
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalivary endotoxin detection using combined mono/polyclonal antibody-based sandwich-type lateral flow immunoassay device†
Daewoo
Han
 a,
Sancai
Xie
b and
Andrew J.
Steckl
*a
a,
Sancai
Xie
b and
Andrew J.
Steckl
*a
aNanoelectronics Laboratory, Department of Electrical and Computer Engineering, University of Cincinnati, Cincinnati, OH 45221, USA. E-mail: a.steckl@uc.edu
bProcter & Gamble Co., Mason, OH 45040, USA
First published on 21st August 2023
Abstract
A point-of-care/use lateral flow assay (LFA) is reported for the detection of P. gingivalis endotoxin, a major saliva biomarker for oral health. Two different approaches of sandwich LFA design using either the combination of mono- and polyclonal antibodies or polyclonal antibody only have been evaluated to detect P. gingivalis endotoxins, having a limit of detection of ∼22 ng mL−1 and 46.5 ng mL−1 for water- and saliva-based samples, respectively. The LFA also exhibits good selectivity to P. gingivalis endotoxin versus other endotoxins and proteins. Saliva pretreatment combining syringe filtration and potato starch successfully inhibits α-amylase activity and provides improved results on LFA devices.
Introduction
Biological fluids, such as blood, urine, saliva, mucus, are maybe the most important source of information regarding our medical status. Historically, and still the case today, blood analysis is the most common tool of diagnosis. Conventional detection of molecular markers found in biofluid specimens is carried out mostly utilizing the well-known polymerase chain reaction (PCR) method, which requires trained personnel and a relatively longer time span in a laboratory setting.1 However, with the ever-growing use of point-of-care (POC) diagnostics other biofluids are gaining in importance. Rapid and low-cost analysis of biomarkers present in various biofluids can provide early indication of various medical conditions, thus improving treatment outcomes while reducing medical costs.2 As demonstrated during the coronavirus disease (COVID) pandemic, lateral flow assay (LFA) is a versatile POC platform with many benefits, such as fast analysis time, low cost, user-friendly colorimetric interface, simple fabrication process, and no need for external instruments.3 Because COVID LFA-based home test kits were widely utilized during the pandemic, a world-wide familiarity with LFA test kits now exists. Furthermore, the use of smartphones has been developed for LFA applications not only to enhance the analysis of test results but also to improve the LFA sensitivity.4One of the most important considerations in LFA development for POC use is the type and nature of sample fluid. Saliva is an attractive biofluid for POC applications, because of stress-free non-invasive sampling and excellent availability in significant sample volume. Saliva is also a versatile medium for analysis as it consists of a wide mixture of proteins, peptides, DNA, cell debris and food particles that contains markers associated with a variety of medical conditions including high stress levels, bacterial and viral infections, presence of drugs and other toxic compounds, etc.3,5 However, this also presents challenges in isolating the specific biomarker of interest for analysis.6
Bacterial endotoxins found in saliva contain lipopolysaccharides (LPS), important macromolecules existing in the outermost part of Gram-negative bacteria that are released after cell death and lysis. Endotoxins mainly consist of lipid and polysaccharide parts as shown in Fig. 1a. Lipid A is the most toxic part and the polysaccharide has diverse configurations depending on different species and strains of the bacteria.7 LPS is a complex heterogeneous biomacromolecule. Its effective molecular weight can range from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to several million Daltons because it normally exists as an agglomeration (e.g. micelles and vesicles) in aqueous solution.8 A large number of LPS molecules is released when bacteria die or are metabolically stressed. For example, a single E. coli bacterium can release ∼2–3 million LPS molecules after death.9,10 Once LPS is released, the toxic lipid A part is exposed to the host's immune system. LPS elicits strong defense reactions, such as immune responses, various inflammations, life-threatening sepsis, and possibly septic shock.11
000 to several million Daltons because it normally exists as an agglomeration (e.g. micelles and vesicles) in aqueous solution.8 A large number of LPS molecules is released when bacteria die or are metabolically stressed. For example, a single E. coli bacterium can release ∼2–3 million LPS molecules after death.9,10 Once LPS is released, the toxic lipid A part is exposed to the host's immune system. LPS elicits strong defense reactions, such as immune responses, various inflammations, life-threatening sepsis, and possibly septic shock.11
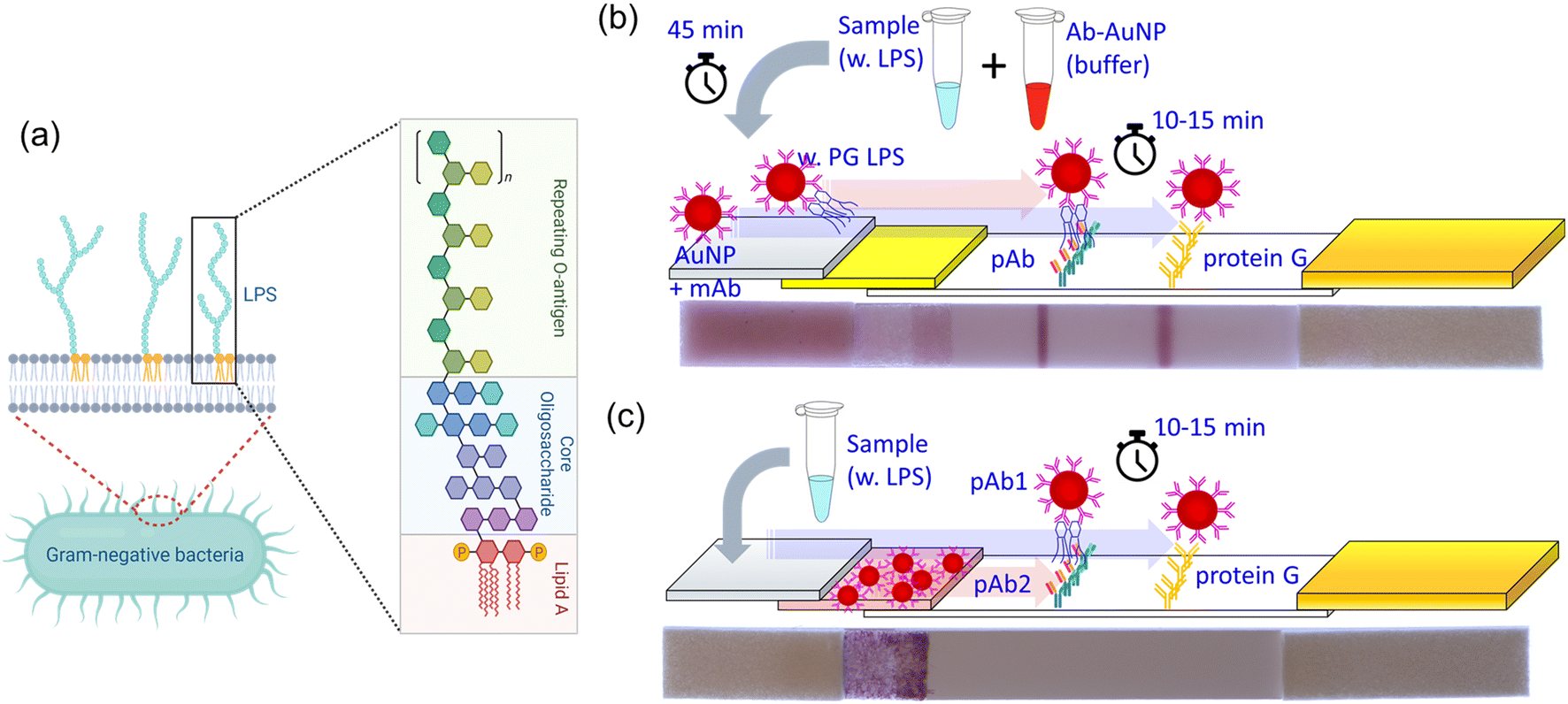 | ||
| Fig. 1 Basic mechanisms and assay designs for combined mono/polyclonal antibody-based lipopolysaccharides (LPS) detection and LFA implementation: (a) structure of LPS released from the Gram-negative bacteria (created with https://Biorender.com); (b) separate premixing of sample and Ab–AuNP solutions (photo taken after the test); (c) simplified approach using polyclonal Ab–AuNPs preloaded on the conjugate pad (photo taken before the test). | ||
Endotoxins induce a strong immune response in hosts, but also serve as important biomarkers useful for diagnosing various bacterial diseases. Porphyromonas gingivalis (P. gingivalis), one of more than 700 bacterial species found in the oral cavity, and one of 500 species present in subgingival plaque12 is a major Gram-negative pathogenic bacterium that is responsible for chronic periodontitis. Periodontal disease presents a chronic inflammation caused by bacterial infection, leading to gum disease and even loss of teeth. LPS is increased in subgingival plaques in gingivitis sites as quantified using TLR4 reporter gene assays and Lumulus Amebocyte Lysate (LAL) test.13,14 LPS from P. gingivalis (PG LPS) induces significant host responses in gingival tissue by increasing the production of inflammatory biomarkers, such as interleukin (IL)-1β, IL-6, IL-8, interferon-γ and tumor necrosis factor alpha (TNF-α) in gingival tissue.15 The salivary presence of this bacteria in a high concentration suggests that the patient has a chronic periodontitis disease. Furthermore, because of the close relationship between P. gingivalis and other important systemic diseases, such as cardiovascular,16,17 rheumatoid arthritis,18 and neurodegenerative (Alzheimer's) diseases19–21 that has emerged in recent years, quantitative evaluation of PG LPS has become a key measure in oral health, as well as in the whole body health system.1,22,23 LAL tests24 have been utilized as a gold-standard for LPS detection, mainly of the lipid A part. Other methods such as enzyme-linked immunosorbent assays (ELISA),25 electrochemical (EC) sensing,26 electrochemical impedance spectroscopy (EIS),27 have been developed.28 However, they have various challenges, such as specificity and portability for POC diagnostics.
We have developed a point-of-care lateral flow assay (LFA) device to detect and quantify PG LPS concentrations in human saliva using the molecular sandwich capture approach. Since LPS is a large biomacromolecule with multiple binding sites, one can beneficially utilize the more sensitive sandwich-type immunoassay rather than the competitive approach.29 The size of LPS aggregates in our buffer solutions were evaluated using dynamic light scattering (DLS) method (Fig. S1†). Interestingly, there is no significant difference in size with relatively tight distribution considering low PDI values. We have evaluated two different designs of sandwich based LFA devices. In the first design (Fig. 1b), two different antibodies are utilized in the device. A monoclonal antibody (mAb) is conjugated to the surface of gold nanoparticles (AuNP) to capture target PG LPS molecules in the sample solution. A polyclonal antibody (pAb) is printed and immobilized on the nitrocellulose (NC) membrane of the LFA device forming a test line that captures the AuNP conjugates bound to PG LPS from the sample solution. When LPS is present in the sample solution, the mAb-conjugated AuNP particles (mAb–AuNP) bind to LPS. Then the combined LPS–mAb–AuNP conjugate particles flow through the porous NC membrane and are captured by the pAb immobilized in the test line stripe on the LFA. Polyclonal antibodies can bind to other available binding sites on the LPS molecules or LPS micelles not occupied by the mAb–AuNP. When sufficient LPS–mAb–AuNP particles are captured a reddish test line is observed on the NC membrane. The control line on the LFA is printed on the NC membrane with protein G, which captures the mAb conjugated on the AuNP. This validates the test by confirming the conjugation status between mAb and AuNP. However, this design is not ideal for POC applications because it needs a relatively long (40 min) incubation time to bind mAb–AuNP conjugates to the target LPS in sample solution. This limitation can be resolved when conjugated Ab–AuNP are pre-loaded into the conjugation pad. In the second approach (Fig. 1c) the polyclonal antibody is utilized both for AuNP conjugation and the test line immobilization. When the sample solution is dispensed on the sample pad and proceeds to hydrate the conjugation pad, the immobilized pAb on AuNP surface quickly binds to the PG LPS in the sample solution. The pAb-conjugated AuNPs are released from the conjugation pad and flow through the NC membrane. In the presence of LPS in the sample, the formed AuNP–pAb–LPS conjugates are captured by the immobilized pAb on the test line. A fraction of the AuNP–pAb that do not bind to LPS are captured by protein G immobilized on the control line. Interestingly, although the same antibody was utilized for both AuNP conjugation and immobilization on the test line, the sandwich structure was successfully formed due to the multiple binding sites of available on the polyclonal antibodies. Apparently, this approach does not require the incubation time to bind the pAb–AuNP conjugates to PG LPS in the sample solution, leading to faster LFA test results.
Experimental section
Materials
The standard gold nanoparticle solution (10 OD, 40 nm diameter) was purchased from Cytodiagnostics (Burlington, ON). Anti-P. gingivalis LPS monoclonal antibody produced in mouse, anti-P. gingivalis polyclonal antibody produced in rabbit, cellulose fiber sample/wicking pad (CFSP001700), Tween-20, Triton™ X-100, bovine serum albumin (lyophilized powder, ≥96%), amylase assay kits were purchased from MilliporeSigma (St. Louis, MO). Ultrapure LPS from P. gingivalis and E. coli were purchased from InvivoGen (San Diego, CA). Tris–HCl 1 M buffer solutions in different pH (7.0, 7.5, 8.0), sodium phosphate tribasic dodecahydrate (Na3PO4·12H2O, 98%, analysis grade), sucrose (certified ACS grade), sodium chloride (certified ACS grade) were all purchased from Fisher Scientific (Waltham, MA). Glass fiber pad (8950) for the conjugate membrane was purchased from Ahlstrom (Helsinki, Finland) and the nitrocellulose membranes (UniSart CN140 and CN95) were kindly provided by Sartorius (Goettingen, Germany). The backing card with dimensions of 60 (W) × 300 (L) mm and 0.01′′ thickness was purchased from DCN Dx (Carlsbad, CA). Human saliva was collected from healthy volunteers using the Pure·SAL saliva collection kit obtained from Oasis Diagnostics (Vancouver, WA). Collected saliva was stored at −20 °C.Conjugation of antibody on gold nanoparticles
The passive adsorption method was used to conjugate gold nanoparticles to antibodies. This binding is “non-specific” and is governed by ionic, van der Waals, and hydrophobic forces. The optical density (OD) of concentrated 40 nm AuNP solution was evaluated using the NanoDrop One microvolume UV-vis spectrometer (Thermo Fisher Scientific) and adjusted to 10 OD. The solution of 200 μL of AuNP (10 OD) was mixed with 195 μL of 2.5 mM Tris–HCl pH 8 buffer with Tween-20 0.05% solution. Then, 5 μL of PG LPS antibody (1 mg mL−1) was added to the solution. The mixed solution was placed in a 4 °C refrigerator and incubated overnight. To remove unbound free antibodies after incubation, the incubated solution was centrifuged at 1300 G for 20 min to form a pellet of Ab–AuNP conjugation at the end of the microtube and the supernatant of unbound antibodies was discarded. Centrifuged Ab–AuNP pellets were redispersed in fresh 2.5 mM Tris–HCl pH 8 buffer solution with 0.05% of Tween-20. This washing step was repeated 4 times. In the 4th washing step, centrifuged Ab–AuNPs were redispersed in 2.5 mM Tris–HCl pH 8 buffer solution with 0.05% of Tween-20 and 0.5% of BSA. The final optical density of conjugated Ab-AuNP solution was adjusted to 5 OD. The prepared solution was stored at 4 °C.Fabrication of lateral flow assay devices
As illustrated in Fig. 1, the LFA device consists of sample pad (cellulose fiber), blocking/conjugation pad (glass fiber), nitrocellulose membrane, and wicking pad (cellulose fiber). Each component was individually prepared before assembling on the adhesive backing card. All pads have the same width of 5 mm, while the length of sample pad, blocking pad, nitrocellulose membrane, and wicking pad are 13, 10, 30, and 17 mm, respectively.Both sample and blocking/conjugation pads are pre-cut into the desired dimensions indicated above and then treated with multiple components before assembling on the backing card. Sample buffer solution for the sample pad was prepared by dissolving 7.5 μL of NaCl 1 M solution, 125 μL of Triton X-100, and 2.5 mL of Tris–HCl 1 M buffer into 50 mL of ultrapure water. Cellulose pads were soaked into the prepared sample buffer solution for 30 min and dried at 50 °C for 90 min. Similarly, the blocking buffer solution for the blocking/conjugation pad was prepared by sequentially dissolving 250 mg of BSA, 500 mg of sucrose, 38.1 mg of Na3PO4, and 12.5 μL of Tween-20 surfactant into 5 mL of ultrapure water. Precut glass fiber pads were fully soaked in blocking buffer solution for 30 min then dried at 50 °C for 90 min.
For loading the Ab–AuNP conjugates on the conjugation pad, 18 μL of Ab–AuNP conjugated solution is dispensed on the conjugation pad loaded with blocking agents, and then dried again in the oven at 50 °C for 90 min.
A sheet of NC membrane (30 mm by 300 mm) and a strip of wicking (17 mm by 300 mm) pad were attached to the adhesive backing card. On the NC membrane, PG LPS antibody for the test line and protein G for the control line were printed using the Biodot AD1500 contactless aspirate dispense printing system (Biodot, Irvine, CA). Different concentrations of antibody were utilized for optimization and sample evaluations, while the concentration of protein G was fixed at 1 mg mL−1 for most cases. After printing both materials on the NC membrane, the membranes are dried in an oven at 50 °C for 10 min and stored in a nitrogen-purged desiccator box overnight. The sheet of prepared cards printed with antibody and protein G is cut into thin strips 5 mm in width using Biodot CM4000 automatic guillotine cutter (Biodot, Irvine, CA). Finally, the treated sample and conjugation pads were assembled on the cut strips. Prepared LFA strips were stored in a nitrogen purged desiccator.
Lateral flow assay testing process
Various concentrations of PG LPS were prepared in nuclease-free ultrapure water. For the structure in Fig. 1b, the sample solution was prepared by mixing 30 μL of mAb–AuNP conjugated solution, 10 μL of 3 M NaCl, 10 μL of PG LPS solution and 40 μL of Tris–HCl buffer solution with 0.05% of Tween-20. The LFA strips were placed on a light panel to obtain a consistent light environment. After 40 min incubation at room temperature, the sample solution was dispensed on the sample pad of the LFA strip. Images of both test and control lines were taken every 30 s for 30 min using a DSLR camera fixed on the tripod. When saliva samples were used, 40 μL of saliva replaces 40 μL of Tris–HCl buffer solution. For the second LFA structure in Fig. 1c, the sample solution was prepared by mixing 20 μL of 3 M NaCl, 10 μL of PG LPS solution and 60 μL of Tris–HCl buffer solution with 0.05% of Tween-20. For the saliva samples, 10 μL of saliva replaces 10 μL of Tris–HCl buffer solution.Pretreatment of saliva samples
Saliva was collected non-invasively from healthy volunteers using Pure·SAL collectors, which removes a high percentage of mucinous materials, cell debris, food particles, etc. The collected saliva was subsequently treated through incubation with potato starch and/or filtration with/without potato starch. For incubation, the collected saliva was vortexed (2000 rpm) with the potato starch in 2:1 weight ratio and incubated at room temperature for the specific time period. After incubation, the mixture was centrifuged at 1.5 kG for 10 min and the supernatant was collected for LFA tests. For syringe filtration, we have used cellulose-based filter membranes with pore size of 0.2 or 0.45 μm. When necessary, potato starch was added to the syringe first, then the saliva was added to the potato starch-loaded syringe. LPS was spiked before the filtration process and the weight ratio of saliva and potato starch was fixed at 2:1 for all cases.
Amylase assay
Saliva was initially diluted 2000× with amylase buffer solution. 10 μL of diluted saliva was dispensed into wells of a 96 well plate followed by addition of 40 μL of amylase assay buffer to each well. Then, 100 μL of the Master Reaction Mix (amylase assay buffer:amylase substrate mix = 50:50) is added in each well. The final solution volume in each well is 150 μL. After 2–3 minutes, the 96-well plate is placed into the reader at 25 °C. The absorbance at 405 nm was recorded every 5 min until the absorbance of the most active sample becomes higher than that of the highest standard (20 nmole/well) for obtaining the linear relationship of results.
Results and discussion
LFA optimizations
Conjugation between antibody and AuNP can be readily accomplished by passive absorption, as described in the Experimental section. While AuNPs are quickly precipitated with polyclonal antibodies even at very low antibody concentrations (Fig. S2a†), monoclonal antibodies formed a stable conjugation with AuNP even at NaCl concentrations as high as 500 mM (Fig. S2b†). Tween-20 (0.05%) surfactant was added to prevent the absorption of Ab–AuNP conjugates on the microtube wall and aggregation of conjugates in solutions. Additionally, the use of buffer solution with proper pH and ionic concentrations provides significant positive effects on the stability of AuNP conjugates. When we used 2 mM Tris–HCl pH 8.0 buffer solution, very stable conjugates were formed even with a polyclonal antibody, as shown in Fig. S2c.† Higher pH is beneficial for stable dispersion of AuNPs because it provides increased negative ion concentration surrounding gold surfaces, which results in stronger repulsion forces between particles.30 Interestingly, when LPS is added to the stable pAb–AuNP conjugate solution, the conjugates are gradually aggregated over time, possibly because multiple pAb–AuNPs can bind to the single LPS (Fig. S3a†).For the first LFA approach (Fig. 1b), we utilized mAb for AuNP conjugation and pAb for the LFA test strip. Monoclonal antibody conjugated on AuNPs can provide good selectivity to PG LPS, while the polyclonal antibody acts as an effective capture probe for LPS bound to AuNP conjugates on the LFA device. As shown in Fig. S3b,† in contrast to the case of pAb-printed LFA device, mAb printed on the LFA does not yield a test line because the mAb-specific binding site of LPS is already occupied by the same mAb present on the conjugated mAb–AuNPs.
Additional optimization procedures were evaluated for enhancing the sensitivity of the assay. A simple mixture of mAb–AuNP conjugates and sample solution without LPS forms the false-positive test line (near the edges of the printed pAb area) (Fig. 2a, top strip). To remove the non-specific binding effectively a combination of concentrated 333 mM NaCl solution, 0.05% of Tween-20, and 0.5% of BSA was utilized. Highly stable mAb–AuNP conjugates are required to avoid aggregation in the presence of high NaCl concentration. Because the mAb–AuNP conjugates that we have synthesized have an excellent stability at 333 mM NaCl concentration, non-specific binding (in absence of LPS) was effectively prevented (Fig. 2a, middle strip) while in the presence of LPS a test line was formed (Fig. 2a, bottom strip). Importantly, the mixtures of mAb–AuNP and sample solutions need to be incubated over a period long enough to provide sufficient time for the mAb–AuNP conjugate to capture LPS molecules. As shown in Fig. 2b, longer incubation times of 30–40 min led to noticeable increases in test line intensity. Therefore, although the test line intensities are similar between 30 and 40 min incubation time, we have incubated all mixtures for 40 min in all subsequent experiments to eliminate any inconsistency of test line intensity caused by potential small variations of the incubation time. The conditions for printing of the polyclonal antibody (pAb) on LFA were also optimized. For the pAb printing, the Biodot automated contactless bioprinter was utilized as described in Experimental section. The test line intensity increases with pAb concentration but begins to saturate at ∼1.0 mg mL−1, as shown in Fig. 2c. Test line intensity increases rapidly with time in the first few minutes after sample dispensing (Fig. 2d) and reaches a steady state level in ∼10–15 min. The pAb concentration of 1.0 mg mL−1 was selected to maximize the test line intensity and to minimize the possible variation from batch to batch of printed pAb (Fig. 2e).
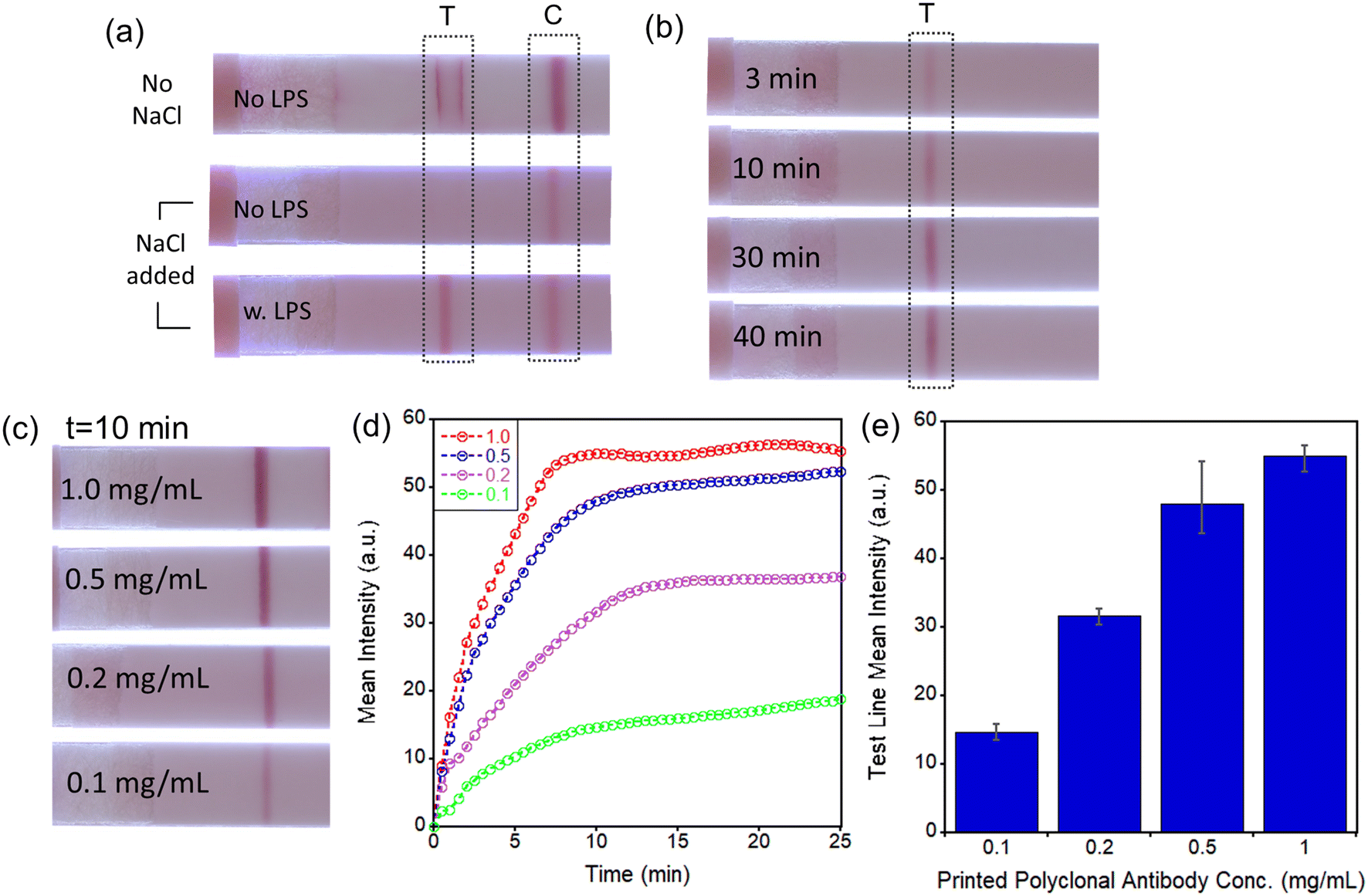 | ||
| Fig. 2 Optimization of test line formation on LFA devices: (a) non-specific binding removal by adding NaCl solution; (b) incubation time after mixing mAb–AuNP and LPS solutions; (c) effect of immobilized polyclonal antibody concentrations from 0.1 to 1.0 mg mL−1 on test line intensity on LFA device; (d) change of test line intensity over time at 4 different pAb concentrations; (e) averaged test line intensities after 10 min (n = 3). All images were taken at 10 min after dispensing sample solutions. | ||
LFA evaluation for LPS in aqueous sample solutions
After the optimization of the antibody conjugation and immobilization was completed, the sensitivity of the LFA assay with different LPS concentrations was evaluated. Sample solutions dispensed onto LFA devices consisted of 30 μL of mAb–AuNP conjugation, 10 μL of sodium chloride 3 M concentration, 10 μL of PG LPS solution (of varying concentrations from 0 to 20 μg mL−1), and 40 μL of ultrapure water. As shown in Fig. 3a, the sandwich immunoassay approach forms clear test and control lines. The test line intensity increases monotonically with LPS concentration, while the control line intensity is approximately constant.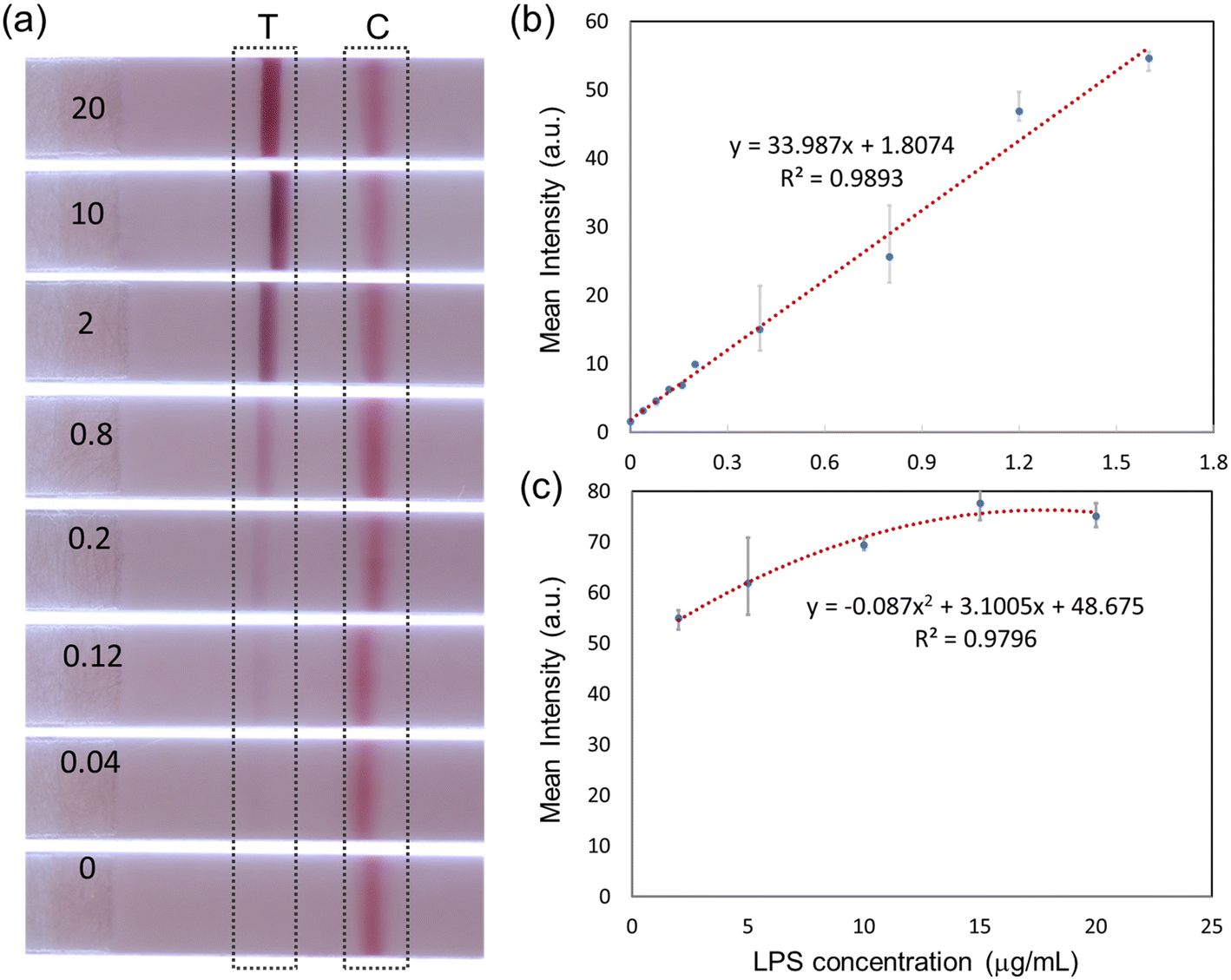 | ||
| Fig. 3 LFA test line intensity for detection of P. gingivalis LPS: (a) quantitative LFA results with different LPS concentrations (μg mL−1); (b) lower concentration regime, below 2 μg mL−1; (c) high concentration regime, up to 20 μg mL−1. (n = 3). | ||
Quantitative results can be extracted using Image J software (National Institute of Health). The mean intensities of the test line area and adjacent empty area (background) were first measured. The final test line intensity was obtained by subtracting the mean background value from the mean value of the test line area. This process is illustrated in Fig. S4.†
Fig. 3b and c show the quantified test line intensity at multiple LPS concentrations. The test line intensity increases linearly with LPS concentration in the low μg mL−1 range (Fig. 3b) and then saturates in the 10–20 μg mL−1 concentration range (Fig. 3c). An effective linear dynamic range of ∼75×, up to ∼1.6 μg mL−1 concentration is obtained. The limit of detection (LOD) of ∼22 ng mL−1 is calculated. LOD is the lowest analyte concentration which can be distinguished from a limit of blank (LOB, no LPS case).31
An initial evaluation of the selectivity of the assay was performed. As shown in Fig. 4, LFA presented the strongest test line intensity for P. gingivalis. The other LPS tested, P. pallens, resulted in a much weaker test line. However, that would not be an issue in the practical use for oral health evaluation because P. pallens LPS is also related to oral disease, especially for gingivitis. Mucin, which is the most abundant protein in saliva, and E. coli LPS did not produce a test line signal.
 | ||
| Fig. 4 Selectivity test with mucin and different endotoxins: E. coli (EC), P. pallens (PP), and P. gingivalis (PG). (n = 3). | ||
Pretreatment of saliva samples for LFA test
After the evaluation of the LFA devices with water-based samples spiked with LPS, we have used LPS-spiked saliva samples from healthy volunteers. Using untreated whole saliva spiked with PG LPS in the LFA device, test and control lines are not formed (Fig. 5a, top strip), most likely due to interference from other biomacromolecules present in saliva. Because α-amylase and mucin are the most abundant biomolecules in saliva, they were tested individually in LFAs, without using saliva (Fig. 5a, middle and bottom strips). Mucin and α-amylase were dissolved into water at concentrations found in saliva of 0.3% and 0.27%, respectively. Interestingly, mucin does not appear to have a negative effect on the P. gingivalis-antibody reactions in the LFA since the test and control lines are clearly visible and well-defined (Fig. 5a, bottom strip). On the other hand, adding α-amylase to the P. gingivalis LPS solution obstructed the immunoreactions, preventing the formation of both test and control lines, similarly to the saliva test result. Mitigating α-amylase from the saliva32 is clearly a very important consideration for salivary endotoxin detection in antibody-based LFA tests.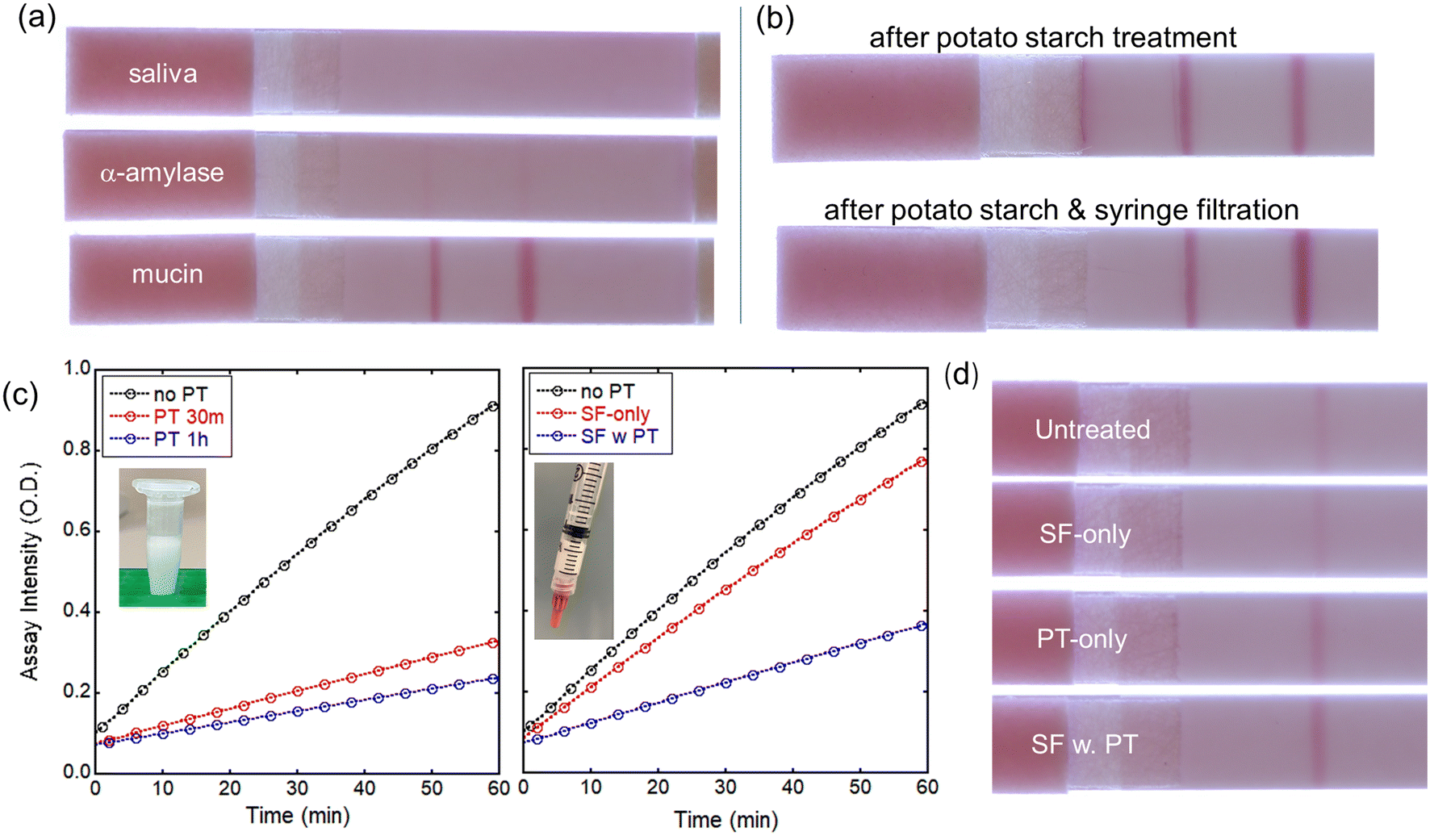 | ||
| Fig. 5 Saliva pre-treatments for P. gingivalis LPS LFA tests: (a) effect of major salivary proteins such as α-amylase and mucin on P. gingivalis LPS detection in LFA tests; (b) saliva treated either with potato starch (top) or potato starch followed by 0.45 μm syringe filtration (bottom); (c) amylase assay results either after incubating with potato starch (left) or one-step syringe filtration with potato starch (right); (d) LFA images with saliva after various pretreatments (SF-only: syringe filtration only; PT-only: potato starch incubation only; SF w. PT: syringe filtration with potato starch). For all samples, LPS was spiked at 2 μg mL−1 concentration. | ||
Amicon ultrafiltration with 100 kDa filter effectively removes most biomolecules including α-amylase. However, because the P. gingivalis LPS exists as a large aggregation >100 kDa, Amicon ultrafiltration cannot be utilized to retain PG LPS while removing α-amylase from the saliva. Instead, other approaches were investigated. Syringe filtering of the saliva was performed using filters with 0.2 or 0.45 μm pore size. Interestingly, after syringe filtering, test and control lines are formed for both 0.2 and 0.45 μm pore sizes although the test line intensity is weaker than that of aqueous solution case.
It has been reported that α-amylase can be removed by using potato starch because of its high affinity to α-amylase.32 Potato starch has been utilized as a substrate for the α-amylase inhibition assay. Saliva was mixed with potato starch in a 2:1 wt. ratio and incubated for 30 min. After incubation, the mixture was centrifuged at 1.5 kG for 10 min and the treated saliva was collected from the supernatant.
After potato starch treatment, test and control lines are formed on the LFA during tests with P. gingivalis in saliva, as shown in Fig. 5b (top). This indicates that the potato starch effectively removes α-amylase while it does not affect the LPS. However, some non-specific binding was observed near the test line, and the flow of the treated saliva on the LFA was not uniform possibly due to the presence of potato starch in saliva samples. To remove any potato starch component and undesired biomolecules in saliva, the treated saliva was filtered prior to dispensing on the LFA using cellulose-based syringe filters with 0.45 μm pore size. This combined treatment reduces the non-specific binding and noticeably improves the line intensities, as shown in Fig. 5b (bottom). To evaluate the effect of potato starch and syringe filtration on α-amylase level, we have utilized the amylase activity assay kit as described in Experimental section. The determination of amylase activity utilizes a coupled enzymatic assay that generates a colorimetric output at 405 nm wavelength. The colorimetric signal is directly proportional to the quantity of cleaved substrate, ethylidene-pNP-G7, by the amylase. As shown in Fig. 5c, amylase level is significantly reduced to ∼30% after 30 min incubation with potato starch. Longer incubation results in minor additional reduction in amylase level, but 30 min incubation seems a good operating condition. Syringe filtration by itself reduces the amylase level only slightly. However, adding potato starch to the syringe yields significant reduction in amylase level even without any time-consuming incubation. For incubation with potato starch, saliva samples were mixed with potato starch in 2:1 wt. ratio, then vortexed at 2000 rpm for desired time. The same amount of potato starch (200 mg) was added to the syringe to filter 400 μL of saliva sample. Clearly, the test line intensity after various pretreatments (Fig. 5d) is related to the level of amylase present.
LFA evaluation for LPS in pre-treated saliva sample solutions
Using the improvements obtained by saliva treatment described above, P. gingivalis LPS detection at different concentrations in saliva was tested on LFA devices. Saliva was first spiked with LPS and then treated with potato starch and syringe filtration. LFA results are shown in Fig. S5† for LPS concentrations from 0.5 to 2 μg mL−1 and for a control experiment without any LPS. Clearly, the LFA assay is sensitive to the presence of LPS in saliva. The test line intensity variation with increasing LPS concentration can provide a quantitative measure of the LPS concentrations in saliva. However, overall test line intensity is still weaker than that of water-based samples (Fig. 3a).As mentioned earlier, the first sandwich LFA design is not very practical for POC application because of the long incubation time. Also, the sensitivity is limited by the currently available monoclonal antibody for PG LPS. To improve the sensitivity and address the incubation time issue, the second LFA design (Fig. 1c) was utilized for treated saliva samples as shown in Fig. 6. Interestingly, all AuNP conjugates are effectively released from the conjugation pad, establishing strong test and control lines, as shown in Fig. 6a. In the first design, a significant amount of AuNP conjugates remains at the sample pad (reddish color, Fig. 5), while no reddish color remains in the conjugation pad of the 2nd design (Fig. 6a), which indicates most of AuNP conjugations were released through the nitrocellulose membrane. Quantitative analysis using ImageJ (Fig. 6b) presented the LOD of ∼46.5 ng mL−1, which is comparable to that of water-based samples (∼22 ng mL−1). The excellent selectivity among other bacterial LPS is obtained as shown in Fig. 6c.
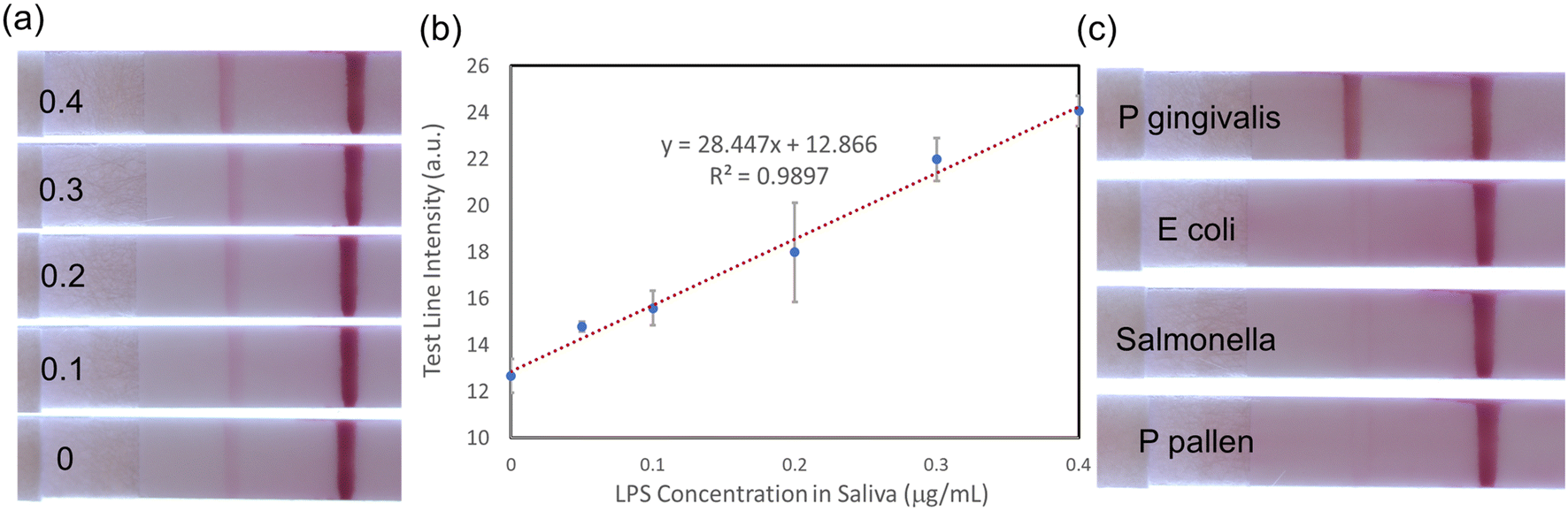 | ||
| Fig. 6 Treated saliva tests with several LPS concentrations: (a) LFA test images of different LPS concentration (μg mL−1) shown in each LFA strip; (b) quantitative analysis using ImageJ (n = 3); (c) selectivity evaluation against various endotoxins. | ||
Summary & conclusion
We have demonstrated an antibody-based sandwich LFA assay to detect P. gingivalis LPS, a major biomarker for oral health. The LFA device has LOD of ∼22 ng mL−1 for aqueous samples and excellent selectivity versus other LPS and salivary proteins. Detection in human saliva was evaluated in combination with potato starch and syringe filtering to reduce the interference from biomolecules in saliva, especially for α-amylase enzymes. LFA immunoassay reactions with saliva samples resulted in comparable results (LOD ∼46.5 ng mL−1) to that of water-based samples.Further developments will be pursued to improve assay sensitivity using saliva samples. The ability to detect multiple LPS molecules related to diseases for more accurate diagnostics of patients' health will also be explored. Finally, because the sensitivity of the current antibody-based detection is significantly affected by the performance of the conjugate antibody, the development of aptamer-based sandwich LFA will be investigated for improved flexibility and performance. The pretreatment for saliva samples discussed above can be also utilized with the aptamer-based sandwich LFA devices.
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The University of Cincinnati authors gratefully acknowledge partial support and technical assistance (Dr. Ryan Haley) by Procter & Gamble Co. The authors also appreciate the use of the Zetasizer instrument at Meridian Bioscience with assistance from Dr. ReJeana Cary and Dr. Dan Siman-Tov. ToC graphic was created with https://Biorender.com.References
- S. R. Lyons, A. L. Griffen and E. J. Leys, J. Clin. Microbiol., 2000, 38, 2362–2365 CrossRef CAS PubMed.
- A. J. Steckl and P. Ray, ACS Sens., 2018, 3, 2025–2044 CrossRef CAS PubMed.
- S. Dalirirad, D. Han and A. J. Steckl, ACS Omega, 2020, 5, 32890–32898 CrossRef CAS PubMed.
- W. Hao, Y. Huang, L. Wang, J. Liang, S. Yang, L. Su and X. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 9665–9674 CrossRef CAS PubMed.
- E. B. Aydın, M. Aydın and M. K. Sezgintürk, Adv. Clin. Chem., 2023, 113, 1–41 Search PubMed.
- T. W. Pittman, D. B. Decsi, C. Punyadeera and C. S. Henry, Theranostics, 2023, 13, 1091–1108 CrossRef CAS PubMed.
- C. R. H. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS PubMed.
- S. Harm, C. Schildböck, K. Strobl and J. Hartmann, Sci. Rep., 2021, 11, 4192 CrossRef CAS PubMed.
- S. H. Rhee, Intest. Res., 2014, 12, 90–95 CrossRef PubMed.
- G. Klein and S. Raina, Int. J. Mol. Sci., 2019, 20, 356 CrossRef PubMed.
- B. S. Park and J.-O. Lee, Exp. Mol. Med., 2013, 45, e66 CrossRef PubMed.
- J. Mysak, S. Podzimek, P. Sommerova, Y. Lyuya-Mi, J. Bartova, T. Janatova, J. Prochazkova and J. Duskova, J. Immunol. Res., 2014, 2014, 476068 Search PubMed.
- M. Klukowska, J. C. Haught, S. Xie, B. Circello, C. S. Tansky, D. Khambe, T. Huggins and D. J. White, J. Clin. Dent., 2017, 28, 16–26 Search PubMed.
- S. Xie, C. Haught, C. Tansky, M. Klukowska, P. Hu, D. Ramsey, B. Circello, T. Huggins and D. White, Am. J. Dent., 2018, 31, 215–224 Search PubMed.
- P.-L. Wang and K. Ohura, Crit. Rev. Oral Biol. Med., 2002, 13, 132–142 CrossRef PubMed.
- J. L. C. Mougeot, C. B. Stevens, B. J. Paster, M. T. Brennan, P. B. Lockhart and F. K. B. Mougeot, J. Oral Microbiol., 2017, 9, 1281562 CrossRef PubMed.
- M. J. Gui, S. G. Dashper, N. Slakeski, Y. Y. Chen and E. C. Reynolds, Mol. Oral Microbiol., 2016, 31, 365–378 CrossRef CAS PubMed.
- B. Rodríguez-Lozano, J. González-Febles, J. L. Garnier-Rodríguez, S. Dadlani, S. Bustabad-Reyes, M. Sanz, F. Sánchez-Alonso, C. Sánchez-Piedra, E. González-Dávila and F. Díaz-González, Arthritis Res. Ther., 2019, 21, 27 CrossRef PubMed.
- C.-K. Chen, Y.-T. Wu and Y.-C. Chang, Alzheimer's Res. Ther., 2017, 9, 56 CrossRef PubMed.
- S. Poole, S. K. Singhrao, L. Kesavalu, M. A. Curtis and S. Crean, J. Alzheimer's Dis., 2013, 36, 665–677 CAS.
- S. S. Dominy, C. Lynch, F. Ermini, M. Benedyk, A. Marczyk, A. Konradi, M. Nguyen, U. Haditsch, D. Raha, C. Griffin, L. J. Holsinger, S. Arastu-Kapur, S. Kaba, A. Lee, M. I. Ryder, B. Potempa, P. Mydel, A. Hellvard, K. Adamowicz, H. Hasturk, G. D. Walker, E. C. Reynolds, R. L. M. Faull, M. A. Curtis, M. Dragunow and J. Potempa, Sci. Adv., 2019, 5, eaau3333 CrossRef PubMed.
- W. He, M. You, W. Wan, F. Xu, F. Li and A. Li, Trends Biotechnol., 2018, 36, 1127–1144 CrossRef CAS PubMed.
- Y. C. Chow, H. C. Yam, B. Gunasekaran, W. Y. Lai, W. Y. Wo, T. Agarwal, Y. Y. Ong, S. L. Cheong and S.-A. Tan, Front. Cell. Infect. Microbiol., 2022, 12, 987683 CrossRef CAS PubMed.
- H. Tamura, J. Reich and I. Nagaoka, Biomedicines, 2021, 9, 536 CrossRef CAS PubMed.
- K. Reither, E. Saathoff, J. Jung, L. T. Minja, I. Kroidl, E. Saad, J. F. Huggett, E. N. Ntinginya, L. Maganga, L. Maboko and M. Hoelscher, BMC Infect. Dis., 2009, 9, 141 CrossRef PubMed.
- M. Cho, L. Chun, M. Lin, W. Choe, J. Nam and Y. Lee, Sens. Actuators, B, 2012, 174, 490–494 CrossRef CAS.
- S. E. Kim, W. Su, M. Cho, Y. Lee and W. S. Choe, Anal. Biochem., 2012, 424, 12–20 CrossRef CAS PubMed.
- R. S. Loreen, M. M. Heather and M. Harshini, Detection Methods for Lipopolysaccharides: Past and Present, IntechOpen, Rijeka, 2017 Search PubMed.
- Y. Ji, Y. Huang, Z. Cheng, W. Hao, G. Liu, Y. Liu and X. Zhang, J. Agric. Food Chem., 2023, 71, 10250–10268 CrossRef CAS PubMed.
- J. Zhu, W. Li, M. Zhu, W. Zhang, W. Niu and G. Liu, AIP Adv., 2014, 4, 031338 CrossRef.
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29, S49–S52 Search PubMed.
- O. Deutsch, Y. Fleissig, B. Zaks, G. Krief, D. J. Aframian and A. Palmon, Electrophoresis, 2008, 29, 4150–4157 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00158j |
| This journal is © The Royal Society of Chemistry 2023 |