
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel strategy for analyzing aptamer dominated sites and detecting AFB1 based on CRISPR–Cas12a†
Chenqi
Niu
a,
Xinhui
Xing
ab and
Chong
Zhang
 *ab
*ab
aMOE Key Laboratory for Industrial Biocatalysis, Institute of Biochemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing, 100084, China. E-mail: chongzhang@mail.tsinghua.edu.cn
bCenter for Synthetic and Systems Biology, Tsinghua University, Beijing, 100084, China
First published on 21st November 2022
Abstract
Profiling of aptamer dominated sites which have a huge impact on aptamer affinity is essential to characterize the relationship between an aptamer and target molecule and show the potential for boosting the sensitivity of aptamer-based biosensors. Here, we report a novel strategy to analyze aptamer dominated sites and detect aflatoxin B1 (AFB1) based on CRISPR–Cas12a. 28 different crRNAs were designed to hybridize with every region of the AFB1 47-nt aptamer, and sites 24–26 from the 5′ to 3′ end in the aptamer (“TGT”) were speculated to be the dominated sites according to the different responses based on the fluorescence intensity inhibition rate. Then, this was verified by replacing the three bases through the BLI assay. Three-dimensional (3D) modeling and molecular docking were also used to laterally validate the results. Moreover, the effects of special structures of DNA on the activated Cas12a trans-cleavage activity were evaluated to enrich our understanding of Cas12a. In addition, a strategy for the detection of AFB1 based on Cas12a was developed. The workflow is convenient, simple, and rapid (37 °C in 20 min) with a detection limit of 0.8 ng mL−1. The strategy presented good specificity over other mycotoxins and aflatoxin analogues and showed potential in complex sample analysis.
1. Introduction
Aflatoxin B1 (AFB1), a highly toxic, secondary metabolite of molds, has been recognized as the most toxic and powerful human carcinogen. Since AFB1 is a serious hazard to human and animal health,1,2 it is of significant importance to develop a sensitive and accurate method for the rapid detection of AFB1. Enzyme-linked immunosorbent assay (ELISA), high-performance liquid chromatography (HPLC) and thin-layer chromatography are the traditional quantitative techniques to detect AFB1.3–5 However, these methods are expensive, time-consuming, or require sophisticated instruments.In recent years, several AFB1 aptamers with high binding affinity have been developed through systematic evolution of ligands by the exponential enrichment assay (SELEX).6–9 Many aptamer-based biosensors were reported accordingly to detect AFB1, including colorimetric assays, electrochemical assays, fluorescence assays, etc.10–14 Compared to the traditional detection methods, aptamer-based methods are easy to perform, have low cost and can improve the stability and sensitivity of AFB1 detection. Recently, Clustered Regularly Interspaced Short Palindromic Repeats associated protein 12a (CRISPR–Cas12a), an RNA guided endonuclease, has been applied for the development of aptamer-based small molecular biosensors.15,16 When the Cas12a–crRNA complex hybridizes with a region with aptamer/analyte dominated sites which have a huge impact on aptamer affinity, the structural change of the aptamer due to analyte binding has a great influence on the hybridization between the Cas12a–crRNA complex and aptamer, leading to a decrease of the trans-cleavage activity of Cas12a. The Cas12a-based competitive biosensor achieved high detection sensitivity without nucleic acid polymerases, and it has more potential for real samples with a complex matrix due to its simple reaction system. Nevertheless, there are still many challenges to be faced. In the competitive binding method with a fluorescence reporter system, crRNAs are designed to hybridize with aptamers and activate the Cas12a trans-cleavage activity, where target molecules act as an inhibitor to decrease the Cas12a trans-cleavage activity. In the presence of the target, the aptamers switching structure and no longer hybridizing with the crRNA–Cas12a complex are the key steps to the assay. However, considering that the hybridization region is 20 bp and the length of excellent aptamers usually ranges from 30–50 bp, the design of crRNAs is not an easy task. In addition, the lack of knowledge of aptamer dominated sites also poses great challenges for crRNA design.
Herein, we report a novel strategy for analyzing AFB1 aptamer dominated sites and detecting AFB1 based on CRISPR–Cas12. 28 different crRNAs were designed to hybridize with every region of the AFB1 47-nt aptamer. The dominated site was analyzed by comparing the repeated site of crRNAs with a high decrease of Cas12a trans-cleavage activity. Sites 24–26 from the 5′ to 3′ end in the aptamer (“TGT”) were speculated to be the dominated sites according to the different responses based on the fluorescence intensity inhibition rate. The results were verified by BLI assays and 3D modeling and molecular docking. In addition, according to the results of the analyzed aptamer dominated sites, a convenient, simple, and rapid method with high sensitivity for detection of AFB1 based on Cas12a was developed with crRNA 24. The strategy presented good specificity over other mycotoxins and aflatoxin analogues and showed potential in complex sample analysis.
2. Materials and methods
2.1. Reagents and materials
An F-Q probe was purchased from Bio-lifesci (Guangzhou, China). NEB CutSmart™ buffer and LbCas12a (Cas12a) were purchased from NEB (USA). crRNAs and aptamers and oligonucleotides (Table S1†) were synthesized and purchased from Genewiz (New Jersey, USA). Aflatoxin B1 (AFB1), ochratoxin A (OTA), ochratoxin B (OTB), fumonisin B1 (FB1), fumonisin B2 (FB2), zearalenone (ZAE), aflatoxin G1 (AFG1) and aflatoxin G2 (AFG2) were purchased from the Ministry of Agriculture Standards Center (Beijing, China). An AFB1 ELISA detection kit was purchased from Solarbio Life Science (Beijing). Red wine, beer and milk for complex matrix tests were purchased from a local supermarket.2.2. Buffer conditions
The buffer for all experiments was NEB CutSmart™ buffer (50 mM KAc, 20 mM Tris-Ac, 10 mM Mg(Ac)2, 100 μg mL−1 BSA, pH 7.9 at 25 °C).2.3. Annealing protocol
All annealing steps in this study were performed with a BIO-RAD T100 thermocycler. The samples were first heated up to 95 °C and then slowly cooled to 20 °C at a constant rate of 1 °C min−1.2.4. Typical trans-cleavage reaction
In the AFB1 aptamer dominated site analysis assay, AFB1 was pre-incubated with the 47-nt aptamer at room temperature for 15 minutes as mixture A, and Cas12a was pre-incubated with crRNA at room temperature for 15 minutes as mixture B. After that, mixture A, mixture B, buffer, F-Q probe and deionized water were mixed evenly to start a trans-cleavage reaction with 80 μL. The final concentration of components: 1× NEB CutSmart™ buffer, 200 nM F-Q probe, 30 nM LbCas12a, 30 nM crRNA, 25 nM aptamer and different concentrations of AFB1 in the experimental group or an equal volume of deionized water in the control group.In the sensitivity test, the concentration of AFB1 ranged from 100 ng mL−1 to 2 μg mL−1.
In the specificity test, AFB1 was replaced with other mycotoxins and aflatoxin analogues with the same concentration.
In the complex matrix sample test, different concentrations of AFB1 were spiked into 5% red wine, 5% beer and 5% milk. The red wine, beer and milk were used directly without pretreatment. 50 μL aliquots of 10 μg mL−1 and 5 μg mL−1 AFB1 were mixed with 950 μL wine to obtain stock solutions of 500 ng mL−1 and 250 ng mL−1 AFB1 in 5% wine, respectively. And, AFB1 was also spiked into beer and milk in the same way as that for the wine to obtain 500 ng mL−1 and 250 ng mL−1 AFB1 in 5% beer and milk, respectively.
2.5. Data collection and processing
The reactants (80 μl, 96-well microplate format) were incubated in a fluorescence plate reader (Tecan M200 pro) for up to 60 minutes at 37 °C with fluorescence measurements taken every 30 seconds (ssDNA F-Q probe substrates = λex: 492 nm; λem: 518 nm).GraphPad Prism 7 graphics software was used to process data and draw images. In this study, the inhibition rate (%), IR (%) was defined to indicate the level of inhibition of trans-cleavage by small molecules. IR (%) = (A − B)/A × 100, where A is the fluorescence intensity in the control group, and B is the fluorescence intensity in the presence of AFB1.
2.6. BLI (biolayer interferometry) assay
The interaction between aptamers and AFB1 was verified by the BLI assay. 10 μM biotinylated aptamer was incubated with a streptavidin sensor (Octet SA Biosensors: 18-5019). Various concentrations of AFB1 from 2 nM to 640 nM were used to associate and dissociate with the biotinylated aptamer using a BLI OctetRED96 instrument.2.7. 3D modeling and molecular docking
The Discovery Studio 2019 package was used for 3D modeling of AFB1 and optimizing the structure to keep the minimum free energy. For 3D modeling of the 47-nt ssDNA aptamer, three steps were needed. Firstly, the secondary structure of the aptamer sequence was predicted using the oligo analysis website Mfold (https://unafold.rna.albany.edu/). Secondly, the RNA 3D modeling website RNA COMPOSER (https://rnacomposer.cs.put.poznan.pl/) was used to obtain the RNA 3D model based on the result of the first step. Thirdly, the result was imported into the Discovery Studio 2019 package and uracil was transformed into thymine. Then, the structure was optimized to reduce the free energy.The AFB1 binding energy and dominated sites of the ssDNA aptamers were estimated with the grid-based ligand docking program AutoDockTools (ADT) 1.5.6 package. The optimized structure of the aptamer was considered to be rigid receptors while AFB1 was considered to be a flexible ligand with only one rotatable bond. The docking simulations used the Lamarckian genetic algorithm and the best complex was selected according to the molecular docking results including binding energy, types of favorable interactions (mainly hydrogen bonds, and hydrophobic and electrostatic interactions) and dominated sites.
3. Results and discussion
3.1. Schematic
A schematic of the strategy is shown in Fig. 1. The reaction system includes the ssDNA aptamer/analyte complex, Cas12a–crRNA complex and F-Q probe (ssDNA labeled with a fluorophore and quencher at both ends). We designed different crRNAs to recognize different regions of the ssDNA aptamer and hybridize with them. | ||
| Fig. 1 Schematic of the principle of the aptamer dominated site analysis and biosensor strategy based on CRISPR–Cas12a. 28 crRNAs were designed to hybridize with the different regions of the aptamer. In the case of the Cas12a–crRNA complex hybridized with the region without an aptamer/analyte dominated site, the structural change of the aptamer due to analyte binding had little influence on the hybridization between the Cas12a–crRNA complex and aptamer. Cas12a was activated to show trans-cleavage activity. The ssDNA fluorescence-reporter probe labeled with a fluorophore and quencher pair at its two ends (denoted as the F-Q probe) was cleaved, resulting in separation of the fluorophore from the quencher, thereby producing an increase in fluorescence signal. And the decrease of trans-cleavage activity (decrease of fluorescence intensity) of Cas12a activated by the ssDNA aptamer was small compared to that of the control group (ssDNA aptamer activated Cas12a–crRNA to cleave the F-Q probe to show fluorescence), which resulted in a low inhibition rate (IR%). In contrast, when the Cas12a–crRNA complex hybridized with the region with aptamer/analyte dominated sites, the structural change has a great influence on the hybridization between the Cas12a–crRNA complex and aptamer. The decrease of trans-cleavage activity of Cas12a activated by the ssDNA aptamer was sharp compared to that of the control group, which resulted in a high inhibition rate (IR%). | ||
In the case of the Cas12a–crRNA complex hybridized with the region without an aptamer/analyte dominated site, the structural change of the aptamer due to analyte binding had little influence on the hybridization between the Cas12a–crRNA complex and aptamer. Cas12a was activated to show trans-cleavage activity. The ssDNA fluorescence-reporter probe labeled with a fluorophore and quencher pair at its two ends (denoted as the F-Q probe) was cleaved, resulting in separation of the fluorophore from the quencher, thereby producing an increase in fluorescence signal. And the decrease of trans-cleavage activity (decrease of fluorescence intensity) of Cas12a activated by the ssDNA aptamer was small compared to that of the control group (ssDNA aptamer activated Cas12a–crRNA to cleave the F-Q probe to show fluorescence), which resulted in a low inhibition rate (IR%).
In contrast, when the Cas12a–crRNA complex hybridized with the region with aptamer/analyte dominated sites, the structural change has a great influence on the hybridization between the Cas12a–crRNA complex and aptamer. The decrease of trans-cleavage activity of Cas12a activated by the ssDNA aptamer was sharp compared to that of the control group, which resulted in a high inhibition rate (IR%).
The dominated sites of the analyte and aptamer can be inferred by analyzing the results of different inhibition rates of different crRNAs.
This not only provides a supplement for our previously reported detection strategy for small molecules based on Cas12a, and further improves the integrity to ensure it can be applied to all kinds of aptamers, but also enriches the application fields of the Cas12a toolbox and fully exploits the potential of CRISPR technologies.
3.2. Evaluation of IR% values of different crRNAs
The dominated sites of AFB1 and the aptamer were first investigated. The 47-nt ssDNA aptamer against AFB1 (5′-GTT![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) TGTTGTCTCTCTGTGTCT
TGTTGTCTCTCTGTGTCT![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) TTCGCTAGGCCC-3′) shows high affinity with a dissociation constant (Kd) of about 12 nM (the underlined bases can form a stem by complementary base pairs) (patent: PCT/CA2010/001292). It has a simple stem-loop structure as obtained from the oligo analysis website Mfold (https://unafold.rna.albany.edu/) (Fig. S1†). We tested several hybridization sizes between the crRNA and aptamer to make the analysis more efficient. Theoretically, the shorter the crRNA, the more accurate and efficient the analysis will be. However, when the hybridization size is below 20 bp, the trans-cleavage activity of Cas12a decreases, which in turn affects the analysis (data not shown). Finally, we designed 28 crRNAs to hybridize with every region of the aptamer with a certain hybridization size of 20 bp (AFB1-crRNA 1 to AFB1-crRNA 28 in Table S1†). For instance, AFB1-crRNA 1 (in short crRNA 1) was designed to hybridize with site 1 to site 20 of the aptamer (5′ to 3′), crRNA 2 was designed to hybridize with site 2 to site 21 of the aptamer and crRNA 28 was designed to hybridize with site 28 to site 47 of the aptamer.
TTCGCTAGGCCC-3′) shows high affinity with a dissociation constant (Kd) of about 12 nM (the underlined bases can form a stem by complementary base pairs) (patent: PCT/CA2010/001292). It has a simple stem-loop structure as obtained from the oligo analysis website Mfold (https://unafold.rna.albany.edu/) (Fig. S1†). We tested several hybridization sizes between the crRNA and aptamer to make the analysis more efficient. Theoretically, the shorter the crRNA, the more accurate and efficient the analysis will be. However, when the hybridization size is below 20 bp, the trans-cleavage activity of Cas12a decreases, which in turn affects the analysis (data not shown). Finally, we designed 28 crRNAs to hybridize with every region of the aptamer with a certain hybridization size of 20 bp (AFB1-crRNA 1 to AFB1-crRNA 28 in Table S1†). For instance, AFB1-crRNA 1 (in short crRNA 1) was designed to hybridize with site 1 to site 20 of the aptamer (5′ to 3′), crRNA 2 was designed to hybridize with site 2 to site 21 of the aptamer and crRNA 28 was designed to hybridize with site 28 to site 47 of the aptamer.
The AFB1 sample used in this study is dissolved in the organic solvent acetonitrile, which may have an effect on the Cas12a trans-cleavage activity. Before evaluating the IR% values of the different crRNAs, the effect of this organic solvent on the Cas12a trans-cleavage activity was evaluated (Fig. S2†). It is indicated that the organic solvent acetonitrile has no significant effect on the Cas12a trans-cleavage activity.
To evaluate the effect of different crRNAs on the inhibition rate of Cas12a trans-cleavage activity in the presence of AFB1, 28 different crRNAs were tested in the Cas12a fluorescence reporter system compared to the respective control groups (Fig. S3†). As shown in Fig. S3,† there were two kinds of IR% vs. time curves. The IR% values of several curves did not change drastically with time, such as crRNA 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 18, 20, 22, 23, 24, 25, 26, 27, and 28. Meanwhile, the IR% values of crRNA 14, 16, 17, 19, and 21 were relatively stable at first, and then dropped suddenly. The second curve was caused by the gradual saturation of the fluorescence intensity of the control group, and the dropped IR% values could not reflect accurate results. Thus, the appropriate data were collected to evaluate the IR% values of different crRNAs (crRNA19 and 21 at 5 min and the others at 10 min) (Fig. 2). According to the schematic, crRNA with high IR% indicated that the dominated site was within the hybridization region. The mean IR% value of crRNA 7–24 was significantly higher than the mean value of the others (One-way ANOVA analysis). This suggested that the repeated site in crRNA 7–24 (sites 24–26 from the 5′ end to the 3′ end in the AFB1 ssDNA aptamer) was speculated to be the dominated site (Fig. S4†). Few reported studies have specifically analyzed the dominated sites between AFB1 and its 47 nt ssDNA aptamer. However, several studies have been reported to develop AFB1 biosensors from different experimental strategies. For example, the fluorescence anisotropy-based AFB1 detection approach was developed by labeling fluorophores on different bases.17 However, the quenching effect of the G base made it hard to be labeled with a fluorophore, greatly limiting the application in aptamer sequences with the G base. Among these studies, we could analyze that the sites causing the maximum change of signal were sites 24 and 26,18 sites 25–27 (ref. 11) and sites 20, 24, 26, and 28.17 Although the results are different, they showed that the dominated sites are near the 3′ end of the loop structure and our results are consistent with the reported results in this study.
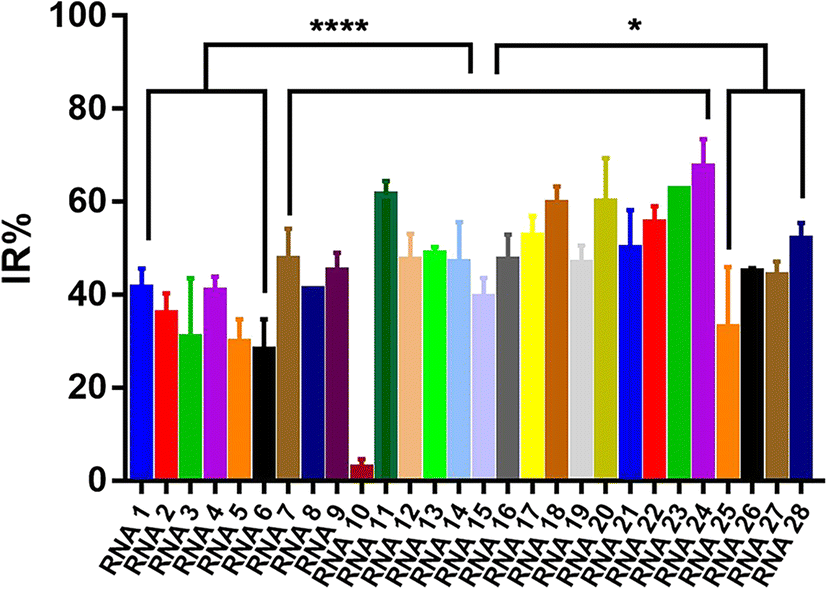 | ||
| Fig. 2 Evaluation of the IR% values of different crRNAs. RNA 1–28(crRNA 1–28): 28 different crRNAs hybridized to the different regions of the 47-nt aptamer in the AFB1/aptamer complex. Error bars in the figures present the standard deviation (SD) based on at least three individual tests. ****: P < 0.0001. *: P < 0.05. | ||
3.3. Verification of the AFB1 dominated site by the BLI (biolayer interferometry) assay
By analyzing the IR% results from different crRNAs, sites 24–26 from the 5′ end to the 3′ end in the 47 nt AFB1 ssDNA aptamer (in this section, also named aptamer-WT) were considered to be the dominated sites for interaction. Sites 24–26 of the aptamer-WT (“TGT”) was replaced with “ACA” and the new aptamer was named aptamer-MT. The BLI assay was employed to test the affinity of the aptamers (aptamer-MT and aptamer-WT) and AFB1 (Fig. 3). Aptamer-WT had a strong affinity with AFB1 and its Kd was 160 nM. Meanwhile, aptamer-MT had no interaction with AFB1 (Fig. 3B). This indicated that sites 24–26 of aptamer-WT (“TGT”) were important for binding with AFB1, and replacing these three bases removes the interaction and disrupts the aptamer affinity. | ||
| Fig. 3 BLI assay to analyze the interaction between AFB1 and aptamers. A) Aptamer-WT. B) Aptamer-MT. 0 to 120 s is the association process and 120 to 240 s is the dissociation process. | ||
3.4. Three-dimensional modeling and molecular docking
Generally, knowledge of the 3D structure of receptor and ligand molecules is crucial for virtual screening. However, the lack of computational tools for the 3D modeling of ssDNA molecules leads to a great challenge for researchers.11 We follow a three-step method previously reported19 to predict the 3D model of ssDNA aptamers. The main steps consist of: (i) building ssDNA secondary structures, (ii) translating the ssDNA secondary structures into equivalent 3D ssRNA models, and (iii) converting the 3D ssRNA models into 3D ssDNA models. Then, the 3D structures of the 47-nt ssDNA aptamer were used for molecular docking studies (Fig. 4A and B).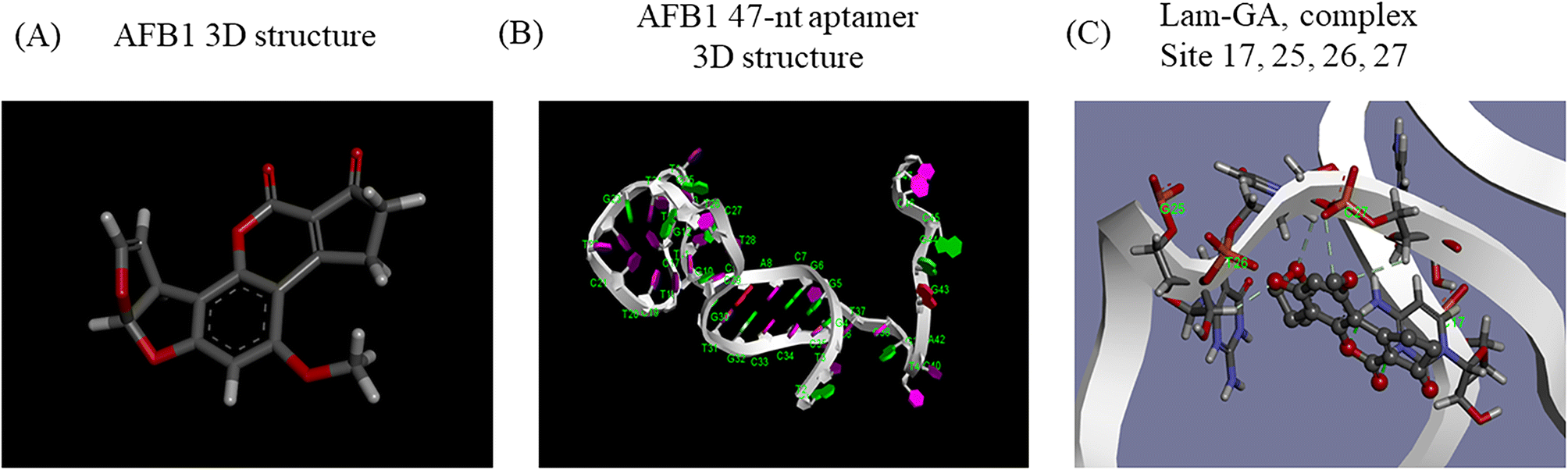 | ||
| Fig. 4 Three-dimensional modeling and molecular docking. A) The predicted 3D structure of AFB1. B) The predicted 3D structure of the 47-nt ssDNA aptamer of AFB1. C) The docking result of AFB1 and the aptamer under the Lamarckian genetic algorithm. Dashes represent conventional hydrogen bonding. | ||
Molecular docking was used to predict the dominated sites and binding energy of interactions between the 47-nt ssDNA aptamer and AFB1. The docking energy is −5.83 kcal mol−1 and the binding residues of the aptamer are C17, G25, T26 and C27 (Fig. 4C). The results showed that these four sites interacted with the furan and coumarin moieties of AFB1 through hydrogen bonding which are consistent with reported results.11,17 This is not completely consistent with our experimental results (sites 24–26), which illustrates that computational tools for 3D modeling still have limitations in predicting accurate molecular interactions under complex conditions, especially in the case of lacking computational tools for 3D modeling of ssDNA molecules. However, it is still good enough as an auxiliary tool and it will achieve excellent performance in analyzing dominated sites of the aptamer together with our experimental strategy.
3.5. Special hybridization region
For crRNA10, the fluorescence signal intensities in the control group and the experimental group were very low (Fig. S3†), which also led to the low IR% (Fig. 2). The low fluorescence intensity of the control group demonstrated that when the crRNA10/Cas12a complex hybridized with the aptamer, the activated trans-cleavage activity was significantly weak. The hybridization region shifting one base to the 5′ end (crRNA 9) or one base to the 3′ end (crRNA 11) could both activate strong trans-cleavage activity. Interestingly, this result is only caused by different regions of crRNA hybridization with the aptamer. Coincidentally, crRNA 10 hybridized to the site 10–29 of aptamer, which is the whole loop in the predicted secondary structure (Fig. S5†).The trans-cleavage activity of Cas12a would be activated in the case of the crRNA/Cas12a complex hybridized with complementary ssDNA or complementary dsDNA with the PAM sequence (TTTN).20,21 However, whether a special DNA structure like a hairpin can activate the trans-cleavage activity of Cas12a has never been reported. Therefore, six DNA structures were designed to evaluate the effects of different special DNA structures on the activated trans-cleavage activity: ssDNA, hairpin, dsDNA-non PAM, triple-complex-non PAM, dsDNA-PAM and triple-complex-PAM (Fig. S6†). In addition, to illustrate that this is not a single case, the crRNA in this design (crRNA-special structure DNA in Table S1†) is different from crRNA 1–28. The annealed six DNA structures were hybridized with the same crRNA sequence and the activated trans-cleavage activities were analyzed through the fluorescence intensity. As shown in Fig. 5, ssDNA and dsDNA-PAM could activate trans-cleavage activity and dsDNA-non PAM could not activate trans-cleavage activity, which were consistent with the reported study mentioned above. Triple-complex-non PAM is a structure between ssDNA and dsDNA-non PAM and crRNA hybridized with the middle ssDNA region. This demonstrated that the process of crRNA hybridization was mainly controlled by the ssDNA structure yet the partial dsDNA at both ends near the middle ssDNA structure affects the trans-cleavage activity. The trans-cleavage activity activated by triple-complex-PAM was weaker than that activated by triple-complex-non PAM. This result supposed that the hybridization efficiency affected due to the PAM sequence is one more step to be verified in the hybridization process. In addition, the trans-cleavage activity activated by hairpin was similar to that activated by triple-complex-non PAM. Partial dsDNA at both ends and a 20-nt middle ssDNA to be hybridized were common characteristics for these two structures. Therefore, the recognition of the hairpin structure could be equivalent to the process shown in Fig. S7.† And the trans-cleavage activity activated by these two structures was weaker than that activated by ssDNA, which could be an explanation for the result of crRNA 10 activated weak trans-cleavage activity.
 | ||
| Fig. 5 Effects of different special DNA structures on activated trans-cleavage activity. Error bars in the figures present the standard deviation (SD) based on at least three individual tests. See the sequence of symbols in Fig. S6.† | ||
3.6. AFB1 biosensor and sensitivity
In the presence of the target molecule AFB1, the aptamer binds to it with high specificity and affinity, resulting in a decrease of the aptamer hybridized with the crRNA–Cas12a duplex. This decrease in activated Cas12a leads to a significant reduction in the fluorescence signal. Thus, an AFB1 biosensor was developed according to the “Molecular Radar” strategy previously proposed by our group.22 Different crRNAs led to different inhibition rates. A higher inhibition rate indicated a larger signal difference compared to the control group in the presence of target molecules. Thus, the crRNAs with high inhibition rates could be applied in the molecular biosensors to obtain better performance. Firstly, we optimized the experimental conditions to obtain the best detection performance (Fig. S8†). 25 nM AFB1 aptamer, 30 nM Cas12a/crRNA complex and a 20 min cutting time were selected for further study. Then, two crRNAs with high IR% (crRNA 11 and crRNA 24), two crRNAs with average IR% (crRNA 13 and crRNA 27) and a crRNA with low IR% (crRNA 10) were applied to establish five biosensors for AFB1 detection with different performances.Quantitative analysis was performed by monitoring the inhibition rate % (IR%) 20 min after the addition of different concentrations of AFB1 (Fig. 6A and S9†). For biosensor-crRNA 24, the resulting plot of IR% vs. AFB1 concentration was linear over the range of 100 ng mL−1 to 2 μg mL−1. The limit of detection (LOD) was calculated to be 0.8 ng mL−1, equivalent to 2.6 nM based on 3*SD/slope, where SD is the standard deviation of nine NC samples (Fig. S9A†). For biosensor-crRNA 11, the linear range was 200 ng mL−1 to 2 μg mL−1 and the LOD was calculated to be 4.2 ng mL−1, equivalent to 13.3 nM (Fig. S9B†). For biosensor-crRNA 13, the linear range was 500 ng mL−1 to 2 μg mL−1 and the LOD was calculated to be 4.5 ng mL−1, equivalent to 14.3 nM (Fig. S9C†). For biosensor-crRNA 27, the linear range was 750 ng mL−1 to 2 μg mL−1 and the LOD was calculated to be 4.7 ng mL−1, equivalent to 15.0 nM (Fig. S9D†). crRNA 10 has no response to AFB1 (Fig. S9E†). The results illustrated that the performance of the different biosensors varied with the different crRNAs, and biosensor-crRNA 24 performed best, reaching a LOD of 2.6 nM within 20 min. The sensitivity of our assay was comparable to those of fluorescence biosensors reported for AFB1 detection (Table S2†).
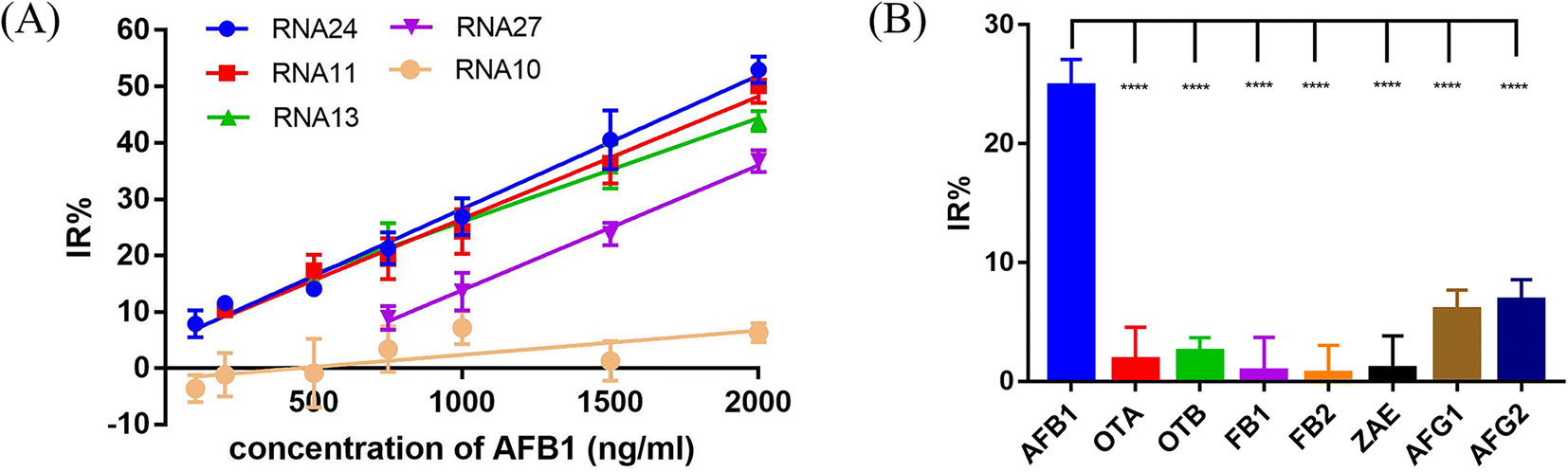 | ||
| Fig. 6 Sensitivity and specificity tests for AFB1 biosensors. (A) The sensitivity test with 100 to 2000 ng mL−1 AFB1. Linear calibration curves are used to show the linear range of 5 different crRNAs. (B) Specificity test with 750 ng mL−1 AFB1 and other mycotoxins and aflatoxin analogues including OTA, OTB, FB1, FB2, AFG1 and AFG2. The error bars indicate the standard deviation. ****: P < 0.0001. | ||
Among the five crRNAs studied in this section, the dominated sites 24–26 (“TGT”) were covered in the hybridization regions of crRNA 24, 11, 13 and 10. Apart from crRNA 10 with a special hybridization region, the biosensors based on crRNA 24, 11, and 13 all had better performance than that based on crRNA 27. This illustrated that our strategy not only analyzed the dominated sites between AFB1 and the aptamer but also developed an AFB1 biosensor with high sensitivity.
3.7. Specificity test and detection in a complex matrix for AFB1
Ochratoxin A (OTA), ochratoxin B (OTB), fumonisin B1 (FB1), fumonisin B2 (FB2), zearalenone (ZAE), aflatoxin G1 (AFG1) and aflatoxin G2 (AFG2) were used to evaluate the specificity of our biosensor-crRNA 24 (Fig. 6B). A slight response was caused by AFG1 and AFG2 and no response was caused by the other mycotoxins. The results demonstrated the good specificity of our assay for these mycotoxins and aflatoxin analogues.The performance of our biosensor in the complex sample matrix was evaluated by spiking with different concentrations of AFB1 in 5% red wine, beer and milk samples.
The estimated recovery of the strategy ranged from 91.6% to 94.47% in red wine, 104.8% to 110% in beer and 87.87% to 90.2% in milk (Table S3†). Further, we tested the AFB1 samples in the complex matrix using the ELISA detection kit method, and the results indicated the accuracy of the method used in the work (Fig. S10†). The above results illustrated the feasibility of our biosensor for detecting AFB1 in a complex sample matrix, and it has the potential to be applied for real sample analysis.
4. Conclusion
This study demonstrated a novel strategy to analyze aptamer dominated sites for small molecules based on CRISPR–Cas12a. By testing the different responses based on the fluorescence intensity inhibition rate of 28 different crRNAs designed to hybridize with every region of the AFB1 47-nt aptamer, sites 24–26 from the 5′ to 3′ end in the aptamer (“TGT”) were speculated to be the dominated sites, and this was verified by replacing the three bases through the BLI assay. 3D modeling and molecular docking were further used to not only obtain the binding energy and binding mode but also laterally validate the experimental results. Moreover, the effects of special structures of DNA on the activated Cas12a trans-cleavage activity were evaluated to enrich our understanding of Cas12a. This is essential to further apply CRISPR–Cas12 to a nucleic acid or small molecule-related biosensor and rationally design related oligonucleotide sequences. In addition, according to the results of the analyzed aptamer dominated sites, a strategy for the detection of AFB1 based on Cas12a was developed. Finally, AFB1 was quantitatively detected at 37 °C in 20 min with one simple step with high sensitivity and specificity, suggesting the potential applications of this strategy in field tests with complex matrix samples. Furthermore, considering that aptamers can bind to a wide range of targets in vitro—from small organic molecules and metal ions to proteins, and even viruses and cells—the universality of our aptamer dominated site analysis strategy and small molecular detection strategy is established. It can not only detect small molecules but also expand the target range. Any target with the corresponding adaptor can be included. In addition to mycotoxins related to food safety, target analytes can also be biomarkers related to medical diagnosis. The strategies revealed here provide a platform that can be adapted for POC and field diagnostic biosensors, expanding the application of the CRISPR–Cas system in the fields of biochemistry and biomedicine.Conflicts of interest
The authors declare that they have no conflicts of interest to this work.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (Grant No. 2019YFA0904800), the Project supported by the Joint Funds of the National Natural Science Foundation of China (Grant U2032210) and the National Natural Science Foundation of China (NSFC) (Grant 21938004).References
- S. Bakırdere, S. Bora, E. Bakırdere, F. Aydın, Y. Arslan, O. Komesli, I. Aydın and E. Yıldırım, Open Chem., 2012, 10, 675–685 CrossRef.
- S. Rawal, J. E. Kim and R. Coulombe, Res. Vet. Sci., 2010, 89, 325–331 CrossRef CAS.
- S. Pradhan and L. Ananthanarayan, J. Planar Chromatogr.--Mod. TLC, 2020, 33, 617–630 CrossRef CAS.
- M. Eslami, Z. Mashak, A. Heshmati, M. Shokrzadeh and A. S. Mozaffari Nejad, Toxin Rev., 2015, 34, 125–128 CrossRef CAS.
- B. Amirkhizi, S. R. Arefhosseini, M. Ansarin and M. Nemati, Food Addit. Contam., Part B, 2015, 8, 245–249 CAS.
- K. Setlem, B. Mondal, S. Ramlal and J. Kingston, Front. Microbiol., 2016, 7, 1909 Search PubMed.
- Z. Zhu, Y. Song, C. Li, Y. Zou, L. Zhu, Y. An and C. J. Yang, Anal. Chem., 2014, 86, 5881–5888 CrossRef CAS.
- X. Ma, W. Wang, X. Chen, Y. Xia, S. Wu, N. Duan and Z. Wang, Eur. Food Res. Technol., 2014, 238, 919–925 CrossRef CAS.
- J. Zhang, Z. Li, S. Zhao and Y. Lu, Analyst, 2016, 141, 4029–4034 RSC.
- L. Chen, F. Wen, M. Li, X. Guo, S. Li, N. Zheng and J. Wang, Food Chem., 2017, 215, 377–382 CrossRef CAS.
- M. Mousivand, L. Anfossi, K. Bagherzadeh, N. Barbero, A. Mirzadi-Gohari and M. Javan-Nikkhah, Anal. Chim. Acta, 2020, 1105, 178–186 CrossRef CAS.
- C. Wang, J. Qian, K. An, C. Ren, X. Lu, N. Hao, Q. Liu, H. Li, X. Huang and K. Wang, Biosens. Bioelectron., 2018, 108, 69–75 CrossRef CAS.
- Y. Seok, J.-Y. Byun, W.-B. Shim and M.-G. Kim, Anal. Chim. Acta, 2015, 886, 182–187 CrossRef CAS PubMed.
- J. Lerdsri, W. Chananchana, J. Upan, T. Sridara and J. Jakmunee, Sens. Actuators, B, 2020, 320, 128356 CrossRef CAS.
- Y. Xiong, J. Zhang, Z. Yang, Q. Mou, Y. Ma, Y. Xiong and Y. Lu, J. Am. Chem. Soc., 2020, 142, 207–213 CrossRef CAS PubMed.
- K. Abnous, N. M. Danesh, M. Ramezani, M. Alibolandi, M. A. Nameghi, T. S. Zavvar and S. M. Taghdisi, Anal. Chim. Acta, 2021, 1165, 338549 CrossRef CAS.
- L. Sun and Q. Zhao, Talanta, 2018, 189, 442–450 CrossRef CAS.
- C. Wang and Q. Zhao, Biosens. Bioelectron., 2020, 167, 112478 CrossRef CAS PubMed.
- I. Jeddi and L. Saiz, Sci. Rep., 2017, 7, 1178 CrossRef.
- M. Asnaashari, R. Esmaeilzadeh Kenari, R. Farahmandfar, S. M. Taghdisi and K. Abnous, Sens. Actuators, B, 2018, 265, 339–345 CrossRef CAS.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- C. Niu, C. Wang, F. Li, X. Zheng, X. Xing and C. Zhang, Biosens. Bioelectron., 2021, 183, 113196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The secondary structure (a simple stem-loop structure) of the 47-nt AFB1 aptamer from oligo analysis website Mfold; effect of organic solvent (acetonitrile) on the trans-cleavage activity of Cas12a; kinetic fluorescence results IR% of 28 RNAs; repeat sites in crRNA 7–24 (sites 24–26 from the 5′ end to the 3′ end in the AFB1 ssDNA aptamer) were speculated to be the dominated sites; RNA 10 hybridization with the whole loop sequence from sites 10–29 of the aptamer; sequences and symbols of six structures; the predicted process of recognition of hairpin structure; sensitivity tests for 5 different crRNAs. See DOI: https://doi.org/10.1039/d2sd00152g |
| This journal is © The Royal Society of Chemistry 2023 |