
Utilizing MOF precursors toward one-step, calcination-free synthesis of MnO2 superstructures for superior lithium storage†
Yang
Fan
a,
Deli
Luo
a,
Yan
Wu
a,
Tianlang
Peng
a,
Qi
Qi
a,
Xubing
Han
a,
Jinxin
Zhou
a,
Yanling
Wang
c,
Bao
Lin
 a,
Qinqin
Xiong
a,
Yongjun
Yuan
a,
Haiying
Qin
a and
Xiaoshi
Hu
*ab
a,
Qinqin
Xiong
a,
Yongjun
Yuan
a,
Haiying
Qin
a and
Xiaoshi
Hu
*ab
aKey Laboratory of Novel Materials for Sensor of Zhejiang Province and New Energy Materials Research Center, College of Materials and Environmental Engineering, Hangzhou Dianzi University, Hangzhou, 310018, PR China. E-mail: xshu@hdu.edu.cn; huxiaoshi.happy@163.com
bState Key Laboratory of Silicon Materials, School of Materials Science & Engineering, Zhejiang University, Hangzhou, 310027, PR China
cColledge of Information Engineering & Art Design, Zhejiang University of Water Resources and Electric Power, Hangzhou, 310018, PR China
First published on 14th November 2022
Abstract
Rationally controlled synthesis of transition metal oxide materials for structure-related applications in diverse areas at room temperature and ambient atmosphere remains a challenge. In this article, we propose a facile one-step calcination-free approach for the rational and green synthesis of 3D hierarchical polyhedron-shaped superstructures of manganese dioxide (MnO2) through the simultaneous ion exchange and oxidation of a preformed Mn-based M2(dobdc) (dobdc = 2,5-dihydroxy-1,4-benzenedicarboxylate) MOF (CPO-27-Mn) template with an adequate open aqueous alkaline solution of a moderate concentration at room temperature, as well as their application in rechargeable lithium cells. Concretely, we proposed that during the solution-phase process, upon exchange of the anionic dobdc4− ligand with OH−, the resultant intermediate manganese hydroxide reacts with dissolved O2 in solution to form MnO2. Electrochemical evaluation showed that the as-synthesized hierarchical MnO2 superstructures exhibited excellent electrochemical performance, including high reversible specific reversibility (866.4 mA h g−1), superior rate capability and long-term cycling stability (797.9 mA h g−1 after 300 cycles at 1000 mA g−1) when serving as anodes. This unique novel MOF-derived protocol provides an alternative perspective on the designed fabrication of advanced transition metal oxide functional nanomaterials.
Introduction
To tackle global warming, with the rapid growth of electric and hybrid vehicles (EVs/HEVs), there is an urgent need for suitable energy storage devices with both high energy and high power densities and good cycle performance in recent years.1–4 As a new generation of energy supply systems, rechargeable lithium-ion batteries (LIBs) have been extensively applied in various portable electronic equipments, and have received extensive attention from researchers.5–8 Commercial graphite has been widely used as the anode material in current LIBs. However, the low energy capacity (theoretically, 372 mA h g−1) and potential safety issues because of a rather low working potential (0.2 V vs. Li/Li+) limit the further development of LIBs.9,10 Therefore, it is highly desirable to replace carbonaceous anodes with high-capacity and high-performance anode materials in order to meet the constantly growing requirements of various electronic products.To address this challenge, various transition metal oxides (TMOs) have been broadly studied as promising anode electrode materials owing to their higher theoretical capacities. Among a number of transition metal oxides, manganese dioxide (MnO2) has a high theoretical reversible capacity (1230 mA h g−1). Besides, its low cost, high abundance and environmental friendliness, as well as moderate output voltage make it one of the most prospective anode candidates for LIBs.11–13 However, MnO2 also suffers from its poor charge storage kinetics and large volume expansion/contraction during lithiation/delithiation cycling, which is similar to other TMOs.14,15 In order to tackle these problems, one method is to fabricate various hierarchically porous nano/micro superstructures assembled from different nanoparticles. It can increase the electrode–electrolyte contact area, shorten the path length for Li+ and electron transport, and reduce volume change during charging and discharging processes.16,17
Metal–organic frameworks (MOFs), a new class of hybrid materials constructed from metal ions/metal–oxygen clusters and organic ligand linkers, have shown great promise for broad applications in heterogeneous catalysis, chemical sensing, gas storage, drug delivery and molecule separation.18–22 In recent years, MOFs were widely used as sacrificial templates or precursors to build porous metal oxides or their complexes with carbon nanostructures owing to their high porosity, large surface area and chemical tunability.23 However, most research use thermal treatment under controlled atmospheres to convert MOFs into metal oxides. For example, Yuan et al. prepared an octahedral CuO wrapped 3D graphene network by calcination in a furnace under a flow of air at 320 °C with a Cu-BTC precursor.24 Hu et al. obtained a mesoporous nanostructured Co3O4 using a Co-BDC template through direct pyrolysis at a relatively low temperature in open air.25 Mn2O3 nanorods and ultrafine MnO@C composites were respectively developed by Gholami-Daghian et al.26 and Chen et al. through thermal decomposition.27 While the traditional high-temperature solid-state technique has been widely used, it has disadvantages of high energy consumption, inefficiency, high pollution and limited synthetic control over morphology. Thus, there remains a need for rational methodologies that may be employed toward the fabrication of oxide nanomaterials.
Herein, we report the rational and controllable fabrication of hierarchically layered mesoporous MnO2 micropolyhedra using a single-step, calcination-free, open alkaline solution-aided solution-phase method involving the simultaneous ion exchange and oxidation of a well-known Mn2(dobdc) MOF (CPO-27-Mn) template. Such a technique was considered to be simple, facile, cost-effective, energy-saving and environmentally friendly compared to the traditional method. Leveraging their favorable structural features, we further tested the application of the as-prepared materials as the anode electrode in lithium ion batteries.
Experimental
Materials
All the chemical reagents used in our experiments were of analytical grade and were used as received without further purification.Preparation of the MOF precursor
The Mn2(dobdc) precursor used here was prepared according to previous literature:28 3 mL ethanol and 3 mL deionized water were mixed completely in 45 mL N,N-dimethylformamide. Then 1.098 g of MnCl2·4H2O and 0.333 g 2,5-dihydroxy 1,4-benzene dicarboxylic acid (H4dobdc) were added into the mixed solution. The resulting solution was then reacted at 135 °C for 24 hours. The obtained yellow precipitate of the precursor was separated by filtration, washed and dried in air thoroughly.Preparation of MnO2 materials
0.32 g NaOH was dissolved in abundant (800 mL) deionized water to obtain NaOH solution (0.01 M). Then powdered Mn2(dobdc) (0.12 g) was added to the system and stirred for 24 hours at room temperature in an open-ambient environment. The product was collected via filtration and multiple washes with deionized water and alcohol to yield targeted MnO2 materials. The settled MnO2 was dried at 110 °C for about 12 h exposed to air.Materials characterization
Powder X-ray diffraction (XRD) patterns were obtained using an X-ray power diffractometer (Ultima IV, SmartLab, Japan Rigaku) using Cu Kα radiation (λ = 0.15418 nm at 3 kW). The micromorphologies and nanostructures of the samples were obtained using a scanning electron microscope (SEM, FEI Quanta 400 FEG, America FEI) and a transmission electron microscope (TEM, FEI Tecnai G2 F20, America FEI operating at 2 kV, 50 pA). The EDS spectrum and elemental maps were also obtained on an FEI Quanta 400 FEG, and the HR-TEM observations, and SAED were also conducted on an FEI Tecnai G2 F20. FTIR spectra were obtained on a Nicolet-Nexus 670 infrared spectrometer. The TG curves were measured in the programmed temperature range from 30 °C to 1000 °C (10 °C min−1) with a thermal analyzer (TGA/DSC 3+, Switzerland Mettler Toledo). XPS examinations were performed by X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250XI, America Thermo) using a monochromatic Al (Kα) X-ray source (1486.6 eV) by referencing the C 1s peak which was corrected to be 284.6 eV. The amounts of Na and Mn in the products were analyzed by atomic absorption and inductively coupled plasma optical emission spectrometry (ICP-OES, Agilent ICP-OES 720). The specific surface areas and pore size distribution were calculated from the results of N2 adsorption/desorption based on Brunauer–Emmett–Teller (BET) theory (3Flex, America Micromeritics).Electrochemical measurements
The working electrodes were prepared according a conventional slurry coating method: firstly, the MnO2 active materials, super P carbon black and sodium carboxymethyl cellulose binder (CMC) were mixed in some water with a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 to obtain a homogeneous slurry of solids. Secondly, the slurry was spread uniformly on pure copper foil and dried in a vacuum at 100 °C for over 12 hours. Lastly, the copper foil was cut into 14 mm diameter electrode discs with active material mass loading in the electrode around 1.5 mg. Coin cells (type-CR2032) were assembled in an Ar-filled glove box (H2O < 0.1 ppm, O2 < 0.1 ppm). To form a cell, pure Li was used as a reference electrode, a Celgard as separator, the copper foil discs carrying active materials as the working electrode. The electrolyte was also used with LiPF6 (1 M) in ethyl methyl carbonate/ethylene carbonate/dimethyl carbonate (1:1:1 by volume) with 5% fluoroethylene carbonate. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted on an electrochemical workstation (Autolab PGSTAT302N). discharge/charge measurements were carried out using a battery tester (LAND CT2001A) in the fixed voltage range of 0.01–3.00 V (vs. Li+/Li) at room temperature.
:2:1 to obtain a homogeneous slurry of solids. Secondly, the slurry was spread uniformly on pure copper foil and dried in a vacuum at 100 °C for over 12 hours. Lastly, the copper foil was cut into 14 mm diameter electrode discs with active material mass loading in the electrode around 1.5 mg. Coin cells (type-CR2032) were assembled in an Ar-filled glove box (H2O < 0.1 ppm, O2 < 0.1 ppm). To form a cell, pure Li was used as a reference electrode, a Celgard as separator, the copper foil discs carrying active materials as the working electrode. The electrolyte was also used with LiPF6 (1 M) in ethyl methyl carbonate/ethylene carbonate/dimethyl carbonate (1:1:1 by volume) with 5% fluoroethylene carbonate. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted on an electrochemical workstation (Autolab PGSTAT302N). discharge/charge measurements were carried out using a battery tester (LAND CT2001A) in the fixed voltage range of 0.01–3.00 V (vs. Li+/Li) at room temperature.
Results and discussion
The simple yet green one-step development of MnO2 is depicted in Scheme 1. | ||
| Scheme 1 Schematic illustration of the one-step alkali solution-assisted synthesis of MnO2 nano/micro superstructures. The precursor was dropped into a 1000 mL open beaker containing 800 mL of 0.01 M NaOH as a corrodent under stirring. | ||
The precursor template was measured by powder X-ray diffraction (XRD) analysis, as shown in Fig. 1a. It confirms that our product is the same as the determined MOF before. Morphology analysis by scanning and transmission electron microscopy (SEM and TEM) (Fig. 1b–d) exhibited a polyhedral solid with sizes ranging from several to several tens of microns. Magnified observations show that these particles are stacked by tightly packed smooth thin layers (thickness about 200 nm, inset in Fig. 1c). The special architecture of MOFs here is different from other reports of the CPO-27-Mn precursor.29,30 As anisotropic structures with nonspherical shapes and complex hierarchical architectures are favorable for Li storage,31,32 such a well-defined layered polyhedral structure of the CPO-27-Mn precursor is attractive as it can lead to the fabrication of anisotropic metal oxide nanostructures through the present solution-based templating approach, as will be discussed below.
 | ||
| Fig. 1 (a) XRD pattern of Mn2(dobdc) precursors. (b) and (c) SEM images; the inset in (c) shows the high magnification image of the precursor surface. (d) TEM image. | ||
The XRD patterns of target MnO2 have been depicted in Fig. 2a. Three obvious peaks in Fig. 2a at about 12.5°, 25.2° and 37.3° can be readily indexed to the (001), (002) and (−111) planes of the monoclinic δ-MnO2 respectively, which agree with the reported values (JCPDS card, no. 80-1098). The broad and weak diffraction suggests that the MnO2 crystallites are small. No characteristic peaks of impurities were observed. We speculate that the chemical mechanism to produce MnO2 could be rationalized as follows: similar to postsynthetic exchange, Mn(OH)2 was firstly formed by exchange of OH− with dobdc4− in Mn2(dobdc) thermodynamically. The released linker in the reaction solution was successfully monitored using the FTIR spectrum (Fig. S1†). Because the Mn(OH)2 intermediate is labile and readily oxidized, in situ MnO2 could be obtained with the presence of dissolved and continuously dissolved oxygen (air) from ambient atmosphere during the self-assembly reaction in solution. The probable course to form MnO2 materials through placing the Mn2(dobdc) precursor in the oxidative basic media could thus be summarized as following. It's worth noting that, the dobdc4− ligand can remain in the solution in the reaction, which might be recycled for the preparation of the Mn2(dobdc) MOF, making this route more economically beneficial.
| Mn2(dobdc) (s) + 4OH− (aq.) → 2Mn(OH)2 + dobdc4− (aq.) |
| 2Mn(OH)2 (s) + O2 (aq.) → 2MnO2 (s) + 2H2O (l) |
 | ||
| Fig. 2 (a) XRD pattern of MnO2 materials, XPS spectra of the (b) survey scan, (c) Mn 2p and (d) O 1s in MnO2. | ||
IR spectra (Fig. S2†) and TG analysis (Fig. S3†) showed that the organic skeleton has been fully depleted. The samples were further measured by X-ray photoelectron spectroscopy (XPS) (Fig. 2b–d). In the survey spectra recorded from 0 to 1200 eV (Fig. 2b), Mn and O signals were clearly observed besides the reference C 1s peak (284.8 eV). In addition, the Na 1s line at 1071.2 eV was also observed, because Na+ ions are prone to embedding into the interlayer during synthesis, which is conducive to the stability of the prepared layered MnO2. In the Mn 2p XPS spectrum (Fig. 2c), the two main peaks at 642.4 and 654.0 eV with a spin-energy separation of 11.6 eV could be assigned to Mn 2p3/2 and Mn 2p1/2, respectively, which are similar to previously reported MnO2, verifying the oxidation of Mn2+ in the precursor (Fig. S4†).33 However, it should be noted that the curves which were deconvoluted into sub-peaks revealed the presence of negligible Mn2+ or Mn3+ valence states, which were due to the presence of a small quantity of electropositive Na+ in the MnO2 interlayer for charge compensation. The composition of the MnO2 phase was determined to be Na0.20MnO2 by chemical analysis, which is similar to those reported in the literature of the monoclinic structure of the birnessite-type δ-MnO2 phase synthesized in solution.34,35 According to the Na0.20MnO2 composition, the average valence state of Mn can be calculated to be +3.8, which is similar to the Mn XPS fitting results. In Fig. 2d, the O 1s spectra show three characteristic oxygen bond contributions. The main peak at ∼529.9 eV refers to the Mn–O–Mn bonds of lattice oxygen. The shoulder peak at ∼531.5 eV is generally related to a number of surface species, for instance surface adsorbed oxygen species and under-coordinated surface lattice oxygen (defects), while the other shoulder peak at ∼533.4 eV is related to the H–O–H bond of water at or near the surface. In summary, the formation of high-purity MnO2 by self-sacrifice of the MOF precursor was confirmed.
It was noted that the alkali liquor concentration and volume are critical influential factors in obtaining a pure nanocrystalline MnO2 product under this synthesis, as manganese has a multifarious valence state (Fig. 3). Obviously, a dilute alkali concentration (0.0025 M) resulted in prephrases or other intermediate phases before transformation to the final high valence product form, monoclinic δ-MnO2, which probably yielded from the oxidation of Mn(OH)2, while when NaOH solution with a concentrated concentration (0.1 M) was used, some Mn(II) was only partially reduced to form a minor Mn3O4 phase (JCPDS card no 24-0734, marked with “*”) in addition to the primary MnO2 phase. Deceasing the volume of reaction solution could also lead to low valence Mn intermediate phases and poor crystal growth of the target MnO2 phase. Thus, it can be concluded that high-purity MnO2 was prepared with an appropriate alkali concentration (0.01 M), probably because the condition of exchange and oxidation kinetics coupling should be well established. An adequate (800 mL) reaction alkali liquor which indicates an enough amount of dissolved oxygen was also needed to make sure that Mn(OH)2 would be completely oxidized and well crystallized to high valence manganese oxide (MnO2) in our scheme.
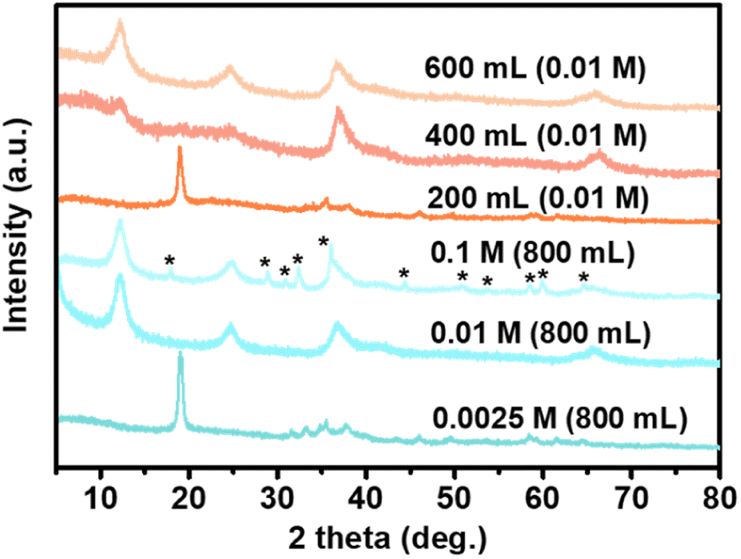 | ||
| Fig. 3 XRD patterns of the samples obtained by reacting Mn2(dobdc) with open aqueous NaOH solution at room temperature for different conditions. | ||
The morphology and structure of the prepared samples were analyzed by SEM and TEM, as is shown in Fig. 4a–d. It is obviously revealed that the three-dimensional (3-D) layered hierarchical MnO2 superstructure is assembled by small ordered (2-D) nanosheets with a thickness of ca. 90 nm and average width of ca. 200 nm. These small sheets form abundant pores within the primeval separate MOF layers, which were clearly displayed in the high magnification SEM image (Fig. 4c). It should be pointed out that the sample basically retained the initial size, polyhedral geometry morphologies, and lamellar stacking characteristic of the Mn2(dobdc) template (Fig. 1b–d).
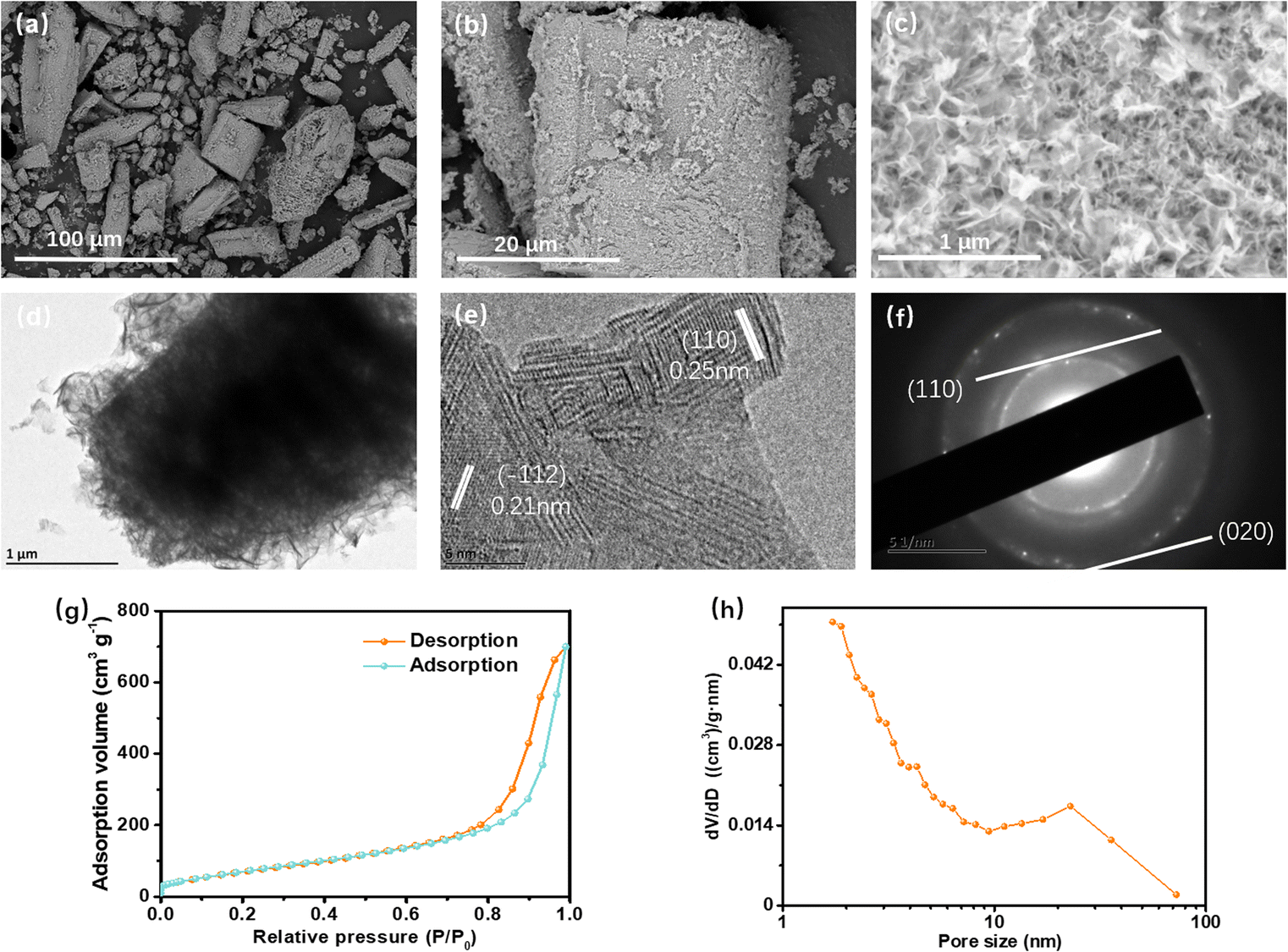 | ||
| Fig. 4 (a–c) SEM images, (d) TEM images, (e) HRTEM images, (f) SAED patterns, (g) N2 sorption isotherms and (h) the corresponding BJH pore size distributions of the as-prepared MnO2 materials. | ||
From the high-resolution TEM (HRTEM) image (Fig. 4e) and its associated selected-area electron diffraction (SAED) pattern (Fig. 4f), it can be definitely evidenced that the polymorph of manganese oxides is the monoclinic δ-MnO2 phase, as corresponding to the XRD pattern (Fig. 2a). The observed d-spacings of ca. 0.21 nm and 0.25 nm in Fig. 4e could be respectively corresponded to the (−112) and (110) planes. Energy dispersive spectroscopy (EDS) analysis shows the existence of Mn and O elements (Fig. S5†) and their uniform distribution throughout the hierarchical polyhedron particle, while the presence of Na in the δ-MnO2 phase should be ascribed to the insertion of Na+ to the δ-MnO2 interlayer. All the above-stated is in good agreement with the result of XPS analysis (Fig. 2b). The porosity characteristics of the hierarchical MnO2 microstructure was further identified by a N2 adsorption–desorption test (Fig. 4g). The nitrogen sorption isotherm shows an approximate type-IV shape in the range of P/P0 from 0.6 to 1.0 suggesting the presence of mainly mesopores with scarce macropores. The Barrett–Joyner–Halenda (BJH) pore size (Fig. 4h) distributes the mesopore structure in 4.3 nm and 22.9 nm, which may come from the inter-nanosheets spaces of MnO2 stacked in situ locations within the framework. Furthermore, the pore volume of MnO2 was found to be 1.09 cm3 g−1 on the basis of the adsorption/desorption isotherm. The Brunauer–Emmett–Teller (BET) specific surface area was measured to be as high as 263.20 m2 g−1. The aplenty open porous structure is supposed to originate from the wastage of ligand linker during the reaction of the Mn2(dobdc) with alkali liquor, and the intrinsic gaps in the lamellar structure. These results show no discrepancy with the aforementioned SEM and TEM observation in Fig. 4a–d. Particularly, the excellent porous structure with high specific surface and nano-sized sheet units can offer more active sites for lithium ion storage, shorten the distance of ion transport and buffer the volume variation during repetitive insertion/extraction of Li+ ions.
To illustrate whether other Mn-based MOF precursors also follow the templating method, nanorods of a manganese 1,3,5-benzenetricarboxylate metal–organic framework (Mn-BTC MOF) was selected as precursors with other reaction parameters unchanged. Obviously, the XRD (Fig. S6†) and SEM images (Fig. S7†) suggested the successful templating of the Mn-BTC MOF in the alkali solution for the formation of unique MnO2 nanorod superstructures.
The electrochemical performance of the prepared hierarchical ordered MnO2 nano–microstructure as the anode material for LIBs is thoroughly evaluated. It can be seen that the general features of cyclic voltammetry (CV) curves and the voltage profiles of the MnO2 electrode are consistent with those extensively reported for the MnO2 electrode based on a conversion-type reaction mechanism.36Fig. 5a shows the CV curves of the MnO2 electrode during initial few cycles recorded at a scan rate of 0.2 mV s−1. During the first cathodic scan, two broad reduction peaks were observed at 1.98 V and 1.20 V, which correspond to the lithium-ion insertion into the crystal, followed by the generation of the MnO phase, and the generation of the solid electrolyte interphase (SEI) layer, respectively.37–39 The two broad features correspond to two sloping voltage regions (2.71–2.42 and 1.74–1.33 V, respectively) in the first discharge curve at a constant current of 200 mA g−1 (Fig. 5b) and degrade during the subsequent cycles. An intense cathodic peak below 0.08 V can be assigned to the reduction of Mn by the conversion reaction. From the second cycle, the peak moves to a higher voltage of ca. 0.25 V, indicating the improved reaction kinetics and increase of surface activation energy owing to the activation of the electrodes.40,41 Similar to the CV behaviour, the corresponding long lithiated plateau region at initial 0.35 V shifts to a higher voltage at 0.40 V in the subsequent cycling. During the charge, an intense anodic peak at ca. 1.17 V and a shoulder at ca. 2.42 V were observed, corresponding to the obvious voltage plateau at about 1.03 V and the following sloping voltage above 2.26 V respectively in the first charge process, which are assigned to the oxidation of Mn from Mn0 to Mn2+ and Mn2+ to a higher oxidation state, respectively. The CV curve tends to overlap from the second cycle, which reflects an excellent electrochemical reversibility. After the formation of the SEI, the coulombic efficiency (CE) achieves 89.97% in the second cycle. However, the first discharge capacity (1923.8 mA h g−1) is much higher than the charge (1018.6 mA h g−1), leading to a low initial CE of 52.95%, which is reasonably attributed to its partially irreversible phase transformation in the first cycle, water escape and irreversible decomposition of electrolyte components owing to the enhanced specific surface area of porous polyhedra.42 The same phenomenon is commonly observed in TMO-based anodes.43,44
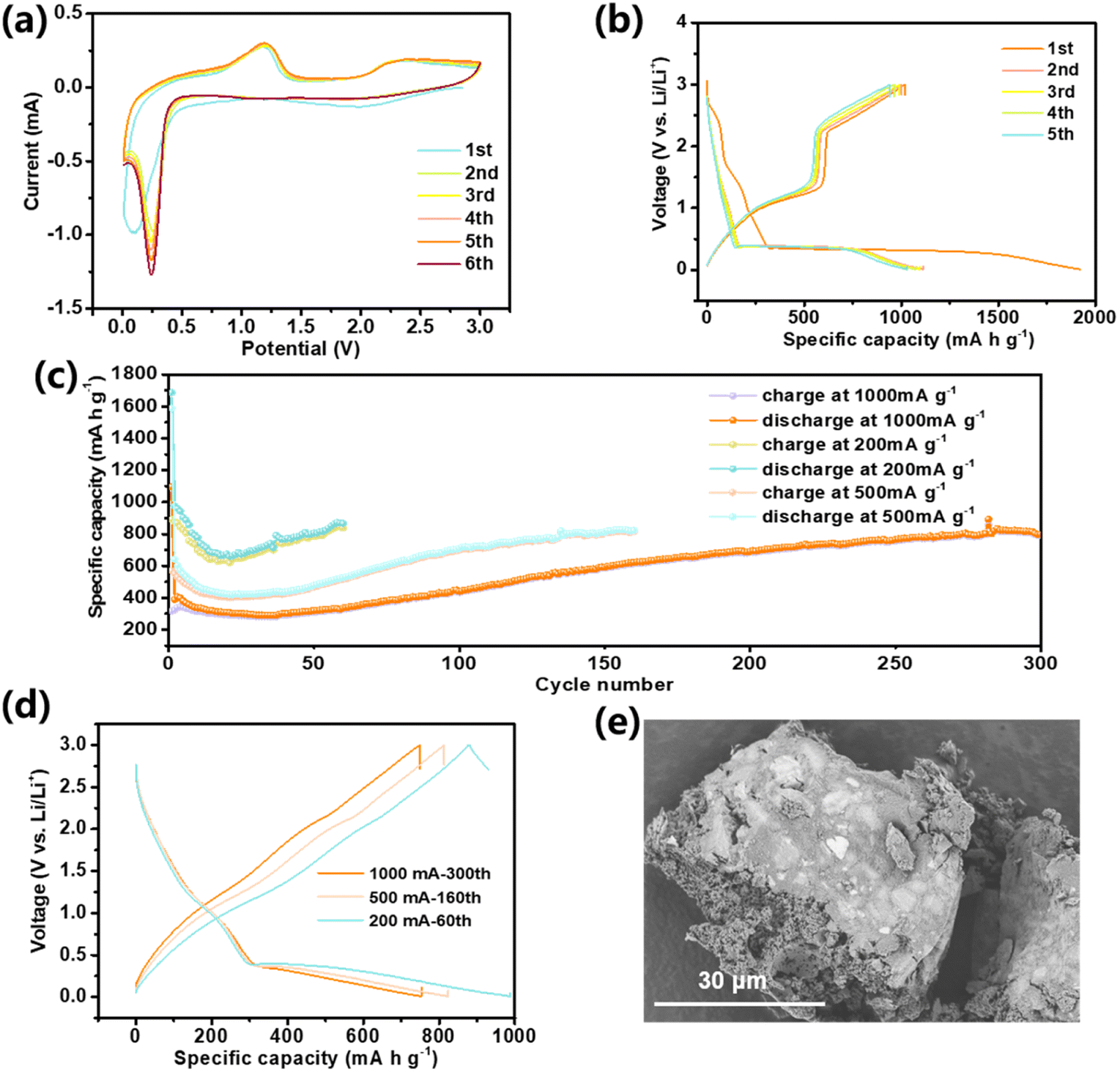 | ||
| Fig. 5 (a) CV curves of the MnO2 electrode tested in the voltage window of 0.001–3 V at a scan rate of 0.2 mV s−1, (b) voltage profiles of the MnO2 electrode for five galvanostatic charge/discharge cycles at a specific current of 200 mA g−1 in the range of 0.01–3 V, (c) cyclability and (d) voltage profiles under different specific current conditions (200, 500 and 1000 mA g−1) and (e) SEM images of MnO2 electrodes at charged state after long-term cycling. | ||
Cycling performance at different specific currents was studied to illustrate the advantage of the MnO2 material in lithium storage, as presented in Fig. 5c. Because of its unique three-dimensional structure, our MnO2 electrodes have better performance than other nanostructures (Table S1†). Under cycling at 200 mA g−1, a specific capacity of 866.4 mA h g−1 was stably delivered over 60 cycles with a high CE of 96.68% (Fig. S8†). When cycled at 500 mA g−1, a stable specific capacity of 823.4 mA h g−1 was delivered after 160 cycles. Even at a high specific current of 1000 mA g−1, it can still deliver a high capacity of 797.9 mA h g−1 after 300 cycles, which is notably higher than that of current commercial graphite (372 mA h g−1). The resemblance of the charge and discharge curves at various specific currents without large voltage hysteresis variations (Fig. 5d) implies limited polarization, which implies a very short Li+ diffusion time in the electrode and hence an excellent rate capability. It should be illustrated that the capacity of the MnO2 electrode declined first and then increased gradually (Fig. 5c). The slight capacity fading in the first few electrochemical cycles should mainly be attributed to the irreversible insertion reaction, loss of water and progressive formation of a complete SEI layer that is common to conversion-type oxide anodes.45 The gradual increasing in their capacity might probably be related to structure evolution, i.e. changes in the lithium environments in the oxide electrode after prolonged cycles, as reflected by the disappearance of charge/discharge plateaus (Fig. 5d).46
To attest the fast kinetics and good reversibility, electrochemical impedance spectra (EIS) and ex situ SEM images after prolonged cycling of the MnO2 electrode are collected. The EIS of the MnO2 electrode during cycling were examined as shown in Fig. S9.† It can be seen that both the charge-transfer resistance (Rct) (reflected by the diameter of the middle-frequency arc) and Li+ ion diffusion in solid (Zw) (reflected by the pitched line of the Z′–Z′′ curves at low frequency) are small during the cycling process, suggesting easy ion and electron transfer to the surface and interior of nanoscale active particles. Based on the EIS results, the lithium ion diffusion coefficient (DLi) was further calculated to be 7.54 × 10−10 cm2 s−1 which was superior to that of many reported TMOs-based materials,47–49 further illustrating the good Li+ diffusion ability of the hierarchical superstructure (Fig. S10†).50 In addition, since no significant increase in the resistance of the SEI layer (RSEI) (indicated by the high-frequency arc) is observed from Fig. S6,† it is proved that the SEI interface formed on the surface of the electrode material is very stable, indicating sustained good cyclability. The good cyclability is also well evidenced by the SEM images obtained from a cycled electrode in Fig. S11† and 5e, which shows a clearly distinguished layered polyhedral structure that retains the original morphology before cycles, validating that our MnO2 superstructure has wonderful mechanical stability during repeated Li+ intercalation/deintercalation processes. Besides, we also performed an XPS test of the cycled electrode after ca. the 100th charge at 1000 mA g−1 (Fig. 6). It can be seen that the valence of Mn can almost maintain after the cyclability test compared to the pristine electrode in Fig. 2c, which are responsible for the good reversibility of the MnO2 electrode. This result might also suggest that the active center, i.e. MnIV was maintained during cycling, which is consistent with those reported in the literature.
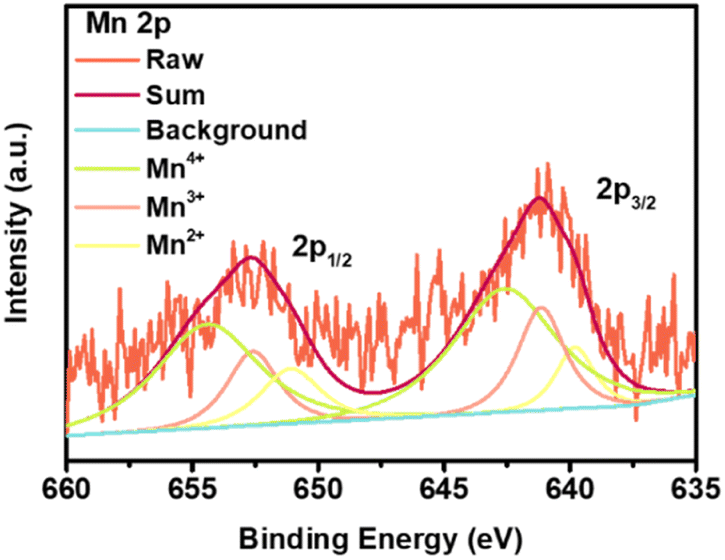 | ||
| Fig. 6 Mn 2p XPS spectrum of the cycled MnO2 electrode after ca. the 100th charge at 1000 mA g−1. | ||
Combining the above analysis, it can be concluded that due to the unique structure and morphological advantages of the MnO2 electrode, it shows excellent lithium storage performance with both high reversible specific capacity and high rate cyclability at a high specific current. On the one hand, its highly porous texture with a high specific surface area enhances the rate of transmission of lithium ions and electrons at the electrode/electrolyte interface. At the same time, the ion movement would be facilitated owing to the unique MnO2 stacked layers, and thus the charge transfer resistance will be further reduced; on the other hand, the small and thin sheet subunits lessen the Li+ diffusion time and electron transport time as well in the electrode which is favorable for reaction kinetics and high utilization efficiency of active materials; moreover, the high porosity arising from the packing of nanosheet building blocks together with the interlayer space is capable of providing free space to ease the volume change during repetitive cycling. Last but not least, the well-organized 3D hierarchical structure with micro-/submicrometer dimensions may also be conducive to the entire structural and morphological stability of the particle. These results may also lead to the good reversibility of the MnO2 conversion reaction at the same time. Therefore, superior electrochemical performance of the MnO2 electrode even at large current density was found to be contributed jointly by the above factors. However, it should be noted that the insertion of the Na+ ion into the MnO2 interlayer might also contribute to the good performance of the layered MnO2 electrode, as the larger-sized Na+ ion might act as a pillar to stabilize the layer structure and enlarge the interlayer space for better Li+ diffusion during Li+ storage, which are to be studied in detail in further work.
Conclusions
In summary, the novel approach of simultaneous ion exchange and oxidation of the Mn2(dobdc) MOF (CPO-27-Mn) in an open beaker containing abundant alkaline NaOH aq. solution with a moderate concentration for the green and facile synthesis of three-dimensional layered MnO2 mesoporous microstructures stacked by nanosheet units has been developed. Concretely, when the Mn2(dobdc) MOF is dropped into NaOH/air solution under stirring, it is supposed to undergo an anion exchange process with OH− to form Mn(OH)2 which will be directly oxidized by dissolved oxygen in the solution to form in situ the MnO2 product. Abundant reaction solution and an appropriate alkaline concentration would ensure the complete oxidation and crystallization of Mn to a pure high oxidation valence (MnIV) product. It is particularly noteworthy that there is neither high energy consumption nor gas emission in the preparation process, which shows great advantages compared with the traditional thermal method. When investigated as anodes in lithium-ion battery applications, our MnO2 sample demonstrated a high charge/discharge capacity (ca. 866.4 mA h g−1), exemplary rate capability (up to 1000 mA h g−1), and excellent cycling behavior that maintained high performances for 300 cycles. This could be mainly attributed to their favorable well-arranged micro-/nano-architectures. We believe that the results presented in this work open a new opportunity for rational design and synthesis of functional hierarchically porous micro-/nano superstructures of transition metal oxides for high-efficient energy storage applications.Author contributions
Y. Fan and X. S. Hu designed the experiments. Y. Fan carried out the sample synthesis and performed materials characterization experiments. Y. Fan, D. L. Luo, Y. Wu and X. B. Han performed the electrochemical testing and analysed the experimental data. Y. Fan and X. S. Hu wrote the manuscript. T. L. Peng, Q. Qi, J. X. Zhou and Y. L. Wang discussed the manuscript. B. Lin, Q. Q. Xiong, Y. J. Yuan and H. Y. Qin provided the help of experiment tools and resources. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China under Grant no. 21902038, the Zhejiang Provincial Natural Science Foundation of China under Grant no. LY22B030009, the Fundamental Research Funds for the Provincial Universities of Zhejiang under Grant no. GK219909299001-414, the Visiting Scholar Fund of State Key Laboratory of Silicon Materials under Grant no. SKL2020-08, and the Zhejiang Provincial Natural Science Foundation of China under Grant no. LQ20E010005.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- X. L. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- C. Li, X. B. Lou, M. Shen, X. S. Hu, Z. Guo, Y. Wang, B. W. Hu and Q. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 15352–15360 CrossRef CAS PubMed.
- S. Hu, W. Chen, J. Zhou, F. Yin, E. Uchaker, Q. F. Zhang and G. Z. Cao, J. Mater. Chem. A, 2014, 2, 7862–7872 RSC.
- D. Deng, Energy Sci. Eng., 2015, 3, 385–418 CrossRef.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 RSC.
- X. Q. Chen, H. B. Lin, X. W. Zheng, X. Cai, P. Xia, Y. M. Zhu, X. P. LI and W. S. Li, J. Mater. Chem. A, 2015, 3, 18198–18206 RSC.
- Y. X. Yu, J. Mater. Chem. A, 2013, 1, 13559–13566 RSC.
- X. Gu, J. Yue, L. J. Li, H. T. Xue, J. Yang and X. B. Zhao, Electrochim. Acta, 2015, 184, 250–256 CrossRef CAS.
- R. B. Wu, X. K. Qian, F. Yu, H. Liu, K. Zhou, J. Wei and Y. Z. Huang, J. Mater. Chem. A, 2013, 1, 11126–11129 RSC.
- G. H. Zhang, S. C. Hou, H. Zhang, W. Zeng, F. L. Yan, C. C. Li and H. G. Duan, Adv. Mater., 2015, 27, 2400–2405 CrossRef CAS PubMed.
- M. Rana, V. Sai Avvaru, N. Boaretto, V. A. de la Peña O'Shea, R. Marcilla, V. Etacheri and J. J. Vilatela, J. Mater. Chem. A, 2019, 7, 26596–26606 RSC.
- S. S. Li, Y. H. Zhao, Z. W. Liu, L. T. Yang, J. Zhang, M. Wang and R. C. Che, Small, 2018, 14, 1801007 CrossRef.
- Q. S. Zhao, J. L. Liu, X. X. Li, Z. Z. Xia, Q. X. Zhang, M. Zhou, W. Tian, M. Wang, H. Hu, Z. T. Li, W. T. Wu, H. Ning and M. B. Wu, Chem. Eng. J., 2019, 369, 215–222 CrossRef CAS.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, Nano Lett., 2009, 9, 1002–1006 CrossRef CAS PubMed.
- X. M. Tian, D. L. Zhao, W. J. Meng, X. Y. Han, H. X. Yang, Y. J. Duan and M. Zhao, J. Alloys Compd., 2019, 792, 487–495 CrossRef CAS.
- Y. R. Zhong, L. W. Su, M. Yang, J. P. Wei and Z. Zhou, ACS Appl. Mater. Interfaces, 2013, 5, 11212–11217 CrossRef CAS PubMed.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- J. Sculley, D. Q. Yuan and H. C. Zhou, Energy Environ. Sci., 2011, 4, 2721–2735 RSC.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS.
- L. E. Kreno, N. G. Greeneltch, O. K. Farha, J. T. Hupp and R. P. Van Duyne, Analyst, 2014, 139, 4073–4080 RSC.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil and G. Férey, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- L. M. Zhang, B. Yan, J. H. Zhang, Y. J. Liu, A. H. Yuan and G. Yang, Ceram. Int., 2016, 42, 5160–5170 CrossRef CAS.
- D. Ji, H. Zhou, Y. L. Tong, J. P. Wang, M. Z. Zhu, T. H. Chen and A. H. Yuan, Chem. Eng. J., 2017, 313, 1623–1632 CrossRef CAS.
- C. Li, T. Q. Chen, W. J. Xu, X. B. Lou, L. K. Pan, Q. Chen and B. W. Hu, J. Mater. Chem. A, 2015, 3, 5585–5591 RSC.
- M. Salavati-Niasari, M. Esmaeili-Zare and M. Gholami-Daghian, Adv. Powder Technol., 2014, 25, 879–884 CrossRef CAS.
- F. C. Zheng, G. L. Xia, Y. Yang and Q. W. Chen, Nanoscale, 2015, 7, 9637–9645 RSC.
- X. S. Hu, X. B. Lou, C. Li, Q. Yang, Q. Chen and B. W. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 14684–14697 CrossRef CAS PubMed.
- H. X. Jiang, Q. Y. Wang, H. Q. Wang, Y. F. Chen and M. H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 26817–26826 CrossRef CAS.
- M. Díaz-García, Á. Mayoral, I. Díaz and M. Sánchez-Sánchez, Cryst. Growth Des., 2014, 14, 2479–2487 CrossRef.
- J. Liu, H. Xia, L. Lu and D. F. Xue, J. Mater. Chem., 2010, 20, 1506–1510 RSC.
- P. Bhattacharya, J. H. Lee, K. K. Kar and H. S. Park, Chem. Eng. J., 2019, 369, 422–431 CrossRef CAS.
- S. J. He, C. X. Hu, H. Q. Hou and W. Chen, J. Power Sources, 2014, 246, 754–761 CrossRef CAS.
- X. Q. Zhang, Z. G. Hou, X. N. Li, J. W. Liang, Y. C. Zhu and Y. T. Qian, J. Mater. Chem. A, 2016, 4, 856–860 RSC.
- X. Q. Shan, F. H. Guo, D. S. Charles, Z. Lebens-Higgins, S. A. Razek, J. P. Wu, W. Q. Xu, W. N. Yang, K. L. Page, J. C. Neuefeind, M. Feygenson, L. F. J. Piper and X. W. Teng, Nat. Commun., 2019, 10, 4975 CrossRef.
- L. Li, A. R. O. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298–6302 CrossRef CAS.
- Z. Y. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. (D. ) Lou, Energy Environ. Sci., 2012, 5, 5252–5256 RSC.
- M. S. Wu, P. C. J. Chiang, J. T. Lee and J. C. Lin, J. Phys. Chem. B, 2005, 109, 23279–23284 CrossRef CAS.
- S. W. Lee, C. W. Lee, S. B. Yoon, M. S. Kim, J. H. Jeong, K. W. Nam, K. C. Roh and K. B. Kim, J. Power Sources, 2016, 312, 207–215 CrossRef CAS.
- G. Q. Jian, Y. H. Xu, L. C. Lai, C. S. Wang and M. R. Zachariah, J. Mater. Chem. A, 2014, 2, 4627–4632 RSC.
- Z. Y. Sui, C. Y. Wang, K. W. Shu, Q. S. Yang, Y. Ge, G. G. Wallace and B. H. Han, J. Mater. Chem. A, 2015, 3, 10403–10412 RSC.
- H. Kim, W. Choi, J. Yoon, E. Lee and W. S. Yoon, Small, 2021, 17, 2006433 CrossRef CAS.
- B. Sun, Z. Chen, H. S. Kim, H. Ahn and G. Wang, J. Power Sources, 2011, 196, 3346–3349 CrossRef CAS.
- X. Gu, L. Chen, Z. C. Ju, H. Y. Xu, J. Yang and Y. T. Qian, Adv. Funct. Mater., 2013, 23, 4049–4056 CrossRef CAS.
- J. Yue, X. Gu, L. Chen, N. Wang, X. L. Jiang, H. Y. Xu, J. Yang and Y. T. Qian, J. Mater. Chem. A, 2014, 2, 17421–17426 RSC.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, J. Power Sources, 2011, 196, 371–378 CrossRef CAS.
- A. A. Voskanyan, C.-K. Ho and K. Y. Chan, J. Power Sources, 2019, 421, 162–168 CrossRef CAS.
- T. R. Penki, S. Shivakumara, M. Minakshi and N. Munichandraiah, Electrochim. Acta, 2015, 167, 330–339 CrossRef CAS.
- X. L. Wang, J. Liu, Y. F. Hu, R. G. Ma and J. C. Wang, Sci. China Mater., 2022, 65, 1421–1430 CrossRef CAS.
- Q. S. Zhao, Z. Z. Xia, T. Qian, X. C. Rong, M. Zhang, Y. F. Dong, J. Q. Chen, H. Ning, Z. T. Li, H. Hu and M. B. Wu, Carbon, 2021, 174, 325–334 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR analysis of the released dobdc4− ligand, comparison of the FTIR spectrum and TGA curves of the Mn2(dobdc) precursor and MnO2 product, Mn 2p XPS of Mn2(dobdc), EDX analysis of the hierarchical MnO2 particle, XRD and SEM images of the Mn-BTC precursor and its derived MnO2 product, the comparison table showing the cycling performance of the synthesized material with recently published reports, coulombic efficiency and Nyquist plots of the MnO2 electrode, relationship between Z′ and ω−1/2 of the electrode, and panoramic SEM images of the cycled MnO2 electrode. See DOI: https://doi.org/10.1039/d2se01224c |
| This journal is © The Royal Society of Chemistry 2023 |