
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn amplification-free, 16S rRNA test for Neisseria gonorrhoeae in urine
Zhenrong
Zheng
a,
Yan
Cao
a,
Sukantha
Chandrasekaran
b,
Jacob J.
Schmidt
c,
Omai B.
Garner
b and
Harold G.
Monbouquette
 *a
*a
aChemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA. E-mail: hmonbouq@ucla.edu
bPathology and Laboratory Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA
cBioengineering, University of California, Los Angeles, Los Angeles, CA 90095, USA
First published on 11th November 2022
Abstract
An amplification-free, nanopore-based nucleic acid detection platform has been demonstrated for rapid, 16S rRNA sequence-specific detection of Neisseria gonorrhoeae at 10–100 CFU mL−1 in human urine against background bacterial flora at 1000 CFU mL−1. Gonorrhea is a very common notifiable communicable disease, antibiotic resistant strains have emerged, and the rate of reported gonococcal infections continues to increase. Since rapid clinical identification of bacterial pathogens in clinical samples is needed to guide proper antibiotic treatment and to control disease spread, it is important to engineer rapid, sensitive, selective, and inexpensive point-of-care (POC) diagnostic devices for pathogens such as N. gonorrhoeae. Our detector technology is based on straightforward conductometric detection of sustained blockage of a glass nanopore. Charge neutral, complementary peptide nucleic acid probes are conjugated to polystyrene beads to capture N. gonorrhoeae 16S rRNA selectively. In the presence of an electric field applied externally through a glass nanopore, the PNA-microbead conjugates that acquire substantial negative charge upon target hybridization are driven to the smaller diameter nanopore. At least partial blockage of the nanopore results in a sustained drop in ionic current that can be measured easily with simple electronics. The ability to detect N. gonorrhoeae over the range of 10 to 100 CFU mL−1 spiked in human urine was demonstrated successfully with estimated sensitivity and specificity of ∼98% and ∼100%, respectively. No false positives were observed for the control group of representative background flora (E. coli, K. pneumoniae, and E. faecalis) at 1000 CFU mL−1. Also, N. gonorrhoeae at 50 CFU mL−1 was successfully detected against 1000 CFU mL−1 of background flora in urine. These results suggest that this amplification-free technology may serve as the basis for rapid, inexpensive, low-power detection of pathogens in clinical samples at the POC.
Introduction
Gonorrhea is the second most common notifiable sexually transmitted infection in the US, and its prevalence has been increasing.1 A total of 677![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 769 cases of gonorrhea were reported to the US Centers for Disease Control and Prevention (CDC) in 2020 out of an estimated 1.6 M new infections that occur each year in the US.1,2 Since the historic low in 2009, reports of gonorrhea infection have increased 111%.1 Alarmingly, strains of the Gram-negative bacterium responsible for gonorrhea infections, Neisseria gonorrhoeae, have developed antibiotic resistance; and about half of all gonorrhea infections reported in 2020 were caused by an antibiotic resistant strain.1 Only one recommended treatment remains: the cephalosporin, ceftriaxone.2 Unfortunately, the infection often is asymptomatic in women, and if untreated, pelvic inflammatory disease leading to ectopic pregnancies and infertility can result.3 Currently, the CDC recommends that all sexually active women under age 25, as well as sexually active gay or bisexual men, be tested each year.3 In order to guide proper usage of antibiotics, to perform timely treatment, and to curtail disease spread, rapid, inexpensive point-of-care (POC) diagnostic tests are needed urgently.
769 cases of gonorrhea were reported to the US Centers for Disease Control and Prevention (CDC) in 2020 out of an estimated 1.6 M new infections that occur each year in the US.1,2 Since the historic low in 2009, reports of gonorrhea infection have increased 111%.1 Alarmingly, strains of the Gram-negative bacterium responsible for gonorrhea infections, Neisseria gonorrhoeae, have developed antibiotic resistance; and about half of all gonorrhea infections reported in 2020 were caused by an antibiotic resistant strain.1 Only one recommended treatment remains: the cephalosporin, ceftriaxone.2 Unfortunately, the infection often is asymptomatic in women, and if untreated, pelvic inflammatory disease leading to ectopic pregnancies and infertility can result.3 Currently, the CDC recommends that all sexually active women under age 25, as well as sexually active gay or bisexual men, be tested each year.3 In order to guide proper usage of antibiotics, to perform timely treatment, and to curtail disease spread, rapid, inexpensive point-of-care (POC) diagnostic tests are needed urgently.
The historical method for N. gonorrhoeae detection in a clinical sample has long entailed culturing; however, at least a day is required for results that must be generated by skilled technicians.4 Microscopic examination of urethral smears also can be used to provide evidence of infection, but the reliability of this diagnostic approach depends strongly on the quality of the specimen and the experience of the microscopist.4 Currently, nucleic acid amplification tests (NAATs) are the preferred methods for gonorrhea diagnostic detection due to sensitivities >90% and very high specificities of ∼99%.5 However, most NAATs are conducted in the clinical laboratory where the turnaround time also is a day or longer, and these complex systems must be operated by trained personnel. POC NAATs have emerged on the market recently (i.e., Binx io, Visby Medical Sexual Health Test),6,7 but an external power source is required for these complex devices that entail nucleic acid (NA) isolation to high purity for removal of polymerase inhibitors; tight control of complex reaction steps involving expensive, perishable reagents (e.g., primers, polymerase, and nucleotides); optical or electrochemical means to detect amplicons; and assay times of ∼15–30 minutes. An ideal NA-based POC diagnostic system would exhibit an assay time of ∼5 minutes, would be battery powered and completely portable, would be inexpensive (∼$20 per test), and would provide competitive sensitivity and specificity of ≥95%.8,9
The need for more optimal POC diagnostic tests has provided impetus for a number of studies focused on simpler, yet powerful, amplification-free, NA-based detection approaches. The “gold standard” limits of detection (LODs) of commercial clinical laboratory-based NAATs available for N. gonorrhoeae in urine in the ∼1–100 CFU mL−1 range are reflective of clinical need and correspond to NA LODs at the single-digit aM (10−18 M) level.10 Over the past 10 years or so, many reports, including ours, have appeared describing NA amplification-free systems exhibiting these very low LODs for NAs of specific sequence.10,11 Our system utilized here relies on the use of peptide nucleic acid (PNA) capture probes.12 PNA is an uncharged polyamide analog to DNA/RNA that we bind covalently to carboxyl-functionalized microbeads to form nearly charge neutral bead-probe conjugates. When the neutral PNA-microbead hybridizes target NA, it gains substantial negative charge thereby making it mobile in an electric field. If the negatively charged PNA-microbeads with hybridized target NA are directed to a glass nanopore of lesser diameter, they will at least partially block it, resulting in a sustained, easily measured drop in ionic current. In previous work, our group has successfully demonstrated the detection of E. coli rRNA at a concentration of 1 aM against a 106-fold background of P. putida RNA and E. coli at 10 CFU mL−1 against a 106-fold background of viable P. putida.11 However, few NA amplification-free detection schemes, including ours, had been tested with clinical, or even mock clinical, specimens.10
Experimental
Materials
Carboxyl-functionalized, 820 nm-dia. Polystyrene microspheres and Vivaspin® 2 mL ultrafiltration devices were purchased from Bangs Laboratories, Inc. (Fishers, IN). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) was obtained from ThermoFisher Scientific (Waltham, MA). 2-(N-Morpholino)ethanesulfonic acid (MES), methoxypolyethylene glycol amine (mPEG-amine) and ethanolamine were purchased from Sigma-Aldrich (St. Louis, MO). Peptide nucleic acid (PNA) probes were synthesized by PNA Bio (Thousand Oaks, CA) and arrived as >95% HPLC-purified, lyophilized powders. E. coli (ATCC 25922), P. putida (ATCC 12633), spy broth and nutrient broth were purchased from American Type Culture Collection (Manassas, VA). Direct-zol RNA Miniprep kits and TRI Reagent were purchased from Zymo Research (Irvine, CA). Two mm-diameter, 4 mm-long Ag/AgCl pellet electrodes were purchased from A-M systems, Inc. (Carlsborg, WA). GE Healthcare Life Sciences Anotop 25 syringe filters (25 mm-diameter, 0.02 μm pore) were supplied by Genesee Scientific (San Diego, CA).Methods
 | ||
| Fig. 1 Glass chip detector assembly. (a) The 1 cm × 1 cm glass chip with ∼1 μm-thick membrane and nanopore in the center. (b) Micrograph of the ∼1 μm-thick membrane at the chip center. (c) Micrograph of the ∼500 nm nanopore at the center of the glass membrane. (d) The glass chip sandwiched between PDMS seals and Teflon chambers (6 mm × 6 mm × 8 mm). The assembly was secured with four bolts (not shown). Fig. 1(a)–(c) reproduced from ref. 11 with permission from the Royal Society of Chemistry. | ||
000 rpm in microfuge tubes. One fourth of the final product was set aside for zeta potential measurements, while the rest of the PNA-beads were stored in hybridization buffer at room temperature (10 mM NaCl, 25 mM Tris-HCL, pH 7). The zeta potential was measured for modified microbeads suspended in the testing buffer (10 nM KCl, 5.5 mM HEPES, 0.01% Tween-80, pH 7) using a Malvern Zetasizer nano ZS (Malvern Instruments Ltd, Worcestershire, England). A zeta potential in the negative single-digit mV range was taken as evidence of successful microbead surface modification.
Results and discussion
Forty-four samples of N. gonorrhoeae spiked in human urine over the 10–100 CFU mL−1 concentration range (14 at 10 CFU mL−1, 10 each at 50 and 100 CFU mL−1, and 10 at 50 CFU mL−1 against representative bacterial flora at 1000 CFU mL−1) were processed and tested as described above. The bacterial flora control representative of microbes frequently present in human urine included E. coli, K. pneumoniae, and E. faecalis. Out of these 44 trials, only one false negative was observed at 50 CFU mL−1. No positive detection results were recorded for any of 10 samples of the background flora alone at a concentration of 1000 CFU mL−1 in human urine, which served as a negative control. Sample data showing easily recognized, reproducible detection events as well as the lack of response to the negative control is presented as Fig. 2, and the complete results are summarized in Table 1.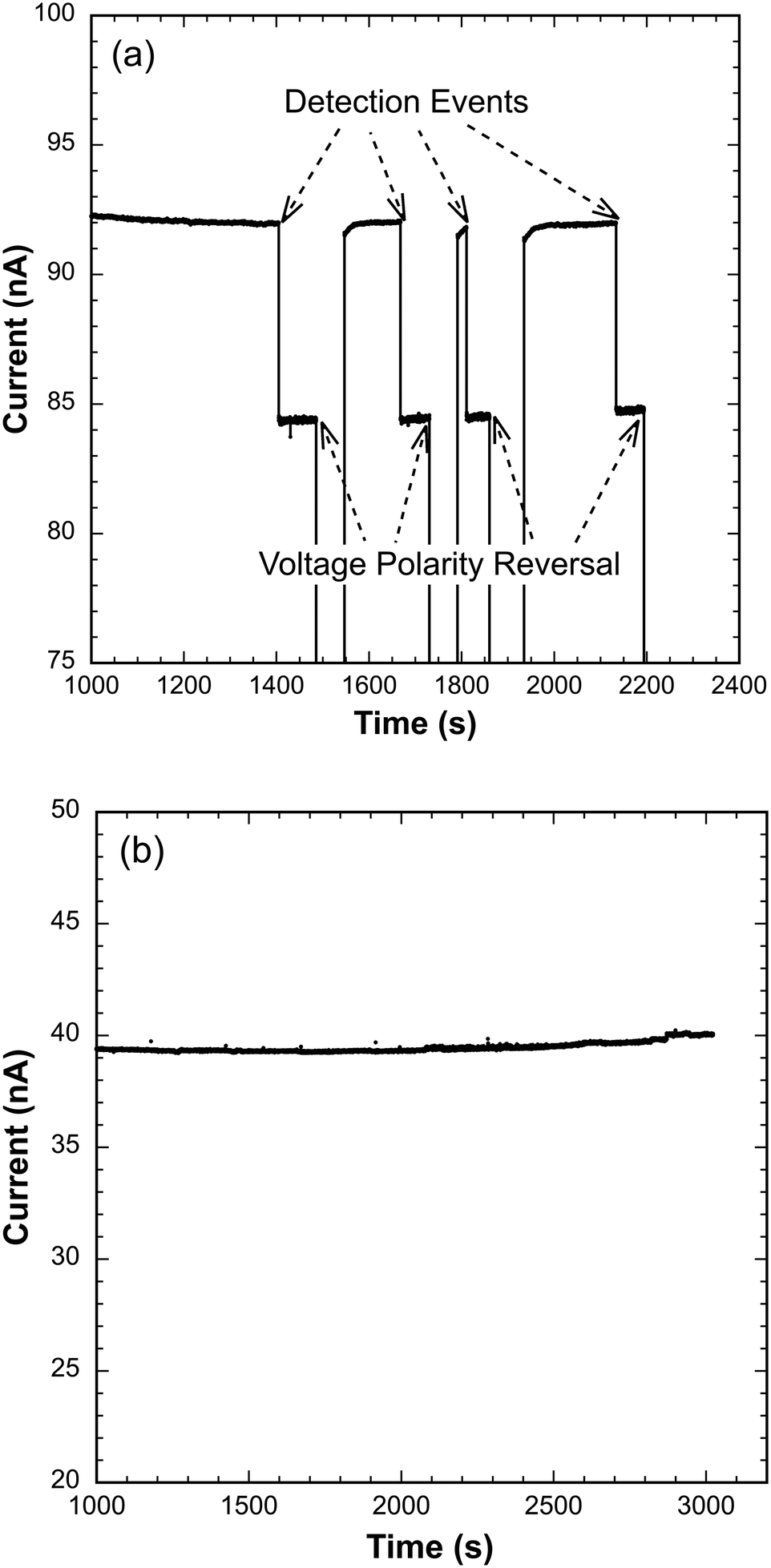 | ||
| Fig. 2 Detector current response to the presence of target N. gonorrhoeae 16S rRNA and the lack of response to RNA extracted from background bacterial flora alone. (a) Reproducible, sustained current signal step reductions in response to the presence of PNA-beads with hybridized target 16S rRNA extracted from N. gonorrhoeae at 10 CFU mL−1 in human urine. The applied voltage for detection events was 1.5 V. After a sustained current signal of ∼50 s, the voltage polarity was reversed temporarily to −1.5 V to drive bead complexes away from the pore before demonstrating signal reversibility by reimposing a potential of 1.5 V. (b) No current signal in response to the presence of PNA-beads hybridized with RNA extracted from background bacterial flora spiked at 1000 CFU mL−1 in human urine (negative control). | ||
| Sample | Agreement with expected resulta |
|---|---|
| a (Number of expected test results)/(number of tests). b Background bacterial flora: E. coli, K. pneumoniae, and E. faecalis. | |
| 10 CFU NG/mL | 14/14 |
| 50 CFU NG/mL | 9/10 |
| 100 CFU NG/mL | 10/10 |
| 50 CFU NG/mL & 1000 CFU florab/mL | 10/10 |
| 1000 CFU florab/mL | 10/10 |
| Total agreement | 53/54 (98.1%) |
The data presented in Table 1 provide the basis for preliminary estimates of sensitivity and specificity of 98% (95% CI: 88–100%) and 100% (95% CI: 69–100%), respectively, for detection of N. gonorrhoeae over the range of 10 to 100 CFU mL−1 when spiked in sterile, pooled human urine. Note that the first detection event for true positive cases, indicated by the first sustained ionic current drop, occurred from 500 seconds to 1500 seconds. The variation in time to signal arises from the random initial distance of beads with hybridized target from the glass chip in the detector assembly (Fig. 1d). Absence of ionic current drop for over 3000 seconds constituted negative detection (likely an extreme waiting period based on many test runs), and only one false negative was observed at 50 CFU mL−1. Since 50 CFU mL−1 is well above the detection limit of our device, we suspect that RNA instability may have played a role since samples were lysed at the off-campus clinical laboratory and subsequently transported on campus for testing. Nevertheless, the preliminary sensitivity and specificity data presented here compare very well to the currently preferred NAATs, both those that are lab-based and those that are designed for the POC.6,7
As an amplification-free test, this technology is considerably less complex than NAATs and avoids the need for perishable reagents (i.e., polymerase, primers, nucleotides). The current lab-based version of our amplification-free test with the glass nanopore detector requires ∼10 minutes for RNA extraction, ∼10 minutes for hybridization, and ∼15 minutes for sample detection, which makes it competitive in overall assay time with many fully automated NAATs. However, the detection chamber in a microfluidic device under development is reduced ∼10× in characteristic dimension thereby reducing the transit time of bead-PNA conjugates with hybridized target to the glass nanopore detector. In the near future, we anticipate reporting on the time savings achieved with our glass chip detectors integrated into a microfluidic system.
Conclusions
A novel amplification-free rRNA test based on a nanopore glass detector was used successfully to assay for N. gonorrhoeae spiked in human urine with high sensitivity (∼98%) and specificity (∼100%). N. gonorrhoeae was detected successfully over the 10–100 CFU mL−1 range and against background bacterial flora at 1000 CFU mL−1. These results suggest that this potentially rapid and inexpensive technology may one day prove advantageous for POC use. Indeed, with further improvement in overall assay time and integration of the glass nanopore detector with automated frontend sample processing, this simple, amplification-free detection technology may lead to rapid, inexpensive, low-power and portable POC pathogen detection devices with minimal reagent requirements.Author contributions
Z. Zheng and Y. Cao: data curation, formal analysis, investigation, methodology, validation, visualization, writing—original draft, writing—review & editing. S. Chandrasekaran: investigation, methodology, writing—review & editing. O. B. Garner: conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, writing—review & editing. J. J. Schmidt and H. G. Monbouquette: conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing—review & editing.Conflicts of interest
H. G. Monbouquette and J. J. Schmidt report holding equity in and Director positions at ElectroNucleics Inc., a company formed to commercialize the technology described in this publication. O. B. Garner serves in an advisory role to ElectroNucleics Inc.Acknowledgements
This research was supported by NIH (1R41AI141290).References
- CDC, Sexually Transmitted Disease Surveillance 2020, https://www.cdc.gov/std/statistics/2020/overview.htm#Gonorrhea, (accessed July 19, 2022).
- K. Schlanger and R. D. Kirkcaldy, Sex. Transm. Dis., 2021, 48, S91–S92 CrossRef.
- CDC, Gonorrhea—CDC Detailed Fact Sheet, https://www.cdc.gov/std/gonorrhea/stdfact-gonorrhea-detailed.htm, (accessed July 19, 2022).
- L.-K. Ng and I. E. Martin, Can. J. Infect. Dis. Med. Microbiol., 2005, 16, 323082 CrossRef.
- J. R. Papp, J. Schachter, C. A. Gaydos and B. Van Der Pol, Morb. Mortal. Wkly. Rep., 2014, 63, 1–19 Search PubMed.
- S. R. Morris, C. C. Bristow, M. R. Wierzbicki, M. Sarno, L. Asbel, A. French, C. A. Gaydos, L. Hazan, L. Mena, P. Madhivanan, S. Philip, S. Schwartz, C. Brown, D. Styers, T. Waymer and J. D. Klausner, Lancet Infect. Dis., 2021, 21, 668–676 CrossRef CAS.
- B. Van Der Pol, S. N. Taylor, L. Mena, J. Lebed, C. J. McNeil, L. Crane, A. Ermel, A. Sukhija-Cohen and C. A. Gaydos, JAMA Netw. Open, 2020, 3, e204819 CrossRef.
- C. A. Gaydos, Y. C. Manabe and J. H. Melendez, Sex. Transm. Dis., 2021, 48, S71–S77 CrossRef.
- L. M. Atkinson, D. Vijeratnam, R. Mani and R. Patel, Int. J. STD AIDS, 2016, 27, 650–655 CrossRef.
- Y. Cao, Z. Zheng and H. G. Monbouquette, Curr. Opin. Biotechnol., 2021, 71, 145–150 CrossRef CAS PubMed.
- B. Koo, A. M. Yorita, J. J. Schmidt and H. G. Monbouquette, Lab Chip, 2018, 18, 2291–2299 RSC.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS.
- L. Chui, T. Chiu, J. Kakulphimp and G. J. Tyrrell, Clin. Microbiol. Infect., 2008, 14, 473–479 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |