
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Passivating quantum dots against histag-displaying enzymes using blocking peptides: salient considerations for self-assembling quantum dot biosensors†
Christopher M.
Green
 *a,
David A.
Hastman
ab,
Kimihiro
Susumu
c,
Joseph
Spangler
a,
David A.
Stenger
a,
Igor L.
Medintz
a and
Sebastián A.
Díaz
*a
*a,
David A.
Hastman
ab,
Kimihiro
Susumu
c,
Joseph
Spangler
a,
David A.
Stenger
a,
Igor L.
Medintz
a and
Sebastián A.
Díaz
*a
aCenter for Bio/Molecular Science and Engineering Code 6900, U.S. Naval Research Laboratory, Washington, D.C. 20375, USA. E-mail: christopher.green@nrl.navy.mil; sebastian.diaz@nrl.navy.mil
bAmerican Society for Engineering Education, Washington, D.C. 20036, USA
cOptical Sciences Division Code 5600, U.S. Naval Research Laboratory, Washington, D.C. 20375, USA
First published on 21st September 2023
Abstract
While tagging proteins and enzymes with metal affinity moieties is a common strategy to facilitate purification, such modifications can be detrimental towards the development of biological assays using nanomaterial reporter agents, especially in cases where such modifications are not indicated by vendors. Particularly, enzymes expressed with polyhistidine tags (histags) may unintentionally and strongly bind to nanoparticles displaying transition metal ions, such as ZnS-coated quantum dots (QDs). Here, we developed a strategy to passivate ZnS-coated QDs using short, histag-containing blocking peptides (BPs), which act to saturate the surface following the addition of functional peptide or nucleic acid substrates. We demonstrated proof of concept for this strategy with the CRISPR-associated LwCas13a enzyme in the presence of QD-based molecular beacons (QD-MBs) for CRISPR-based diagnostics. The BP loading capacity and efficiency of QD surface passivation against dye-labeled peptides and a secondary protein were then characterized for BPs of varied lengths and charges using fluorescence and biophysical approaches. We further found that blocking peptides improved the colloidal stability of QDs in certain environments while having no observable effect on the sterics. These findings suggest that blocking peptides could be a beneficial addition to QDs in a variety of experimental conditions, and they are a crucial variable to consider when designing QD-based biosensors. Importantly the addition of blocking peptides does not require any re-design or modification of the original system and can be tested ad hoc when QD biosensors are not performing as intended.
Introduction
The addition of peptidyl functional moieties to proteins and enzymes, often called tagging, provides enzymes with secondary functionalities.1,2 These tags can be peptides, enzymes, or other molecules recombinantly added to the C- and N-terminus of the enzyme sequence. One commonly used tag is the polyhistidine tag, known as the histag, which is composed of five or more consecutive histidines with lengths of up to 10 histidines reported, although six histidines is the most common length.3,4 The histag exploits the fact that histidine and cysteine are relatively rare in most enzyme sequences, but they have the highest affinity for transition metal ions.5 Histags are often used to facilitate enzyme purification using immobilized metal affinity chromatography (IMAC).3 For example, Ni2+–nitrilotriacetic acid (NTA) gels and columns have been used to purify enzymes for nearly 40 years.4 The histag is so widely used that, while most commercial vendors utilize it, it is often not clearly mentioned in product descriptions for such enzymes. Therefore, while we cannot determine the exact percentage of enzymes expressed with histags for purification, it is clearly a considerable proportion.The utilization of histags has expanded beyond enzyme purification to encompass a versatile strategy for conjugation to various molecules and nanomaterials. Through the use of Ni2+–NTA functionalized dyes, which share chemistry with many IMAC columns, peptides or proteins containing histags can be selectively labeled.6,7 Additionally, histags have been successfully utilized for conjugation of enzymes and peptides to the surface of colloidal nanoparticles (NPs), particularly semiconductor quantum dots (QDs) and gold NPs.8–10 For ZnS-coated QDs, the histag binds directly to the surface by metal affinity coordination with Zn2+, a strong but non-covalent interaction with a dissociation constant (KD) of around 1 nM (see Fig. 1a).9,11 Such QDs are used in a broad array of biosensing applications to detect peptides, proteins, antibodies, nucleic acids, viruses, small molecules, and a plethora of other targets, as well as having numerous applications in fluorescence imaging (cellular and in vivo), cell-free biosynthesis, enzyme kinetics enhancement, bioluminescent imaging, and energy harvesting.12–26 In some of these applications, histag-modified enzymes are employed advantageously to selectively bind to QDs. However, it is important to recognize that most histag-enzymes are modified solely for IMAC purification and are not intended for conjugation to nanomaterials. If these enzymes are used in conjunction with QDs, as is the case with many peptide- or nucleic acid-conjugated QDs for applications in biosensing,27–29 the unintended conjugation of enzymes to QDs could hinder the functionality of the sensors. Even in cases where enzyme activity is increased by conjugation to the surface of QDs, as observed in multiple enzyme systems,25 the unintended change in activity could skew the results and lead to over or underestimating the presence of a target.
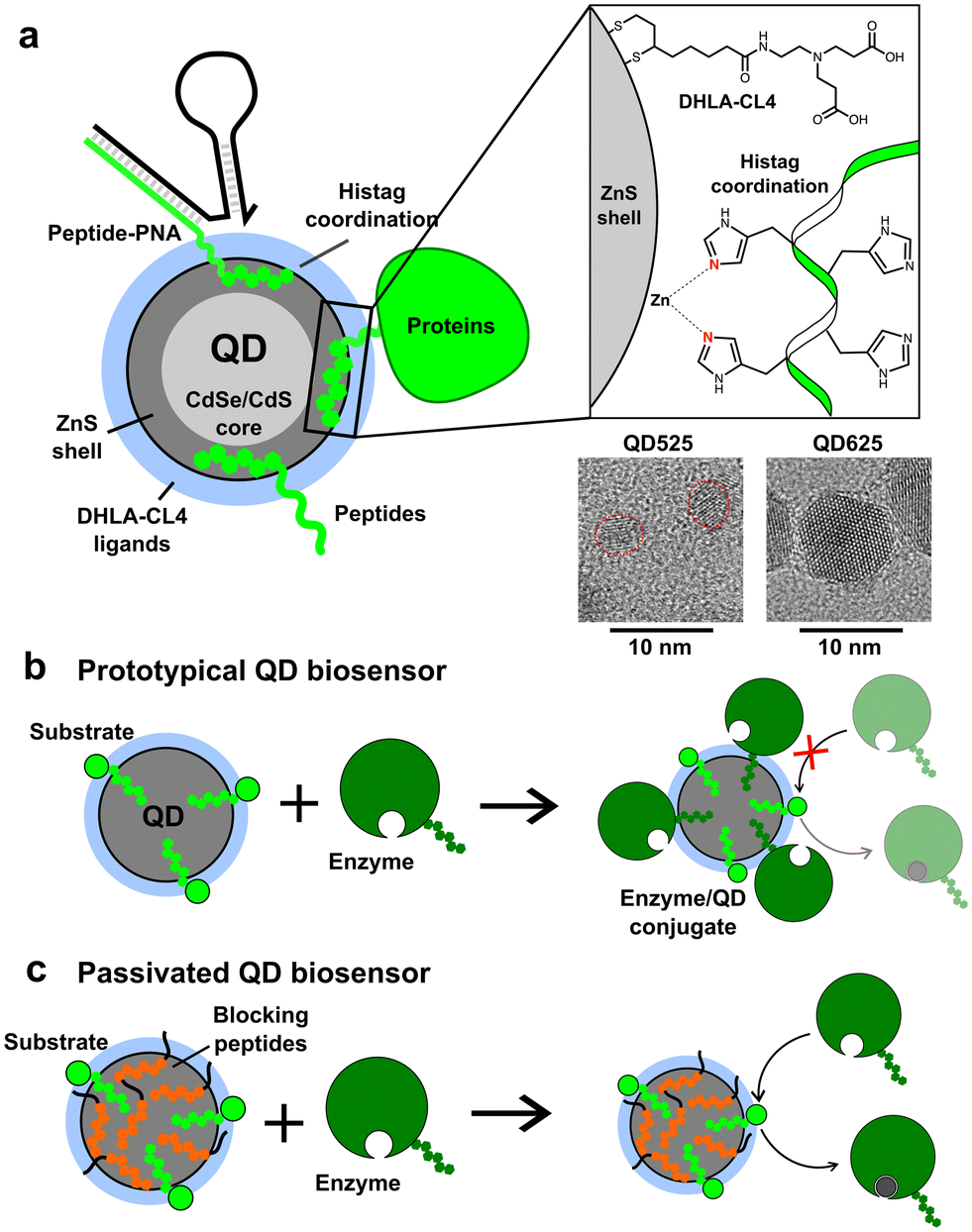 | ||
| Fig. 1 Polyhistidine conjugation of biomolecules to ZnS-coated quantum dots. (a) Schematic depicting a ZnS-coated QD with a peptide, protein, and peptide–PNA complex bound to the ZnS surface via a terminal histag. The enlarged inset schematic depicts the coordination of histidines with Zn on the surface, as well as the structure of the dihyrolipoic acid (DHLA) derived CL4 ligands on the surface. Transmission electron microscope images of the QDs utilized in this study are shown below the schematic. (b and c) Depictions of QD-based biosensors with immobilized substrates to detect the presence of active enzymes possessing a histag. Unintended conjugation of the enzyme to the exposed surface of the QD in (b) may prevent the functionality of the sensor. Passivation of the QD surface with blocking peptides, as depicted in (c), prevents conjugation of the enzyme to the QD surface and restores functionality of the sensor. | ||
We first realized the need for passivation of QDs against histag-containing enzymes during the development of QD-based molecular beacons (QD-MBs) for CRISPR-based diagnostics,30 and we have since found that this hypothesis can be retroactively applied to account for previously unexplained results. CRISPR-based diagnostics typically rely on the CRISPR-associated (Cas) enzymes Cas12a and Cas13a, endonucleases which utilize a guide RNA (gRNA) to search for and bind any desired target nucleic acid specified by the guide. The gRNA contains an RNA motif that is recognized and bound by Cas proteins and a second, arbitrary domain that encodes the complement to the target of interest.31 The Cas12a and Cas13a enzymes are initially inert, but upon binding to the target specified by the gRNA, they transform into non-specific DNA or RNA nucleases, respectively.32–34 In CRISPR-based diagnostics, the unlocked nuclease activity is then used to cleave a molecular beacon, producing a fluorescent change as an indicator of the target.30,33,35 When incorporating QD-MBs for readout in RNA detection assays with Cas13a, we discovered that the enzymes were completely deactivated by the presence of QD-MBs despite robust nuclease activity in identical conditions excluding the QDs.30 It was subsequently found that the Cas13a enzyme contained a histag for purification that was not indicated by the vendor, and thus we speculated that the enzyme was binding to available sites on the QD surface through histag coordination. Further experimentation has shown that electrostatics likely play a role as well (vide infra).
To passivate QDs against histag-containing enzymes, we devised a method utilizing short, histag-containing “blocking peptides” (BPs) to saturate any remaining surface available for histag binding following the conjugation of substrates to the QDs. Initial proof of concept was performed on the CRISPR/Cas system with a 9-residue peptide containing a six-histidine tag, and the addition of the peptide successfully restored the activity of LwCas13a in the presence of QD-MBs, enabling the development of QD-MBs for CRISPR/Cas-based diagnostics.30 To confirm the robustness and generalized applicability of this strategy, we designed and tested a range of BPs with varied lengths and charge to determine the loading capacity of BPs on QDs and the efficiency of surface passivation. For characterization, Förster resonance energy transfer (FRET)36 and gel electrophoresis mobility shift assays were performed against dye-labeled peptides and a model enzyme. We also investigated the effects of BPs on the colloidal stability and fluorescence signal of QDs. While in most cases the addition of BPs had little effect on stability, we found that colloidal stability could be significantly improved in scenarios where QDs were prone to precipitate. Overall, our study demonstrates the versatility of BPs for passivating QDs against histag-enzymes and suggests potential benefits of their use in other applications.
Results and discussion
Initial proof of concept was performed using small CdSe/CdS/ZnS QDs (QD525, 4.1 ± 0.5 nm diameter, 525 nm emission peak) produced in-house that were made colloidally stable with short, zwitterionic surface ligands called CL4.37 The CL4 ligand, shown in Fig. 1a, is composed of a bidentate dihydrolipoic acid (DHLA) anchor group with a zwitterionic character provided by a tertiary amine which is terminated by two alkyl carboxyl groups. The QD525-CL4 combination has been used extensively in the literature, with over 50 registered unique publications and a half-dozen patents, due to the combination of maximal colloidal stability with minimal steric hindrance.38–41 Within our group alone it has been used as a biosensor scaffold for endopeptidases, exopeptidases, lipases, nucleases, restriction enzymes, and kinases.12,13,15,22,40,42–48 The general design was the self-assembly of a dye-labeled peptide substrate onto the QD surface through a histag, allowing the system to function as a FRET-based biosensor. In all cases it was important that the enzyme approach the QD, interact with the substrate, but not bind irreversibly.16,46,49–51 This QD-ligand combination was also the basis of the QD-MB employed for CRISPR-based diagnostics (Fig. S1†), in which a dye-labeled RNA or DNA hairpin (RHP/DHP) was immobilized on the surface of QD525 through hybridization to a chimeric peptide/peptide nucleic acid (peptide–PNA) which contained a histag to bind the ZnS surface.3,8,52 A larger, commercially acquired QD (Qdot625 ITK, 9.4 nm diameter, 625 nm emission peak) was also utilized to study the effects of QD size on the loading capacity of BPs. TEM images of both QDs are available in Fig. 1, and absorbance and emission spectra are provided in Fig. S1.† The BPs tested in this study are presented in Table 1 and vary in length and charge, with GSWH6 initially chosen for CRISPR/Cas studies and the other peptides synthesized to determine the effects of the size and charge of BPs on surface passivation and QD colloidal stability. All BPs consisted of two distinct domains, the six-histidine histag and a shorter domain of 3–5 residues with varying numbers of neutral or charged residues. The naming convention consists of the shorthand, single letter peptide sequence provided in Table 1.| BP sequence | Length (aa)/Mw [Da] | Residual length (nm) | pI | Charge @ pH 7.0, 8.0 |
|---|---|---|---|---|
| pI, pH-dependent charges were calculated by accessing https://www.novoprolabs.com/tools/calc_peptide_property | ||||
| [NH2] GSWH6 [NH2] | 9/1171 | 1.2 ± 0.3 nm | 8.2 | 0.6, 0 |
| [NH2] H6GWD [COOH] | 9/1199 | 1.2 ± 0.3 nm | 6.7 | −0.4, −1 |
| [NH2] H6GWD2 [COOH] | 10/1314 | 1.5 ± 0.3 nm | 6.3 | −1.4, −2 |
| [NH2] H6GWD3 [COOH] | 11/1429 | 1.8 ± 0.3 nm | 6.0 | −2.4, −3 |
| [NH2] H6G2WD [COOH] | 10/1256 | 1.5 ± 0.3 nm | 6.7 | −0.4, −1 |
| [NH2] H6G3WD [COOH] | 11/1313 | 1.8 ± 0.3 nm | 6.7 | −0.4, −1 |
To demonstrate surface passivation of QD525 with BPs and determine the amount of BP needed to fully passivate the surface against other histag-containing species, a blocking assay was performed against peptide–PNA/DHP-Cy3 complexes (Fig. S1†) which bind to ZnS-coated QDs through a terminal histag on the peptide–PNA. Upon binding of peptide–PNA/DHP-Cy3 complexes to the surface of QD525, Cy3 are immobilized 4.9 ± 0.1 nm from the center of the QD, enabling efficient FRET from QD525 to Cy3.30 This energy transfer can be observed as quenching of donor photoluminescence (QD525, IDA) and sensitization of acceptor photoluminescence (Cy3, IAD). The ratio of acceptor to donor photoluminescence (PL ratio, IAD/IDA) increases approximately linearly with the ratio of Cy3 bound per QD (A/D ratio) as a result of increasing FRET efficiency, thus the PL ratio can be used to roughly estimate the average number of DHP-Cy3 complexes bound per QD525 in the presence of varying ratios of BP/QD525.36 To more accurately determine the A/D ratio from measured PL ratios, a calibration curve was generated from titrations of peptide–PNA/DHP-Cy3 with QD525 (Fig. S2†).
For the blocking assay, each BP was titrated against QD525 at ratios of BP/QD525 ranging from 0–50 in 10 mM Tris-HCl (pH 8.0), 10 mM MgCl2, and 50 mM NaCl. The BP/QD mixtures were incubated for 1 h at 20 °C, then peptide–PNA/DHP-Cy3 complexes were added to each solution at a ratio of 6 Cy3/QD525 and immediately loaded into a plate reader to monitor the PL ratio of Cy3 and QD525 at 604 nm and 528 nm over 4 h (Fig. S3†). The emission spectra of the samples were acquired after 4 h with excitation at 380 nm (Fig. S4 and S5†), and the spectra for QD525/Cy3 complexes passivated by H6GWD2 are shown in Fig. 2b as a representative sample. The PL ratios of each sample at 4 h were then used to determine the average A/D ratios by interpolation from the calibration curve in Fig. S2.† The A/D ratio as a function of BP/QD525 is provided in Fig. 2c for all samples.
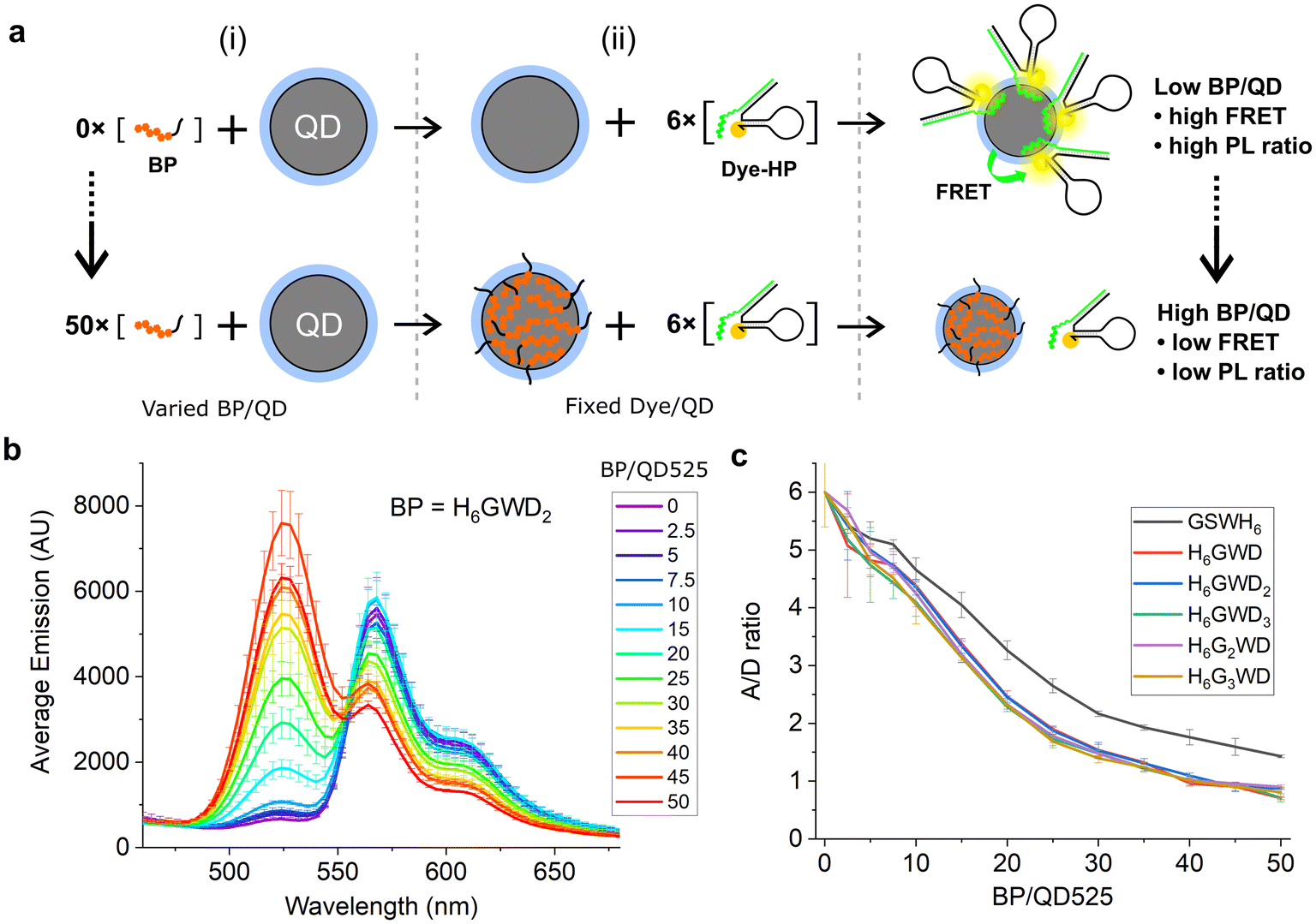 | ||
| Fig. 2 (a) QD525–Cy3 FRET complexes assembled after (i) mixing with varied ratios of blocking peptides and subsequent (ii) addition of six FRET acceptors per QD. (b) Fluorescence spectra of QD525–Cy3 complexes with fluorescence peaks at 528 nm from QD525 and at 572 nm from FRET sensitization of Cy3. QD donor emission increased while Cy3 acceptor sensitization decreased with greater concentrations of blocking peptide. All spectra can be found in Fig. S4.† (c) A/D ratios for varying ratios of BP/QD525, interpolated from PL ratios of Cy3 and QD525 at 604 nm and 528 nm, respectively. | ||
As shown in Fig. 2c, all blocking peptides hindered the conjugation of Cy3 complexes to QD525, and the degree of passivation increased throughout the range of BP/QD ratios tested. Cy3-QD conjugation was reduced 50% by approximately 20 BP/QD and approached an asymptote by 50 BP/QD for all BPs. GSWH6 appeared slightly less effective at passivating QD525 against Cy3 complexes, though this is likely due to the additional effects of electrostatic repulsion between the negative BPs and the large DNA hairpins. We note that, though the QD–histag interaction is quite strong, it is not covalent and would thus be expected to approach a dynamic equilibrium between blocking peptides and other histag-containing species competing for the QD surface. The dynamic equilibrium in a system with many components is quite complex and falls outside the scope of the current study. However, by focusing on specific materials and interactions within a narrow concentration range, as is the case with the QD-based biosensors discussed here, the insights gained can be qualitatively applied to similar systems. For example, QDs are typically used for biosensing at concentrations on the order of 100 nM, while histag-substrates (e.g., peptides or DNA hairpins) and enzymes are typically used at ∼10-fold excess or less to QDs. In these conditions, we speculated that BPs could be added in relative abundance to other components and achieve similarly efficient passivation, and we confirmed this with two distinct enzymes in subsequent experiments.
To confirm that this passivation strategy was compatible with QD-MBs for CRISPR-based diagnostics, an RNA detection assay was performed with the LwCas13a enzyme, a ∼139 kDa monomeric Cas13a enzyme from L. wadei.33 RNaseAlert v2 (RA), a commercially available RNA-based fluorophore–quencher molecular beacon, was used for readout of LwCas13a activity in the presence of a 35 nucleotide (nt) target RNA strand.53–55 When bound to a target RNA, LwCas13a transforms into an RNA nuclease and cleaves the RA molecular beacon along with any other free RNA in solution. Previously, we found that bare ZnS-coated QDs interfered with LwCas13a-based RNA detection assays, and we speculated that this effect was due to conjugation of LwCas13a to QDs as depicted in Fig. 3. To demonstrate this effect with QD625, and to determine the extent to which the addition of BPs would restore LwCas13a activity by preventing conjugation, two master mixes of LwCas13a were prepared: an activated LwCas13a solution with target RNA, and an inactive control lacking the target RNA. Both mixes contained 200 nM LwCas13a and 200 nM gRNA in buffer (10 mM Tris-HCl pH 8.0, 10 mM MgCl2, and 50 mM NaCl), and the active Cas solution contained 2 nM of the 35 nt target RNA (methods provided in ESI†). Each master mix was divided into four samples and diluted 2× for the assay: (1) control without QD625, (2) QD625 in buffer, (3) QD625 with 100 GSWH6/QD, and (4) QD625 with 100 H6GWD2/QD. For samples 2–4, QD625 was added at a ratio of 1 Cas/QD, and all samples were prepared in triplicate and incubated at RT for 30 min prior to initiating the assays. The RNA detection assays were initiated by the addition of the reporter, 200 nM RA, to every sample, and the samples were immediately transferred to a fluorescent plate reader at 37 °C to monitor the progress of LwCas13a cleavage of RA.
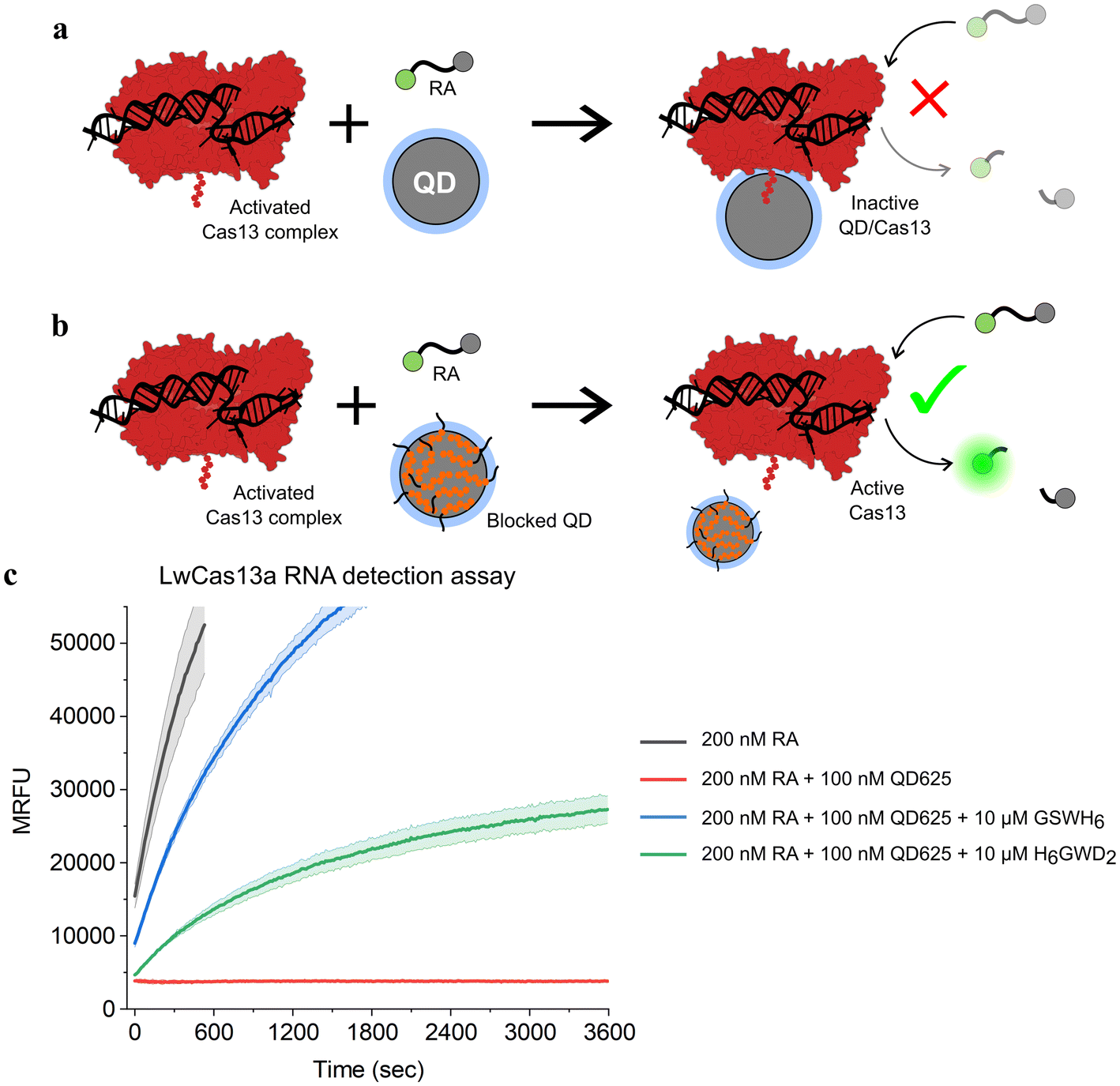 | ||
| Fig. 3 Schematic of CRISPR-based RNA detection in the presence of (a) bare and (b) blocked QDs. Conjugation of the LwCas13a enzyme (AF-U2PSH1-F1)56,57 to the surface of ZnS-coated QDs results in the complete deactivation of the enzyme's ribonuclease activity. (c) Time series fluorescence measurements of LwCas13a-based RNA detection assays with samples containing a commercially available molecular beacon, RNaseAlert (RA), for readout of Cas nuclease activity. QD625 was added to a subset of samples without BPs (red) or with 100 BP/QD of GSWH6 (blue) and H6GWD2 (green). Samples were excited at 485 nm and fluorescence was measured at 525 nm over 1 h to measure Cas activity. For all samples shown, LwCas13a was pre-activated with target RNA in a single mixture prior to the addition of QDs and RA, ensuring an equivalent initial concentration of active Cas nucleases in each sample. The results for samples containing inactive LwCas13a are provided in Fig. S6.† | ||
The results of the CRISPR/Cas assays are shown in Fig. 3. The effect of the presence of bare QD625 (red) was clearly demonstrated; no LwCas13a activity was observed for samples containing bare QD625 despite confirmation of LwCas13a activity in control samples (black). For samples containing QD625 passivated by the neutral BP (GSWH6, blue) or negative BP (H6GWD2, green), Cas activity was restored. Interestingly, GSWH6 restored LwCas13a activity considerably better than H6GWD2. We speculated that this was due to the high net positive charge of LwCas13a and the frequency of basic amino acids on its surface (Fig. S6†);33 the addition of negatively-charged BPs may have promoted QD–Cas interactions due to electrostatics despite the prevention of histag binding. This result suggests a need to tune the properties of BPs for specific applications, though simply swapping out the terminal residue for a basic or acidic amino acid appears to suffice, allowing users to easily adjust to their particular system. We note that more positively charged BPs were not feasible in this study (limited as it was to the CL4 ligand and CRISPR/Cas buffer conditions) due to decreases in colloidal stability of the QD.
To further quantify the effects of blocking peptides on protein binding to QDs, binding assays were performed with alkaline phosphatase (AlkP), a homodimeric enzyme that readily binds to the ZnS surface of QDs and is amenable to characterization by agarose gel mobility shift assays.58,59 AlkP–QD binding assays were performed on QDs of 4.1 nm (QD525) and 9.4 nm diameter (QD625) with H6GWD2. QD525 was titrated with AlkP at ratios ranging from 0–8 AlkP/QD in the presence (+) or lack of (−) 40 H6GWD2/QD (Fig. 4 left-top). QD525 was also titrated with H6GWD2/QD at ratios ranging from 0–40 prior to the addition of 8 AlkP/QD (Fig. 4 bottom left). All blocked QDs were incubated at 18 °C for 1 h prior to the addition of AlkP, and QDs were incubated for an additional 2 h after adding AlkP to promote binding to QDs. These experiments were repeated for QD625 (Fig. 4 right) though with a 4-fold reduction in the concentration of QD625, effectively increasing the ratios of AlkP/QD and H6GWD2/QD by 4-fold relative to the experiments with QD525. It was found that conjugation of AlkP to QDs was strongly pH dependent due to the net negative charge of AlkP dimers at pH > 8 (Fig. S7†), thus experiments were run in HEPES buffer at pH ∼ 7.5.
 | ||
| Fig. 4 Gel mobility shift assay of AlkP conjugation to QD525 (left) and QD625 (right). QD525 concentration was fixed at 250 nM, while QD625 concentration was fixed at 62.5 nM. The concentrations of all other components, including BP H6GWD2, were identical between experiments. Electrophoresis was performed in 1.5% agarose gel with 50 mM HEPES at 7 V cm−1 for 45 min (left) and 60 min (right), and gel images were pseudocolored to distinguish the green-emitting (QD525) and red-emitting (QD625) QDs. | ||
The results of the AlkP–QD binding electrophoretic mobility shift assay (EMSA) for QD525 and QD625 are provided in Fig. 4, and the gel images clearly demonstrate the complete passivation of QDs to AlkP binding at high BP/QD ratios (gel images at earlier time points are provided in Fig. S8 and S9†). For bare QDs (0 BP/QD), QDs displayed decreasing mobility with increasing quantities of AlkP, indicating the assembly of multiple AlkP dimers on the surface of QDs. In the presence of blocking peptides at 40 BP/QD525 and 160 BP/QD625, the mobilities of both QDs were unaffected by the presence of AlkP, indicating complete passivation of the surface to the enzyme. Titrations of blocking peptides from 0–40 BP/QD525 prior to the addition of 8 AlkP demonstrated that approximately 30 H6GWD2/QD525 was sufficient to fully passivate the surface to AlkP. Similar titrations with QD625 showed that approximately 80 BP/QD625 was sufficient to fully passivate the larger QD surface to AlkP. Interestingly, the data suggest that the amount of BP needed to passivate QDs does not scale proportionally with surface area. This discrepancy is hypothesized to arise from variations in the completeness of CL4 ligand exchange, as well as the effects of QD curvature,60 though differences in the ligand exchange process and uncertainty regarding the full composition of QD625 limit this to speculation.
The results of an early EMSA assay performed with an aged stock of QD625, which had compromised colloidal stability, provided motivation to further study the effects of BPs on the colloidal stability of QDs. In the EMSA assay of note, shown in Fig. S10,† QD625 did not migrate out of the gel wells unless the BP was added at ratios of 60 BP/QD or higher. This observation suggested that the addition of the BP could significantly improve QD colloidal stability in some scenarios. Supplemental characterization of colloidal stability and physical properties of the BP/QD complexes for each blocking peptide were performed with fluorescence spectroscopy, dynamic light scattering (DLS), and zeta (Z)-potential measurements. QD525 was prepared with each blocking peptide at 40 BP/QD and allowed to assemble to QDs for 30 minutes. The QDs were then characterized with DLS and Z-potential.52 Samples were diluted to 50 nM in pure water, diluting the buffer by 20-fold, and filtered through 0.2 μm filters before measuring. For all negatively charged BPs, no statistically significant difference was observed between bare QDs and QDs conjugated with negatively charged BPs; all had hydrodynamic diameters within 8 ± 2 nm and a Z-potential of −5 ± 9 mV (data available in Fig. S11†). The only samples that were distinct were QD complexes with GSWH6. The measured hydrodynamic diameters were not divergent from the other samples, but QD complexes with GSWH6 had a Z-potential of 25 ± 11 mV. This confirms that the BPs are small and do not increase the steric considerations of molecules presented on the QD surface; in line with the residual length of the BP (see Table 1) being smaller than the extension of the CL4 from the QD surface. We further confirmed this through agarose gels, where the mobility of the bare QDs and those saturated with BPs was very similar (see first columns of top gels in Fig. 4).
We then looked at the stability of the BP/QD complexes in varying pH from 4.5 to 9.0; all samples were prepared in deionized H2O and then incubated at RT for 1 h prior to pH adjustment. BP/QD complexes were diluted in 50 mM solutions of common buffers (sodium acetate, HEPES, phosphate, TRIS, carbonate) that covered the pH range and transferred to a 384-well microplate for fluorescence characterization. For the sake of simplicity we will refer to the BPs that have a negative charge at pH 7, i.e. all except GSWH6, as ‘negative BPs’ even if at lower pH such as 5.0 all BPs have positive charges. The spectra were then obtained (excitation 400 nm) every ∼9 min over a 3 h period; the results are presented in Fig. 5.
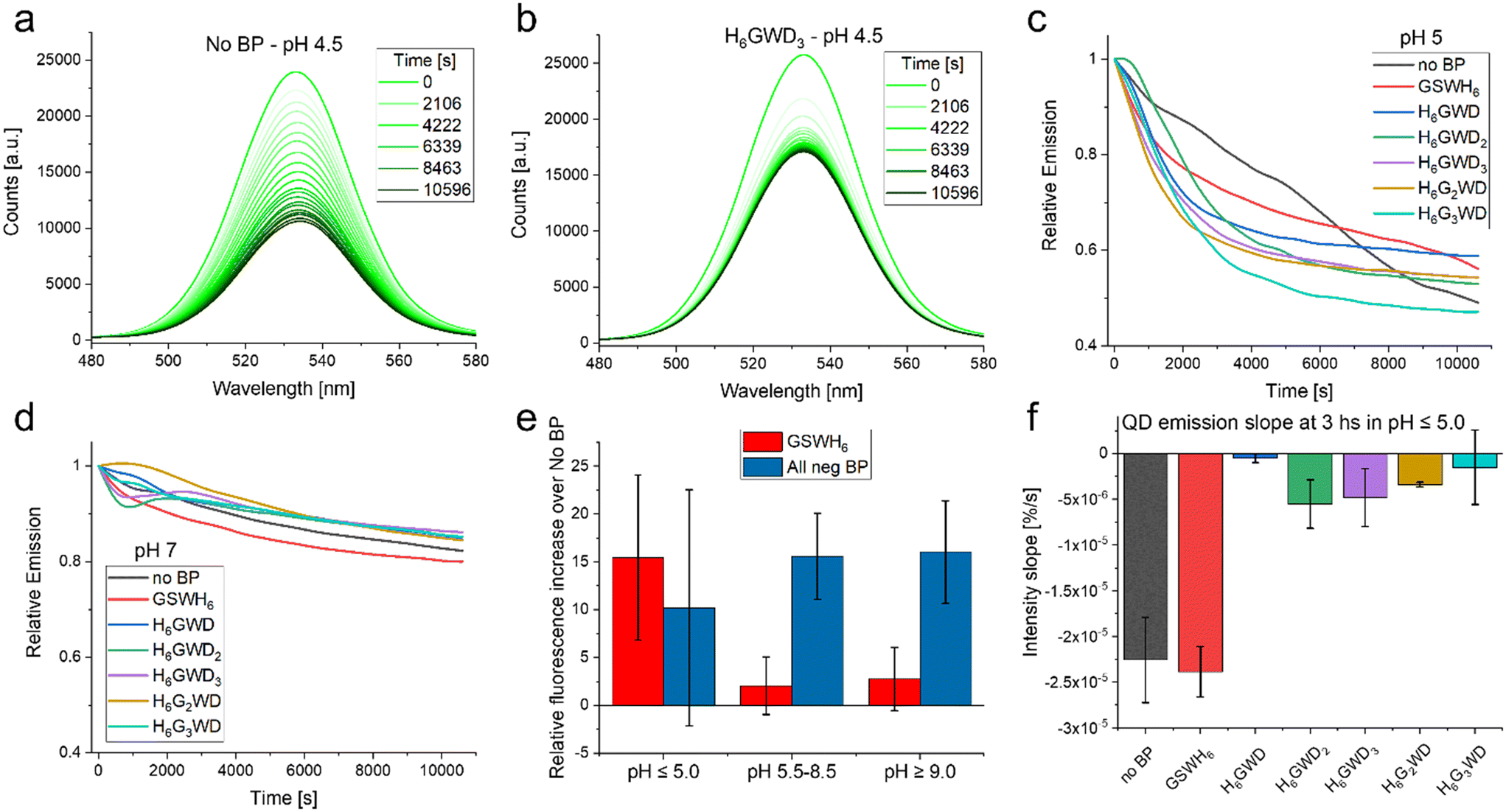 | ||
| Fig. 5 Stability observations. (a) Emission of QD with no BP (control sample) over 3 hours in acidic pH 4.5. This is the lower end of the QD-CL4 stability range. (b) Emission of QD with BP H6GWD3 over 3 hours in acidic pH 4.5. (c) Relative emission, t = 0 is set to 1.0, of the varying QD samples at pH 5.0 as a function of time. (d) Relative emission, t = 0 is set to 1.0, of the varying QD samples at pH 7.0 as a function of time. (e) Increase in overall intensity of QD emission (integrated over the 3 hours) as a function of the chosen BP as well the pH condition. (f) Slope of curves observed in C as well as equivalent data at pH 4.5 at the 3 hours end point. For further details see Fig. S12 in the ESI.† | ||
It was observed that BPs generally increased the colloidal stability and overall intensity of the QD emission, though the effect in many cases was minimal. Peptides have been reported as aqueous solubilization ligands in the past, though were generally less successful than chemical or polymeric approaches.61 In our case the BPs had very small effects in conditions in which the QD-CL4 system are designed to be most efficient, i.e. common aqueous buffers in pH 5.5–9.0.37 In these pH ranges we observed that the stability of the QDs was not modified in any statistically significant way by addition of the BP as determined by the slope over the 3 hours of observation. The intensity of the emission was increased at pH > 5.0 when considering the negative BPs as seen in Fig. 5E. This can be a measure of QD stability but could also be a matter of slight increases in the fluorescence quantum yield, this could be summarized as a preservation of the optical properties, including the minimization of spectral drift within the plate-reader. Interestingly in the limits of the QD system stability, i.e. below pH 5.0, there was no statistically significant increase in fluorescence emission. One important observation is that in the lower pH the negative BPs increased the colloidal stability of the QD in comparison to the no BP QD as well as the QD with GSWH6. Though the QD seemed to decrease quicker with the negative BPs it stabilized at around an hour while the QD without BP and with GSWH6 continued decreasing as observed in Fig. 5F. This trend continued even for extended times; samples were stored at 4 °C overnight and remeasured the next day, at which point the QD with negative BPs had almost twice the intensity of the No BP and GSWH6 samples. Clarification on the quantification methodology used in Fig. 5 is available in the caption of Fig. S12.† To summarize under optimal conditions the BPs seem to have a slight beneficial effect on stability and QY, independent of the sequence of the BPs, with this effect becoming more noticeable under more stressful conditions, e.g. extreme pH.
Conclusion
We have shown the applicability of using short histag peptides, which we have referred to as blocking peptides (BPs), to inhibit conjugation of competing histag moieties to QDs. This work is meant to be viewed as a proof-of-concept of the utility of the BP as a tool to optimize NP based biosensors. As a specific example, we have shown how the popular CRISPR/Cas biosensing system can be inactivated through histag conjugation to QDs found in solution. Yet, this case is not unique, looking back at some previous work with kallikrein proteolytic reporters we similarly observed inactivity in the presence of QDs.43 At the time the reason was not clear, yet the data supports the hypothesis of inactivation of the enzyme through histag conjugation to the QD. Furthermore, preliminary work with the β-secretase enzyme, a key target in Alzheimer's disease drug developing pipeline,62 has shown similar results; the addition of BPs was required for optimal functionality of a biosensor designed to test the enzyme's activity. Surprisingly the β-secretase (Sigma-Aldrich, # S4195) did not contain a histag (though it did contain a FLAG-tag),1 so it is likely that the enzyme's endemic aa and/or electrostatics were driving the interaction. As seen in the LwCas13a example electrostatics may also play a role. This merely expands the application space in which the BPs might prove beneficial for biosensor design.The use of BPs can also be extended to other NP-biological interactions. Cell-free biosynthesis is a growing field of interest and as stated in the introduction NPs have been shown to modify the kinetics of enzyme activity as well as being capable of creating stochastic enzyme channeling through formation of NP-enzyme clusters.63 While this can be very beneficial for improving the flux through enzyme cascades, as we have demonstrated some enzymes can be inhibited upon conjugation to NPs.30,44,64 One could imagine coupled enzyme reactions that benefited with conjugation (due to changes in kinetics and channeling) but subsequently added BPs to the clusters to block the surface from capturing a particular enzyme within the system. This enzyme would be a key step of the entire cascade, and as such required in solution, but the activity is starkly diminished by histag binding to the NPs. Thus the BPs would allow for the inclusion of the QDs for the enzymes that demonstrated benefits without the deleterious effect on the particular enzyme.
As can be seen in Fig. 4 and 5, the inclusion of the BP also results in tighter bands in agarose gels and increased colloidal stability in buffers with different pHs. Though we stressed the QDs stability through changes in pH, we believe this same effect could transfer to other stresses such as inclusion of organic solvents or other contaminants. We further suggest and have seen initial evidence that BPs would be beneficial in interacting QDs with other biological moieties such as antibodies or DNA nanostructures as well.
We have presented a new consideration and/or tool when trying to optimize NP, particularly QD, based biosensors. While they will not be necessary in all cases, BPs have demonstrated the capability to save otherwise non-functional biosensors in a simple, relatively economic, self-assembly approach. This is particularly salient when enzymes have been expressed with the histag or are naturally rich in histidines and cysteines. Importantly the addition of BPs does not require any re-design or modification of the original system and can be tested ad hoc when NP biosensors are having issues.
Author contributions
CMG, DAH, KS, JS, SAD: experimental results and sample preparation (investigation). CMG, SAD: data curation and formal analysis. CMG, DS, ILM, SAD: designed experimental protocols (methodology and conceptualization). CMG, SAD: original draft. All authors: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Office of Naval Research, the U.S. Naval Research Laboratory (NRL), and the NRL's Nanoscience Institute for programmatic funding.References
- X. Zhao, G. Li and S. Liang, J. Anal. Methods Chem., 2013, 2013, 581093 CrossRef PubMed.
- E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentzi and D. Stuber, Nat. Biotechnol., 1988, 6, 1321–1325 CrossRef CAS.
- J. Crowe, H. Dobeli, R. Gentz, E. Hochuli, D. Stiiber and K. Henco, in Protocols for gene analysis, ed. A. J. Harwood, Humana Press, Totowa, NJ, 1994, pp. 371–387 Search PubMed.
- S.-Y. Tsai, S.-C. Lin, S.-Y. Suen and W.-H. Hsu, Process Biochem., 2006, 41, 2058–2067 CrossRef CAS.
- J. L. Michalek, A. N. Besold and S. L. J. Michel, Dalton Trans., 2011, 40, 12619–12632 RSC.
- C. Zhao, L. M. Hellman, X. Zhan, W. S. Bowman, S. W. Whiteheart and M. G. Fried, Anal. Biochem., 2010, 399, 237–245 CrossRef CAS PubMed.
- W. P. Klein, S. A. Díaz, M. Chiriboga, S. A. Walper and I. L. Medintz, ACS Appl. Nano Mater., 2019, 2, 7459–7465 CrossRef CAS.
- C. M. Green, D. Mathur, K. Susumu, E. Oh, I. L. Medintz and S. A. Díaz, in Bioluminescence: Methods and protocols, ed. S.-B. Kim, Springer US, New York, NY, 2022, vol. 2, pp. 61–91 Search PubMed.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, J. Phys. Chem. C, 2007, 111, 11528–11538 CrossRef CAS.
- V. Palomo, P. A. Cistrone, N. Zhan, G. Palui, H. Mattoussi and P. E. Dawson, Bioconjugate Chem., 2018, 29, 3144–3153 CrossRef CAS PubMed.
- D. E. Prasuhn, J. B. Blanco-Canosa, G. J. Vora, J. B. Delehanty, K. Susumu, B. C. Mei, P. E. Dawson and I. L. Medintz, ACS Nano, 2010, 4, 267–278 CrossRef CAS PubMed.
- D. E. Prasuhn, A. Feltz, J. B. Blanco-Canosa, K. Susumu, M. H. Stewart, B. C. Mei, A. V. Yakovlev, C. Loukou, J.-M. Mallet, M. Oheim, P. E. Dawson and I. L. Medintz, ACS Nano, 2010, 4, 5487–5497 CrossRef CAS PubMed.
- K. E. Sapsford, J. Granek, J. R. Deschamps, K. Boeneman, J. B. Blanco-Canosa, P. E. Dawson, K. Susumu, M. H. Stewart and I. L. Medintz, ACS Nano, 2011, 5, 2687–2699 CrossRef CAS PubMed.
- X. Li, D. Deng, J. Xue, L. Qu, S. Achilefu and Y. Gu, Biosens. Bioelectron., 2014, 61, 512–518 CrossRef CAS PubMed.
- S. A. Díaz, A. P. Malonoski, K. Susumu, R. V. Hofele, E. Oh and I. L. Medintz, Anal. Bioanal. Chem., 2015, 407, 7307–7318 CrossRef PubMed.
- M. Wu and W. R. Algar, ACS Appl. Mater. Interfaces, 2015, 7, 2535–2545 CrossRef CAS PubMed.
- S. Tsuboi and T. Jin, ChemBioChem, 2017, 18, 2231–2235 CrossRef CAS PubMed.
- F. Ma, C.-C. Li and C.-Y. Zhang, J. Mater. Chem. B, 2018, 6, 6173–6190 RSC.
- H. Bui, C. W. Brown III, S. Buckhout-White, S. A. Díaz, M. H. Stewart, K. Susumu, E. Oh, M. G. Ancona, E. R. Goldman and I. L. Medintz, Small, 2019, 15, 1805384 CrossRef PubMed.
- C. Léger, A. Yahia-Ammar, K. Susumu, I. L. Medintz, A. Urvoas, M. Valerio-Lepiniec, P. Minard and N. Hildebrandt, ACS Nano, 2020, 14, 5956–5967 CrossRef PubMed.
- K. Gorshkov, K. Susumu, J. Chen, M. Xu, M. Pradhan, W. Zhu, X. Hu, J. C. Breger, M. Wolak and E. Oh, ACS Nano, 2020, 14, 12234–12247 CrossRef CAS PubMed.
- D. Mathur, M. Thakur, S. A. Díaz, K. Susumu, M. H. Stewart, E. Oh, S. A. Walper and I. L. Medintz, ACS Synth. Biol., 2022, 11, 4089–4102 CrossRef CAS PubMed.
- C. Grazon, M. Chern, P. Lally, R. C. Baer, A. Fan, S. Lecommandoux, C. Klapperich, A. M. Dennis, J. E. Galagan and M. W. Grinstaff, Chem. Sci., 2022, 13, 6715–6731 RSC.
- S. A. Díaz, P. Choo, E. Oh, K. Susumu, W. P. Klein, S. A. Walper, D. A. Hastman, T. W. Odom and I. L. Medintz, ACS Catal., 2021, 11, 627–638 CrossRef.
- J. C. Breger, J. N. Vranish, E. Oh, M. H. Stewart, K. Susumu, G. Lasarte-Aragonés, G. A. Ellis, S. A. Walper, S. A. Díaz, S. L. Hooe, W. P. Klein, M. Thakur, M. G. Ancona and I. L. Medintz, Nat. Commun., 2023, 14, 1757 CrossRef CAS PubMed.
- S. Hooe, J. Breger, S. Dean, K. Susumu, E. Oh, S. Walper, G. A. Ellis and I. L. Medintz, ACS Appl. Nano Mater., 2022, 5, 10900–10911 CrossRef CAS.
- J. A. Hansen, J. Wang, A.-N. Kawde, Y. Xiang, K. V. Gothelf and G. Collins, J. Am. Chem. Soc., 2006, 128, 2228–2229 CrossRef CAS PubMed.
- J. Shu and D. Tang, Chem. – Asian J., 2017, 12, 2780–2789 CrossRef CAS PubMed.
- A. Dif, F. Boulmedais, M. Pinot, V. Roullier, M. Baudy-Floc'h, F. M. Coquelle, S. Clarke, P. Neveu, F. Vignaux, R. L. Borgne, M. Dahan, Z. Gueroui and V. Marchi-Artzner, J. Am. Chem. Soc., 2009, 131, 14738–14746 CrossRef CAS PubMed.
- C. M. Green, J. Spangler, K. Susumu, D. A. Stenger, I. L. Medintz and S. A. Díaz, ACS Nano, 2022, 16, 20693–20704 CrossRef CAS PubMed.
- A. Ramachandran and J. G. Santiago, Anal. Chem., 2021, 93, 7456–7464 CrossRef CAS PubMed.
- A. East-Seletsky, M. R. O'Connell, S. C. Knight, D. Burstein, J. H. Cate, R. Tjian and J. A. Doudna, Nature, 2016, 538, 270–273 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- D. Zhang, Y. Yan, H. Que, T. Yang, X. Cheng, S. Ding, X. Zhang and W. Cheng, ACS Sens., 2020, 5, 557–562 CrossRef CAS PubMed.
- P. T. Snee, TrAC, Trends Anal. Chem., 2020, 123, 115750 CrossRef CAS.
- K. Susumu, E. Oh, J. B. Delehanty, J. B. Blanco-Canosa, B. J. Johnson, V. Jain, W. J. T. Hervey, W. R. Algar, K. Boeneman, P. E. Dawson and I. L. Medintz, J. Am. Chem. Soc., 2011, 133, 9480–9496 CrossRef CAS PubMed.
- I. L. Medintz, J. N. Vranish, M. Ancona, K. Susumu and S. A. Diaz, US Pat., 11512305, 2022 Search PubMed.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- J. C. Breger, K. Susumu, G. Lasarte-Aragonés, S. A. Díaz, J. Brask and I. L. Medintz, ACS Sens., 2020, 5, 1295–1304 CrossRef CAS PubMed.
- I. Medintz, J. A. Breger, K. Susumu, S. A. Diaz and J. Brask, US Pat., 17161889, 2021 Search PubMed.
- W. R. Algar, A. Malonoski, J. R. Deschamps, J. B. Blanco-Canosa, K. Susumu, M. H. Stewart, B. J. Johnson, P. E. Dawson and I. L. Medintz, Nano Lett., 2012, 12, 3793–3802 CrossRef CAS PubMed.
- J. C. Breger, K. E. Sapsford, J. Ganek, K. Susumu, M. H. Stewart and I. L. Medintz, ACS Appl. Mater. Interfaces, 2014, 6, 11529–11535 CrossRef CAS PubMed.
- S. Díaz, J. Breger, A. Malanoski, J. Claussen, S. Walper, M. Ancona, C. Brown, M. Stewart, E. Oh, K. Susumu and I. Medintz, presented in part at the SPIE Nanoscience + Engineering, San Diego, California, United States, 2015 Search PubMed.
- V. Palomo, S. A. Díaz, M. H. Stewart, K. Susumu, I. L. Medintz and P. E. Dawson, ACS Nano, 2016, 10, 6090–6099 CrossRef CAS PubMed.
- S. A. Díaz, S. Sen, K. Boeneman Gemmill, C. W. Brown, III, E. Oh, K. Susumu, M. H. Stewart, J. C. Breger, G. Lasarte Aragonés, L. D. Field, J. R. Deschamps, P. Král and I. L. Medintz, ACS Nano, 2017, 11, 5884–5896 CrossRef PubMed.
- S. A. Díaz, G. Lasarte-Aragones, R. G. Lowery, Aniket, J. N. Vranish, W. P. Klein, K. Susumu and I. L. Medintz, ACS Appl. Nano Mater., 2018, 1, 3006–3014 CrossRef.
- A. Sangtani, E. Petryayeva, M. Wu, K. Susumu, E. Oh, A. L. Huston, G. Lasarte-Aragones, I. L. Medintz, W. R. Algar and J. B. Delehanty, Bioconjugate Chem., 2018, 29, 136–148 CrossRef CAS PubMed.
- Y.-Q. Yu, W.-Q. Chen, X.-H. Li, M. Liu, X.-H. He, Y. Liu and F.-L. Jiang, Langmuir, 2023, 39, 3967–3978 CrossRef CAS PubMed.
- E. Petryayeva, T. Jeen and W. R. Algar, ACS Appl. Mater. Interfaces, 2017, 9, 30359–30372 CrossRef CAS PubMed.
- T. Jeen and W. R. Algar, Bioconjugate Chem., 2018, 29, 3783–3792 CrossRef CAS PubMed.
- C. M. Green, D. A. Hastman, D. Mathur, K. Susumu, E. Oh, I. L. Medintz and S. A. Díaz, ACS Nano, 2021, 15, 9101–9110 CrossRef CAS PubMed.
- J. R. Spangler, T. A. Leski, Z. Schultzhaus, Z. Wang and D. A. Stenger, Sci. Rep., 2022, 12, 13953 CrossRef CAS PubMed.
- T. A. Leski, J. R. Spangler, Z. Wang, Z. Schultzhaus, C. R. Taitt, S. N. Dean and D. A. Stenger, Sci. Rep., 2023, 13, 6506 CrossRef CAS PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2021, 50, D439–D444 CrossRef PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. E. Coleman, Annu. Rev. Biophys. Biomol. Struct., 1992, 21, 441–483 CrossRef CAS PubMed.
- J. C. Claussen, A. Malanoski, J. C. Breger, E. Oh, S. A. Walper, K. Susumu, R. Goswami, J. R. Deschamps and I. L. Medintz, J. Phys. Chem. C, 2015, 119, 2208–2221 CrossRef CAS.
- E. Gonzalez Solveyra, D. H. Thompson and I. Szleifer, Polymer, 2022, 14, 739 CAS.
- M. Zhou and I. Ghosh, Pept. Sci., 2007, 88, 325–339 CrossRef CAS PubMed.
- R. Nicsanu, C. Cervellati, L. Benussi, R. Squitti, R. Zanardini, V. Rosta, A. Trentini, C. Ferrari, C. Saraceno, A. Longobardi, S. Bellini, G. Binetti, O. Zanetti, G. Zuliani and R. Ghidoni, J. Alzheimer's Dis., 2022, 87, 433–441 CAS.
- G. A. Ellis, S. A. Díaz and I. L. Medintz, Curr. Opin. Biotechnol., 2021, 71, 77–90 CrossRef CAS PubMed.
- Z. Wu, B. Zhang and B. Yan, Int. J. Mol. Sci., 2009, 10, 4198–4209 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes: Additional schematics, methods, and analysis; additional kinetic spectra, raw titration data, additional EMSA, DLS and Z-potential data. See DOI: https://doi.org/10.1039/d3sd00149k |
| This journal is © The Royal Society of Chemistry 2023 |