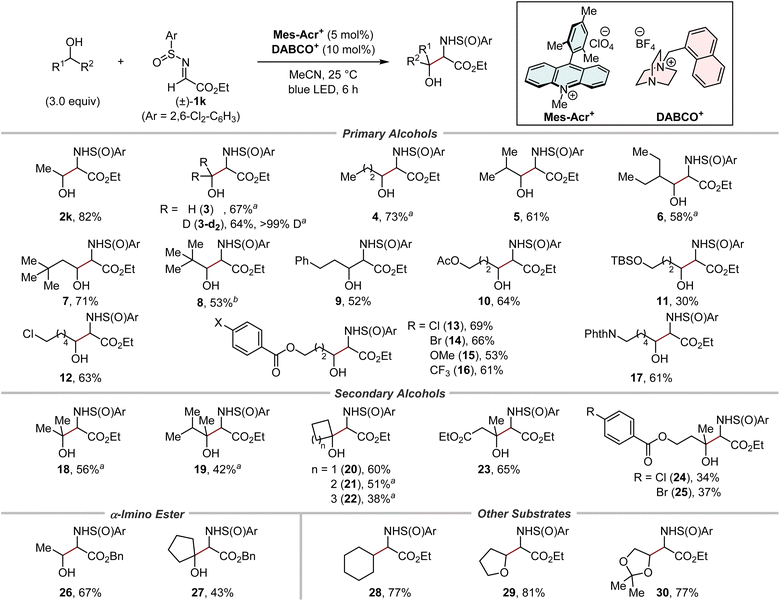 |
| | Fig. 5 Substrate scope. Combined yields of isolated products were shown. Unless otherwise noted, syn/anti selectivity of the 1,2-aminoalcohol moiety was <1.8:1 (see the ESI for details†). a 6.0 Equivalents of alcohols were used. bSyn/anti selectivity was 2.6:1. | |
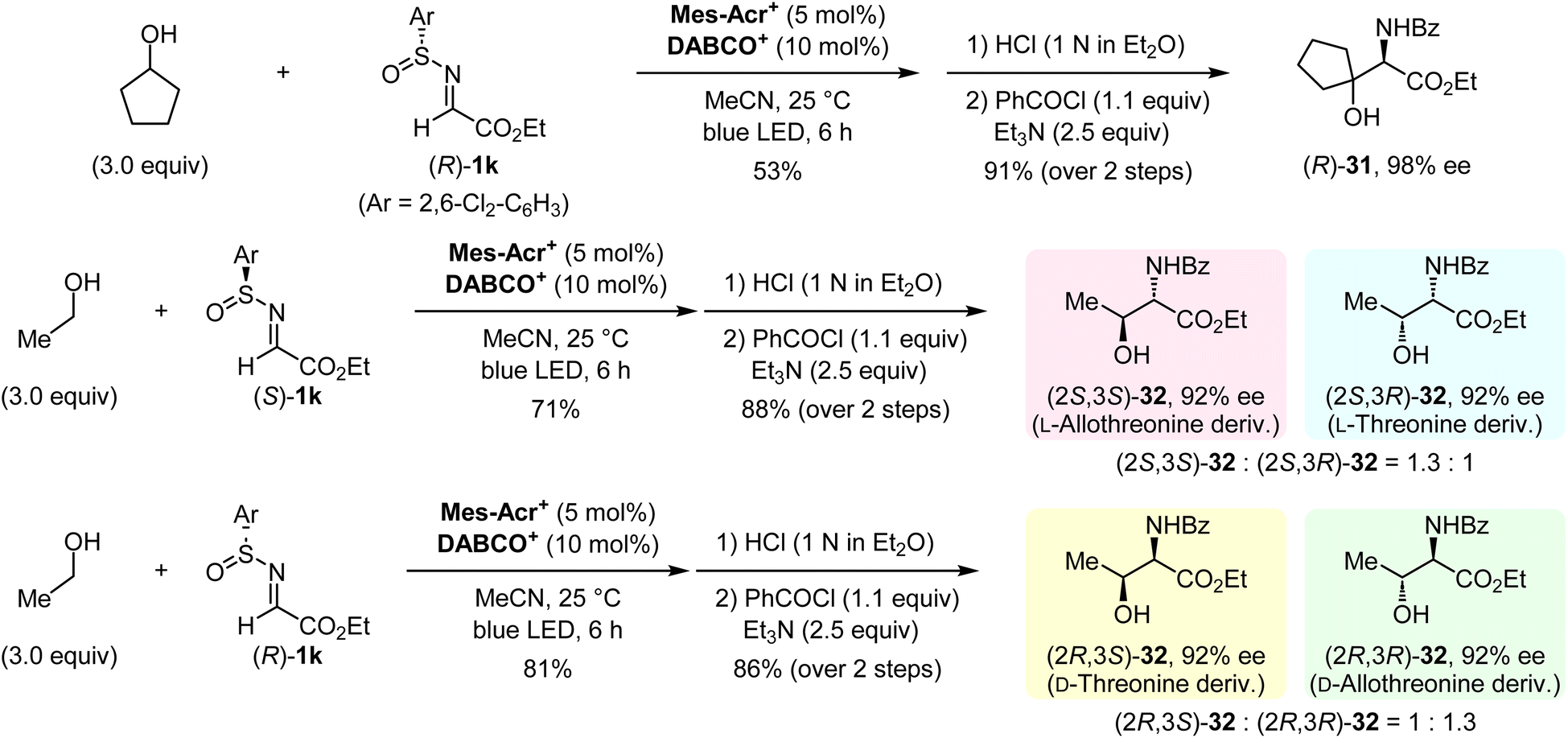
Having established our protocol for the facile construction of 1,2-aminoalcohols, an asymmetric synthesis was conducted by using optically active N-sulfinyl α-iminoesters (Fig. 6). Both enantiomers of 1k were prepared according to Senanayake's method (see the ESI†).13 The α-C–H aminoalkylation of cyclopentanol with (R)-1k under optimal conditions, followed by the deprotection of the sulfinyl moiety and N-benzoylation, afforded the enantiomerically enriched 1,2-aminoalcohol (R)-31 with 98% ee. We then demonstrated the synthesis of all possible isomers of threonine and allothreonine derivatives. The use of ethanol and (S)-1k in this protocol provided a mixture of (2S,3S)-32 and (2S,3R)-32 as derivatives of L-allothreonine and L-threonine, respectively. Similarly, the reaction of ethanol with (R)-1k gave a mixture of (2R,3S)-32 and (2R,3R)-32, which are derivatives of D-threonine and D-allothreonine, respectively. These results indicate that the present method can be used for rapid access to the desired stereoisomers of chiral 1,2-aminoalcohols starting from an aliphatic alcohol.
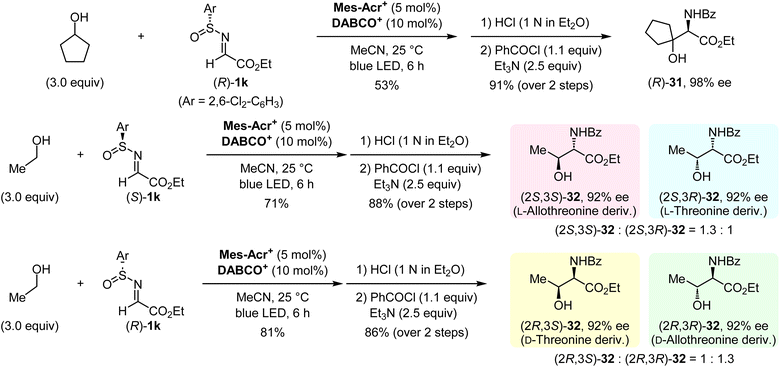 |
| | Fig. 6 Asymmetric synthesis of 1,2-aminoalcohols. | |
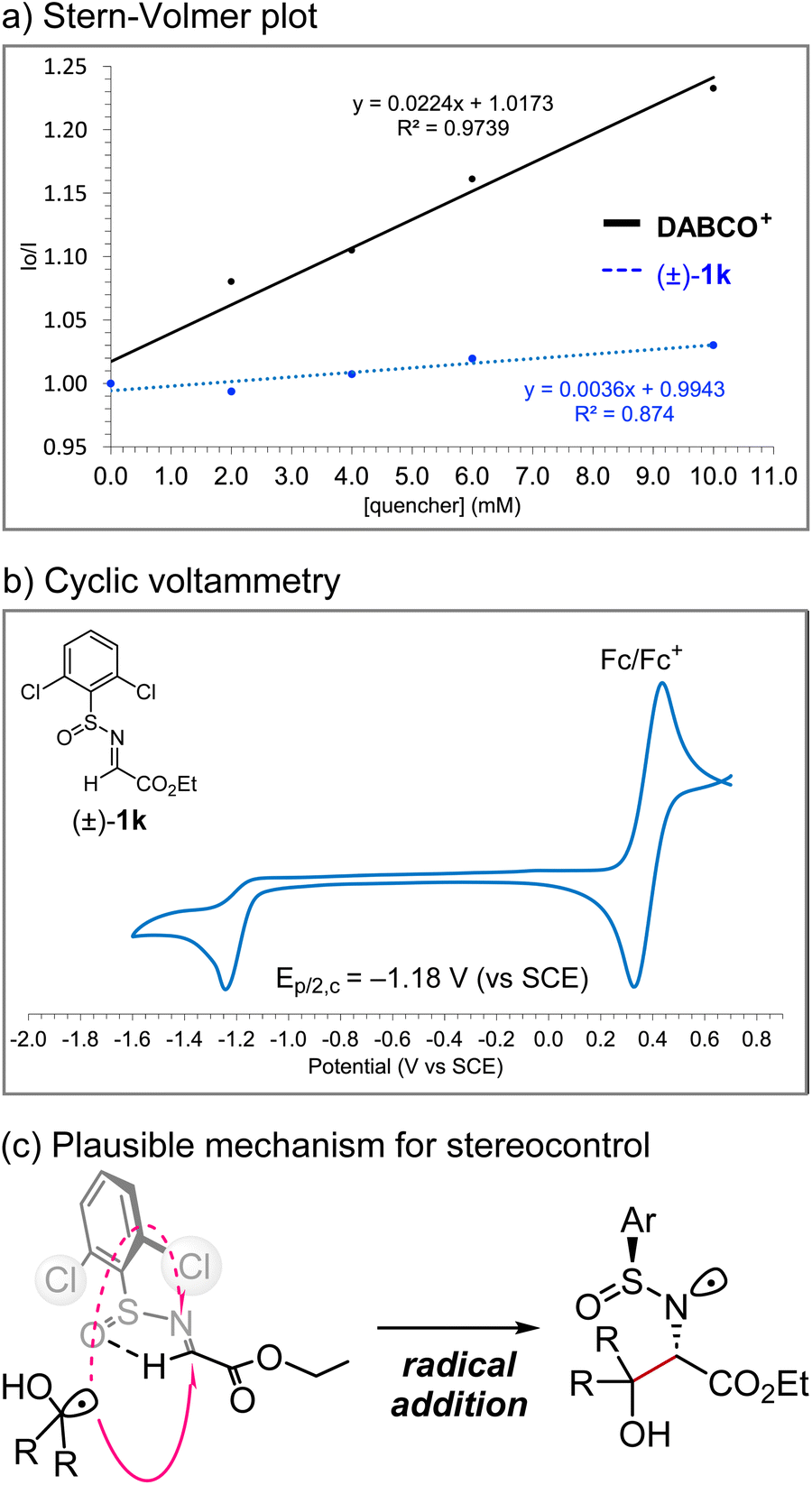
We performed analytical studies to better understand the reaction mechanism (Fig. 7). Stern–Volmer fluorescence quenching studies revealed that DABCO+ quenches the excited state of photocatalyst Mes-Acr+*, whereas quenching by (±)-1k is negligible (Fig. 7a). The reduction potential of (±)-1k was −1.2 V vs. SCE according to the cyclic voltammetry measurement, indicating that the single-electron reduction of (±)-1k by acridine radical Mes-Acr˙ (E1/2[Mes-Acr+/Mes-Acr˙] = −0.57 V vs. SCE) would not be favoured (Fig. 7b).11 Therefore, a radical–radical coupling between an α-hydroxy radical and an α-amino radical generated in situ from the alcohol and N-sulfinyl α-iminoester, respectively, would be unlikely. On the other hand, the efficient diastereocontrol at the α-carbon relative to the sulfur centre supports the radical addition pathway to the s-cis form of the N-sulfinyl α-iminoester, which is conformationally locked due to internal hydrogen bonding, as described by Kärkäs et al. (Fig. 7c).12b Overall, these results are consistent with the proposed mechanism shown in Fig. 2.
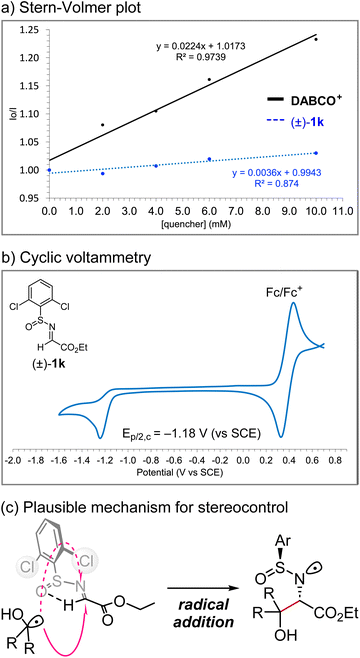 |
| | Fig. 7 Mechanistic studies. | |
3 Conclusions
In summary, we have developed a straightforward method for the synthesis of 1,2-aminoalcohols from readily available alcohols and imine derivatives. A dual photocatalytic system consisting of an acridinium organophotoredox catalyst and a cationic DABCO-based catalyst enabled an efficient and site-selective HAT from the α-C–H bond of various primary and secondary alcohols. In addition, a newly designed N-sulfinyl α-iminoester 1k was found to be a suitable imine acceptor for the in situ-generated α-hydroxy carbon-centred radical, which successfully expanded the scope of C–H aminoalkylation via a photoinduced HAT process. The asymmetric synthesis of 1,2-aminoalcohols was also achieved by using an enantiomerically enriched N-sulfinyl α-iminoester.
Author contributions
A. M. conceptualized the research. A. M. and J. C. performed the experiments and prepared the manuscript. K. M. supervised the project and edited the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
K. M. gratefully acknowledges financial support via JSPS KAKENHI grant numbers JP21H05026 and JP23H04910. A. M. is thankful for financial support via JSPS KAKENHI grant number JP22K14680. We thank Natsumi Maeda for technical assistance with fluorescence quenching experiments.
Notes and references
-
(a) P. Gupta and N. Mahajan, New J. Chem., 2018, 42, 12296–12327 RSC;
(b) T. Sehl, Z. Maugeri and D. Rother, J. Mol. Catal. B: Enzym., 2015, 114, 65–71 CrossRef CAS;
(c) D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS;
(d) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2012, 10, 4311–4326 RSC;
(e) M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed;
(f) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561–2576 CrossRef CAS.
-
(a)
Q. Zhou, Privileged Chiral Ligands and Catalysts, John Wiley & Sons Inc., New York, 2011 CrossRef;
(b) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–876 CrossRef CAS PubMed.
-
(a) B. N. Hemric, Org. Biomol. Chem., 2021, 19, 46–81 RSC;
(b) M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, RSC Adv., 2018, 8, 6634–6659 RSC;
(c) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem.–Euro. J., 2011, 17, 58–76 CrossRef CAS PubMed;
(d) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed;
(e) O. N. Burchak and S. Py, Tetrahedron, 2009, 65, 7333–7356 CrossRef CAS;
(f) B. Shrestha, B. T. Rose, C. L. Olen, A. Roth, A. C. Kwong, Y. Wang and S. E. Denmark, J. Org. Chem., 2021, 86, 3490–3534 CrossRef CAS PubMed;
(g) S. Kobayashi, H. Ishitani and M. Ueno, J. Am. Chem. Soc., 1998, 120, 431–432 CrossRef CAS;
(h) J. Meng, H. Lv and X. Zhang, Org. Lett., 2015, 17, 1842–1845 CrossRef CAS PubMed;
(i) M. Shimizu, K. Tsukamoto, T. Matsutani and T. Fujisawa, Tetrahedron, 1998, 54, 10265–10274 CrossRef CAS For recent examples via radical intermediates, see:;
(j) T. Patra, M. Das, C. G. Daniliuc and F. Glorius, Nat. Catal., 2021, 4, 54–61 CrossRef CAS;
(k) J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, J. Am. Chem. Soc., 2020, 142, 2168–2174 CrossRef CAS PubMed;
(l) S. M. Rafferty, J. E. Rutherford, L. Zhang, L. Wang and D. A. Nagib, J. Am. Chem. Soc., 2021, 143, 5622–5628 CrossRef CAS PubMed;
(m) H. Hu and Z. Wang, J. Am. Chem. Soc., 2023, 145, 20775–20781 CrossRef CAS PubMed.
-
(a) B. Spielmann, M. Xiang, L. A. Schwartz and M. J. Krische, J. Am. Chem. Soc., 2019, 141, 14136–14141 CrossRef PubMed;
(b) W. Zhang, W. Chen, H. Xiao and M. J. Krische, Org. Lett., 2017, 19, 4876–4879 CrossRef CAS PubMed.
-
(a)
Hydrogen-Transfer Reactions, ed. J. T. Hynes, J. P. Klinman, H.-H. Limbach and R. L. Schowen, Wiley-VCH, Weinheim, 2007 Search PubMed;
(b) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
-
(a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed;
(b) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS;
(c) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC;
(d) L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed.
-
(a) S. Yang, S. Zhu, D. Lu and Y. Gong, Org. Lett., 2019, 21, 2019–2024 CrossRef CAS PubMed;
(b) V. I. Supranovich, V. V. Levin and A. D. Dilman, Org. Lett., 2019, 21, 4271–4274 CrossRef CAS PubMed;
(c) Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016–1026 CrossRef CAS;
(d) W. K. Weigel, H. T. Dang, H.-B. Yang and D. B. C. Martin, Chem. Commun., 2020, 56, 9699–9702 RSC;
(e) J. Jia, R. Kancherla, M. Rueping and L. Huang, Chem. Sci., 2020, 68, 42–61 Search PubMed;
(f) F. Babawale, K. Murugesan, R. Narobe and B. König, Org. Lett., 2022, 24, 4793–4797 CrossRef CAS PubMed;
(g) A. Pulcinella, S. Bonciolini, F. Lukas, A. Sorato and T. Noël, Angew. Chem., Int. Ed., 2023, 62, e202215374 CrossRef CAS PubMed;
(h) A. Jorea, C. Raviola, M. Giustiniano and D. Ravelli, ChemCatChem, 2023, 15, e202300042 CrossRef CAS.
- Selected examples:
(a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed;
(b) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed;
(c) S. Kamijo, G. Takao, K. Kamijo, T. Tsuno, K. Ishiguro and T. Murafuji, Org. Lett., 2016, 18, 4912–4915 CrossRef CAS PubMed;
(d) T. Fukuyama, K. Yamada, T. Nishikawa, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Lett., 2018, 47, 207–209 CrossRef CAS;
(e) X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed;
(f) H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed;
(g) V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed;
(h) K. Ohmatsu, R. Suzuki, Y. Furukawa, M. Sato and T. Ooi, ACS Catal., 2020, 10, 2627–2632 CrossRef CAS;
(i) K. Sakai, K. Oisaki and M. Kanai, Adv. Synth. Catal., 2020, 362, 337–343 CrossRef CAS;
(j) H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS PubMed;
(k) B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. Mccabe, L. Clark, R. Young, J. Pu and Y. Deng, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS;
(l) M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS PubMed;
(m) M. Yoshida, M. Sawamura and Y. Masuda, ChemCatChem, 2022, 14, e202200744 CrossRef CAS.
- A. Matsumoto, M. Yamamoto and K. Maruoka, ACS Catal., 2022, 12, 2045–2051 CrossRef CAS.
-
(a) A. Ruffoni, R. C. Mykura, M. Bietti and D. Leonori, Nat. Synth., 2022, 1, 682–695 CrossRef;
(b) F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem, 2021, 5, 486–499 CrossRef CAS PubMed.
-
(a) S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed;
(b) K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
-
(a) W. Huang, J.-L. Ye, W. Zheng, H.-Q. Dong and B.-G. Wei, J. Org. Chem., 2013, 78, 11229–11237 CrossRef CAS PubMed;
(b) A. Shatskiy, A. Axelsson, E. V. Stepanova, J.-Q. Liu, A. Z. Temerdashev, B. P. Kore, B. Blomkvist, J. M. Gardner, P. Dinér and M. D. Kärkäs, Chem. Sci., 2021, 12, 5430–5437 RSC;
(c) J. L. M. Matos, S. Vásquez-Céspedes, J. Gu, T. Oguma and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 16976–16981 CrossRef CAS PubMed;
(d) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560–14565 CrossRef CAS PubMed;
(e) A. F. Garrido-Castro, H. Choubane, M. Daaou, M. C. Maestro and J. Alemán, Chem. Commun., 2017, 53, 7764–7767 RSC.
-
(a) Z. Han, D. Krishnamurthy, P. Grover, Q. K. Fang and C. H. Senanayake, J. Am. Chem. Soc., 2002, 124, 7880–7881 CrossRef CAS PubMed;
(b) Z. S. Han, A. M. Meyer, Y. Xu, Y. Zhang, R. Busch, S. Shen, N. Grinberg, B. Z. Lu, D. Krishnamurthy and C. H. Senanayake, J. Org. Chem., 2011, 76, 5480–5484 CrossRef CAS PubMed;
(c) T. Ramachandar, Y. Wu, J. Zhang and F. A. Davis, Org. Synth., 2006, 83, 131–140 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization for all relevant compounds. See DOI: https://doi.org/10.1039/d3sc05305a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Akira
Matsumoto
a,
Akira
Matsumoto