DOI:
10.1039/D3SD00080J
(Paper)
Sens. Diagn., 2023,
2, 938-947
Ion-transfer electroanalytical detection of perfluorooctanoic acid at a liquid–liquid micro-interface array†
Received
6th April 2023
, Accepted 22nd June 2023
First published on 23rd June 2023
Abstract
Per- and polyfluoroalkyl substances (PFAS) are synthetic materials that bioaccumulate and are environmentally persistent. Due to their serious health threats, including to humans, new methods for the sensing of PFAS are needed. Here, we report the electrochemical behaviour of perfluorooctanoic acid (PFOA) via ion transfer voltammetry at an array of microinterfaces between two immiscible electrolytic solutions (μITIES). Investigation of electrochemical sensing of PFOA at the μITIES array by voltammetry showed that differential pulse stripping voltammetry (DPSV) with a 300 s preconcentration time was the most sensitive approach. This enabled a limit of detection (LOD) of 1.2 nM. Matrix effects of different types of samples, like tap water, and aqueous phases spiked with a protein (bovine serum albumin) or complexing reagent (β-cyclodextrin), on the analytical performance of PFOA were evaluated. The findings presented here provide a basis for sensing of PFOA using ion transfer voltammetry.
Introduction
Per- and polyfluoroalkyl substances (PFAS) are synthetic pollutants that are highly persistent in the environment.1–3 They have been widely used in firefighting foams, in paper and packaging industries, in clothing, in personal care products, in carpet manufacture as stain- and water-resistant coatings, and on various non-stick cookware.1,2,4–8 PFAS are resistant to biodegradation, are distributed everywhere, and are persistent in the environment due to the strong fluorine-carbon bond.9,10 Firefighting training sites,11–15 landfill sites,16,17 PFAS manufacturing sites,14,18 and wastewater purification plants14,19 are common zones of PFAS contamination. PFAS are also present in vegetables,20,21 human breast milk22,23 and blood serum,24 and in drinking water.14,25,26 Recently Whitehead et al. reported PFAS in various cosmetic products purchased in Canada and the United State of America (USA).8
PFAS can enter into living entities via contaminated drinking water, dust particles, foods (especially seafood), and various consumer items.5,25,27–30 The lipophilic nature of PFAS causes bioaccumulation and toxicity.31,32 The toxicity of PFAS to different aquatic (e.g. Daphnia magna and Moina macrocopa) and terrestrial (e.g. earthworm) species was investigated.33,34 The toxicity of PFAS has been reported, for example DNA damage in earthworms34 and Escherichia coli,35 their effect on the neuroendocrine system of rats,36 their effect on developmental toxicity and hepatotoxicity in monkeys37 and their immunotoxicity in mice.38 The toxicity and bioaccumulating nature of PFAS raise important environmental and human health concerns. PFAS has a negative effect in reproduction, immune function, endocrine function, and neurological function in humans.32,39 Bartell and Vieira undertook a meta-analysis review of the role of one PFAS, perfluorooctanoic acid (PFOA), in kidney cancer and testicular cancer in humans, and suggested that the average cancer (kidney cancer and testicular cancer) risk is increased if the serum PFOA concentration is increased.40 Since PFAS are directly implicated in toxic effects in different biological species, including humans, the availability of suitable methods for their analysis is important.
Methods based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas chromatography-mass spectrometry (GC-MS) are widely used for the determination of PFAS.41–44 Even if the analytical performances of these techniques are suitable, common drawbacks include difficulty in on-site measurements, costs, instrumentation and sample pretreatment needs, and the requirement for highly-trained experts. Spectroscopic methods such as turn-on or turn-off fluorescence, and surface-enhanced Raman scattering are available for PFAS detection.45–50 However, these still have some limitations like low selectivity, reasonable reproducibility, and the need for pretreatment steps.50
Direct redox electrochemistry of PFAS is difficult to use for monitoring due to their high stability; despite this, electroanalytical approaches have received some attention.51 For example, some researchers have detected PFAS by indirect redox electrochemistry using molecularly imprinted polymer (MIP) electrodes.52–55 Karimian et al. (limit of detection (LOD) = 0.04 nM; ca. 20 ng L−1)53 and Kazemi et al. (LOD = 0.05 nM; ca. 25 ng L−1)55 reported MIP-modified electrochemical sensors for detection of perfluorooctane sulfonate (PFOS). Clark and Dick also detected PFOS (LOD = 3.4 pM; ca. 1.7 ng L−1) present in river water using a MIP-modified carbon electrode.52 Glasscott et al. reported a LOD of 250 fM (ca. 87 pg L−1) for GenX using a MIP-modified microelectrode.54 Ranaweera et al. used a bubble-nucleation approach on a nanoelectrode for the detection of PFOA and PFOS, yielding LODs of 72 nM (ca. 30 μg L−1) and 160 nM (ca. 80 μg L−1), respectively. This group also improved the LOD for PFOS to 0.08 nM (ca. 40 ng L−1) by coupling with solid phase extraction.56 Gong et al. developed a photoelectrochemical-based fluorescent sensor for the detection of PFOA, resulting in a LOD of 24 pM (ca. 0.01 μg L−1).57 Fang et al. also investigated PFOA detection using a MIP electrode, yielding a LOD of ∼0.1 μM (ca. 41 μg L−1).58
Similarly, electrochemistry at the interface between two immiscible electrolyte solutions (ITIES), which enables the non-redox detection of ionised or ionisable species, also opens a strategy for the detection of PFAS.31 Ion transfer electrochemistry at the ITIES is used for detection of many ionisable analytes.59 Problems with traditional electrochemical cells for measurements at the ITIES (e.g. mechanical instability, large capacitance) can be overcome by miniaturisation of the ITIES to the microscale.60,61 This also has the added advantage of increasing mass transport flux to the ITIES.62 Mechanical stability of the ITIES can be achieved by using a porous membrane or gelled organic phase.63–65 Amemiya's group investigated the lipophilic nature of some PFAS at the ITIES31 and also developed a method for the determination of PFAS using an organogel-based ITIES supported on a modified gold electrode; they reported a LOD of 50 pM (ca. 25 ng L−1) for PFOS.66 Viada et al. used a μITIES array to study PFOS and achieved a LOD of 30 pM (ca. 15 ng L−1).67 A recent study assessed the prospects for selectivity in the detection of PFAS mixtures by ion transfer electrochemistry at a micropipette-based μITIES.68 Whilst PFOS can be detected sensitively at a μITIES array,67 no studies were reported on PFOA detection at low concentrations.
The aim of the work reported here was to evaluate the electrochemical behaviour and detection of PFOA at a μITIES array. Different voltammetric techniques were employed, and the electroanalytical performances were assessed. Furthermore, matrix effects on the electrochemical response of PFOA were examined. The results show that PFOA sensing using electrochemistry at a μITIES array is viable approach.
Experimental section
Reagents
All reagents were purchased from Sigma-Aldrich Australia and used as received, unless otherwise indicated. Organic electrolyte salt bis(triphenylphosphoranylidene)ammonium tetrakis(4-chlorophenyl)borate (BTPPATPBCl) was synthesized by metathesis of equimolar quantities of bis(triphenylphosphoranylidene)ammonium chloride (BTPPACl) and potassium tetrakis(4-chlorophenyl)borate (KTPBCl) (STREM Chemicals).69 The suitability of the prepared salt BTPPATPBCl was assessed by voltammograms in pure electrolyte solutions (i.e. no analyte present in either phase). The absence of peaks in these blank voltammograms indicated the absence of impurities and hence the suitability of the salt for these experiments. Pentadecafluorooctanoic acid (PFOA, purity: 98%) (STREM Chemicals) solutions were prepared in 10 mM lithium chloride (LiCl) solution unless otherwise indicated. Aqueous phase electrolyte solutions were LiCl (10 or 1 mM) or lithium sulfate (10 mM), and the organic phase electrolyte solution was 10 mM BTPPATPBCl or 10 mM tetradodecylammonum tetrakis(4-chloropheny)borate (TDDATPBCl) in 1,2-dichloroethane (1,2-DCE). All aqueous solutions were prepared using purified water with a resistivity of 18.2 MΩ cm (Milli-Q, Millipore Pty. Ltd., Australia). All experiments were conducted at ambient temperature.
Apparatus
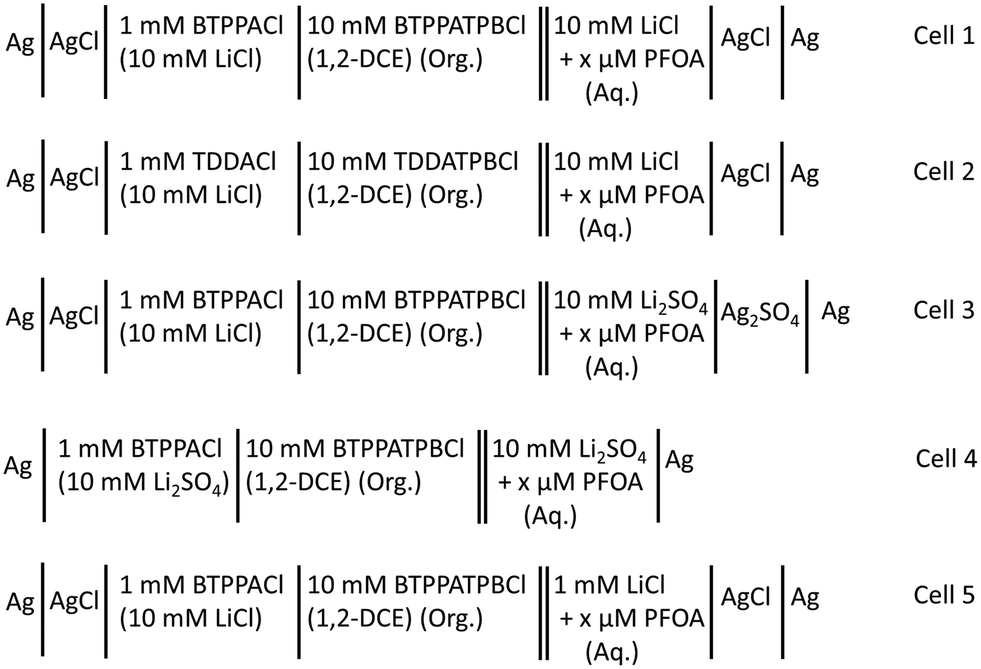
Electrochemical measurements were conducted using an AUTOLAB PGSTAT302N electrochemical workstation (Metrohm, The Netherlands) controlled with the supplied NOVA software. These experiments were performed within a Faraday cage. All electrochemical measurements were performed using μITIES arrays prepared with glass membranes having 100 micropores (10 × 10 square array) made by laser ablation in a ∼130 μm thick borosilicate substrate.70 Due to the laser ablation process, the pores in the glass membranes were wider on the laser entry side (diameter: 42 μm) than on the laser exit side (diameter: 21 μm). One surface (laser exit side) of the glass membranes and the inner walls of the pores were made hydrophobic by silanisation with trichloro(1H,1H,2H,2H-perfluorooctyl)silane, while the other side (laser entry side) remained hydrophilic.70 The glass membrane was sealed to the mouth of a glass cylinder using silicone sealant (Selley, Australia and New Zealand) such that the hydrophobic side faced towards the glass cylinder so the organic phase can be placed inside and subsequently into the pores. The sealant was allowed to cure for 24 h and the entire assembly was cleaned with acetone and dried in air before experiments. For each electrochemical cell set-up (Scheme 1), ∼200 μL of organic electrolyte solution was placed into the glass cylinder. Then, the organic reference solution was placed on top of the organic phase (Scheme 1). By inserting this assembly into the aqueous phase and adding a pair of in-house prepared electrodes, which were either silver/silver chloride, silver/silver sulfate or silver, a two-electrode cell was prepared. All electrochemical cells used during these experiments are summarised in Scheme 1. The pH of the aqueous phases was measured with a CP-511 Elmetron laboratory pH meter with combination pH electrode (Ionode, Queensland, Australia). This was calibrated with three pH buffer solutions (pH 4, 7, and 10) before measurements.
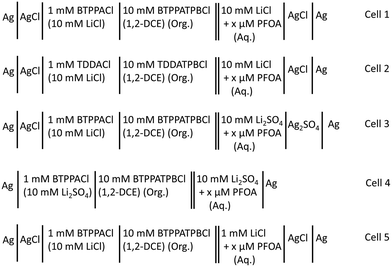 |
| | Scheme 1 Schematic summary of electrochemical cell compositions used for the electrochemical study of PFOA. x = concentration of PFOA in the aqueous phase. Org. = organic phase and Aq. = aqueous phase. | |
Electrochemical measurements
Cyclic voltammetry (CV), differential pulse voltammetry (DPV) and differential pulse stripping voltammetry (DPSV) were employed to study PFOA. The CV scan rate was 10 mV s−1. The waveform parameters for DPV were: step potential: 0.005 V, modulation amplitude: 0.025 V, modulation time: 0.05 s, and interval time: 0.5 s. For each DPSV measurement, a 0.1 V preconcentration step (unless otherwise stated; time indicated in results) and a 0.5 V preconditioning potential (for 40 s) were applied.
Results and discussion
Cyclic voltammetry of PFOA
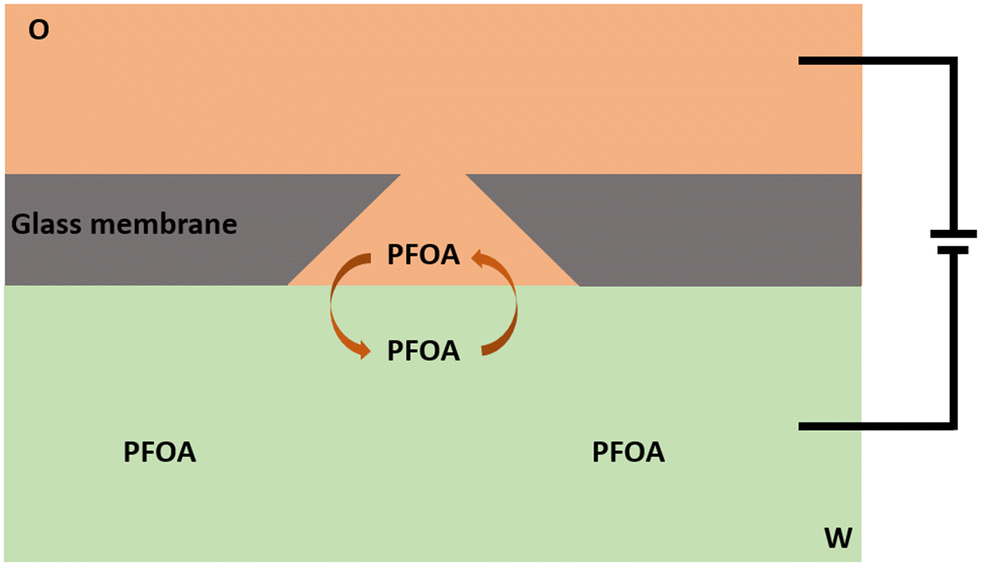
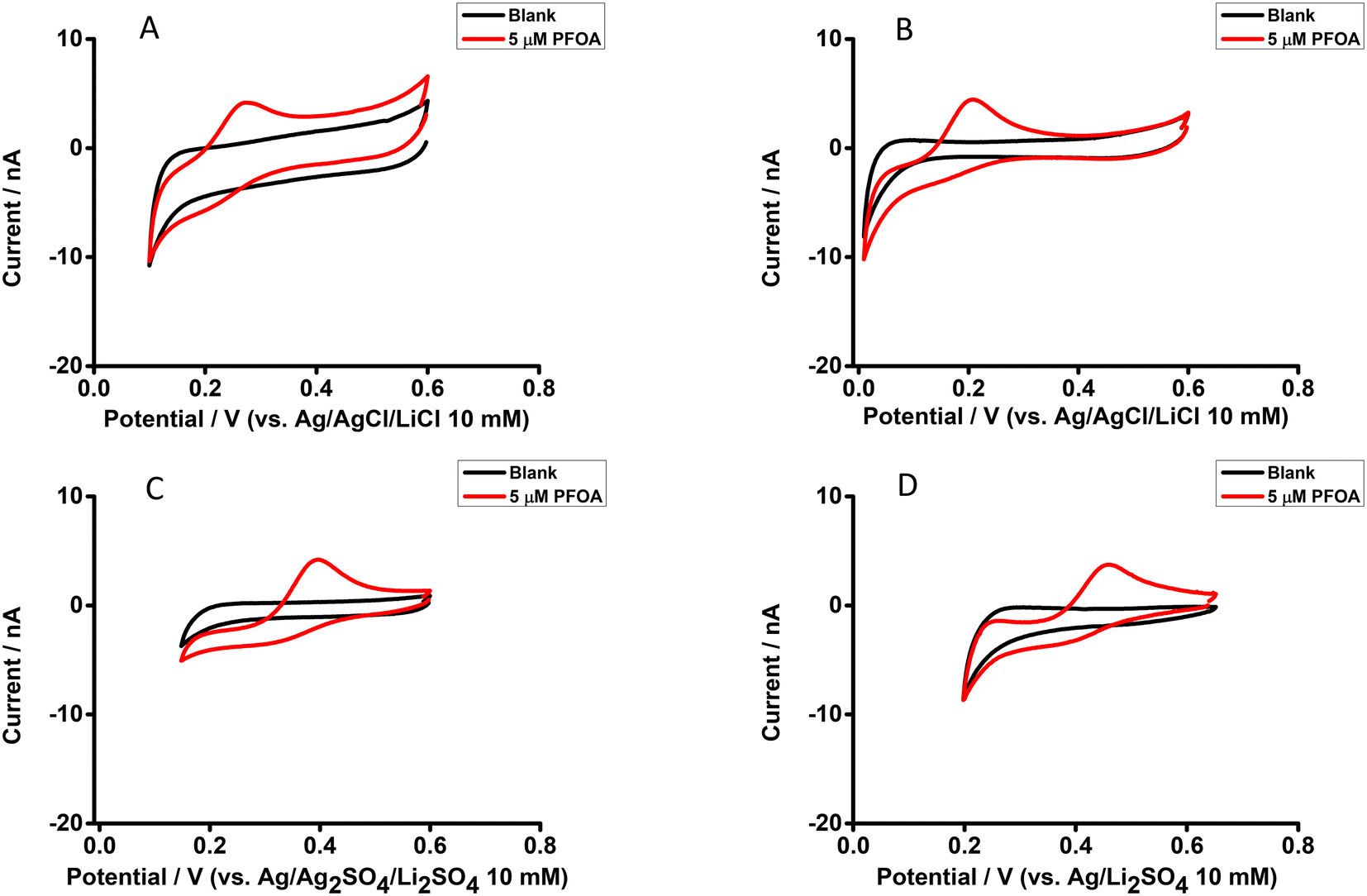
The electrochemical behaviour of PFOA at the μITIES array was investigated by CV in different electrochemical cell compositions (Scheme 1). Fig. 1 shows the principle of ion transfer electrochemical behaviour. Under the influence of an applied potential at the aqueous-organic interface, ionised PFOA will transfer and be detected. For CV, the transfer from water phase to organic phase occurs on the forward scan, and the back-transfer from organic to water phase occurs on the reverse scan. The study of different cell compositions was undertaken to understand the transfer behaviour of PFOA in the presence of different electrolyte concentrations and species, as these control the available potential window for voltammetric experiments. While chloride salts are usually used in the aqueous phase, and the transfer of chloride to the organic phase limits the negative side of the potential window at the ITIES, use of a sulfate salt in the aqueous phase might enable a wider potential window, due to its greater hydration energy, and hence might improve the ability to detect PFOA. Typical CVs are shown in Fig. 2. Asymmetric CVs were obtained in all cases, with steady-state voltammograms on the forward sweep (due to radial diffusion to the pores holding the μITIES), scanning from positive towards negative potentials, while the reverse-direction scans are peak shaped (due to linear diffusion within the pores), in agreement with previous studies.67,68 CV also indicated no adsorption, emulsification, or instability of the interface.67 In all cases, the reverse peak currents increased linearly with the PFOA concentration in the studied ranges (Fig. S1†). Similarly, calibration graph slope (sensitivity) of the current response to PFOA in the various cells and the LOD (defined as 3σ/m, where σ is standard deviation of the blank (intercept) and m is the calibration graph slope) for PFOA in the various cells were also determined. Cell 1 displayed the greatest sensitivity (ca. 1.25 nA μM−1) amongst these. Calculated sensitivity of cells 2, 3, and 4 were ca. 0.78, ca. 0.87, and ca. 0.85 nA μM−1, respectively. Also, cells 3 (LOD = 0.3 μM) and 4 (LOD = 1.1 μM) gave lower LODs than cells 1 (LOD = 3.5 μM) and 2 (LOD = 2.3 μM). The lower LODs of cells 3, 4 are due to the presence of more hydrophilic aqueous supporting electrolyte anion, i.e. SO42−, which produced a wider potential window compared to cells with Cl− (which is less hydrophilic than SO42−) as the aqueous phase electrolyte anion. The LODs found in all cells show that CV is not sufficient for the detection of the lower concentrations of PFOA required for most applications. Although the sensitivities and LODs in SO42−-based electrolytes are slightly lower, Cl−-based electrolytes were selected for further studies because of the presence of Cl− in most environmental or biological samples.
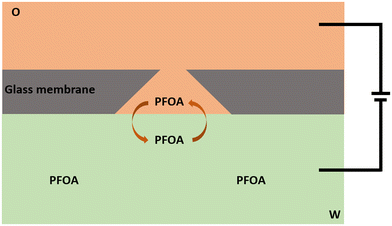 |
| | Fig. 1 Sketch representing the principle of ion transfer across the liquid–liquid interface using a μITIES array, illustrated here at a single conical-pore-based μITIES. O = organic phase; W = aqueous phase, containing PFOA. Arrows indicate movement of ionised PFOA between aqueous and organic phases under the influence of applied potential. Entire electrochemical cell compositions are presented in Scheme 1. | |
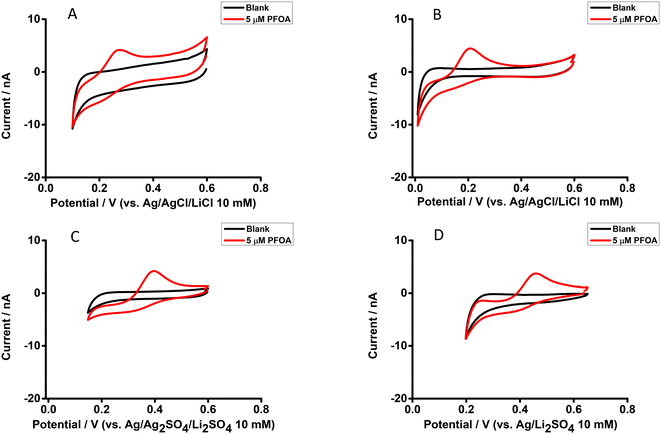 |
| | Fig. 2 CVs without background-subtraction in the presence and absence (black) of PFOA in the aqueous phase. (A) 10 mM LiCl as the aqueous phase (pH = 6.0) and 10 mM BTPPATPBCl (1,2-DCE) as the organic phase using cell 1. (B) 10 mM LiCl as the aqueous phase (pH = 6.0) and 10 mM TDDATPBCl as the organic phase using cell 2. (C) and (D) 10 mM Li2SO4 as the aqueous phase (pH = 6.3) and 10 mM BTPPATPBCl (1,2-DCE) as the organic phase using cell 3 and cell 4, respectively. Scan rate: 10 mV s−1. | |
Influence of pH on the electrochemical behaviour of PFOA
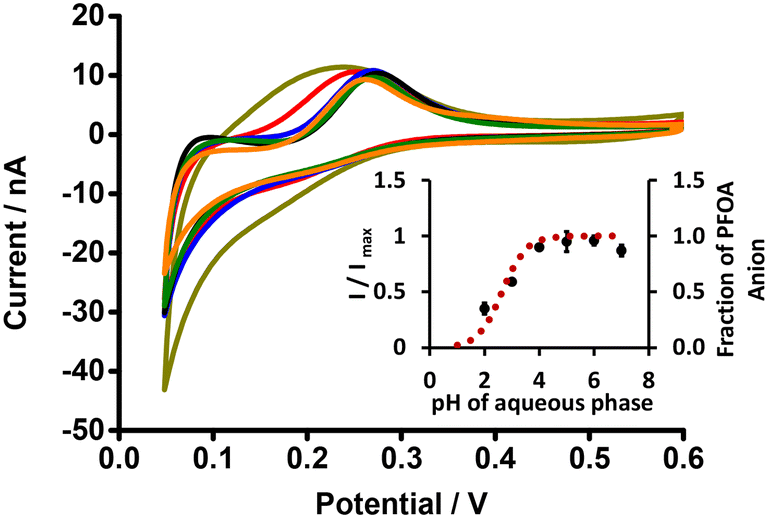
The electrochemical behaviour of PFOA at different aqueous phase pH values was explored using cell 1 (Scheme 1). Fig. 3 shows CVs of 10 μM PFOA at different aqueous phase pH. As can be seen, an indistinct reverse peak current was seen at pH ≤ 3; however, distinct current signals were observed at higher pH. Since the pKα of PFOA is 2.8,71 the lower peak current and poor shape of the voltammogram at lower pH is due to incomplete dissociation of PFOA and hence less anionic PFOA is available to transfer. Similarly, the higher peak current at pH ≥ 4 is due to availability of more anionic PFOA for transfer due to complete dissociation of PFOA. This is also supported by the fit of experimental data to the theoretical relationship between the pH and fraction of anionic PFOA (inset, Fig. 3). These results demonstrate that pH ≥ 4 is best for the electrochemical study of PFOA at the ITIES.
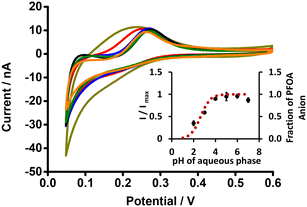 |
| | Fig. 3 CVs of PFOA at different pH of aqueous phase (dark yellow, red, blue, black, olive, and orange lines respectively indicate CVs at pH 2, 3, 4, 5, 6, and 7) (without background-subtraction). 10 mM BTPPATPBCl (1,2-DCE) as the organic phase. Inset: Plot of I/Imax (black dots) or fraction of PFOA anion (red dotted line) versus pH of the aqueous phase. Concentration of PFOA: 10 μM, scan rate: 10 mV s−1. Error bars represent ±1 standard deviation from three independent experiments. | |
Differential pulse voltammetry of PFOA
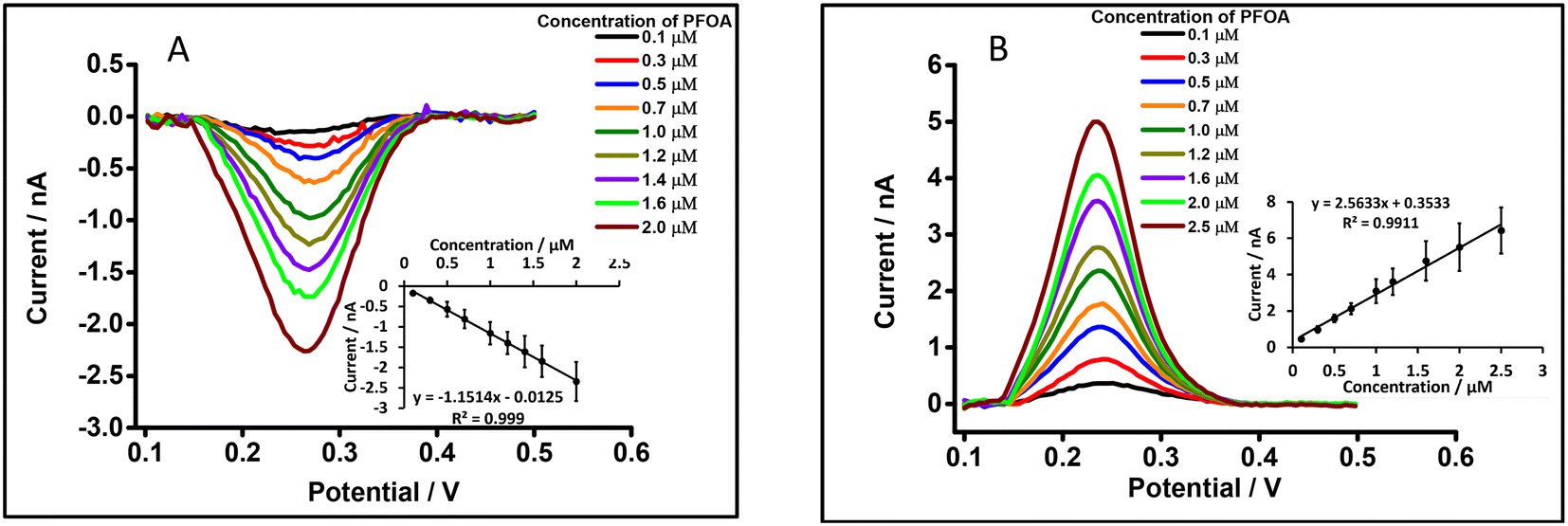
The μITIES array was used to conduct DPV, which is well-known72 to achieve the detection of lower concentrations of the analyte compared to CV. Background-subtracted followed by baseline-corrected DPVs at different concentrations are presented in Fig. 4(A and B) using cell 1. Calibration curves between peak current and concentration of PFOA for all DPVs indicate that the peak current linearly increased with PFOA concentration. Five electrochemical cell compositions, including cell 1, were used for the DPV detection of PFOA (voltammograms are shown in Fig. S2† for cells 2, 3, 4 and 5).
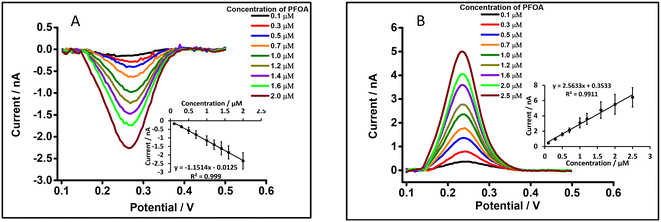 |
| | Fig. 4 DPVs at different concentrations of PFOA at the μITIES array using cell 1. (A) and (B) represent DPVs for forward and backward scan, respectively, using 10 mM LiCl as the aqueous phase (pH = 6.0) and 10 mM BTPPATPBCl (1,2-DCE) as the organic phase. Insets: Calibration curves between current and concentration of PFOA. Error bars represent ±1 standard deviation from three different independent measurements. All DPVs were background-subtracted then baseline-corrected. | |
Table 1 summarises the DPV analytical characteristics for PFOA. Higher sensitivities and lower LODs were obtained in the backward-scan direction compared to the forward-scan direction using cell 1. Amongst all cells, the highest sensitivity was found in cell 2 (Table 1), the lowest LOD (0.01 μM) was found in cell 5, while the highest LOD (0.12 μM) was found in cell 3. LODs from DPV were lower than those from CV, as expected. As described in the literature, further lowering of LODs could be achieved by the application of a systematic preconcentration step.73–75 Since the detection capability of PFOA using cell 5 (Scheme 1) was the best, this cell composition was used for further improvement of the LOD for PFOA by stripping voltammetry.
Table 1 Comparison of sensitivity and limit of detection for PFOA using DPV in different electrochemical cells
| Cell used |
Scan direction |
Calibration graph slope/nA μM−1 |
LOD/μM |
| Cell 1 |
Forward |
1.2 |
0.06 |
| Cell 1 |
Backward |
2.6 |
0.05 |
| Cell 2 |
Backward |
4.0 |
0.05 |
| Cell 3 |
Backward |
2.1 |
0.12 |
| Cell 4 |
Backward |
2.2 |
0.09 |
| Cell 5 |
Backward |
2.9 |
0.01 |
Differential pulse stripping voltammetry of PFOA
DPSV combines the advantages of an inherent preconcentration of the analyte with detection by differential pulse voltammetry,76 and so offers the prospect for improved analyte detection relative to DPV and CV. In DPSV, a preconcentration step at a fixed potential is followed by a voltammetric scan, providing the two steps involved in this analytical approach. LODs are improved by choosing an appropriate preconcentration step. Based on the CV behaviour (Fig. S3†), 0.1 V was chosen as the preconcentration potential. At this potential, ionised PFOA is driven from the aqueous phase into the organic phase where it is accumulated. Additionally, the geometry of the pores in the membrane used to form the μITIES array (Fig. 1) also affects the accumulation of PFOA in this analytical strategy. If the pores of the membrane are tapered towards the organic phase side, diffusion of PFOA through the pores into the bulk organic phase is restricted, and hence the concentration of PFOA in the organic phase within the pores is enhanced.70 At the end of the preconcentration period, DPV is applied as the detection step to strip the accumulated PFOA from the organic phase back to the aqueous phase and produce a peak response that is dependent on both PFOA concentration and the preconcentration time.
Effect of preconcentration time in detection of PFOA using DPSV
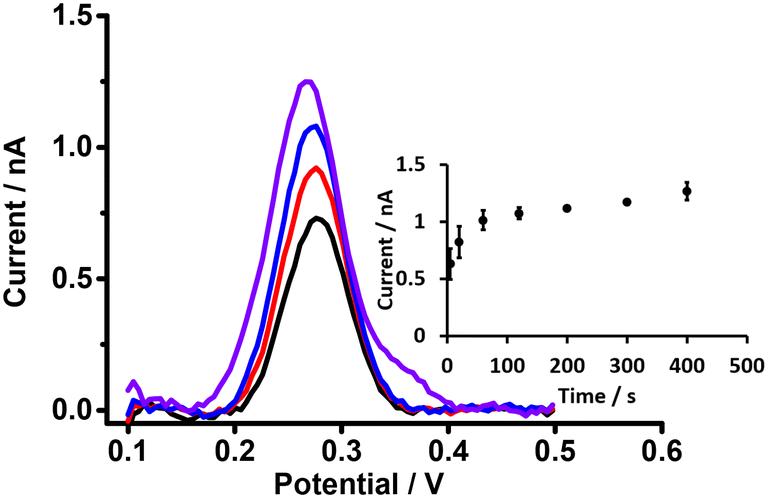
Preconcentration time is an important parameter in the detection of analytes by DPSV. Here, based on the DPV results, cell 5 (Scheme 1) was used to study the effects of preconcentration time (at 0.5 μM PFOA), without stirring of the aqueous phase, because mass transport by radial diffusion is achieved by use of a μITIES array. Fig. 5 shows DPSVs of PFOA at different preconcentration times. The results show that the peak response increased with preconcentration time and reached a plateau after 200 s (inset, Fig. 5). The plateau indicates a saturation effect, as described for a range of other analytes.67,75,77–79 Therefore, a preconcentration time of 300 s was chosen as suitable for the further investigation of PFOA detection by DPSV.
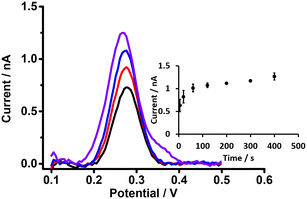 |
| | Fig. 5 DPSVs of 0.5 μM of PFOA at different preconcentration times using the μITIES array with cell 5. 10 mM BTPPATPBCl (1,2-DCE) as the organic phase. Black, red, blue, and violet lines respectively indicate CVs at 5, 20, 60, and 400 s, respectively. 1 mM LiCl as the aqueous phase (pH = 6.4), unstirred. Preconcentration times: 5–400 s. Inset: calibration curve between stripping peak currents and preconcentration times. Error bars are ±1 standard deviation obtained from three different experimental trials. All DPSVs were background-subtracted followed by baseline-corrected. | |
Detection of PFOA using DPSV
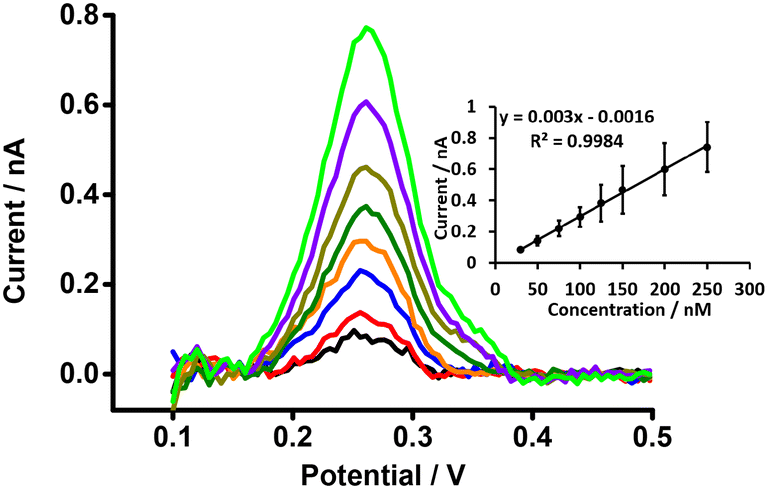
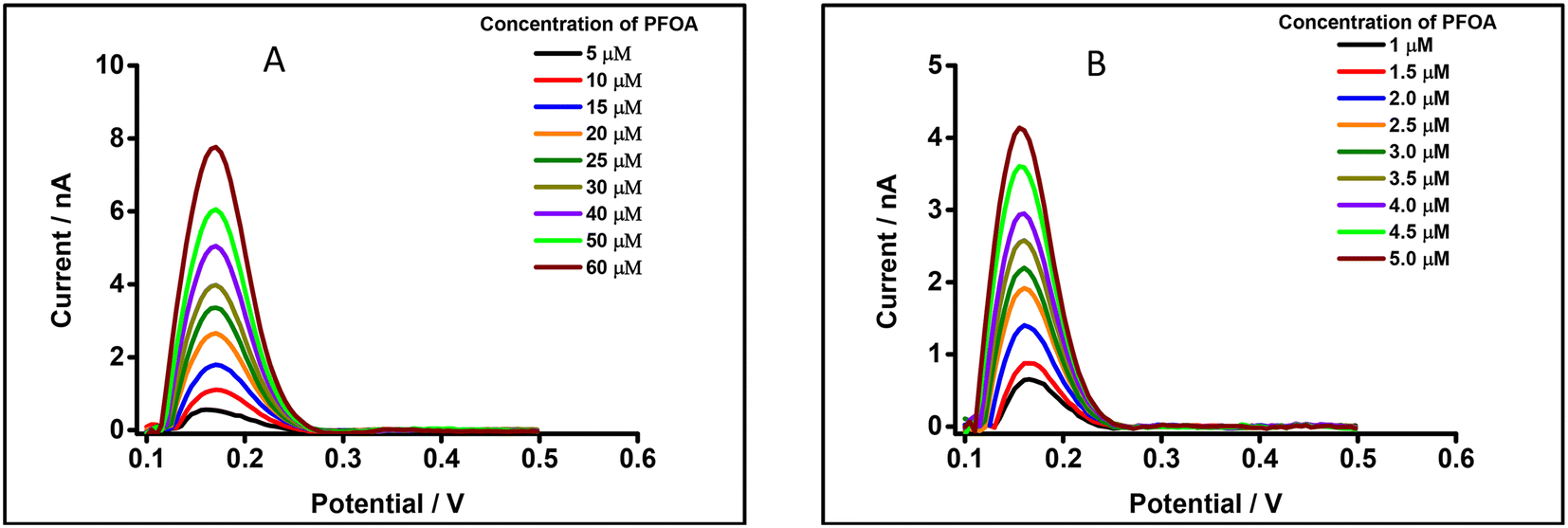
DPSV at the μITIES array was further tested to detect PFOA at lower concentrations using cell 5 (Scheme 1) with a 300 s preconcentration time. The results show that the stripping peak current was concentration-dependent in the range 30–250 nM (Fig. 6). Linear behaviour was obtained (inset, Fig. 6), yielding LOD (3σ/m) and sensitivity values of 1.2 nM (0.5 μg L−1) and 0.003 nA nM−1, respectively. However, Viada et al. achieved a picomolar LOD for PFOS using a μITIES array with DPSV and a 300 s preconcentration time.67 Since the carboxylate group in PFOA possesses stronger hydration than the sulfonate of PFOS, PFOA has lower lipophilicity than PFOS.66,80 As reported in the literature, the preconcentration step is more favourable for more lipophilic species, like PFOS.81 As a result, the higher LOD for PFOA compared to PFOS might be due to its lower lipophilicity. Many other investigations also detected lower concentrations of PFOS than PFOA.82–84 Nevertheless, the LOD obtained here for PFOA is the lowest obtained for ion-transfer stripping voltammetry of PFOA at the μITIES array and shows that this detection approach was better than those of Ranaweera et al., using a Pt nanoelectrode (LOD = 72 nM; 30 μg L−1),56 Cheng et al., using a fluorescent sensor for the detection of PFOA (LOD = 11.8 nM; ca. 5 μg L−1),45 Shanbhag et al., using hafnium-doped tungsten oxide as electrode material (LOD = 18.3 nM (7.6 μg L−1),85 Fang et al., using a MIP electrode (LOD = ∼100 nM; ca. 41 μg L−1),58 and Sahu et al., using screen-printed electrodes (LOD = 15 nM; 6.5 μg L−1).86 Table S1† compares the analytical performances for PFOA with these different analytical approaches. However, many chromatographic-mass spectrometric approaches (e.g. HPLC-MS, GC-MS, and LC-MS-MS) are widely used for the detection of PFAS, and these can detect picomolar or even lower concentrations, showing lower LODs than our approach. But these methods have several drawbacks (e.g. need for expert users, time consuming, sample preparation step, expense, difficulty in on-site measurement).50,87–89 The LOD achieved here is comparable to the recommended limit of 1.4 nM (ca. 0.56 μg L−1) for drinkable water set by the Department of Health, Australia,90 but significantly higher than the level established by the USA Environmental Protection Agency (EPA) (10 fM; ca. 0.004 ng L−1).91 Nevertheless, the analytical capability is lower than the recommended level of 24 nM (ca. 10 μg L−1) for recreational waters (Department of Health, Australia),90 suggesting that it could potentially be used to pre-screen environmental samples for PFOA contamination, with potential benefits in both time and money terms.
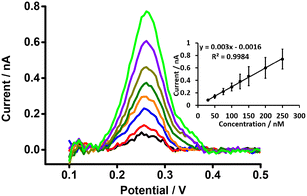 |
| | Fig. 6 DPSVs at different concentrations of PFOA at a μITIES array using cell 5. Organic phase: 10 mM BTPPATPBCl (1,2-DCE). 1 mM LiCl as the aqueous phase (pH = 6.4), concentration range (30–250 nM). Black, red, blue, orange, olive, dark yellow, violet and green lines respectively indicate CVs at 30, 50, 75, 100, 125, 150, 200, and 250 nM, respectively. Inset: calibration curve between stripping current and concentration of PFOA. Data points are average of three different trials, and error bars are ±1 standard deviation. All DPSVs were background-subtracted followed by baseline-corrected. | |
Study of matrix effects on electrochemical response of PFOA
For appropriate monitoring of the concentration of PFAS pollutants, knowledge of any matrix effects is important because the matrix components might alter the electrochemical signal of the analyte.92–94 These impacts may be due to one or more of the following reasons: a) interaction between the analyte and other components of a sample which sequester the analyte making it unavailable for detection, b) interaction of matrix components with the sensing interface, which makes it more difficult to sense the analyte, and c) overlap of transfer potential of matrix components and the analyte of interest. Accordingly, for the development of a PFOA sensor, a matrix effect study is very important.
Matrix effect of different types of water
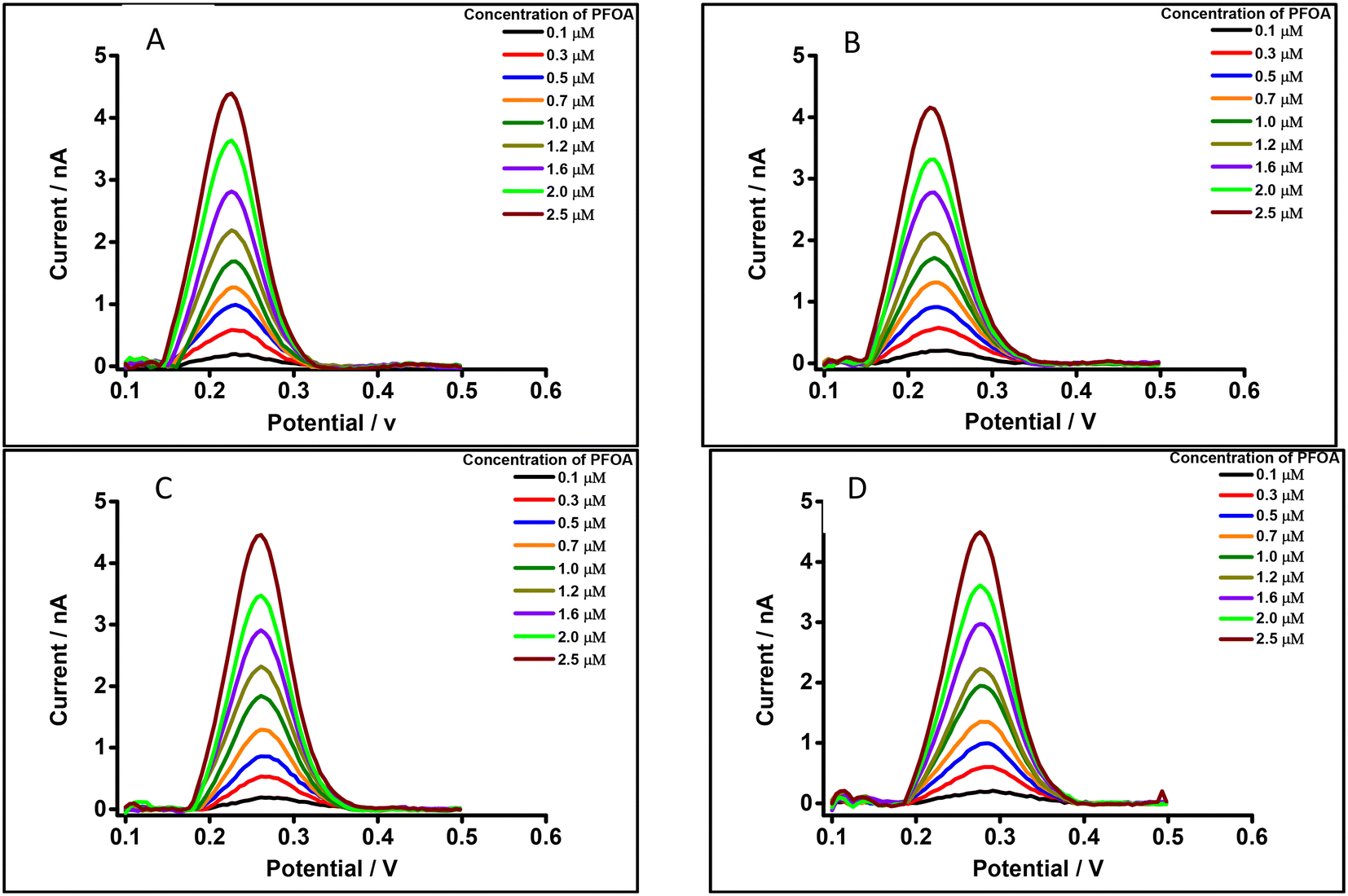
Different water types were analysed to see if there was a matrix effect on the PFOA response. Fig. 7A and B show background-subtracted followed by baseline corrected DPVs of PFOA using drinking water (with 10 mM LiCl) and laboratory tap water (with 10 mM LiCl) (water taken from local supplies at Curtin University), respectively, as the aqueous phase. For comparison, Fig. 7C and D show the corresponding water samples without added 10 mM LiCl. These results show that there is a slight shift in the peak potential of PFOA depending on whether added 10 mM LiCl is present, which is attributed to differences in chloride composition of the water and the dependence of the aqueous phase reference electrode on this chloride content.95 Likewise, the peak responses of PFOA in both matrices were slightly decreased compared to the aqueous phase comprised of 10 mM LiCl in high purity water (Fig. 4B). However, in simple matrices like clean water, there is little impact of the sample matrix on the detection capability.
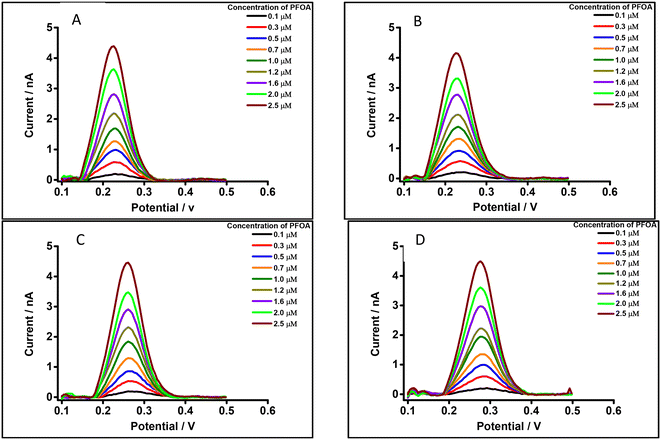 |
| | Fig. 7 DPVs of PFOA using 10 mM BTPPATPBCl (1,2-DCE) as the organic phase, cell 1. Concentration range: 0.1–2.5 μM. (A) Drinking water with 10 mM LiCl as the aqueous phase (pH = 7.4), (B) laboratory tap water with 10 mM LiCl as the aqueous phase (pH = 7.2), (C) drinking water without LiCl as the aqueous phase (pH = 7.4), (D) laboratory tap water without LiCl as the aqueous phase (pH = 7.2). All DPVs were background-subtracted followed by baseline corrected. | |
Matrix effects of bovine serum albumin (BSA) and β-cyclodextrin (β-CD)
Studies have shown that PFAS such as PFOA and PFOS interact with serum albumin to form complexes.96,97 Likewise, β-CD forms a complex with PFAS, e.g. PFOA.98,99 In order to understand possible matrix effects of BSA and β-CD on the detection of PFOA at a μITIES array, experiments were undertaken using DPV in the presence of a physiological concentration of BSA (i.e. 42 mg mL−1; ca. 0.6 mM)100 in 10 mM LiCl as the aqueous phase. Fig. 8A displays DPVs of different concentrations of PFOA in the presence of this concentration of BSA. The results reveal that the detection of PFOA in the presence of the physiological concentration of BSA is significantly attenuated compared to without BSA (Fig. 4B). However, once PFOA was detected, the DPV current increased linearly with PFOA concentration. Similarly, the effect of β-CD on the electrochemical response of PFOA was also investigated, in the presence of 0.6 mM β-CD in 10 mM LiCl aqueous phase. Fig. 8B shows DPVs of different concentrations of PFOA at fixed concentration of β-CD. At higher PFOA concentrations, there is linearity between the peak current and PFOA concentration. In the presence of β-CD, detection of PFOA is easier than in the presence of the same concentration of BSA. However, the detection of PFOA in the presence of 0.6 mM β-CD is lower than in absence of β-CD (Fig. 4B). These results show that BSA or β-CD have a significant influence on the electrochemical response of PFOA. This is attributed to the complexation of PFOA by BSA or by β-CD; this complexation removes free ionised PFOA from solution and makes it unavailable for transfer across the ITIES. BSA and β-CD are used here as model complexants for PFOA, illustrating the possible impact of matrix components that sequester PFOA in samples and consequently alter the detection sensitivity.
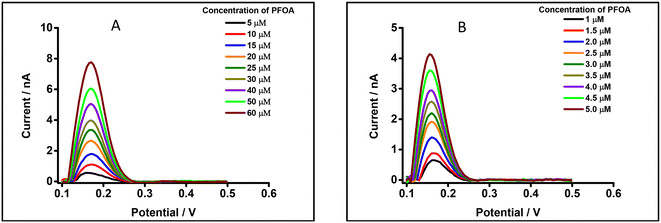 |
| | Fig. 8 DPVs of PFOA using 10 mM BTPPATPBCl (1,2-DCE) as the organic phase. (A) 0.6 mM BSA in 10 mM LiCl as the aqueous phase (pH = 6.8). Concentration range: 5–60 μM of PFOA. (B) 0.6 mM β-CD in 10 mM LiCl as the aqueous phase (pH = 7.3). Concentration range: 1–5 μM of PFOA. All DPVs were background-subtracted followed by baseline corrected. | |
Conclusions
PFOA is an anthropogenic environmental contaminant which is persistent and bioaccumulative in nature. In this study, the electrochemical behaviour of PFOA and the sensitivity of different voltammetric techniques for the detection of PFOA at the μITIES array were investigated. CV revealed that steady state current and peak current were obtained, respectively, for water to organic and organic to water transfers. A pH study revealed that the pH of the aqueous phase pH ≤ 3 is unsuitable for the detection of PFOA, due to its acid–base properties. Among the techniques studied, DPSV was the most sensitive. The lowest LOD for PFOA was 1.2 nM (0.5 μg L−1), following preconcentration at 0.1 V for 300 s without stirring. Furthermore, the matrix study revealed that the electrochemical response of PFOA was impacted by the matrix component of the studied sample. Overall these results show that PFOA electrochemistry at the μITIES array may be useful for screening and detection of PFOA in environmental samples.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank Curtin University for provision of a PhD Scholarship to HBL. We thank the South Australian and OptoFab nodes of the NCRIS-enabled Australian National Fabrication Facility (ANFF-SA and ANFF-OptoFab, respectively) for fabrication of the glass microporous membranes.
References
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
-
A. Falk-Filipsson, S. Fischer, J. Ivarsson, J. Forsberg and M. Delvin, Occurrence and use of highly fluorinated substances and alternatives, The Swedish Chemical Agency (KEMI), Stockholm, 2015 Search PubMed.
-
E. Kissa, Fluorinated Surfactants and Repellents, Marcel Dekker, New York, 2001 Search PubMed.
- G. W. Curtzwiler, P. Silva, A. Hall, A. Ivey and K. Vorst, Integr. Environ. Assess. Manage., 2021, 17, 7–12 CrossRef CAS PubMed.
- J. R. Lang, B. M. Allred, G. F. Peaslee, J. A. Field and M. A. Barlaz, Environ. Sci. Technol., 2016, 50, 5024–5032 CrossRef PubMed.
- X. Trier, K. Granby and J. H. Christensen, Environ. Sci. Pollut. Res., 2011, 18, 1108–1120 CrossRef CAS PubMed.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- H. D. Whitehead, M. Venier, Y. Wu, E. Eastman, S. Urbanik, M. L. Diamond, A. Shalin, H. Schwartz-Narbonne, T. A. Bruton, A. Blum, Z. Wang, M. Green, M. Tighe, J. T. Wilkinson, S. McGuinness and G. F. Peaslee, Environ. Sci. Technol. Lett., 2021, 8, 538–544 CrossRef CAS.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed.
- N. A. Fernandez, L. Rodriguez-Freire, M. Keswani and R. Sierra-Alvarez, Environ. Sci.: Water Res. Technol., 2016, 2, 975–983 RSC.
- J. L. Guelfo and D. T. Adamson, Environ. Pollut., 2018, 236, 505–513 CrossRef CAS PubMed.
- S. Banzhaf, M. Filipovic, J. Lewis, C. J. Sparrenbom and R. Barthel, Ambio, 2017, 46, 335–346 CrossRef CAS PubMed.
- X. Dauchy, V. Boiteux, C. Bach, C. Rosin and J. F. Munoz, Chemosphere, 2017, 183, 53–61 CrossRef CAS PubMed.
- X. C. Hu, D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum, S. A. Balan, C. P. Higgins and E. M. Sunderland, Environ. Sci. Technol. Lett., 2016, 3, 344–350 CrossRef CAS PubMed.
- V. Boiteux, C. Bach, V. Sagres, J. Hemard, A. Colin, C. Rosin, J. F. Munoz and X. Dauchy, Int. J. Environ. Anal. Chem., 2016, 96, 705–728 CrossRef CAS.
- H. Yan, I. T. Cousins, C. Zhang and Q. Zhou, Sci. Total Environ., 2015, 524–525, 23–31 CrossRef CAS PubMed.
- C. Gallen, D. Drage, G. Eaglesham, S. Grant, M. Bowman and J. F. Mueller, J. Hazard. Mater., 2017, 331, 132–141 CrossRef CAS PubMed.
- C. Bach, X. Dauchy, V. Boiteux, A. Colin, J. Hemard, V. Sagres, C. Rosin and J.-F. Munoz, Environ. Sci. Pollut. Res., 2017, 24, 4916–4925 CrossRef CAS PubMed.
- T. L. Coggan, D. Moodie, A. Kolobaric, D. Szabo, J. Shimeta, N. D. Crosbie, E. Lee, M. Fernandes and B. O. Clarke, Heliyon, 2019, 5, e02316 CrossRef PubMed.
- M. S. Lal, M. Megharaj, R. Naidu and M. M. Bahar, Environ. Technol. Innovation, 2020, 19, 100863 CrossRef.
- J. Bao, C. L. Li, Y. Liu, X. Wang, W. J. Yu, Z. Q. Liu, L. X. Shao and Y. H. Jin, Environ. Res., 2020, 188, 109751 CrossRef CAS PubMed.
- M. K. So, N. Yamashita, S. Taniyasu, Q. Jiang, J. P. Giesy, K. Chen and P. K. S. Lam, Environ. Sci. Technol., 2006, 40, 2924–2929 CrossRef CAS PubMed.
- G. Zheng, E. Schreder, J. C. Dempsey, N. Uding, V. Chu, G. Andres, S. Sathyanarayana and A. Salamova, Environ. Sci. Technol., 2021, 55, 7510–7520 CrossRef CAS PubMed.
- L. M. L. Toms, A. M. Calafat, K. Kato, J. Thompson, F. Harden, P. Hobson, A. Sjödin and J. F. Mueller, Environ. Sci. Technol., 2009, 43, 4194–4199 CrossRef CAS PubMed.
- J. Thompson, G. Eaglesham and J. Mueller, Chemosphere, 2011, 83, 1320–1325 CrossRef CAS PubMed.
- O. Quiñones and S. A. Snyder, Environ. Sci. Technol., 2009, 43, 9089–9095 CrossRef PubMed.
- S. Hansen, R. Vestergren, D. Herzke, M. Melhus, A. Evenset, L. Hanssen, M. Brustad and T. M. Sandanger, Environ. Int., 2016, 94, 272–282 CrossRef CAS PubMed.
- P. A. Fair, B. Wolf, N. D. White, S. A. Arnott, K. Kannan, R. Karthikraj and J. E. Vena, Environ. Res., 2019, 171, 266–277 CrossRef CAS PubMed.
- F. Xiao, Water Res., 2017, 124, 482–495 CrossRef CAS PubMed.
- C. Tang, J. Tan, C. Wang and X. Peng, J. Chromatogr. A, 2014, 1341, 50–56 CrossRef CAS PubMed.
- P. Jing, P. J. Rodgers and S. Amemiya, J. Am. Chem. Soc., 2009, 131, 2290–2296 CrossRef CAS PubMed.
- L. J. L. Espartero, M. Yamada, J. Ford, G. Owens, T. Prow and A. Juhasz, Environ. Res., 2022, 212, 113431 CrossRef PubMed.
- K. Ji, Y. Kim, S. Oh, B. Ahn, H. Jo and K. Choi, Environ. Toxicol. Chem., 2008, 27, 2159–2168 CrossRef CAS PubMed.
- D. Xu, C. Li, Y. Wen and W. Liu, Environ. Pollut., 2013, 174, 121–127 CrossRef CAS PubMed.
- G. Liu, S. Zhang, K. Yang, L. Zhu and D. Lin, Environ. Pollut., 2016, 214, 806–815 CrossRef CAS PubMed.
- M. E. Austin, B. S. Kasturi, M. Barber, K. Kannan, P. S. MohanKumar and S. M. J. MohanKumar, Environ. Health Perspect., 2003, 111, 1485–1489 CrossRef CAS PubMed.
- A. M. Seacat, P. J. Thomford, K. J. Hansen, G. W. Olsen, M. T. Case and J. L. Butenhoff, Toxicol. Sci., 2002, 68, 249–264 CrossRef CAS PubMed.
- Q. Yang, M. Abedi-Valugerdi, Y. Xie, X. Y. Zhao, G. Möller, B. D. Nelson and J. W. DePierre, Int. Immunopharmacol., 2002, 2, 389–397 CrossRef CAS PubMed.
-
M. Kirk, K. Smurthwaite, J. Bräunig, S. Trevenar, C. D'Este, R. Lucas, A. Lal, R. Korda, A. Clements, J. Mueller and B. P. Armstrong, The PFAS Health Study: Systematic Literature Review, The Australian National University, Canberra, 2018 Search PubMed.
- S. M. Bartell and V. M. Vieira, J. Air Waste Manage. Assoc., 2021, 71, 663–679 CrossRef CAS PubMed.
- M. Trojanowicz and M. Koc, Microchim. Acta, 2013, 180, 957–971 CrossRef CAS PubMed.
- M. Takino, S. Daishima and T. Nakahara, Rapid Commun. Mass Spectrom., 2003, 17, 383–390 CrossRef CAS PubMed.
- S. Poothong, S. K. Boontanon and N. Boontanon, J. Hazard. Mater., 2012, 205–206, 139–143 CrossRef CAS PubMed.
- C. Gremmel, T. Frömel and T. P. Knepper, Anal. Bioanal. Chem., 2017, 409, 1643–1655 CrossRef CAS PubMed.
- Z. Cheng, L. Du, P. Zhu, Q. Chen and K. Tan, Spectrochim. Acta, Part A, 2018, 201, 281–287 CrossRef CAS PubMed.
- Z. Cheng, H. Dong, J. Liang, F. Zhang, X. Chen, L. Du and K. Tan, Spectrochim. Acta, Part A, 2019, 207, 262–269 CrossRef CAS PubMed.
- Y. Wang and H. Zhu, Anal. Methods, 2014, 6, 2379–2383 RSC.
- Q. Liu, A. Huang, N. Wang, G. Zheng and L. Zhu, J. Lumin., 2015, 161, 374–381 CrossRef CAS.
- C. Fang, M. Megharaj and R. Naidu, RSC Adv., 2016, 6, 11140–11145 RSC.
- H. Ryu, B. Li, S. De Guise, J. McCutcheon and Y. Lei, J. Hazard. Mater., 2021, 408, 124437 CrossRef CAS PubMed.
- H. B. Lamichhane and D. W. M. Arrigan, Curr. Opin. Electrochem., 2023, 40, 101309 CrossRef CAS.
- R. B. Clark and J. E. Dick, ACS Sens., 2020, 5, 3591–3598 CrossRef CAS PubMed.
- N. Karimian, A. M. Stortini, L. M. Moretto, C. Costantino, S. Bogialli and P. Ugo, ACS Sens., 2018, 3, 1291–1298 CrossRef CAS PubMed.
- M. W. Glasscott, K. J. Vannoy, R. Kazemi, M. D. Verber and J. E. Dick, Environ. Sci. Technol. Lett., 2020, 7, 489–495 CrossRef CAS.
- R. Kazemi, E. I. Potts and J. E. Dick, Anal. Chem., 2020, 92, 10597–10605 CrossRef CAS PubMed.
- R. Ranaweera, C. Ghafari and L. Luo, Anal. Chem., 2019, 91, 7744–7748 CrossRef CAS PubMed.
- J. Gong, T. Fang, D. Peng, A. Li and L. Zhang, Biosens. Bioelectron., 2015, 73, 256–263 CrossRef CAS PubMed.
- C. Fang, Z. Chen, M. Megharaj and R. Naidu, Environ. Technol. Innovation, 2016, 5, 52–59 CrossRef.
- P. Vanýsek and L. B. Ramírez, J. Chil. Chem. Soc., 2008, 53, 1455–1463 Search PubMed.
- A. A. Stewart, G. Taylor, H. H. Girault and J. McAleer, J. Electroanal. Chem. Interfacial Electrochem., 1990, 296, 491–515 CrossRef CAS.
- G. Taylor and H. H. J. Girault, J. Electroanal. Chem. Interfacial Electrochem., 1986, 208, 179–183 CrossRef CAS.
- M. D. Scanlon and D. W. M. Arrigan, Electroanalysis, 2011, 23, 1023–1028 CrossRef CAS.
- G. Herzog, Analyst, 2015, 140, 3888–3896 RSC.
- T. Kakutani, T. Ohkouchi, T. Osakai, T. Kakiuchi and M. Senda, Anal. Sci., 1985, 1, 219–225 CrossRef CAS.
- T. Osakai, T. Kakutani and M. Senda, Bunseki Kagaku, 1984, 33, E371–E377 CrossRef CAS.
- M. B. Garada, B. Kabagambe, Y. Kim and S. Amemiya, Anal. Chem., 2014, 86, 11230–11237 CrossRef CAS PubMed.
- B. N. Viada, L. M. Yudi and D. W. M. Arrigan, Analyst, 2020, 145, 5776–5786 RSC.
- G. J. Islam and D. W. M. Arrigan, ACS Sens., 2022, 7, 2960–2967 CrossRef CAS PubMed.
- A. J. Olaya, M. A. Méndez, F. Cortes-Salazar and H. H. Girault, J. Electroanal. Chem., 2010, 644, 60–66 CrossRef CAS.
- E. Alvarez de Eulate, J. Strutwolf, Y. Liu, K. O'Donnell and D. W. M. Arrigan, Anal. Chem., 2016, 88, 2596–2604 CrossRef CAS PubMed.
- N. O. Brace, J. Org. Chem., 1962, 27, 4491–4498 CrossRef CAS.
-
A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2022, p. 369 Search PubMed.
- M. M. Hossain, S. H. Lee, H. H. Girault, V. Devaud and H. J. Lee, Electrochim. Acta, 2012, 82, 12–18 CrossRef CAS.
- H. R. Kim, C. M. Pereira, H. Y. Han and H. J. Lee, Anal. Chem., 2015, 87, 5356–5362 CrossRef CAS PubMed.
- G. Herzog and V. Beni, Anal. Chim. Acta, 2013, 769, 10–21 CrossRef CAS PubMed.
- C. Ariño, C. E. Banks, A. Bobrowski, R. D. Crapnell, A. Economou, A. Krolicka, C. Perez-Rafols, D. Soulis and J. Wang, Nat. Rev. Methods Primers, 2022, 2, 62 CrossRef.
- J. Strutwolf, M. D. Scanlon and D. W. M. Arrigan, J. Electroanal. Chem., 2010, 641, 7–13 CrossRef CAS.
- M. D. Scanlon, G. Herzog and D. W. M. Arrigan, Anal. Chem., 2008, 80, 5743–5749 CrossRef CAS PubMed.
- X. Jiang, K. Gao, D. Hu, H. Wang, S. Bian and Y. Chen, Analyst, 2015, 140, 2823–2833 RSC.
- S. Kihara, M. Suzuki, M. Sugiyama and M. Matsui, J. Electroanal. Chem. Interfacial Electrochem., 1988, 249, 109–122 CrossRef CAS.
- Y. Kim, P. J. Rodgers, R. Ishimatsu and S. Amemiya, Anal. Chem., 2009, 81, 7262–7270 CrossRef CAS PubMed.
- L. D. Chen, C. Z. Lai, L. P. Granda, M. A. Fierke, D. Mandal, A. Stein, J. A. Gladysz and P. Bühlmann, Anal. Chem., 2013, 85, 7471–7477 CrossRef CAS PubMed.
- Z. Zheng, H. Yu, W. C. Geng, X. Y. Hu, Y. Y. Wang, Z. Li, Y. Wang and D. S. Guo, Nat. Commun., 2019, 10, 5762 CrossRef CAS PubMed.
- J. Wang, Y. Shi and Y. Cai, J. Chromatogr. A, 2018, 1544, 1–7 CrossRef CAS PubMed.
- M. M. Shanbhag, N. P. Shetti, S. S. Kalanur, B. G. Pollet, M. N. Nadagouda and T. M. Aminabhavi, Chem. Eng. J., 2022, 434, 134700 CrossRef CAS.
- S. P. Sahu, S. Kole, C. G. Arges and M. R. Gartia, ACS Omega, 2022, 7, 5001–5007 CrossRef CAS PubMed.
- R. F. Menger, E. Funk, C. S. Henry and T. Borch, Chem. Eng. J., 2021, 417, 129133 CrossRef CAS.
- C. A. Huset and K. M. Barry, MethodsX, 2018, 5, 697–704 CrossRef PubMed.
- K. L. Rodriguez, J. H. Hwang, A. R. Esfahani, A. H. M. A. Sadmani and W. H. Lee, Micromachines, 2020, 11, 667 CrossRef PubMed.
- Department of Health, Health Based Guidance values for Per- and Poly-fluoroalkyl substances (PFAS), https://www.health.gov.au/sites/default/files/documents/2022/07/health-based-guidance-values-for-pfas-for-use-in-site-investigations-in-australia_0.pdf, (accessed 05/March/2023)..
- Drinking Water Health Advisories for PFOA and PFOS, 2022 Interim Updated PFOA and PFOS Health Advisories, https://www.epa.gov/sdwa/drinking-water-health-advisories-pfoa-and-pfos, (accessed 01/January/2023).
- Y. Liu, J. Bao, X. M. Hu, G. L. Lu, W. J. Yu and Z. H. Meng, Microchem. J., 2020, 155, 104673 CrossRef CAS.
- J. Cheng, C. D. Vecitis, H. Park, B. T. Mader and M. R. Hoffmann, Environ. Sci. Technol., 2008, 42, 8057–8063 CrossRef CAS PubMed.
- C. E. Schaefer, C. Andaya, A. Burant, C. W. Condee, A. Urtiaga, T. J. Strathmann and C. P. Higgins, Chem. Eng. J., 2017, 317, 424–432 CrossRef CAS.
- K. R. Temsamani and K. L. Cheng, Sens. Actuators, B, 2001, 76, 551–555 CrossRef CAS.
- X. Han, T. A. Snow, R. A. Kemper and G. W. Jepson, Chem. Res. Toxicol., 2003, 16, 775–781 Search PubMed.
- P. D. Jones, W. Hu, W. De Coen, J. L. Newsted and J. P. Giesy, Environ. Toxicol. Chem., 2003, 22, 2639–2649 CrossRef CAS PubMed.
- M. J. Weiss-Errico, J. Miksovska and K. E. O'Shea, Chem. Res. Toxicol., 2018, 31, 277–284 Search PubMed.
- A. H. Karoyo, A. S. Borisov, L. D. Wilson and P. Hazendonk, J. Phys. Chem. B, 2011, 115, 9511–9527 CrossRef CAS PubMed.
- L. R. S. Barbosa, M. G. Ortore, F. Spinozzi, P. Mariani, S. Bernstorff and R. Itri, Biophys. J., 2010, 98, 147–157 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*