
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of functional surface and molecular dynamics at Pt-rGO by spacer 1,6-hexanediamine for precise detection of biomolecules: uric acid as a specimen†
Mohammad Razaul
Karim
 *a,
Mohammad
Jayed
a,
Md. Zakariya Rahman
Laskar
a,
Md Murshed
Bhuyan
b,
Md. Saidul
Islam
c,
Shinya
Hayami
c and
Mohammed M.
Rahman
d
*a,
Mohammad
Jayed
a,
Md. Zakariya Rahman
Laskar
a,
Md Murshed
Bhuyan
b,
Md. Saidul
Islam
c,
Shinya
Hayami
c and
Mohammed M.
Rahman
d
aDepartment of Chemistry, School of Physical Sciences, Shahjalal University of Science and Technology, Sylhet-3114, Bangladesh. E-mail: krazaul@yahoo.com; Tel: +8801750200968
bDepartment of Mechanical Engineering, Gachon University, South Korea
cDepartment of Chemistry, GSST, Kumamoto University, Japan
dDepartment of Chemistry, King Abdulaziz University, Saudi Arabia
First published on 1st September 2023
Abstract
The enhancement of a functional surface and molecular migration through reduced graphene oxide (rGO) with consequent detection of low-concentration uric acid (UA) is presented. We observed excellent molecular migration in an organic–inorganic composite, in which 1,6-hexanediamine (HA) was incorporated in rGO-platinum (Pt) matrices to obtain HA-rGO-Pt. The rGO, Pt and HA precursor played the roles of conductive template, electrocatalyst, and spacer group. As a sensing probe on a glassy carbon electrode (surface area 0.0316 cm2), the material afforded precise uric acid (UA) detection with a linear dynamic range (LDR) and sensitivity of 0.1 nM–0.1 mM UA and 24.2089 μA μM−1 cm−2. The signal to noise ratio was 3, and the lower limit of detection (LOD) was 39.22 ± 1.96 pM. The high sensitivity and rapid detection of UA was possible due to the improved hydronium ion conductivity (∼10−5–10−4 S cm−1) and moderate electron conductivity. This strategy of widening the molecular migration track might be considered as a facile way to adopt graphene-based composites for various applications, including sensing and catalysis.
1. Introduction
Facile molecular dynamics through layered materials is necessary to achieve their successful application in catalysis, sensing, ion conduction and so on. Due to the stacking nature of graphene materials during solid state processing, the optimum benefit of the high surface area of graphene is overwhelmed.1 In stacked form, the molecular entity fails to penetrate through the narrow layers of graphene oxide (GO) or partially reduced graphene oxide (rGO). This drawback becomes serious in the case of biomolecular detection, as previously we found that an organic molecular entity can pass through the nanopore and junction but becomes blocked when migrating through the interlayer channels of stacked GO or rGO.2 The present work is designed to solve this issue. Precise quantitative detection of biomolecules through electrochemical assay is of emerging interest, as these techniques can be utilized to generate portable sensing kits for healthcare, industry, and pharmaceuticals.3,4 In addition to electrical signal generation, digitalization and display, the assembly of nanomaterials on electrode tips is a vital task. However, fixing the composition of the electrode tip is the most vulnerable part. The design of the electrode is stipulated by the need to confirm both the electron mediation property and catalysis of the sensing process (redox, degradation or catalytic reactions) to detect the target molecules.5 Even when these essential criteria are achieved by developing highly conductive and reaction-selective sensing electrode tips, sometimes the detection of bulk-sized analytes presents difficulties, as the analytes cannot easily migrate around the active sites of the electrode. Even much smaller and hydrophilic analyte materials in the solvated form fail to penetrate the stacked active material in some aqueous systems. For graphene- (G) or rGO-based electrode assemblies, another serious drawback arises from their hydrophobic character. After losing the extended functional groups, the layers of rGO tend to be stacked with shrinking of the interlayer distance. Thus, they reduce the migration of the analyte.6 To solve this problem, in the present report we considered increasing both the hydrophilicity and flexibility of the composite sensing probe. We introduced Pt to GO, which was then treated with 1,6-hexanediamine (HA). The HA-GO-Pt was then reduced to a HA-rGO-Pt composite. The fabrication of this composite system was designed to address some technical issues. First, rGO matrices possessing excellent electron conductivity have been reported to function as electron mediators. Second, Pt nanoparticles can function as a catalyst and are also capable of increasing the stability, electron mediation property and electrode scaffolding capacity. Finally, the spacer group HA was introduced to increase both the hydrophilicity and flexibility of the rGO interlayer.7,8 It was assumed that the conductive Pt-rGO matrices might show association with the electrode tips due to the flat structure of rGO and tiny spherical shape of Pt. Thus, an excellent electron mediation process was expected. The migration of biomolecules through the gap between two neighboring rGO nanosheets might be expedited by a more flexible interlayer. We choose uric acid (UA) as a typical biomolecular specimen to investigate the efficiency of the HA-rGO-Pt sensor.UA acts as a very active reducing and anti-oxidizing agent in human blood serum and sweat.9 Due to the metabolism of purine, uric acid is produced in humans and animals and excreted renally by means of blood circulation. In the case of poor kidney function, the concentration of UA in human blood serum is gradually enriched, and physical syndromes such as perinatal asphyxia, hyperuricemia, cerebral ischemia, Parkinson's disease, atherosclerosis, leukemia, pre-eclampsia, high blood lipids, gout, high blood pressure, tumors, and kidney disease are observed in humans.10–12 Therefore, the primary and early diagnosis of high UA in human blood and urine serum can save thousands of lives. For this purpose, tiny smart devices are used for their comfortable and facile application. Research into the analytical detection of UA through easy and more inexpensive methods is thus are carried out worldwide. Presently, the analytical methods used for this purpose include high-performance liquid chromatography, electrochemical methods, fluorescence probes23 and enzymatic detection.13–16 Among these, the electrochemical detection technique, specifically, the monitoring of current with respect to voltage (I–V method) represents the easiest and most frequent method. To date, a list of sensing probes composed of nanomaterials, including SnO2/V2O5, Ag2O/TiO2/ZrO2, etc., have been reported to function in I–V-method-based UA sensor probes. The related reports indicated excellent sensitivity values of 17.0918 and 56.3892 μA μM−1 cm−2 with a linear dynamic range of 0.1 nM–0.01 mM. The respective detection limits were 95.40 and 91.70 pM.17,18 In other studies, CuO·Fe2O3 and ZnO/Ag2O/Co3O4 based UA sensors exhibited wider detection ranges of 0.1 nM–0.01 mM and 100.0 pM–100.0 mM, respectively. The respective sensitivities were 82.3323 and 3.16 × 10−5 μA μM−1 cm−2.19,20 The detection of UA using G or GO composite electrodes is not so common. In recent years, a linear detection response for UA in the range of 0.200–22.0 μM was observed by Baig et al.21 Additionally, there exist other carbon-templated nanocrystalline materials for the precise detection of UA.22,23
Even when rGO was used as the electrode material, both the detection limit and sensitivity were comparatively lower than those of previous nanocomposite-based sensors.24 We propose that the reason for this disadvantage is the absence of a transition metal electrocatalytic precursor, as well as the difficulty of the migration of UA around the active sites of the electrode tip. However, the aim of the present study is to solve these issues. We introduced Pt nanoparticles and HA to overcome the drawbacks. This article represents not only a method for the detection of UA, but also a possible way of improving the sensing capability of rGO-based composites by expediting molecular migration through the interlayers. As a broader concept, this strategy of alkyl group incorporation for interlayer distance widening can be applied for other applications including catalysis and sensing. Although rGO nanocomposites are widely used in biosensors, herein, we first introduced a spacer group in such a hybrid to make the interlayer channel more flexible. This mechanistic advantage through influencing the molecular migration of the analyte through the interlayer of rGO is still not known. Hence, the present report clearly represents a novel strategy to enhance the sensing functionality of HA-rGO-Pt compared with that of rGO-Pt. The finding implies that an adjustable interlayer made by incorporating a suitable spacer group in the active materials of a sensing device can ensure better performance.
2. Experimental
2.1 Materials and methods
All the reagents, which included graphite powder, sulfuric acid, sodium nitrate, potassium permanganate, HA, hydrogen peroxide, HPtCl4, hydrazine solution, ammonia, UA, dopamine, glutathione, ascorbic acid, choline, cholesterol, testosterone, L-aspartic acid, lactic acid, dopamine, glucose, xanthine, Nafion, and mono- and disodium phosphate, were obtained from Merck, Germany. The HA-rGO-Pt composite was synthesized via a three-step synthetic route, which included GO synthesis from graphite powder by a modified Hummers' method, the physicochemical conversion of GO into HA-GO-Pt, and finally, the chemical conversion of HA-GO-Pt into HA-rGO-Pt. Here, rGO functions as the conductive dispersion matrix. HA was expected to play the roles of spacer group and N-doping agent. The Pt nanoparticles were grown to contribute stability, binding capacity and catalytic ability.2.2 Synthesis of HA-rGO-Pt
Hummers' method for graphite oxidation was followed to prepare GO.25 In short, graphite powder (0.3 g), H2SO4 (15 ml, 97%), and NaNO3 (0.3 g) in meshed form were mixed in a round bottom flask with stirring for 45 minutes using an ice bath. 1 g KMnO4 powder then was added and stirred to keep the temperature at around 15 °C for 2 h. The flask was then heated to 40 °C for 40 min. Subsequently, 60 ml water was then poured slowly. The temperature was allowed to increase to 95 °C for 3 hours. Then, the mixture was poured into 120 ml water in a large beaker. Afterward, 4 mL H2O2 (30%) solution was then added, and the mixture was then centrifuged at 4200 rpm for 25 min. The residue was cleaned with 10% HCl solution (×1), and water (×3). The solid mass obtained was dried for 25 h at 60 °C. The as-prepared solid GO was exfoliated in distilled water by sonication for 3 h, and the resultant GO dispersion (0.1 g L−1) was centrifuged to remove undispersed GO. Then, 80 ml of the GO solution was mixed with 80 ml of a 5 mM HPtCl4 solution in DMF with continuous stirring for 3 days. After that, 5 mg HA was added and the mixture was stirred for 2 more days. Then, 20 ml hydrazine solution was added, and the whole mixture was stirred for a further 2 days. Thus, after a total reaction time of 7 days, the resultant black residues were washed with distilled water and methanol and hexane. The synthetic route is illustrated in Fig. 1.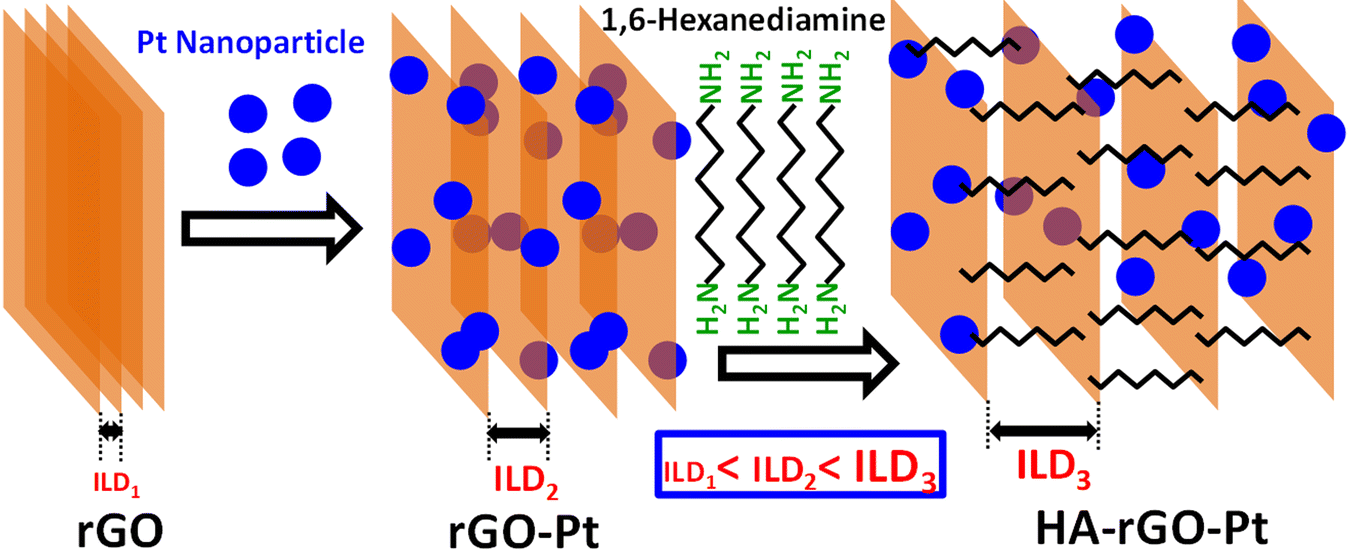 | ||
| Fig. 1 Scheme of the synthesis of the HA-rGO-Pt composite. Two principal steps were followed to synthesize the material. In the first step, GO was mixed with HPtCl4 in an aqueous system. In the second step, the GO-Pt was mixed with HA and subsequently reduced using hydrazine solution. The ILD gradually increases from rGO to rGO-Pt to HA-rGO-Pt. | ||
2.3 Characterization
The dispersion of Pt nanoparticles was studied using SEM (Hitachi High Tech SU-8000) and TEM analysis. The carbon hybridization state was studied using Raman spectroscopy (Raman spectrometer, JASCO Japan, NRS-3100 micro) with a 532 nm excitation source at room temperature. The ILD of the materials was studied using powder X-ray diffraction (PXRD). A Rigaku RAD-2A X-ray diffractometer equipped with a 2.0 kW Cu Kα X-ray source was used. In the PXRD sampling, the materials were collected by scratching the surface of a sample pellet that was prepared at a pressure of 40 Pa for 30 min. The chemical structures of the samples were investigated using FT-IR and XPS analysis. For XPS, a Thermo Scientific, Sigma Probe instrument with a monochromatized X-ray source (Al Kα, hν = 1486.6 eV) and a discharge source (He I, hν = 21.2 eV) was used. A vacuum of at least 10−7 Pa was ensured during the measurement.2.4 Measurements of molecular dynamics
The molecular dynamics were analyzed by studying the hydronium ion migration. Based on some previous reports, we determined the proton conductivity (PC) through pellets of GO, rGO, rGO-Pt and HA-rGO-Pt. The measurements were accomplished using a four-probe impedance (at 1 to 106 Hz) analyzing system employing a Solartron 1260/1296. A 120 mg solid sample was mechanically pressed into a coin-shaped pellet under a pressure of 40 Pa for 30 minutes.26 The faces were connected by 50 μm of gold using gold paste. Any connection between the two faces was carefully avoided. The impedance of PC was measured at room temperature and under various humidity conditions.2.5 Fabrication of HA-rGO-Pt-GCE
The GCE probe was prepared via some modifications. First, a slurry of 1 mg HA-rGO-Pt in 1 ml ethanol was prepared by ultrasonication for 5 min. The GCE top with a surface area of 0.0316 cm2 was then covered with the slurry using the drop-casting method. A thin layer was formed, which was further firmly attached using one drop of Nafion. The electrode was then incubated in an oven at 35 °C for an hour to obtain the HA-rGO-Pt-GCE electrode. A Keithley electrometer was used to investigate the desired electrochemical sensing performance. For this, the modified HA-rGO-Pt-GCE and a Pt wire were connected to the electrometer in series to function as working and counter electrodes, respectively. Solutions of the targeted analyte solution UA with concentrations of 0.1 nM to 1.0 mM were prepared by diluting UA in deionized water. The mono- and disodium phosphate solutions in deionized water were used to stabilize the buffer system with distinct pH values (5.7–8.0). For each measurement, 10.0 mL of the buffer solution was taken in the analyzing cell.3. Results and discussion
Fig. 2(a and b) presents the SEM image of the substance. The Pt nanoparticles can be clearly observed as bright spots. The TEM image of the substance is presented in Fig. 2(c). These microscopic images show that the Pt precursors are ∼5–12 nm apart from each other and are randomly dispersed on the rGO nanosheet. The EDS results and particle distances are presented in Fig. S1 of the ESI.†Fig. 2(b) depicts that HA-rGO-Pt retains a layered nature. The material seems to obtain a sandwich-like pattern in which HA precursors are located within the rGO-Pt layers. The Pt ionic precursors are at first trapped within the rGO matrix by electrostatic force. After reduction, the Pt precursors are encapsulated within the carbon sieve of rGO. Fig. 2(d) presents the PXRD spectra of GO, rGO-Pt and HA-rGO-Pt. The intercalation of the Pt nanoparticles and quantification of the interlayer distance (ILD) were analyzed using the peak position. Graphite, in general, presents a semi-crystalline structure. The synthesized GO exhibited a peak at ≈11.08° (2θ value). This value is associated with the 001 plane of graphite semi-crystals. Applying Bragg's law (1.54 Å), the ILD was calculated to be 7.98 Å. This value is typical for pristine GO.27 In rGO-Pt, the ILD is decreased to 3.52 Å (2θ = 25.28°, for the 002 graphitic semicrystalline plane). The shrinkage of the ILD is due to the removal of the extended functional groups of GO during its reduction to rGO. The PXRD of HA-rGO-Pt also shows three sharp peaks at 2θ values of 39.9°, 46.3° and 67.5°. These values correspond to the 111, 200 and 220 planes of the face-centered Pt crystal (JCPDS no. 03-065-2868).28 The HA-rGO-Pt exhibits an ILD of 4.10 Å (2θ = 21.65° for the 002 plane). Due to the intercalation of HA, the ILD is clearly increased in HA-rGO-Pt (compared with that in rGO-Pt). The graphitic peak broadening in the lower-angle region (for rGO) is due to the irregular distance between two neighboring rGO nanosheets. In fact, there is the possibility that some HA precursors do not necessarily stay perpendicular to the graphitic plane. In addition, the thermal motion of the nanosheets is also responsible for this irregular ILD. The irregular shape and size of the Pt nano-crystalline state is responsible for the widening of the diffraction peaks in the higher-angle region.29 The conversion of GO to conductive HA-rGO-Pt was also analyzed using the Raman spectra presented in Fig. 2(e). The relative sizes of the sp3 and sp2 carbon domains indicate the chemical change of GO into HA-rGO-Pt composites. The G band at ∼1595 cm−1 and D band at ∼1350 cm−1 in the graphitic materials arise due to the existence of the E2g photon of the sp2 carbon sites and the photon around the A1g symmetry κ-point for the breathing mode of the sp3 carbon atom. The peak intensity ratios of the D band to the G band (D/G) are found to be 0.92 and 0.83 for GO and HA-rGO-Pt, respectively. The value of D/G quantifies the extent of sp2 domain by an inverse proportion.30 Thus, the lowering of the D/G ratio during the conversion of GO into HA-rGO-Pt implies an increase in the conductive sp2 domain size. In addition to the destruction of epoxy sp3 sites, the association of the Pt precursor is responsible for the softening of the D/G ratio. Pt is deposited and grows in the rGO matrices mainly via the coulombic-attraction-oriented seeding process that we described in our previous report.31Fig. 2(f) presents the FT-IR spectra of GO and HA-rGO-Pt. The presence of hydrophilic groups in GO and disappearance of those in rGO agree with some previous reports.32 In GO, the typical FT-IR peaks at ∼3100–3300 cm−1 and ∼3300–3500 cm−1 arise from N–H and O–H bonds. The peak at around 1950 cm−1 arises from the existence of weak aromatic systems or![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H functional groups. The peak at ∼1080–1360 cm−1 representing C–N bonds is pronounced in HA-rGO-Pt, which indicates that due to the hydrazine-induced reduction, HA-rGO-Pt contains some carbon–nitrogen bonded systems.33 The existence of Pt in the HA-rGO-Pt composite is primarily indicated by the peaks below 1000 cm−1. The peaks at around 1640, 1730 and 1180 cm−1 arise from the stretching of –C–O–H, –CO and –C–O–C– (hydroxyl, carbonyl and carboxylic) bonds. These peaks have also nearly disappeared in HA-rGO-Pt. The peak at around 2500 cm−1 arises due the appearance of residual –OH functional groups, which are generated by the decomposition of the epoxy sites. However, the existence of some residual polar sites was confirmed later from the XPS analysis.
C–H functional groups. The peak at ∼1080–1360 cm−1 representing C–N bonds is pronounced in HA-rGO-Pt, which indicates that due to the hydrazine-induced reduction, HA-rGO-Pt contains some carbon–nitrogen bonded systems.33 The existence of Pt in the HA-rGO-Pt composite is primarily indicated by the peaks below 1000 cm−1. The peaks at around 1640, 1730 and 1180 cm−1 arise from the stretching of –C–O–H, –CO and –C–O–C– (hydroxyl, carbonyl and carboxylic) bonds. These peaks have also nearly disappeared in HA-rGO-Pt. The peak at around 2500 cm−1 arises due the appearance of residual –OH functional groups, which are generated by the decomposition of the epoxy sites. However, the existence of some residual polar sites was confirmed later from the XPS analysis.
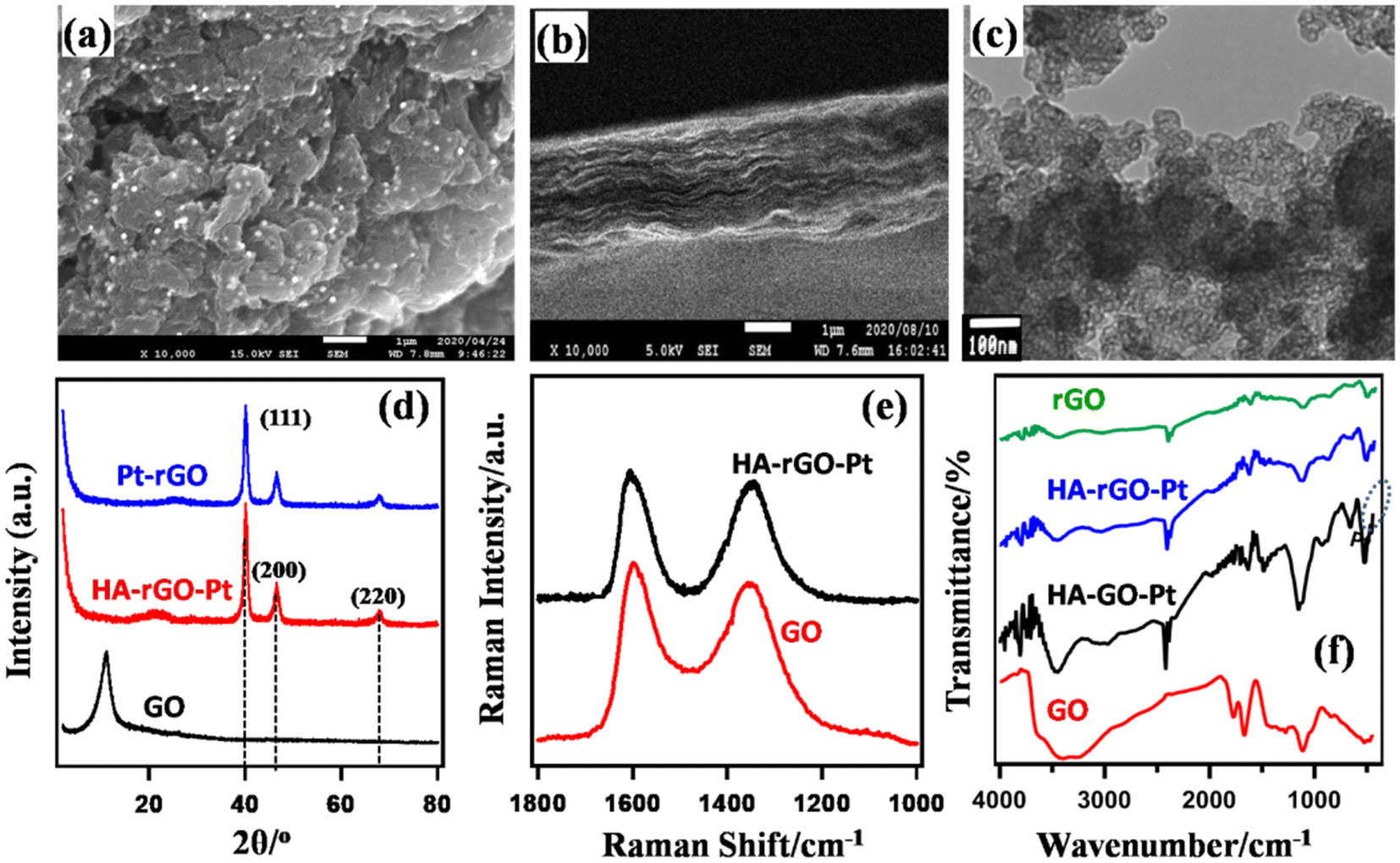 | ||
| Fig. 2 Characterization of HA-rGO-Pt composite system using SEM (a and b) and TEM (c) imaging, and PXRD (d), Raman (e), and FT-IR (f) spectra. | ||
Fig. 3 shows the XPS spectra of HA-rGO-Pt. The XPS study confirmed the association of both the doped nitrogen atoms and Pt ions with the rGO precursor. The individual spectra of the relevant materials are presented in Fig. S2.† In Fig. 3(a), the overall survey confirms the presence of Pt4f, N1s and O1s components at binding energies of ∼74, ∼284 and ∼768 eV, respectively. The presence of XPS peaks for oxygen-containing functional groups in GO and some residual oxygenated sites, along with the presence of nitrogen-containing groups, in rGO is well reported.34 Hence, herein, we have studied the XPS peaks associated with the Pt4f precursor in detail (Fig. 3(b)). The XPS spectra confirm that the composite contains platinum (Pt) along with platinum ions in the form of Pt2+ and Pt4+. The Pt4f spectrum was analyzed by splitting it into doublets, which are labeled as 4f5/2 and 4f7/2. In case of Pt4f5/2, the binding energies associated with Pt, Pt2+ and Pt4+ are found to be ∼74.7, ∼75.9, and ∼77.2 eV, respectively. However, for Pt4f7/2, the respective binding energies are ∼71.5, ∼72.6, and ∼73.8 eV. The higher peak intensity for Pt2+ (around 75.9 and 72.6 eV) indicates the existence of a greater amount of Pt2+ compared with the other components. The relative amounts of Pt and Pt ions were found to be 18.0 and 82.0%, respectively. The XPS spectra clearly provided information on the ionic states of Pt on the GO nanosheets. The XPS analysis thus confirmed the presence of Pt ions and metallic Pt in the composite system.
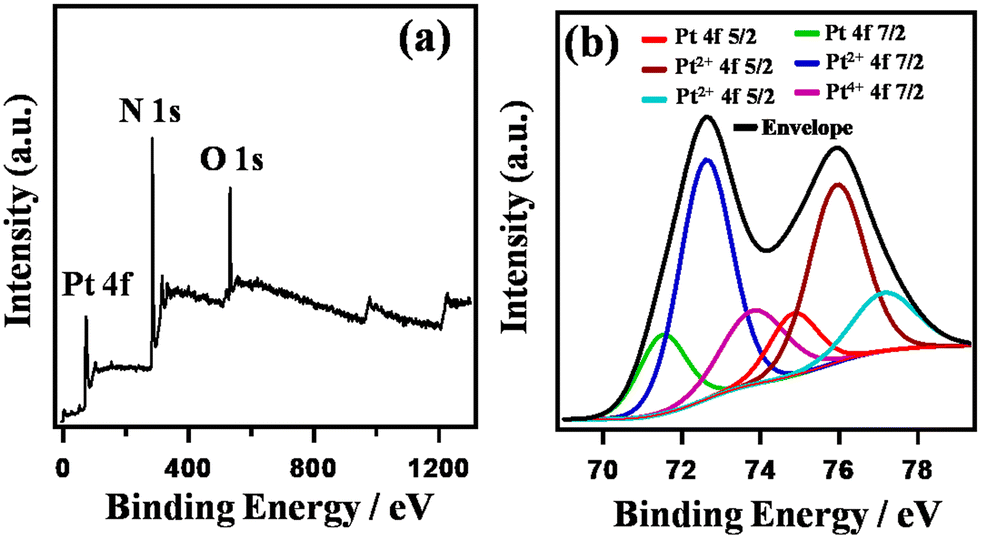 | ||
| Fig. 3 XPS spectra of HA-rGO-Pt. Overall survey (a) and Pt4f component (b). | ||
Fig. 4 presents the results of UA detection using the HA-rGO-Pt-GCE electrode assembly. First, a series of biomolecules, including UA, dopamine, glutathione, ascorbic acid, choline, cholesterol, testosterone and L-aspartic acid, were subjected to I–V analysis by applying the assembled sensor in pH 7.0 buffer. The obtained I–V datasets are presented in Fig. 4(a). The displayed currents gradually increase with voltage. Although all samples exhibit some response, UA shows the maximum current when scanning the voltage over 0–1.5 V. At 1.5 V, the current exhibited by the UA sample is 272.68 μA, which is much higher than the displayed current for testosterone (107.18), xanthine (92.99), L-aspartic acid (69.88), lactic acid (59.37), ascorbic acid (53.59), dopamine (47.81), and choline (32.05). Thus, the HA-rGO-Pt-GCE sensor was concluded to fit with the optimized selectivity for UA during the electrochemical detection. It is necessary for the assembled sensor to show similar I–V activity in distinct buffer solutions of various pH values. Therefore, we accomplished the detection of a 0.1 μM UA sample in buffer solutions of various pH values ranging from 5.7 to 8.0. The relevant results presented in Fig. 4(b) show that the current changes as a function of potential. The maximum response current of 270.33 μA is observed at pH 7.0. The observed current for detection in other buffer solutions of pH 7.5, 6.5, 5.7 and 8.0 was 226.15, 217.58, 175.38, and 151.65 μA, respectively. Fig. 4(c) presents the results of I–V detection using UA solutions of various concentrations (1.0 mM, 0.1 mM, 0.01 mM, 1.0 μM, 0.1 μM, 0.01 μM, 1.0 nM, 0.1 nM) at pH 7.0. In general, the displayed current increases with both increasing the concentration of UA and increasing the scan voltage. At 1.5 V, the optimized current was observed for 1.0 mM solution. However, the current in μA followed the trend 362.32 (1.0 mM) > 336.23 (0.1 mM) > 313.05 (0.01 mM) > 294.20 (1.0 μM) > 271.02 (0.1 μM) > 255.80 (0.01 μM) > 239.86 (1.0 nM) > 223.92 (0.1 nM). This direct proportional relationship between the I–V responses and UA concentrations indicates the proper functioning of the sensor and matches with some previous reports.35,36
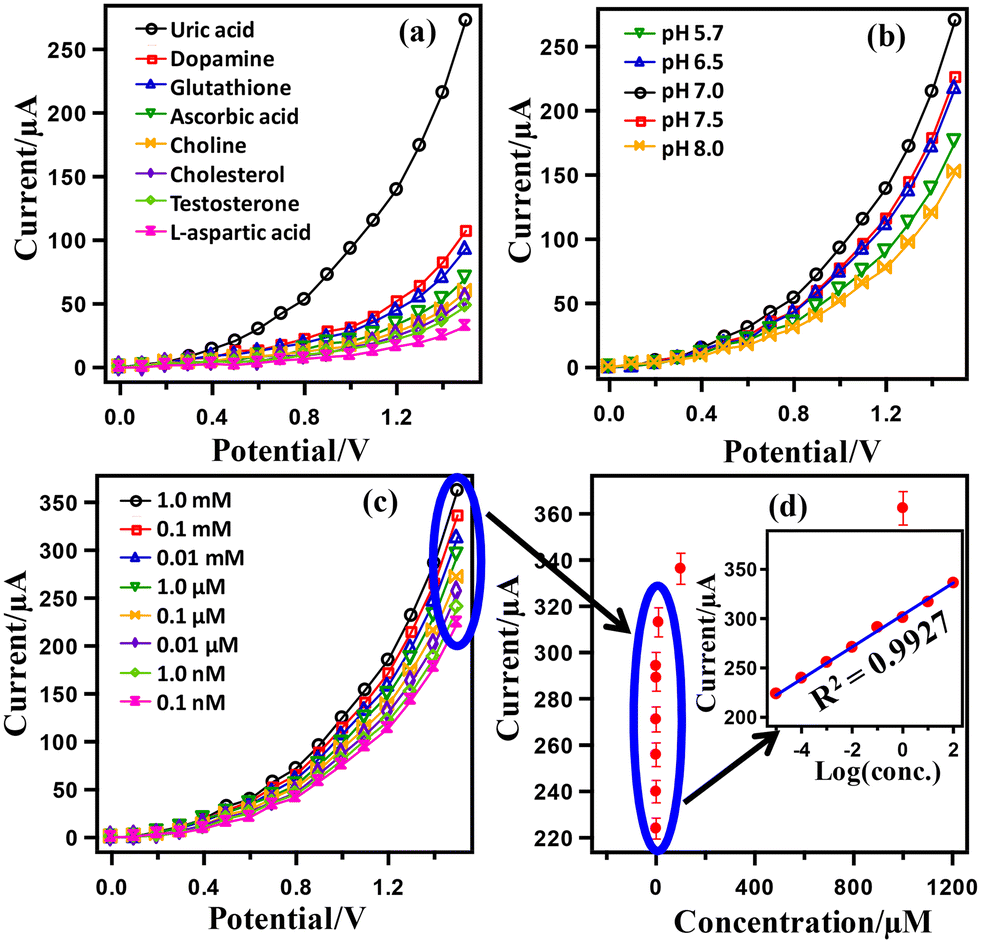 | ||
| Fig. 4 The electrochemical detection of UA using the HA-rGO-Pt-GCE sensor probe. Analysis of a series of biomolecules to identify selectivity (a); detection of 0.1 μM UA at various pH values (b); I–V responses for various concentrations of UA at pH 7 (c); current vs. concentration of UA at a fixed potential of 1.5 V (d); respective current vs. log (conc.) profile (inset of (d)). | ||
Fig. 4(d) presents a typical calibration curve obtained from the linear relationship between the displayed current and the UA concentration. As the calibration curve was almost linear for the concentration range of 0.1 mM–0.1 nM, this range may be defined as the linear dynamic range (LDR) for UA detection. The inset of Fig. 4(d) presents the plot of log (conc.) versus the displayed current, which has a regression co-efficient of R2 = 0.9927. This value supports the excellent linearity of LDR. From the slope of the LDR in Fig. 4(d), a sensor sensitivity of 24.2089 μA μM−1 cm−2 was calculated using the relationship sensitivity (μA μM−1 cm−2) = slope (μA μM−1)/surface area of the electrode (0.0316 cm2). In addition, using a signal/noise ratio (S/N) of 3, the limit of detection (LOD) was calculated as 39.22 ± 1.96 pM.
A control experiment was performed to observe the I–V responses using bare GCE and coated rGO-Pt-GCE, HA-rGO-Pt-GCE, BA-rGO-Pt-GCE and nHA-rGO-Pt-GCE (BA = butyl amine and nHA = n-hexyl amine) in 0.1 μM UA (buffer pH = 7) (Fig. 5(a)). A notable and optimum current response was observed using the HA-rGO-Pt-GCE electrode assembly. No current response was observed in the case of the bare GCE electrode. An inferior current response was displayed by BA-rGO-Pt-GCE and rGO-Pt-GCE. Although nHA-rGO-Pt-GCE exhibited some current response, it is inferior to the current generated by HA-rGO-Pt-GCE. This control experiment implies that the HA spacer group plays some vital role in the sensing reaction. The results for the measurements of the response time using 0.1 μM UA solution are presented in Fig. 5(b).
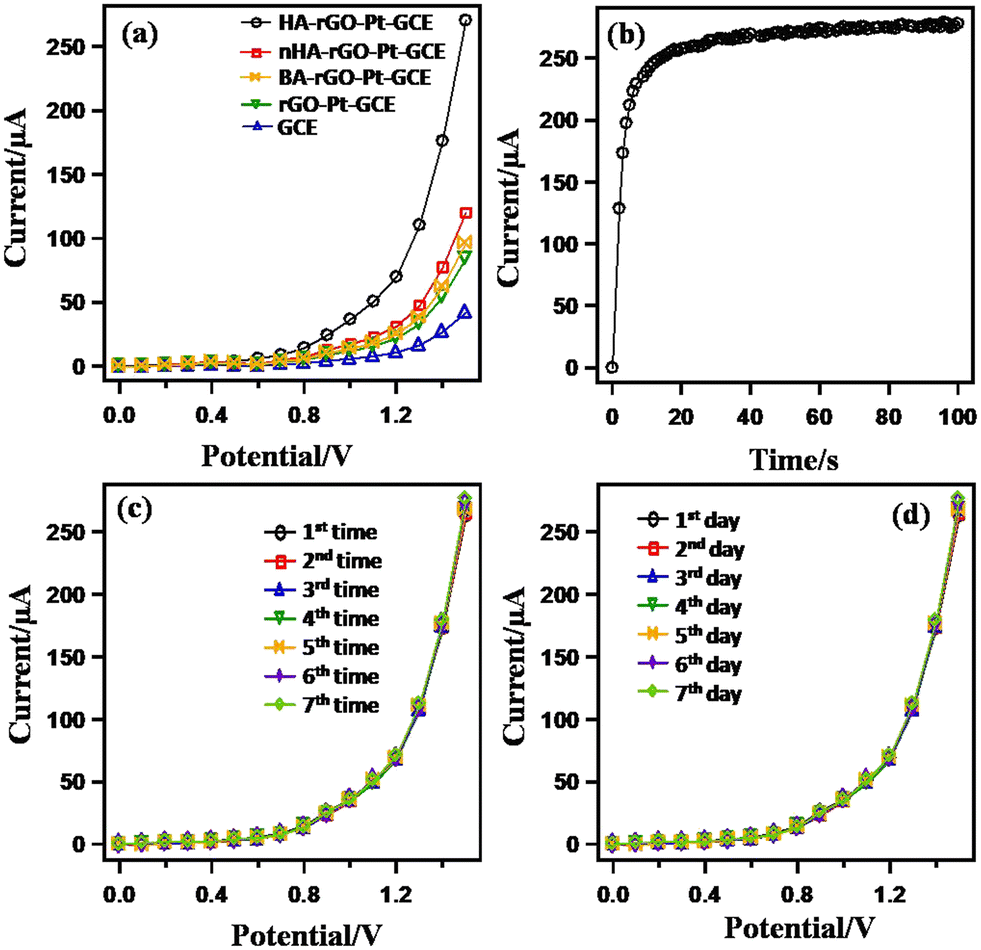 | ||
| Fig. 5 Control experiments for the UA sensor through monitoring the I–V response in 0.1 μM UA (7.0 pH buffer). Bare and modified GCE (a); varied response time (b); reusability (c); and (d) stability. | ||
The plot shows that the current becomes steady and constant beyond 20 s. Thus, an incubation time of 20 s is needed to execute the I–V analysis. The reliability of the UA sensor was studied by observing the reusability parameter. For this, the experiment was repeated successively up to seven times, and the data is plotted in Fig. 5(c). It is noted that the current is similar for all the repeated experiments. No appreciable changes in I–V response were noted, even when the working electrode was washed after each run. The repeatability parameter was estimated by measuring the relative standard deviation (RSD) of the current at +1.5 V and found to be 1.31% (RSD), which provided evidence of high precision. The long-term stability of the working electrode is also significant. This issue was investigated by repeating the experiments for seven consecutive days, and the results are presented in Fig. 5(d). The results were similar for all the test runs executed over seven days.
The necessity of the presence of Pt nanoparticles and the spacer HA groups to fabricate the electrode assembly was investigated by assembling a series of electrodes with non-Nafion conducting polymers, namely, poly(3,4-ethylenedioxythiophene) (PEDOT) and its complex with poly(styrene sulfonate) (PEDOT-PSS), and rGO along with other components. For this, the GCE was modified with the single element of HA-rGO-Pt-GCE-PEDOT-PSS and was subjected to I–V analysis using 0.1 μM UA and a buffer solution of pH 7.0. The resultant I–V curves are illustrated in Fig. 6(a).
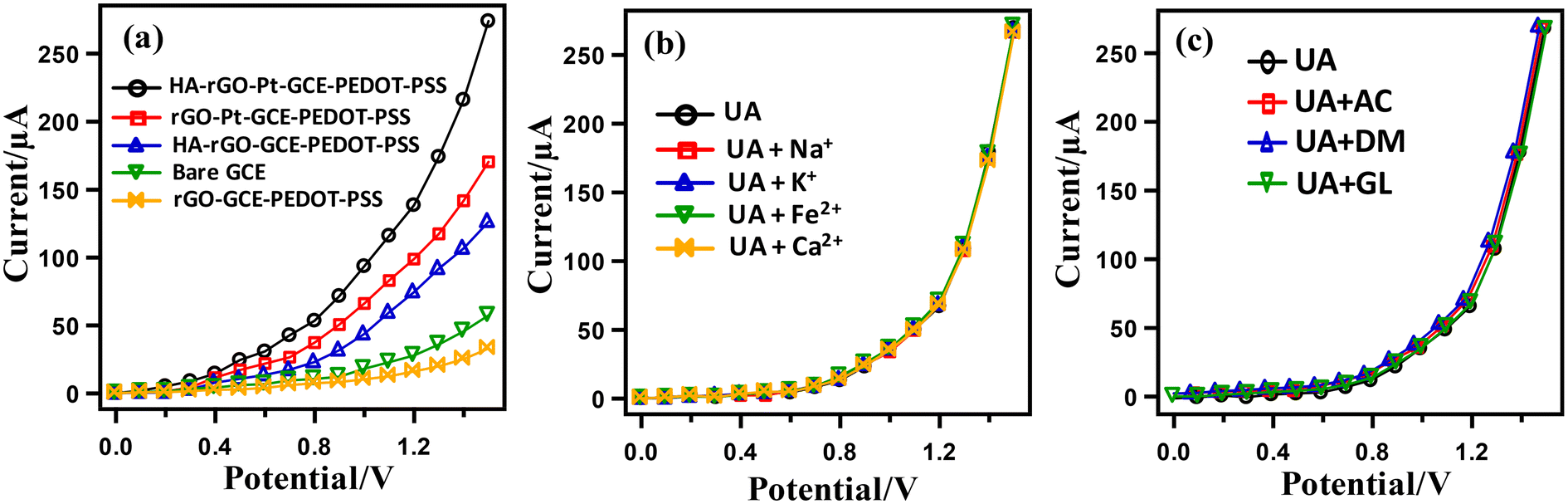 | ||
| Fig. 6 Effect of the substrate and interfering species. Current response of HA-rGO-Pt-GCE and other substrates in 0.1 μM UA solution (a); interference of common electrolytes (b); and interference of organic species including glucose (GL), ascorbic acid (AC) and dopamine (DM) (c). | ||
The HA-rGO-Pt-GCE-PEDOT-PSS composite exhibited the maximum output current response, which was attributed to the presence of the HA, rGO, Pt and GCE components. Next, we applied the sensor probe to detect UA in presence of various cations. Human serum (blood and urine) contains various electrolytes, including Na+, K+, Ca2+, Fe2+, etc. For the clinical diagnosis of UA, it is necessary to investigate the interferences of these electrolytes during the I–V analysis. For this, a control experiment was performed using 0.1 μM UA in presence of these electrolytes (Fig. 6(b)). It was observed that the I–V responses in the analysis of UA with and without electrolytes were repeatably similar. Hence, it was confirmed that the presence of common electrolytes does not influence the resulting current response. Similarly, in Fig. 6(c), the almost identical I–V response for UA alone and UA in presence of GL, AC and DM indicates that the modified electrode can detect UA without any interference from these common bio-impurities.
The catalytic UA sensing performance can be explained by studying the hydration dynamics of the material. The first step in UA detection is its affinity toward the electrode. Additionally, it is essential for the analyte to penetrate through the rGO walls to stay in close proximity to the catalytic ingredients, which are the Pt nanoparticles. Although the rGO walls slow the migration of UA to the active center, we cannot prepare a hybrid system without rGO, as it functions as an excellent electron mediator. In fact, a better solution is to ensure sufficient space at the rGO interlayer to support facile movement of the UA molecules. In this regard, HA was found to exhibit excellent functionality. We thus observed the extent of PC through the samples. As PC is directly associated with water adsorption, we also compared the amount of water adsorbed by the samples through TGA analysis. Before TGA analysis, the materials were humidified at 80% relative humidity (RH) for four hours at ambient conditions. The PC was evaluated by measuring the resistance when the hydrogen ion passes through the pellet samples. Details of the PC measurement are reported elsewhere.26 In the Cole–Cole plot (obtained from the real and imaginary part of the impedance study), the appearance of distorted semi-circular curves with a second circle confirmed the hydronium-ion-oriented conduction.25 Also, in a previous report, we observed a related isotope effect that confirmed the hydronium-ion-driven conductivity in GO-based materials.26Fig. 7(a) shows the PC value (σ) of the sample measured at 30 °C. For all the samples, σ increases with increasing relative humidity. In general, σ followed the trend GO (∼10−3–10−3 S cm−1) > HA-GO (∼10−5–10−3 S cm−1) > HA-rGO-Pt (∼10−5–10−4 S cm−1) > rGO-Pt (∼10−6 S cm−1). As the material accommodated an increased amount of adsorbed water at high relative humidity, the σ value increased gradually with humidification. Fig. 7(b) presents the results of the TGA data analysis. The gradual weight losses up to 100 °C indicated the existence of adsorbed water in the samples. The weight losses in the range of 100–200 °C indicated the decomposition of the functional groups. The hydrophilic GO exhibited the maximum weight loss. The overall weight loss profiles follow the trend GO > HA-GO > HA-rGO-Pt > rGO-Pt. The water-adsorbing capacity of GO gradually decreased due to the modification. With the attachment of the Pt metallic precursor, the weight loss profile decreased significantly. This observation implies that the metal nanoparticles reduce the hydrophilicity. However, the insertion of HA could recover the water adsorption capacity significantly. The electron conductivity measured using the pellet samples (which were used to investigate the PC) is presented in Fig. 7(c). The electron conduction values increased with the voltage biasing. At 1 V, the conductivities followed the trend rGO-Pt > HA-GO > HA-rGO-Pt > rGO-Pt. Investigation of the electron conduction property is essential, as the derivative detection of UA molecule is correlated to the extent of electron conductivity.
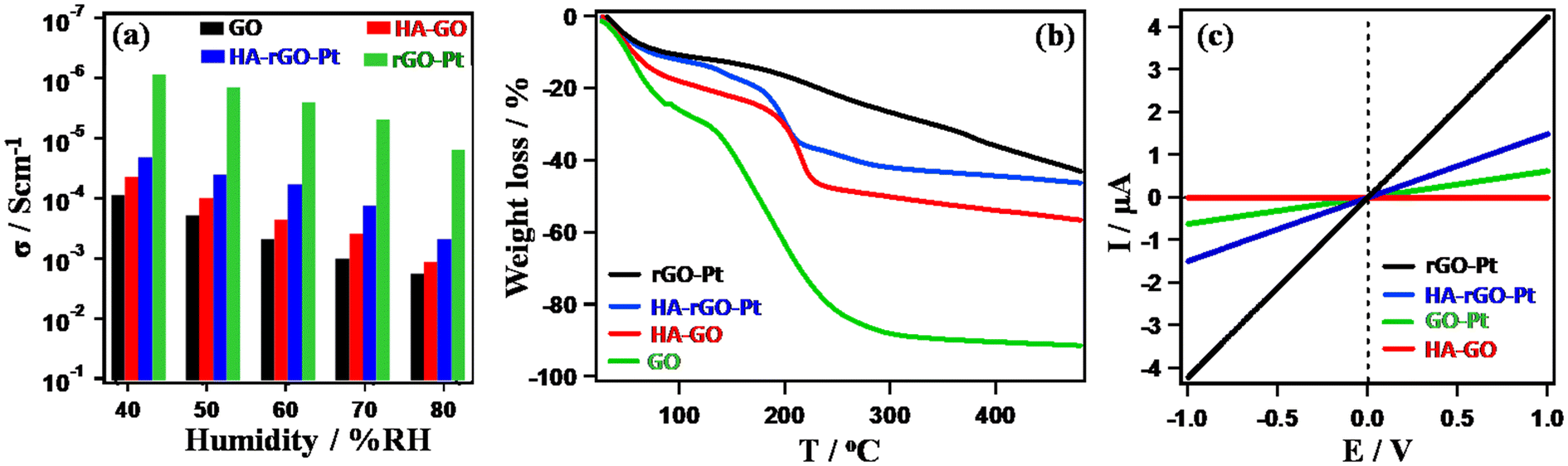 | ||
| Fig. 7 Study of hydronium ion migration. Relative-humidity-dependent PC (a); thermogravimetric weight loss profile (b); and electron conductivity (c). | ||
Table 1 presents a comparison of some of the analytical parameters, including the LDR, LOD, and sensitivity of various UA sensors. It seems that the performances achieved in this study in terms of LDR, sensitivity, and LOD are substantially superior compared with those of previous studies. Other parameters, including sensor stability, reproducibility, response time and the utility for analyzing samples in a real environment, were also competitive. Next, using the HA-rGO-Pt electrode probe, we performed quantitative detection of UA in various real samples including human blood serum, human urine serum, rabbit serum and mouse serum. The resultant data is listed in Table 2. The analysis was performed by applying the recovery method. Three repeated measurements for every sample were carried out, and the results were found to be satisfactory.
| Modified electrode | LOD | LDR | Sensitivity | Ref. |
|---|---|---|---|---|
| a LOD (detection limit). b LDR (linear dynamic range), pM (picomole), mM (millimole). | ||||
| SnO2/V2O5NMs/GCE | 95.40 pM | 1.0 nM–0.01 mM | 17.0918 μA μM−1 cm−2 | 20 |
| CuO·Fe2O3 NCs/GCE | 10.20 pM | 100 pM–100 mM | 3.16 × 10−5 μA μM−1 cm−2 | 19 |
| PVA-GCE | 0.6 μM | 2.0–50.0 μM | — | 42 |
| Co3O4 nano-berries/GCE | 2.4 mM | 5–3000 mM | 206 μA mM−1 cm−2 | 43 |
| PPy-CB-Co3O4/GCE | 0.46 μM | 0.75–305 μM | 0.8786 μA μM−1 | 44 |
| MOF-71/GCE | 15.61 μM | 50–1000 μM | 0.4811 mA M−1 cm−2 | 45 |
| PPy/FCN/Fe | 23 μM | 100–5000 μM | 0.0046 μA μM−1 | 46 |
| DBCMT-CoPc/CNT/GCE | 0.03 μM | 0.1–18 μM | 131.85 μA μM−1 | 47 |
| Mesoporous NiO | 0.005 μM | <0.374 mM | 0.7562 μA μM−1 cm−2 | 48 |
| Pristine graphene | 4.82 μM | 6–1330 μM | 0.1029 μA μM−1 | 49 |
| HA-rGO-Pt/GCE | 39.22 pM | 0.1 nM–0.1 mM | 24.2089 μA μM−1 m−2 | — |
| Sample | Added UA conc. (μM) | Measured UA conc.a using HA-rGO-Pt (μM) | Average recovery (%) | RSDb (%) (n = 3) | ||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| a Mean of three repeated determinations (signal to noise ratio 3) with HA-rGO-Pt. b Relative standard deviation value indicates precision among three repeated measurements (R1, R2, R3). | ||||||
| Human blood serum | 0.100 | 0.1421 | 0.1397 | 0.1400 | 140.6 | 0.93 |
| Human urine serum | 0.1000 | 0.1744 | 0.1821 | 0.1792 | 179.57 | 2.18 |
| Rabbit serum | 0.100 | 0.0962 | 0.0965 | 0.0964 | 96.37 | 0.16 |
| Mouse serum | 0.100 | 0.0945 | 0.0976 | 0.0973 | 96.47 | 1.77 |
We further accomplished electrochemical investigation through impedance analysis, cyclic voltammetry and differential pulse voltammetry to investigate the selectivity and efficiency of the composite electrode. The reversibility of the composite electrode was investigated by correlating the anodic and cathodic peak currents (in Fe2+/Fe3+ couple) with the root of the scan rate (Fig. S3†). The cyclic voltammogram (CV) data in Fig. 8(a) presents the current–voltage plots of the Pt-GCE, rGO-Pt-GCE and HA-rGO-Pt-GC working electrode in H2SO4 solution (1.0 m) measured at a 100 mV s−1 scan rate. Pt-GCE was prepared through a reported procedure.37 In general, the curves display two peaks in both the forward and reverse biasing of potential between −0.2 and +0.2 V. The CV curves match the typical hydrogen desorption–adsorption pattern on the working electrode surface.38 The active electrochemical surface area (EA) of the electrodes was calculated by considering the area under the hydrogen adsorption–desorption peaks present in the CV curves. Considering the deposition of a chemisorbed hydrogen monolayer on the Pt electrode, the respective surface area was estimated using the relationship EA = AHA/APt where AHA and APt stand for the charge calculated for the HA-rGO-Pt and Pt working electrode, respectively. In the case of the Pt electrode, the charge needed was assumed to be 210 μC cm−2 as per a previous report.39 Using this reference value, the active surface areas were thus found to be 1.51, 1.93, and 6.28 cm2 for the Pt-GCE, rGO-Pt-GCE and HA-rGO-Pt-GCE and electrodes, respectively. The multiplication of the calculated active surface area with respect to the geometrical surface area is well reported.40 However, a clear three-fold multiplication of the active surface area in HA-rGO-Pt was achieved due to the intercalation of HA groups in the Pt-rGO matrices. In Fig. 8(b), the charge transfer resistance of the HA-rGO-Pt electrode (<100 W) is significantly smaller than that of the rGO-Pt electrode (>300 W). Consequently, the electron transfer process is much easier in the HA-rGO-Pt electrode. The selectivity of the fabricated electrode toward UA detection was investigated by performing differential pulse voltammetry (DPV) analysis. Fig. 8(c) displays the DPV curves for UA in the presence of AC and DM. The voltametric response peak intervals of DM to AC and UA of 190 mV and 155 mV, respectively, match with some previous reports.41 The separated peaks signify that the HA-rGO-Pt electrode can distinguish and quantify the analyte UA even in the presence of potentially interfering chemical species.
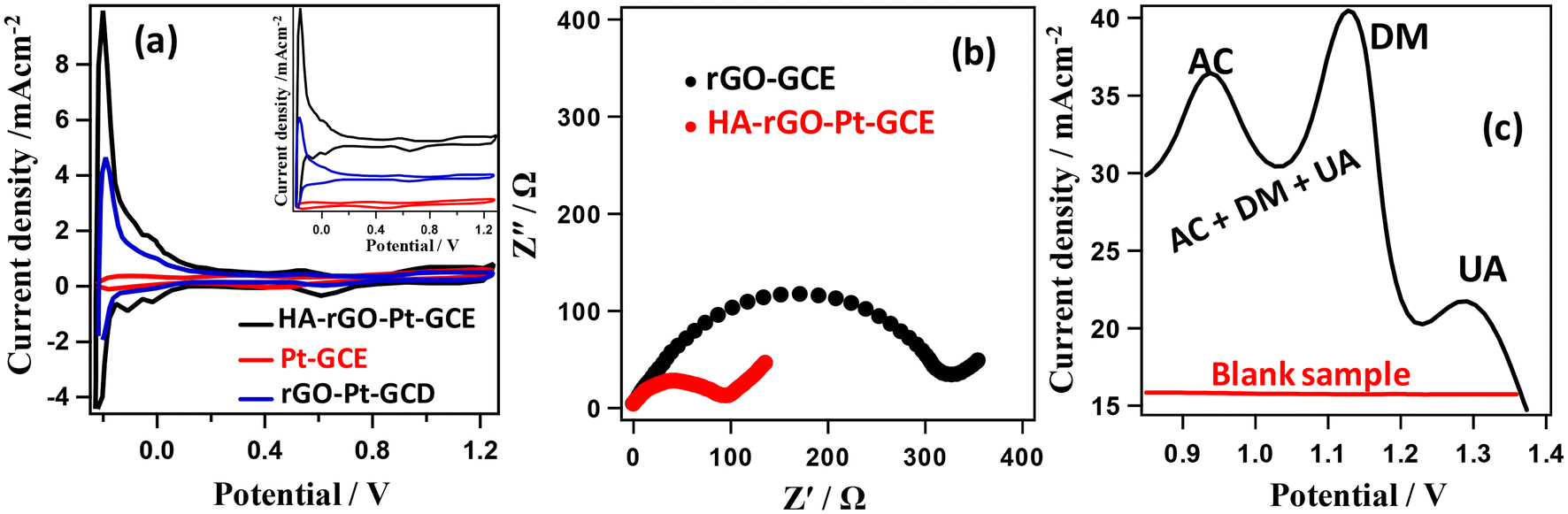 | ||
| Fig. 8 Electrochemical characterization of the electrodes. Cyclic voltammograms for the Pt-GCE, rGO-Pt-GCE and HA-rGO-Pt-GCE working electrodes in 1 M H2SO4 solution at a scan rate of 100 mV s−1 (a); impedance curves for the rGO-GCE and HA-rGO-Pt-GCE working electrodes in a mixture of 1.0 mM K3Fe(CN)6 and 1.0 mM K4Fe(CN)6 with supporting 0.1 M KCl solution (b); and differential pulse voltammogram of the HA-rGO-Pt-GCE working electrode in a blank sample solution and a mixed solution containing 0.1 mm AC, 0.005 mm DM, and 0.1 mm UA (c). | ||
The selection of materials for detecting biomolecules in the present report was considered on the basis of some scientific facts. The major reason behind the use of a G or rGO substrate/template in the composite system was their enormous surface properties and the scaffolding capacity of G/rGO toward functional ingredients. Although GO can form various composite systems, the developed materials suffer from some disadvantages in terms of the proliferation of the expected applications. These disadvantages arise due to the stacking tendency of nanosheets, which reduces the surface exposure of the rGO precursors. Single-layer GO nanosheets, being negatively charged, repulse each other in the pristine form. The surface properties of GO are hence optimized. After being reduced or forming composites, GO loses its functional groups and polarity. The materials thus become stacked. Thus, even when Pt nanoparticles were successfully loaded and dispersed on rGO, it was not possible to avoid the stacking tendency of rGO-Pt. The stacking is much denser, because of which the migration of the reactant molecule, hydronium ion, or solvated ingredients becomes difficult. This situation is unfavorable for precise biomolecular detection. To solve this problem, we achieved widening of the space between two neighboring rGO-Pt walls by intercalating HA.
HA functions not only widen the interlayer distance, but also endow the composite with polarity. The attachment of HA to a graphene precursor in a previous report by Ren et al. was investigated for an ethanol oxidation catalyst, in which the alkyl groups functioned as a support for a Pd catalyst.50 However, in the present report, we incorporated the HA group as a spacer agent to support molecular migration through the rGO layer. The composite system could thus admit reactant molecules or hydrated precursors. The association of Pt was confirmed from PXRD spectra, EDS, SEM and TEM images and XPS analysis. The intercalation of HA and the widening of the interlayer distance were confirmed from the PXRD and XPS spectra. The hydronium ion migration and electron conductivity of the materials confirmed the regulation of the material for both the molecular migration and catalytic electron transfer process. In the graphene material, the electron conductivity and hydronium ion conductivity were found to be reciprocal. In our previous study, we found that the loading of any metal precursor or the reduction of GO into rGO results in an increase in electron conductivity and reduction in hydronium ion migration. On the other hand, the loading of any polar precursor or increasing the oxygenated sites could increase the extent of molecular dynamics. A balanced optimization of electron conductivity and hydronium ion conductivity seems to exist in HA-rGO-Pt. Thus, among the series, only HA-rGO-Pt could successfully detect UA. The insertion of other molecules, including nHA or BA, did not result in a similar sensing response. Fig. 9(a and b) presents the schemes for the modification of sensor probe using HA-rGO-Pt as the active ingredient and the electrochemical detection of UA. The UA molecules are absorbed on the surface of the modified GCE, and during voltage biasing, it is oxidized to form allantoin (Fig. 9(c)). The oxidation process of UA also generates protons and electrons.
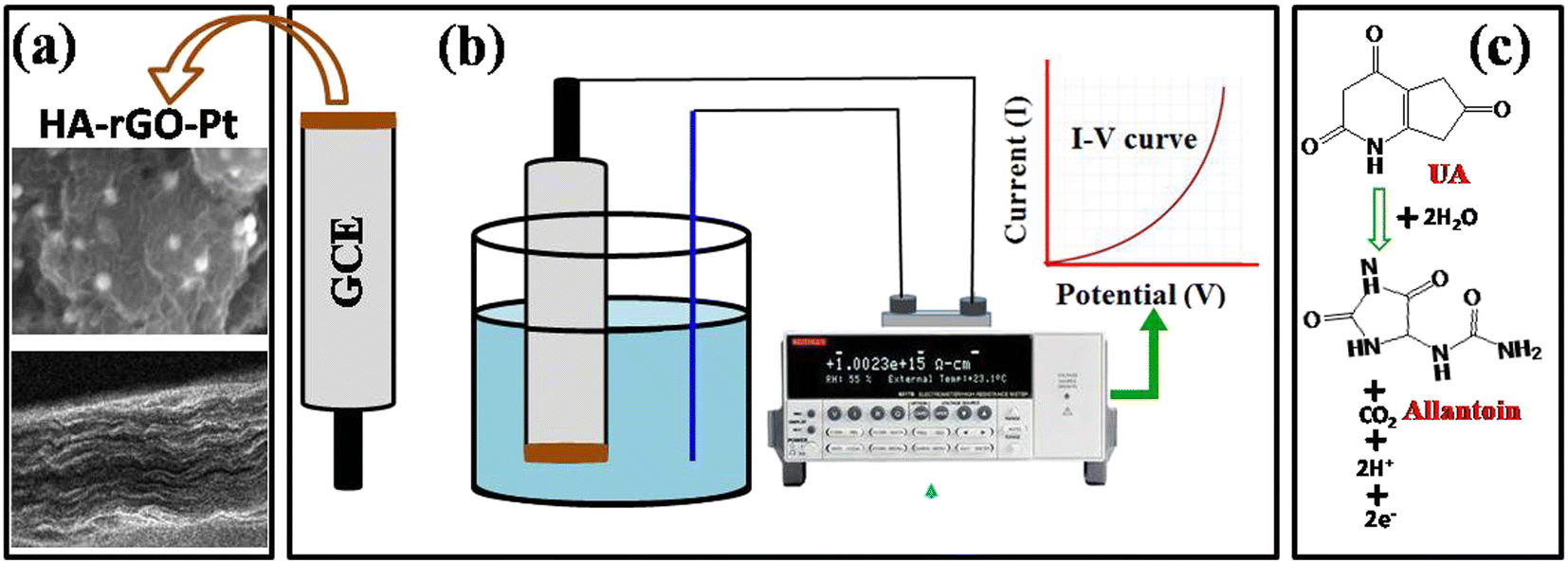 | ||
| Fig. 9 Schemes of the sensor probe modification and detection of UA via electrochemical investigation. The modification of GCE with HA-rGO-Pt includes layered deposition of the composite, which was confirmed from the SEM image (a); the instrumentation involves the assembly of a two-electrode system for I–V analysis (b); the detection of UA involves signal generation due to its electron transfer reaction to produce allantoin (c). | ||
The respective I–V curve was presented as the displayed result. The oxidation of UA to allantoin during electrochemical detection was reported previously.51 Obviously, for rapid and precise detection, the movement of UA and allantoin through the rGO interlayer must be easy. In addition, the oxidative transformation of UA to allantoin might be enhanced due to the transmutation of generated electrons through the rGO conductive matrices. As explained above, due to the stacking nature of rGO, an entire portion of the possible surface area of rGO-based composites is difficult to utilize. Therefore, any application associated with the necessity of surface exposure cannot be investigated properly. The electrochemical detection of biomolecules using rGO-based materials is one such application. However, we focused on reducing the stacking of rGO by intercalating HA spacer groups, and we used UA as a representative biomolecule analyte specimen. The rGO matrices could function as the dispersion medium for the active metallic ingredient, which is the Pt nanoparticle. In electrochemical detection, one UA molecule undergoes a chemical change to produce two electrons, two protons, and one allantoin molecule. Hence, the choice of HA-rGO-Pt was perfect, as it can transport electrons and can also support the propagation of protons. Once the electrons and protons are generated from the dissociation of some UA molecules, they must be removed from the proximity of the reaction, as new incoming UA molecules need to find room. In this perspective, the HA-rGO-Pt could function nicely because of its dual conduction ability. The platinum nanoparticles dispersed on the rGO matrices participated as the catalyst for the electrochemical UA sensing reaction. The dispersed Pt nanoparticle array also supported faster electron conduction properties. The HA precursor played a vital role as the spacer group. The active electrode surface area was exposed to the UA precursor to the optimized extent due to the availability of multiple layers. This was possible due to the presence of the HA spacer group. The HA molecules seem to pass through all the layers of rGO attached to the electrode. The entire surface area and the deposited Pt nanoparticles could thus take part in the sensing reaction. This situation was not possible to achieve in the absence of the HA.
The HA molecules consist of two terminal polar groups, which can combine with two neighboring GO nanosheets. We propose that the structure takes the shape of a ladder. The alkyl chain of HA contains six carbon–carbon single bonds, whereas the UA molecule contains a maximum of 4 C–C/N bonds in total along the longest dimension. The spacing that the HA molecule can induce within rGO thus is expected to be larger than the dimensions of UA molecules. The translation of UA molecules through the channel is thus theoretically facile. In fact, using nHA or BA as the spacer group, no impressive results for UA sensing were observed (Fig. 5(a)). Thus, only the HA molecule can play the perfect role of a spacer group to permit the UA molecules access to the catalytic ingredient. The schematic for the translation of UA molecules in the channels and the conduction of electrons at the surface is presented in Fig. 10. The electron-conducting π-network system of rGO transports the electrons, whereas the HA-loaded interlayer permits the passage of protons as well as the UA molecules. This work represents a facile technique to utilize the optimized surface area of rGO or G. Without this strategy, it was not possible to achieve the benefits of applying G/rGO-based materials in the present report. Thus, we propose that the present work will open the door to the adoption of GO materials for a wide range of applications in which the surface properties are a predominating factor.
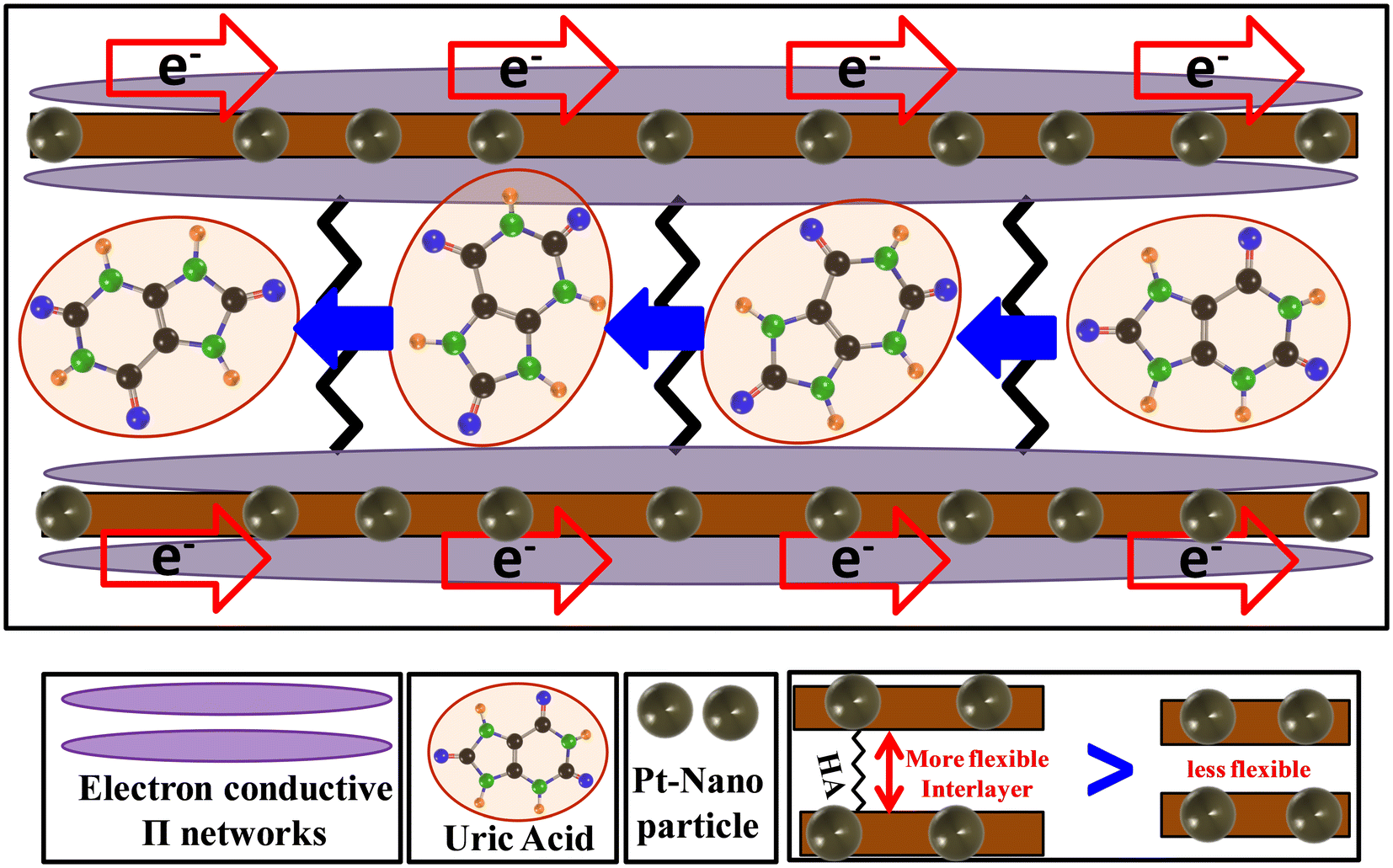 | ||
| Fig. 10 Scheme for the electron and UA conduction process through the channels of rGO in the HA-rGO-Pt composites. | ||
4. Conclusion
The 1,6-hexanediamine (HA) spacer group was intercalated within platinum-nanoparticle-loaded rGO matrices to obtain a stable HA-rGO-Pt composite. The material was characterized using surface morphology studies (SEM, EDS and TEM imaging) and IR, PXRD, Raman, and XPS spectra. The material exhibited moderate electron conductivity and proton conductivity (∼10−5–10−4 S cm−1). The HA precursor bonded with the residual oxygenated sites of rGO to generate sufficient interlayer free space to permit the passage of the uric acid (UA) molecule. Therefore, excellent electrochemical sensing was possible using the composite as an active electrode material. The UA sensor displayed excellent performance with a sensitivity and linear dynamic range (LDR) of detection of 24.2089 μA μM−1 cm−2 and 0.1 nM–0.1 mM UA, respectively. With a signal-to-noise ratio of 3, the lower limit of detection (LOD) was calculated to be 39.22 ± 1.96 pM. The sensor exhibited excellent accuracy, reproducibility, response time, and long-term working ability. Similar efficient detection was not possible using a Pt-rGO or Et-rGO-Pt composite. The Pt ingredient could support the electrocatalytic conversion of UA by releasing both protons and electrons. The flexible channels of the material supported the facile propagation of UA and its degradation product. This work might show the way toward adopting GO-based composite materials for a range of applications in which enhancement of the surface properties and the passage of molecules through the material are essential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge SUST Research Center for the grant PS/2023/1/14.References
- S. Zheng, Q. Tu, J. J. Urban, S. Li and B. Mi, ACS Nano, 2017, 11, 6440–6450 CrossRef CAS PubMed.
- M. R. Karim, M. N. Uddin, M. A. Shaikh, M. S. Rahaman, I. A. Siddiquey, M. A. Arafath, M. S. Islam, S. Hayami, K. A. Alamry, A. M. Asiri and M. M. Rahman, J. Taiwan Inst. Chem. Eng., 2021, 120, 325–335 CrossRef CAS.
- A. Sharma, A. Singh, V. Gupta and S. Arya, Sens. Diagn., 2022, 1, 387–404 RSC.
- W. Ma, L. Liu, Y. Xu, L. Wang, L. Chen, S. Yan, L. Shui, Z. Wang and S. Li, Analyst, 2020, 145, 4204–4211 RSC.
- A. Sharma, A. Singh, V. Gupta, A. K. Sundramoorthy and S. Arya, Trends Environ. Anal. Chem., 2023, e00200 CrossRef CAS.
- M. R. Karim, M. S. Islam, N. N. Rabin, H. Takehira, K. Wakata, M. Nakamura, R. Ohtani, K. Toda and S. Hayami, ChemistrySelect, 2017, 2, 4248–4254 CrossRef CAS.
- M. R. Karim, M. M. Rahman and A. M. Asiri, New J. Chem., 2021, 45, 383–393 RSC.
- M. R. Karim, K. Hatakeyama, M. Koinuma and S. Hayami, J. Mater. Chem. A, 2017, 5, 7243–7256 RSC.
- A. Singh, A. Sharma and S. Arya, Diamond Relat. Mater., 2022, 130, 109518 CrossRef CAS.
- S. R. J. Maxwell, H. Thomason, D. Sandler, C. Leguen, M. A. Baxter, G. H. G. Thorpe, A. F. Jones and A. H. Barnett, Eur. J. Clin. Invest., 1997, 27, 484–490 CrossRef CAS PubMed.
- D. I. Feig, D.-H. Kang and R. J. Johnson, N. Engl. J. Med., 2008, 359, 1811–1821 CrossRef CAS PubMed.
- A. So and B. Thorens, J. Clin. Invest., 2010, 120, 1791–1799 CrossRef CAS PubMed.
- J. Gupta, S. Arya, S. Verma, A. Singh, A. Sharma, B. Singh and R. Sharma, Mater. Chem. Phys., 2019, 238, 121969 CrossRef CAS.
- C.-L. Sun, H.-H. Lee, J.-M. Yang and C.-C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef CAS PubMed.
- A. Fang, Q. Wu, Q. Lu, H. Chen, H. Li, M. Liu, Y. Zhang and S. Yao, Biosens. Bioelectron., 2016, 86, 664–670 CrossRef CAS PubMed.
- J. Galbán, Y. Andreu, M. J. Almenara, S. de Marcos and J. R. Castillo, Talanta, 2001, 54, 847–854 CrossRef PubMed.
- M. M. Alam, M. T. Uddin, A. M. Asiri, M. M. Rahman and M. A. Islam, Arabian J. Chem., 2020, 13, 5406–5416 CrossRef CAS.
- M. M. Rahman, M. M. Alam and A. M. Asiri, J. Mater. Sci.: Mater. Electron., 2020, 31, 15422–15433 CrossRef CAS.
- M. M. Alam, A. M. Asiri, M. T. Uddin, M. A. Islam, M. R. Awual and M. M. Rahman, New J. Chem., 2019, 43, 8651–8659 RSC.
- M. M. Rahman, M. M. Hussain and A. M. Asiri, New J. Chem., 2020, 44, 19581–19590 RSC.
- N. Baig, A.-N. Kawde and A. Elgamouz, Biosens. Bioelectron.: X, 2022, 11, 100205 CAS.
- L. Yang, A.-J. Wang, X. Weng and J.-J. Feng, Microchem. J., 2023, 187, 108421 CrossRef CAS.
- T.-Q. Xu, Q.-L. Zhang, J.-N. Zheng, Z.-Y. Lv, J. Wei, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2014, 115, 109–115 CrossRef CAS.
- A. Ahmed, A. Singh, S.-J. Young, V. Gupta, M. Singh and S. Arya, Composites, Part A, 2022, 107373 Search PubMed.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura and S. Noro, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS PubMed.
- M. R. Karim, M. S. Islam, K. Hatakeyama, M. Nakamura, R. Ohtani, M. Koinuma and S. Hayami, J. Phys. Chem. C, 2016, 120, 21976–21982 CrossRef CAS.
- M. R. Karim, M. M. Rahman and A. M. Asiri, ACS Appl. Electron. Mater., 2020, 2, 1304–1312 CrossRef CAS.
- A. Bolhan, N. A. Ludin, M. S. Suait, M. A. Mat-Teridi, K. Sopian, N. M. Mohamed and H. Arakawa, Malay. J. Catal., 2018, 3, 29–31 Search PubMed.
- M. Faraji, H. Gharibi and M. Javaheri, J. Nanostruct., 2016, 6, 156–166 Search PubMed.
- L. G. Cançado, K. Takai, T. Enoki, M. Endo, Y. A. Kim, H. Mizusaki, A. Jorio, L. N. Coelho, R. Magalhães-Paniago and M. A. Pimenta, Appl. Phys. Lett., 2006, 88, 163106 CrossRef.
- M. R. Karim, H. Shinoda, M. Nakai, K. Hatakeyama, H. Kamihata, T. Matsui, T. Taniguchi, M. Koinuma, K. Kuroiwa and M. Kurmoo, Adv. Funct. Mater., 2013, 23, 323–332 CrossRef CAS.
- M. R. Karim, M. M. Rahman and A. M. Asiri, J. Taiwan Inst. Chem. Eng., 2020, 112, 87–96 CrossRef CAS.
- H.-L. Ma, Y. Zhang, Q.-H. Hu, D. Yan, Z.-Z. Yu and M. Zhai, J. Mater. Chem., 2012, 22, 5914–5916 RSC.
- M. Koinuma, H. Tateishi, K. Hatakeyama, S. Miyamoto, C. Ogata, A. Funatsu, T. Taniguchi and Y. Matsumoto, Chem. Lett., 2013, 42, 924–926 CrossRef CAS.
- M. M. Alam, M. Z. Bin Mukhlish, A. Tazrin, N. A. Jui, A. M. Asiri, M. M. Rahman, M. A. Islam and M. T. Uddin, RSC Adv., 2020, 10, 32532–32547 RSC.
- M. M. Rahman, M. M. Alam, A. M. Asiri and F. A. D. M. Opo, Sci. Rep., 2020, 10, 20248 CrossRef CAS PubMed.
- F. Cao, Z. Zang, S. Sun, X. Sun, X. Li, T. Liu and J. Wu, RSC Adv., 2017, 7, 25429–25436 RSC.
- Y. Xu and X. Lin, J. Power Sources, 2007, 170, 13–19 CrossRef CAS.
- C. Chaiburi and V. Hacker, Energy Procedia, 2017, 138, 229–234 CrossRef CAS.
- X. Xie and R. Holze, Batteries, 2022, 8, 146 CrossRef CAS.
- C.-L. Sun, C.-T. Chang, H.-H. Lee, J. Zhou, J. Wang, T.-K. Sham and W.-F. Pong, ACS Nano, 2011, 5, 7788–7795 CrossRef CAS PubMed.
- Y. Li and X. Lin, Sens. Actuators, B, 2006, 115, 134–139 CrossRef CAS.
- V. Nagal, T. Tuba, V. Kumar, S. Alam, A. Ahmad, M. B. Alshammari, A. K. Hafiz and R. Ahmad, New J. Chem., 2022, 46, 12333–12341 RSC.
- J. Ahmed, M. Faisal, S. A. Alsareii and F. A. Harraz, Adv. Compos. Hybrid Mater., 2022, 5, 920–933 CrossRef CAS.
- S. A. Abrori, N. L. W. Septiani, F. N. Hakim, A. Maulana, I. Anshori and B. Yuliarto, IEEE Sens. J., 2020, 21, 170–177 CrossRef PubMed.
- D. Oukil, L. Benhaddad, R. Aitout, L. Makhloufi, F. Pillier and B. Saidani, Sens. Actuators, B, 2014, 204, 203–210 CrossRef CAS.
- M. Pari and K. R. V. Reddy, Anal. Bioanal. Electrochem., 2019, 11, 1383–1397 CAS.
- W. Huang, Y. Cao, Y. Chen, Y. Zhou and Q. Huang, Appl. Surf. Sci., 2015, 359, 221–226 CrossRef CAS.
- S. Qi, B. Zhao, H. Tang and X. Jiang, Electrochim. Acta, 2015, 161, 395–402 CrossRef CAS.
- F. Ren, K. Zhang, D. Bin, B. Yang, H. Wang, P. Yang, Z. Fei and Y. Du, ChemCatChem, 2015, 7, 3299–3306 CrossRef CAS.
- M. M. I. Khan, A.-M. J. Haque and K. Kim, J. Electroanal. Chem., 2013, 700, 54–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00135k |
| This journal is © The Royal Society of Chemistry 2023 |