
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new detection mode for gold nanoparticle-linked immunosorbent assay (GNLISA) based on a clock reaction: instrument- and enzyme-free visual quantitative detection of prostate-specific antigen (PSA)†
Tianxiang
Wu
 * and
Xiao-Yuan
Li
* and
Xiao-Yuan
Li
Department of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong S.A.R, The People's Republic of China. E-mail: twuas@connect.ust.hk
First published on 10th August 2023
Abstract
The enzyme-linked immunosorbent assay (ELISA) has been serving as both the workhorse and the gold standard in immunoassays due to its high specificity, sensitivity, and accuracy, despite its known shortcomings and limitations. In this work, we report our effort to improve the current ELISA protocol significantly by achieving the following four goals all together: first of all, we achieved enzyme-free signal generation in ELISA by replacing the conventional enzyme-based color generation step with an enzyme-free pure chemical color change process from a clock reaction. Secondly, we achieved the use of time lapse characteristically associated with a clock reaction as the quantitative readout signal for analyte concentration. Thirdly, gold nanoparticles (AuNPs) were employed for dual functions, both as a secondary antibody carrier and a catalytic regulator for the clock reaction. And last but not least, the clocking time associated with the colorimetric detection in our modified ELISA can be read out either by the naked eye assisted with a stopwatch free from any benchtop instrument or more accurately on any benchtop instrument with a built-in timer and with pre-set absorbance associated with the characteristic absorption wavelength of the clock reaction. The viability of our method is demonstrated by the quantitative detection of PSA in human serum and validated by both instrument-based measurements and commercial ELISA kits. The naked-eye detection range for PSA was found to be from 1.00 to 100 ng ml−1 with a limit of detection (LOD) of 0.96 ng ml−1, which is lower than the typical threshold value (4 ng ml−1), and a sensitivity of ca. 3.1 ng mL−1 min−1 for the early diagnosis of prostate cancer.
1. Introduction
The enzyme-linked immunosorbent assay (ELISA) has been considered the gold standard in immunoassays due to its high specificity, sensitivity, and accuracy.1 ELISA has found extensive applications in medical diagnosis,2,3 immunology,4,5 vaccine development,4,6 toxicology,7,8 drug monitoring and the pharmaceutical industry,9,10 transplantation,11,12 and the food industry,13–15 among others. The conventional ELISA has been known to exhibit several shortcomings and limitations, including but not limited to (i) the tedious and laborious procedure, (ii) the lack of multiplexing options, (iii) the dependence on instruments for signal readout and quantitation, (iv) the requirement for a relatively high sample volume,16 and (v) the need for skilled and trained operating personnel. Therefore, there have been extensive efforts to modify the current ELISA protocol with the aim of improving or even overcoming its shortcomings.17–21 In the meantime, for its dominant applications in medical and clinical practices, the improvements on ELISA are expected to meet the World Health Organization (WHO)'s ASSURED requirements for bio-analytical platforms especially for the developing and under-developed areas, namely, (i) affordable, (ii) sensitive, (iii) specific, (iv) user-friendly, (v) rapid and robust, (vi) equipment-free, and (vii) deliverable to those in need for such technologies.22The signal generation and readout in the conventional ELISA is often based on a colorimetric method consisting of two main steps.23,24 The signal generation step employs an enzyme-based color change process of a chemical dye, while the signal readout and quantitation step often relies on a benchtop instrument to measure either absorbance or fluorescence.25 The quantitative measurement by the naked eye, the most convenient, desirable and instrument-free detection, cannot be achieved and implemented on the conventional ELISA. On the other hand, metallic nanoparticles, especially Au and Ag nanoparticles, display substantial color changes when the nanoparticles' size, shape, or local environment changes associated with the changes of their localized surface plasmon resonance (LSPR).26–28 There have been extensive reports on the use of various nanoparticles, especially gold nanoparticles (AuNPs) in ELISA to improve its signal generation and readout.29 For example, ELISA combined with LSPR is named plasmonic ELISA (pELISA).30 It appears that the more colors that can be read out in the concentration range comparable to that of the analyte, the higher the accuracy of the detection method when detected by the naked eye. By far, many multi-color detection schemes have been developed, including but not limited to wine red → colorless → yellow,31 red → blue or blue → red,32–35 blue → purple,36 blue → mauve,37 colorless → blue → red,38 colorless → yellow,39 and brownish → gray → cyan → green → blue → violet → red → colorless → yellow,21 to name just a few. However, without exception, these reported efforts still rely on instruments for quantitative detection. Recently, Qin's group reported a ruler-based method that reads the height associated with the air pressure in a modified ELISA to achieve enzyme-free and visual quantitative detection.19,20,40,41
Clock reactions belong to a special class of reactions that have been known for more than a century.42 The definition of a clock reaction is a system presenting an induction period (well-defined time lag) followed by an abrupt change in the concentration of some chemical species participating in the reaction. One unique feature of certain clock reactions is that it allows the establishment of quantitative relationships between the induction time (or clocking time) and the concentration of involved chemical species. The quantitative readout of time lapse requires only a stopwatch, offering a quantitative analytical method free from any benchtop instrument. In addition, certain clock reactions are known to involve the color change of reacting species, making them viable choices for quantitative detection via visual colorimetry. The use of clock reactions in quantitative chemical and biochemical detection has been rare.43–45 There were two clock reaction-based immunoassays to achieve instrument-free visual quantitative detection. The first work was reported in 1987, and the height of the chromogenic substance was used as the readout signal for detection.46 The second work was reported in 1997 where the time lapse was used as the readout signal for detection.47 However, both studies still utilized enzymes in their detection protocol. In this work, we report the combination of a modified ELISA (gold nanoparticle-linked immunosorbent assay or GNLISA) with a clock reaction to achieve instrument- and enzyme-free visual quantitative detection of prostate-specific antigen (PSA).
2. Experimental section
2.1 Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O), hexadecyltrimethylammonium bromide (CTAB), sodium borohydride (NaBH4, powder), methylene blue (MB+, chloride salt), mercury(II) chloride, cadmium(II) chloride, lead(II) chloride, iron(III) chloride, di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O), potassium phosphate monobasic (KH2PO4), sodium chloride (NaCl), potassium chloride (KCl), Tween 20, bovine serum albumin (BSA), and human serum were all from Sigma Aldrich and were used as received without further purification. Sodium citrate (Fisher Scientific), sodium thiosulfate solution (Fluka), coating prostate-specific antibodies (Monoclonal, Linc-Bio), prostate-specific antigen (Linc-Bio), labeling prostate-specific antibodies (Monoclonal, Linc-Bio), and a human kallikrein 3/PSA Quantikine ELISA kit (R&D System) were purchased from respective suppliers and were used without any further treatment.2.2 Instruments
All UV-vis absorption spectra were recorded on a UV-1800 spectrometer (Shimadzu, Japan). All cycle times of clock reaction were recorded using a stopwatch. The morphology and size distribution of the as-prepared AuNPs were measured with a JEOL 2010F analytical transmission electron microscope (TEM). Ultrapure deionized water from a NANOpure (Barnstead) source was used throughout the experiments. A microplate reader (Thermo Scientific) was used when using the commercial ELISA kits for antigen detection. Excessive labeling antibodies not modified on the AuNPs were removed using a centrifuge 5418 R (Eppendorf). The measurements were conducted at room temperature of 22 °C, while the constant temperature condition of 37 °C for certain steps was obtained on an incubator (Memmert).2.3 Preparation and characterization of AuNPs
The preparation of AuNPs was based on the well-established method using citrate reduction of HAuCl4 in water.48 Briefly, a 72.7 mL aqueous solution of 1 mmol L−1 HAuCl4 was added into a round-bottom flask equipped with a reflux condenser and then heated to boil under gentle stirring. 38.8 mmol L−1 sodium citrate (7.3 mL) was added rapidly to the solution. The solution was heated under reflux for another 15 min. The color changed from pale yellow to deep red. The solution was stirred continuously and cooled down to room temperature. Finally, the AuNPs were obtained and stored at 4 °C for further use. The as-prepared AuNPs were analyzed with TEM. As can be seen from the TEM image (Fig. S1†), the as-synthesized AuNPs are quite uniform in size and shape with an average size of ca. 13 nm.2.4 Preparation of the washing and blocking buffers
Washing buffer: 0.20 g KH2PO4, 1.44 g Na2HPO4·2H2O, 8.00 g NaCl, 0.20 g KCl, and 0.5 mL Tween 20 were dissolved in ultrapure deionized water. Then, the solution was transferred into a 1000 ml volumetric flask, and ultrapure water was added to 1000 ml.Blocking buffer: 2.00 g BSA was dissolved in 100 ml washing buffer.
2.5 Preparation of AuNPs loaded with the labeling antibodies
55 μL of 0.20 M NaOH was added into 5.0 mL of 3.0 nM AuNPs under stirring at 400 rpm. 50 μL of 1 mg mL−1 labeling antibody was then added into the mixture at room temperature and kept still for 30 min. The as-prepared mixture was then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 30 min. After that, 4.1 mL supernatant was discarded, and 100 μL of 10% BSA solution was added into the mixture to incubate at room temperature for 30 min. After the incubation, 4.4 mL washing buffer was added to the mixture and centrifuged at 13000 rpm at 4 °C for 30 min (this process was repeated three times). Finally, the precipitate was re-dissolved in 5.0 mL washing buffer and stored in a 4 °C refrigerator for further use.
000 rpm at 4 °C for 30 min. After that, 4.1 mL supernatant was discarded, and 100 μL of 10% BSA solution was added into the mixture to incubate at room temperature for 30 min. After the incubation, 4.4 mL washing buffer was added to the mixture and centrifuged at 13000 rpm at 4 °C for 30 min (this process was repeated three times). Finally, the precipitate was re-dissolved in 5.0 mL washing buffer and stored in a 4 °C refrigerator for further use.
2.6 The procedure of GNLISA for PSA detection
1 mg mL−1 coating antibody was diluted 200 times with 50 mM bicarbonate of pH 9.60 to obtain 5 μg mL−1 coating antibody solution. Then, 100 μL of 5 μg mL−1 coating antibody solution was added into the wells of a 96-well plate and incubated at 4 °C overnight. After removing the coating antibody, the plate wells were washed three times by filling the wells with 300 μL washing buffer. 200 μL blocking buffer was then added into the plate wells to block the remaining protein binding site for 2 h at room temperature. After three times washing with 300 μL washing buffer, 100 μL of different concentrations of PSA antigen were added into each well. After 1 hour incubation at 37 °C, the plate wells were washed three times with 300 μL washing buffer. 100 μL AuNPs loaded with the labeling antibodies were added to each well and the plate was incubated for 1 hour at 37 °C. After three times washing with 300 μL washing buffer, the plate wells were washed with ultrapure water three times as well. 75 μL of pH 7.96 Britton–Robinson buffer (B–R buffer), 10 μL of 0.10 M Na2S2O3, 10 μL of 1 μM Cd2+, 5 μL of 0.10 M CTAB, and 3.5 μL of 0.45 mM MB+ were added into each well for 40 min to leach the AuNPs into Au(S2O3)23−. Then, 20 μL of ω = 1.00% NaBH4, was added to the plate wells, and the timer was then started. After the color changed from navy blue to colorless and back to light blue (0.68 μM MB+, absorbance was at ca. 0.05), the timer was stopped to record the cycle time.3. Results and discussion
3.1 The procedure of the method
Fig. 1 shows the mechanism of the proposed GNLISA and clock reaction for the instrument- and enzyme-free visual quantitative detection of PSA. Briefly, a typical sandwich-type ELISA is used and further modified. The coating antibody immobilized on the surface of the wells of a 96-well plate was utilized for the capture of the antigen. Next, AuNPs loaded with the labeling antibodies were combined with the antigen to form an antibody–antigen–second antibody sandwich complex on the well surface. The AuNP leaching reaction in the scheme is given below:| Leaching Reaction: 4Au + 8S2O32− + O2 + 2H2O → 4[Au(S2O3)2]3− + 4OH− |
 | ||
| Fig. 1 Scheme of detection of PSA based on the GNLISA and clock reaction. | ||
3.2 Optimization of the key assay conditions
When the concentration of Na2S2O3 is lower than 0.05 M after NaBH4 is added, the color cannot become colorless, or the time for the color to become colorless is too long (at least 6 min) to be applied as a cycle time for 100 ng ml−1 PSA. As for 0 ng ml−1 PSA, when the concentration of Na2S2O3 is lower than 0.1 M, it shows the same phenomenon. As shown in Fig. 2A, when the concentration of Na2S2O3 is in the range from 0.05 to 1.00 M for 100 ng ml−1 PSA, the cycle time is positively correlated with the concentration of Na2S2O3. For 0 ng ml−1 PSA, the same phenomenon occurs in the concentration range from 0.10 M to 1.00 M. Due to the higher concentration of Na2S2O3, more AuNPs will be leached into Au(S2O3)23− in 40 min leaching time, which results in a higher concentration of Aux/Pby nanoalloy. The higher concentration of Aux/Pby nanoalloy leads to a longer cycle time. Finally, 0.10 M Na2S2O3 was selected for the experiment after considering the sensitivity (the cycle time difference between 0 and 100 ng ml−1 PSA antigen divided by (100–0) ng ml−1) and effectiveness (the length of cycle time and leaching time).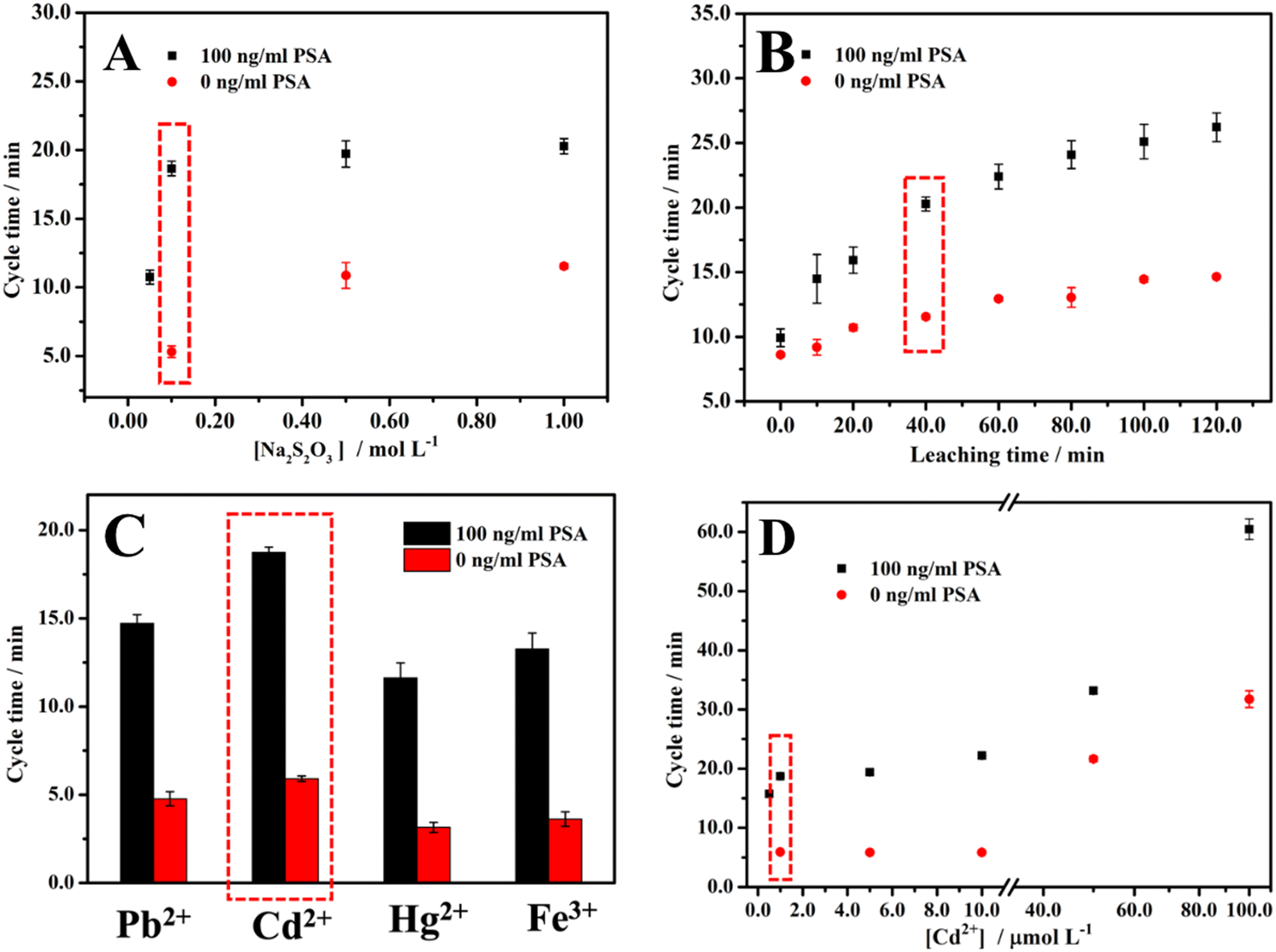 | ||
| Fig. 2 The effect of (A) the concentration of the leaching agent Na2S2O3, (B) the leaching time of AuNPs, (C) the different AuNP passivating metal ions at 1 μM concentration, and (D) Cd2+ at different concentrations on the cycle time. See text for the detailed conditions. | ||
Since it has been reported that some metal ions will accelerate the leaching reaction, and different Aux/My nanoalloys show different effects on the catalytic clock reaction, we used Pb2+ ions (Cd2+ ions display a similar trend but higher activity) when optimizing the leaching time.45,50–53 As shown in Fig. 2B, the cycle times for 0 and 100 ng ml−1 PSA are both positively correlated with the leaching time. According to the scheme of the method, the cycle time for 0 ng ml−1 PSA is unrelated to the leaching time, which is inconsistent with the result. The possible reason is the non-specific adsorption of the AuNPs loaded with the labeling antibodies. The reason for the positive correlation between the cycle time and the leaching time is that a longer leaching time means that more AuNPs will be leached to produce more Au(S2O3)23−, thereby leading to a higher concentration of Aux/Pby nanoalloy particles. Considering the balance between sensitivity and effectiveness in the measurements, we chose 40 min leaching time for all the experiments in measuring PSA.
It was reported that the leaching reaction could be accelerated in the presence of Pb2+ or other divalent metal ions.50–53 The effect of the concentration of Pb2+ on the cycle time was explored (Fig. S2†). When the concentration of Pb2+ is lower than 0.50 μM after NaBH4 is added, either the color of the solution remains unchanged or the time for the color to become colorless is too long (>6 min) to be practically used as a cycle time for 100 ng ml−1 PSA. As for 0 ng ml−1 PSA, when the concentration of Pb2+ is lower than 1.0 μM, the same phenomenon occurs. As shown in Fig. S2,† when the concentration of Pb2+ was in the range from 0.50 to 100 μM for 100 ng ml−1 PSA, the cycle time was in a positive correlation with the concentration of Pb2+. When the Pb2+ concentration was in the range from 1.0 to 100 μM, the same relationship between the cycle time and the concentration of Pb2+ was obtained for 0 ng ml−1 PSA. This is because the higher the concentration of Pb2+, the more AuNPs will be leached into Au(S2O3)23−, resulting in a higher concentration of Aux/Pby nanoalloy particles. The higher Aux/Pby nanoalloy particle concentration would result in a longer cycle time. We chose 1.0 μM Pb2+ concentration as the optimal experiment condition considering the balance between the sensitivity and effectiveness.
Different metal ions have different effects on the leaching and catalytic reactions by forming different types of Aux/My nanoalloy particles. It has been shown that Pb2+, Cd2+, Hg2+, and Fe3+ all have a noticeable effect on the cycle time.40 Therefore, the effects of Pb2+, Cd2+, Hg2+, and Fe3+ at a concentration of 1.0 μM on the cycle time were comparatively studied and the results are shown in Fig. 2C. Since Cd2+ is more relatively less toxic than Pb2+, the effect of Cd2+ at different concentrations on the cycle time was also studied and the results are shown in Fig. 2D. Balancing all the considerations, we selected Cd2+ at 1.0 μM concentration for the following experiments.
3.3 Instrument-free visual quantitative detection of PSA
Quantitative PSA detection using time lapse as the readout both by the naked eye/stopwatch (Fig. 3A) and on a benchtop UV-vis spectrophotometer (Fig. 3B) was conducted. The video of the color change corresponding to different concentrations of PSA is shown as Video S1 in the ESI.† The cycle times were 5.9, 6.6, 7.0, 9.0, 13.4, 15.9, and 18.7 min with the naked eye for 0, 1.00, 5.00, 10.0, 25.0, 50.0, and 100 ng ml−1 PSA, respectively. For the same set of samples with the measurements on a UV-vis spectrophotometer in time mode at the absorbance of 0.05, the cycle times were 5.5, 6.3, 7.9, 9.2, 13.1, 15.9, and 18.8 min, respectively, in quite good consistency with the cycle times obtained by the naked eye/stopwatch method (Fig. 3A). This demonstrates that the cycle times observed with the naked eye are reliable. The limit of detection (LOD: the PSA concentration corresponding to a signal that is three times the standard deviation above the zero calibrator) of this assay was measured, where the concentration of PSA was increased from 0 to 100 ng ml−1 and the cycle time ranged from 5.9 to 18.7 min. The calibration curve of the cycle time against the PSA concentration was linear from 1 to 100 ng ml−1 (r2 = 0.992, n = 3) with a LOD of 0.96 ng ml−1 (Fig. 3C). The threshold value was 4 ng ml−1 for the early diagnosis of prostate cancer,54 indicating that this method, when optimized, can be employed in the early diagnosis of prostate cancer. We also explored the cycle times observed by different observers and the results are listed in Table S1.† Different observers did show variation in the judgment of the endpoint color, and the typical deviation is less than 0.7 min. Table S2† summarizes the advantages (instrument- and enzyme-free visual quantitative detection) of this method in comparison with other reported methods. The relevant literature studies are also given in the ESI.† | ||
| Fig. 3 (A) The times of color change of MB+ in the clock reaction for different concentrations of PSA, (B) the changes of absorbance at 665 nm (a characteristic peak of MB+) with time for different concentrations of PSA. The points at a–g denote the times when the absorbance at 665 nm is 0.05, and (C) the corresponding plot of time observed by the naked eye vs. the concentration of PSA. Inset: calibration plot of PSA. | ||
3.4 Selectivity of the method
The selectivity of our GNLISA method in the PSA detection was examined and is shown in Fig. 4. We investigated the cycle times in the presence of various possible interferents and then compared them with the cycle time for PSA alone, including uric acid (UA), glucose, ascorbic acid (AA), immunoglobulin G (IgG), carcinoembryonic antigen (CEA), and alpha-fetoprotein (AFP). We also studied the cycle time of PSA mixed with interferents and then compared it with the cycle time for PSA alone. Fig. 4A shows the colors of 1000 ng ml−1 interfering substances and 25 ng ml−1 PSA after 0, 3.0, 6.5, and 13.5 min of reaction. The corresponding observed cycle times are listed in Fig. 4B and Table S3.† In the presence of an interfering substance at a concentration 40 times that of PSA, the cycle time did not show appreciable change. These results illustrate that the presence of individual interfering substances does not affect the PSA detection. To further illustrate the high selectivity of this method, we also explored a set of binary systems and compared the cycle time for PSA alone with the cycle time for PSA mixed with individual interfering substances. The results are listed in Fig. 4C and D, and Table S3,† respectively. Fig. 4C shows the colors of 25 ng ml−1 PSA and 25 ng ml−1 PSA + 1000 ng ml−1 interfering substances after 0, 3.0, 14.0, and 17.0 minutes of reaction. Fig. 4D and Table S3† show the cycle times for 25 ng ml−1 PSA and 25 ng ml−1 PSA + 1000 ng ml−1 interfering substances. The cycle times for 25 ng ml−1 PSA in the absence and presence of a 1000 ng ml−1 interfering substance are very close to each other, demonstrating that even the coexistence of interfering substances at a concentration of 40 times higher than that of PSA will not affect the test results. Put together, these results demonstrate that our method has a high selectivity. | ||
| Fig. 4 (A) The color changes of MB+ in the clock reaction along with time for 1000 ng ml−1 UA (a), 1000 ng ml−1 glucose (b), 1000 ng ml−1 AA (c), 1000 ng ml−1 IgG (d), 1000 ng ml−1 CEA (e), 1000 ng ml−1 AFP (f), and 25 ng ml−1 PSA (g). (B) Histogram of the cycle time towards 1000 ng ml−1 UA (a), 1000 ng ml−1 glucose (b), 1000 ng ml−1 AA (c), 1000 ng ml−1 IgG (d), 1000 ng ml−1 CEA (e), 1000 ng ml−1 AFP (f), and 25 ng ml−1 PSA (g). (C) 1000 ng ml−1 UA + 25 ng ml−1 PSA (h), 1000 ng ml−1 glucose + 25 ng ml−1 PSA (i), 1000 ng ml−1 AA + 25 ng ml−1 PSA (j), 1000 ng ml−1 IgG + 25 ng ml−1 PSA (k), 1000 ng ml−1 CEA + 25 ng ml−1 PSA (l), 1000 ng ml−1 AFP + 25 ng ml−1 PSA (m), and 25 ng ml−1 PSA (g). (D) Histogram of the cycle time towards 1000 ng ml−1 UA + 25 ng ml−1 PSA (h), 1000 ng ml−1 glucose + 25 ng ml−1 PSA (i), 1000 ng ml−1 AA + 25 ng ml−1 PSA (j), 1000 ng ml−1 IgG + 25 ng ml−1 PSA (k), 1000 ng ml−1 CEA + 25 ng ml−1 PSA (l), 1000 ng ml−1 AFP + 25 ng ml−1 PSA (m), and 25 ng ml−1 PSA (g). | ||
3.5 Validation of this method by the commercial ELISA kit for the PSA detection
After having obtained the standard calibration curve and confirmed the high selectivity of this method, to demonstrate its viability in actual applications, we used the method to detect PSA in human serum and compared the results with those obtained from the commercial ELISA kit method. The standard curve of the commercial ELISA kit was measured first and is shown in Fig. 5. The PSA in human serum samples and human serum alone (the blank) was detected by our method. The results from our method and those from the commercial ELISA kit are listed in Table 1. The recovery is in the range of 85.3% to 96.9%, demonstrating that our method is viable in potential applications in the detection of PSA in human serum. The typical assay times of the commercial ELISA kit and this method are comparable, depending on the concentration range of PSA. | ||
| Fig. 5 The top photograph shows the detection of PSA in different concentrations using the commercial ELISA kit. The bottom graph is the calibration curve employing a set of standard solutions with known PSA concentrations. | ||
| Added (ng ml−1) | Found by the commercial ELISA kit (ng ml−1) | Standard deviation for the commercial ELISA kit | Found by this method (ng ml−1) | Standard deviation for this method | Recovery (%) |
|---|---|---|---|---|---|
| 0 | 0.914 | 0.0150 | 0.850 | 0.130 | 93.0 |
| 1 | 1.09 | 0.151 | 0.960 | 0.174 | 88.1 |
| 4 | 4.98 | 0.233 | 4.25 | 0.328 | 85.3 |
| 10 | 11.9 | 0.325 | 11.4 | 0.435 | 95.8 |
| 60 | 58.2 | 0.268 | 56.4 | 0.816 | 96.9 |
It should be pointed out, however, that this method has its own drawbacks and limitations for further improvement. For example, the clocking time of the employed clock reaction is intrinsically dependent on both the temperature (Fig. S3†) and the oxygen partial pressure of the atmosphere. This would eventually require an optimized and facile calibration protocol for viable applications in underdeveloped areas. The use of a relatively time-consuming leaching reaction is another issue calling for further improvement. Last but not least is the desire to replace cadmium ions with other less toxic ions such as Fe(III) or Cu(II). Future work may focus on solving or improving these limitations.
4. Conclusion
In conclusion, we report the development of a new mode of detection to significantly improve the current ELISA protocol. Our method has four main features. First of all, we achieved enzyme-free signal generation in ELISA by replacing the conventional enzyme-based color generation step with an enzyme-free pure chemical color change process from a clock reaction. Secondly, we achieved the use of the clocking time characteristically associated with a clock reaction as the quantitative readout signal for analyte concentration. Thirdly, AuNPs were employed for dual functions, both as a secondary antibody carrier and a catalytic regulator for the clock reaction. And last but not least, the characteristic time associated with the colorimetric detection in our modified ELISA can be read out either by the naked eye assisted with a stopwatch free from any benchtop instrument or more accurately on any benchtop instrument with a built-in timer and with pre-set absorbance associated with the characteristic absorption wavelength of the clock reaction. The feasibility of our method is demonstrated by the quantitative detection of PSA in human serum and validated by both instrument-based measurements and commercial ELISA kits. The naked-eye detection range for PSA is from 1.00 to 100 ng ml−1 with a LOD of 0.96 ng ml−1, which is lower than the threshold value (4 ng ml−1) for the early diagnosis of prostate cancer.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was supported by the Department of Chemistry of HKUST and the Grant from Research Grant Council of Hong Kong (No. 16307721).References
-
S. Hosseini, P. Vázquez-Villegas, M. Rito-Palomares and S. O. Martinez-Chapa, Enzyme-linked Immunosorbent Assay (ELISA) From A to Z, Springer Singapore, Singapore, 2018 Search PubMed
.
- J. N. Roberts, T. M. Byrem and D. L. Grooms, Reprod. Domest. Anim., 2015, 50, 651–658 CrossRef CAS PubMed
- H. Ye, K. Yang, J. Tao, Y. Liu, Q. Zhang, S. Habibi, Z. Nie and X. Xia, ACS Nano, 2017, 11, 2052–2059 CrossRef CAS PubMed
- N. Sharma, S. Hanif, D. Upadhyay and M. K. Chhikara, J. Immunol. Methods, 2019, 473, 112634–112640 CrossRef CAS PubMed
- G. Mameli, D. Cossu, E. Cocco, J. Frau, M. G. Marrosu, M. Niegowska and L. A. Sechi, J. Neuroimmunol., 2015, 280, 66–68 CrossRef CAS PubMed
- Q. Pan, J. Wang, Y. Gao, H. Cui, C. Liu, X. Qi, Y. Zhang, Y. Wang, K. Li, L. Gao and X. Wang, Appl. Microbiol. Biotechnol., 2020, 104, 853–859 CrossRef CAS PubMed
- Y. Xiong, K. Pei, Y. Wu and Y. Xiong, Food Control, 2017, 78, 317–323 CrossRef CAS
- M. Laloup, G. Tilman, V. Maes, G. De Boeck, P. Wallemacq, J. Ramaekers and N. Samyn, Forensic Sci. Int., 2005, 153, 29–37 CrossRef CAS PubMed
- B. D. Hock, L. K. Stamp, M. W. Hayman, P. E. Keating, E. T. Helms and M. L. Barclay, Ther. Drug Monit., 2016, 38, 32–41 CrossRef CAS PubMed
- E. M. H. Schmitz, D. Kerkhof, D. Hamann, J. L. Dongen, P. H. M. Kuijper, L. Brunsveld, V. Scharnhorst and M. A. C. Broeren, Clin. Chem. Lab. Med., 2016, 54, 1211–1219 CrossRef CAS PubMed
- M. H. L. Christiaans, F. Nieman, J. P. Hooff and E. M. Berg-Loonen, Transplantation, 2000, 69, 917–927 CrossRef CAS PubMed
- G. Fernández-Fresnedo, J. M. Pastor, M. López-Hoyos, J. C. Ruiz, J. A. Zubimendi, J. Gonzalez-Cotorruelo, E. Rodrigo, A. L. M. Francisco and M. Arias, Nephrol., Dial., Transplant., 2003, 18, 990–995 CrossRef PubMed
- B. Mariager, M. Sølve, M. Eriksen and C. H. Brogren, Food Agric. Immunol., 1994, 6, 73–83 CrossRef CAS
- X. Jiang, Q. Rao, K. Mittl and Y. H. P. Hsieh, Food Control, 2020, 110, 107045–107052 CrossRef CAS
- K. O. Ivens, J. L. Baumert and S. L. Taylor, J. Food Sci., 2016, 81, T1871–T1878 CrossRef CAS PubMed
- Ref. 1, pp. 68–69.
- H. D. Hill and C. A. Mirkin, Nat. Protoc., 2006, 1, 324–336 CrossRef CAS PubMed
- S. I. Stoeva, J. S. Lee, C. S. Thaxton and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 3303–3306 CrossRef CAS PubMed
- Y. Li, J. Xuan, Y. Song, W. Qi, B. He, P. Wang and L. Qin, ACS Nano, 2016, 10, 1640–1647 CrossRef CAS PubMed
- Y. Song, X. Xia, X. Wu, P. Wang and L. Qin, Angew. Chem., Int. Ed., 2014, 53, 12451–12455 CAS
- X. Ma, Z. Chen, P. Kannan, Z. Lin, B. Qiu and L. Guo, Anal. Chem., 2016, 88, 3227–3234 CrossRef CAS PubMed
- M. Urdea, L. A. Penny, S. S. Olmsted, M. Y. Giovanni, P. Kaspar, A. Shepherd, P. Wilson, C. A. Dahl, S. Buchsbaum, G. Moeller and D. C. Hay Burgess, Nature, 2006, 444, 73–79 CrossRef PubMed
- Z. Yu, H. Gong, M. Li and D. Tang, Biosens. Bioelectron., 2022, 218, 114751–114759 CrossRef CAS PubMed
- R. Ren, G. Cai, Z. Yu, Y. Zeng and D. Tang, Anal. Chem., 2018, 90, 11099–11105 CrossRef CAS PubMed
- P. Peng, C. Liu, Z. Li, Z. Xue, P. Mao, J. Hu, F. Xu, C. Yao and M. You, TrAC, Trends Anal. Chem., 2022, 152, 116605–116617 CrossRef CAS
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed
- G. Cai, Z. Yu, R. Ren and D. Tang, ACS Sens., 2018, 3, 632–639 CrossRef CAS PubMed
- S. Lv, K. Zhang, Q. Zhou and D. Tang, Sens. Actuators, B, 2020, 310, 127874–127880 CrossRef CAS
- M. S. Tabatabaei, R. Islam and M. Ahmed, Anal. Chim. Acta, 2021, 1143, 250–266 CrossRef CAS PubMed
- J. Satija, N. Punjabi, D. Mishra and S. Mukherji, RSC Adv., 2016, 6, 85440–85456 RSC
- L. Guo, S. Xu, X. Ma, B. Qiu, Z. Lin and G. Chen, Sci. Rep., 2016, 6, 1–7 CrossRef PubMed
- Z. Zhang, Z. Chen, S. Wang, F. Cheng and L. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27639–27645 CrossRef CAS PubMed
- D. Cecchin, R. De La Rica, R. E. S. Bain, M. W. Finnis, M. M. Stevens and G. Battaglia, Nanoscale, 2014, 6, 9559–9562 RSC
- R. De La Rica and M. M. Stevens, Nat. Nanotechnol., 2012, 7, 821–824 CrossRef CAS PubMed
- R. De La Rica and M. M. Stevens, Nat. Protoc., 2013, 8, 1759–1764 CrossRef PubMed
- J. Liang, C. Yao, X. Li, Z. Wu, C. Huang, Q. Fu, C. Lan, D. Cao and Y. Tang, Biosens. Bioelectron., 2015, 69, 128–134 CrossRef CAS PubMed
- C. Yao, S. Yu, X. Li, Z. Wu, J. Liang, Q. Fu, W. Xiao, T. Jiang and Y. Tang, Anal. Bioanal. Chem., 2017, 409, 1093–1100 CrossRef CAS PubMed
- X. Huang, R. Chen, H. Xu and W. Lai, Anal. Chem., 2016, 88, 1951–1958 CrossRef CAS PubMed
- Z. Xuan, M. Li, P. Rong, W. Wang, Y. Li and D. Liu, Nanoscale, 2016, 8, 17271–17277 RSC
- Y. Song, Y. Zhang, P. E. Bernard, J. M. Reuben, N. T. Ueno, R. B. Arlinghaus, Y. Zu and L. Qin, Nat. Commun., 2012, 3, 1–9 Search PubMed
- Y. Li, J. Xuan, T. Xia, X. Han, Y. Song, Z. Cao, X. Jiang, Y. Guo, P. Wang and L. Qin, Anal. Chem., 2015, 87, 3771–3777 CrossRef CAS PubMed
- A. K. Horváth and I. Nagypál, ChemPhysChem, 2015, 16, 588–594 CrossRef PubMed
- L. Zheng, H. Yu, Y. Yue, F. Wu and Y. He, ACS Appl. Mater. Interfaces, 2017, 9, 11798–11802 CrossRef CAS PubMed
- T. Wu and Z. Ma, Microchim. Acta, 2017, 184, 577–582 CrossRef CAS
- T. Wu, J. Shan and Z. Ma, ACS Sustainable Chem. Eng., 2017, 5, 4976–4981 CrossRef CAS
- R. Chen, T. M. Li, H. Merrick, R. F. Parrish, V. Bruno, A. Kwong, C. Stiso and D. J. Litman, Clin. Chem., 1987, 33, 1521–1525 CrossRef CAS
- R. Muller, Biotechnol. Appl. Biochem., 1997, 26, 73–78 CAS
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
- T. Wu and X. Y. Li, Anal. Methods, 2023, 15, 48–55 RSC
- X. Fu, T. Lou, Z. Chen, M. Lin, W. Feng and L. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 1080–1086 CrossRef CAS PubMed
- Y. Y. Chen, H. T. Chang, Y. C. Shiang, Y. L. Hung, C. K. Chiang and C. C. Huang, Anal. Chem., 2009, 81, 9433–9439 CrossRef CAS PubMed
- G. Deschenes, R. Lastra, J. R. Brown, S. Jin, O. May and E. Ghali, Miner. Eng., 2000, 13, 1263–1279 CrossRef CAS
- D. Feng and J. S. J. Van Deventer, Hydrometallurgy, 2002, 64, 231–246 CrossRef CAS
- D. A. Healy, C. J. Hayes, P. Leonard, L. McKenna and R. O'Kennedy, Trends Biotechnol., 2007, 25, 125–131 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00058c |
| This journal is © The Royal Society of Chemistry 2023 |