
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new pyridoxal-derived gelator for selective recognition of CN− and F− under different conditions†
Subhasis
Ghosh
 ,
Nabajyoti
Baildya
and
Kumaresh
Ghosh
*
,
Nabajyoti
Baildya
and
Kumaresh
Ghosh
*
Department of Chemistry, University of Kalyani, Kalyani-741235, India. E-mail: ghosh_k2003@yahoo.co.in; kumareshchem18@klyuniv.ac.in; Fax: +913325828282; Tel: +913325828750
First published on 4th April 2023
Abstract
A pyridoxal-based new supramolecular gelator 1 is designed and synthesized. Its gelation properties have been tested in various solvents. The gelator 1 forms gels in DMSO/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and DMF/H2O (10:1, v/v). The DMF/H2O (10:1, v/v) gel shows a highly cross-linked fibrous morphology and good viscoelastic properties. The gel matrix has been utilized for efficient ‘naked eye’ detection of CN− and F− ions through a phase transition with a colour change. While the DMF:H2O gel of 1 is broken in the presence of CN−, the gel-to-gel colour change takes place upon contact with F− and AcO− ions. The use of Ca2+ distinguishes F− from AcO−. In solution, ratiometric changes in absorbance for 1 with CN− and F− over other anions are noted with a distinct colour change. Even in emission, a considerable decrease in the intensity of 1 is observed with both the ions. Importantly, the changes are pronounced with CN− compared to F−. The anion responsive nature of 1 in the solid state is verified using Merrifield resin-hooked bead 1a. A theoretical study is done to correlate with the experimental findings.
:1, v/v) and DMF/H2O (10:1, v/v). The DMF/H2O (10:1, v/v) gel shows a highly cross-linked fibrous morphology and good viscoelastic properties. The gel matrix has been utilized for efficient ‘naked eye’ detection of CN− and F− ions through a phase transition with a colour change. While the DMF:H2O gel of 1 is broken in the presence of CN−, the gel-to-gel colour change takes place upon contact with F− and AcO− ions. The use of Ca2+ distinguishes F− from AcO−. In solution, ratiometric changes in absorbance for 1 with CN− and F− over other anions are noted with a distinct colour change. Even in emission, a considerable decrease in the intensity of 1 is observed with both the ions. Importantly, the changes are pronounced with CN− compared to F−. The anion responsive nature of 1 in the solid state is verified using Merrifield resin-hooked bead 1a. A theoretical study is done to correlate with the experimental findings.
Sustainability spotlightThe research and development of new supramolecular gelators from biological molecules are important as they are utilized a lot in sensing and other various applications. The gel phase of these molecules is weakened (gel-to-sol transition) or restored (sol-to-gel transition) in the presence of suitable stimuli. An anion-induced change is of practical utility for naked eye detection without assistance of any sophisticated instrument. In this regard, the use of pyridoxal-based gelators has not been reported excluding examples from our laboratory. So herein we report a new pyridoxal-based gelator that recognizes CN− and F− under different conditions. Recognition of CN− and F− that have a significant effect on the environment and biosystems has drawn attention. |
Introduction
Design and synthesis of new molecular architectures built on biologically relevant compounds has garnered continued interest in molecular recognition research.1–3 Of the different biologically important compounds, pyridoxal (vitamin B6 cofactor) has attracted attention owing to its good coordinating and photophysical properties. Its use as a metal ion binder in recognition studies is well discussed through various designs in the literature.4,5 In comparison, the anion interaction behavior is less explored. Importantly, anion interaction features of pyridoxal-based supramolecular gelators in both gel and solution phases are not known in the literature except a few from our laboratory.6–8Supramolecular gelators are a class of compounds which form viscoelastic soft materials through aggregation involving several weak forces such as hydrogen bonding, π–π stacking, hydrophobic and van der Waals interactions. 9–12 Tuning of these weak forces using different stimuli (heat, light, redox, pH, ions, etc.) results in either weakening (gel-to-sol transition) or restoration (sol-to-gel transition) of the aggregated form.12–15 Such a phase transformation is a novel approach for detection of relevant anions by the naked eye.16–19
Among various anions, CN− and F− have attracted attention owing to their biological and environmental relevance. CN− is a toxic anion to the human body. It causes damage to health on accumulation and ensures death within a few minutes.20 Importantly, this ion is used in many industries for metal ion extraction and other processes.21 The fluoride ion is also biologically important. While it prevents the dental cavities and osteoporosis, it invites fluorosis being present in excess.22
Therefore, the recognition and detection of these ions are essential despite the presence of various fluorogenic and chromogenic probes.23–29 In this regard, the use of soft gels is limited, especially low molecular gelators (LMWGs) for CN− ions.30
During our ongoing work on developing stimuli-responsive gelators cum chemical sensors,8,31 we desire to disclose here a pyridoxal-based new gelator 1 for selective detection of CN− and F− ions under different conditions (Fig. 1). Compound 1 forms a strong yellow gel in DMF:H2O (10:1, v/v) which undergoes gel-to-sol transformation with a color change in the presence of CN−. In contrast, F− encourages a sharp gel-to-gel change under identical conditions. Further, these two ions are sensed and distinguished spectroscopically in DMF:H2O (10:1, v/v). In the design, pyridoxal is linked to the naphthalene motif which acts as a stacking group for π–π interaction. The phenolic –OH groups serve as the anion interaction sites. The attachment of the sensory system 1 with Merrifield resin produced the polymer bead 1a which conveniently detected CN− and F− with distinguishable features in organic solvent DMF. The beads are reusable.
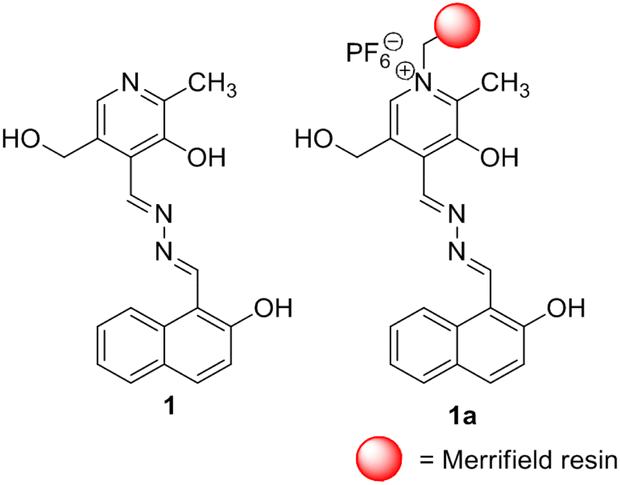 | ||
| Fig. 1 Structures of compounds 1 and 1a. | ||
Results and discussion
Synthesis
Scheme 1 depicts the synthesis pathway to achieve compound 1. Initially, on formylation, β-naphthol 2 gave aldehyde 332 which on reaction with hydrazine in ethanol afforded compound 4. Finally, the reaction of 4 with pyridoxal gave the Schiff base 1 in good yield. Then compound 1 was heated to reflux with chloromethylated polystyrene (Merrifield resin) in dry DMF for 6 h to furnish the resin-bound sensor as the chloride salt. Chloride exchange was done using NH4PF6 in DMF–CH3OH mixture solvent to have the resin-bound sensor 1a. FTIR analysis revealed the attachment of 1 to the resin. The characteristic signals of 1 were noted in the FTIR spectrum of 1a. The characterization of the rest of the compounds was done by usual spectroscopic methods.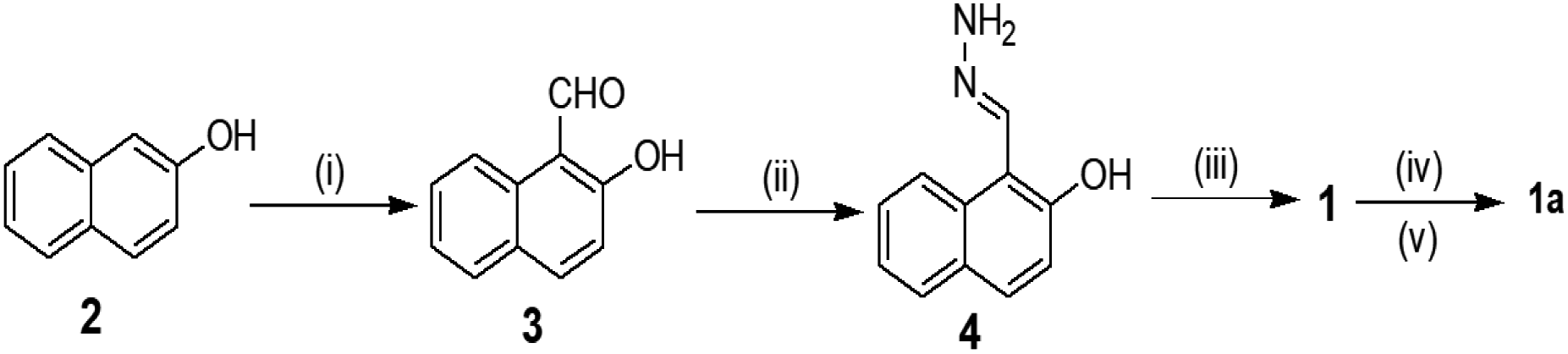 | ||
| Scheme 1 (i) CHCl3, NaOH; (ii) N2H4, EtOH, 60 °C, 10 min; (iii) pyridoxal hydrochloride, Et3N, MeOH, rt, 30 min; (iv) Merrifield resin (chloromethylated polystyrene), dry DMF, reflux, 6 h; (v) NH4PF6 (aq. solution), DMF–CH3OH, reflux, 1 h. | ||
Aggregation, morphology and rheological studies of compound 1
Owing to the occurrence of naphthalene having a reasonable π-surface and phenolic –OH, alcoholic –OH, pyridine and imine nitrogens as the possible H-bonding sites, compound 1 exhibited aggregation behaviour in aqueous organic solvents (Table 1S†). Compound 1 gave a stable yellow gel (tested by the inversion of vial method) in DMF–H2O (10:1, v/v). The gel was thermoreversible (Tg = 59 °C; Fig. 2a). The critical gelation concentration (cgc) in this solvent was determined to be 7 mg mL−1. With the increase in the concentration of gelator, Tgel increases and the time taken for the formation of gel decreases in a regular manner (Fig. 1S†).
 | ||
| Fig. 2 Photograph showing (a) the thermoreversible nature of 1 in DMF:H2O (10:1,v/v), (b) SEM image of the gel and (c) confocal microscopic image of the gel (λex = 405 nm). | ||
The DMF–H2O (10:1, v/v) gel of 1 is composed of nanofibres as confirmed from SEM (Fig. 2b) as well as confocal microscopic images (Fig. 2c). The fibres are highly cross-linked.
The FTIR spectrum of 1 in the amorphous state was compared with the gel state to highlight the role of functional groups in the aggregation (Fig. 2S†). The stretching signal for –OH at 3154 cm−1 in 1 in the amorphous state changed its position in the gel state. A decrease in stretching by 19 cm−1 interpreted the participation of –OH in H-bonding during aggregation. The stretching frequency at 1628 cm−1 for the imine group in the amorphous state was changed to 1668 cm−1 in the gel state. Such an increase in stretching is associated with hardening of the imine bond resulting from its involvement in weak interaction.
In UV-vis spectroscopy, the absorbance at 397 nm in solution moved to 406 nm with a shoulder at 446 nm in the gel state (Fig. 3a). Such a red-shift of the absorbance in UV-vis spectroscopy suggests J-type aggregation33 during gel formation. In fluorescence, the emission intensity at 522 nm in the solution is increased with a red-shift to 577 nm in the aggregated state (Fig. 3b). The increase in emission is ascribed to the aggregation-induced enhancement emission (AIEE)34 of 1 in the gel state. Under UV exposure the gel shows a brown colour.
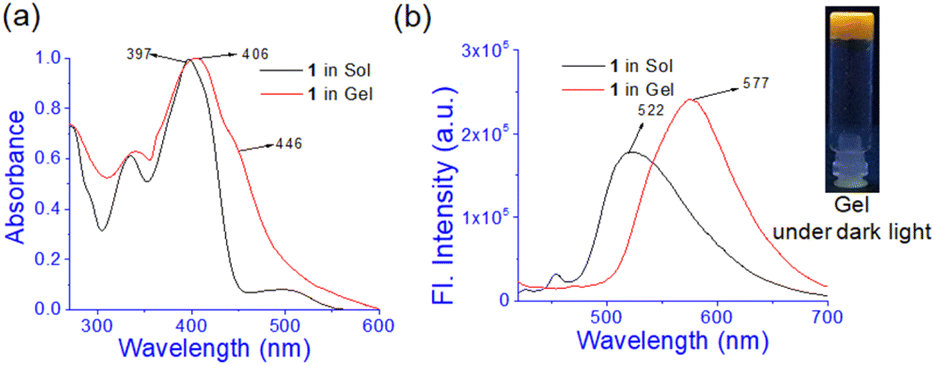 | ||
| Fig. 3 Comparison of (a) normalized UV-vis and (b) fluorescence spectra of 1 (λex = 400 nm) in sol and gel states. | ||
Density functional theory (DFT) was adopted to predict the probable mode of aggregation of 1 in aqueous DMF.35 The geometry of 1 was optimized alone and then its water assisted tetramer was optimized in DMF (Fig. 4). Without water the aggregation was impossible (Fig. 3S†). The occurrence of a small amount of water enhances the aggregation as reflected in Fig. 4b and c. During gelation, parallel π-stacking interaction between the aromatic moieties occurs within 3.35–3.63 Å. Water molecules bridge the pyridine moieties with a distance ranging from 1.91 to 2.20 Å. Fig. 4d and e represent the H → L and H-1 → L+2 electronic transitions for 1 and the aggregated form of 1, respectively. The UV-vis spectra of 1 and its aggregated form are shown in Fig. 4f. The geometry of 1 is confirmed from the UV-vis spectrum with λmax at 421 nm which is quite satisfactory with the experimental λmax at 397 nm. The geometry of the suggested aggregated form is also confirmed from λmax at 427 nm which is in concordance with the experimental value of 406 nm. The major electronic transitions along with their respective oscillator strengths are tabulated in Table 2S.†
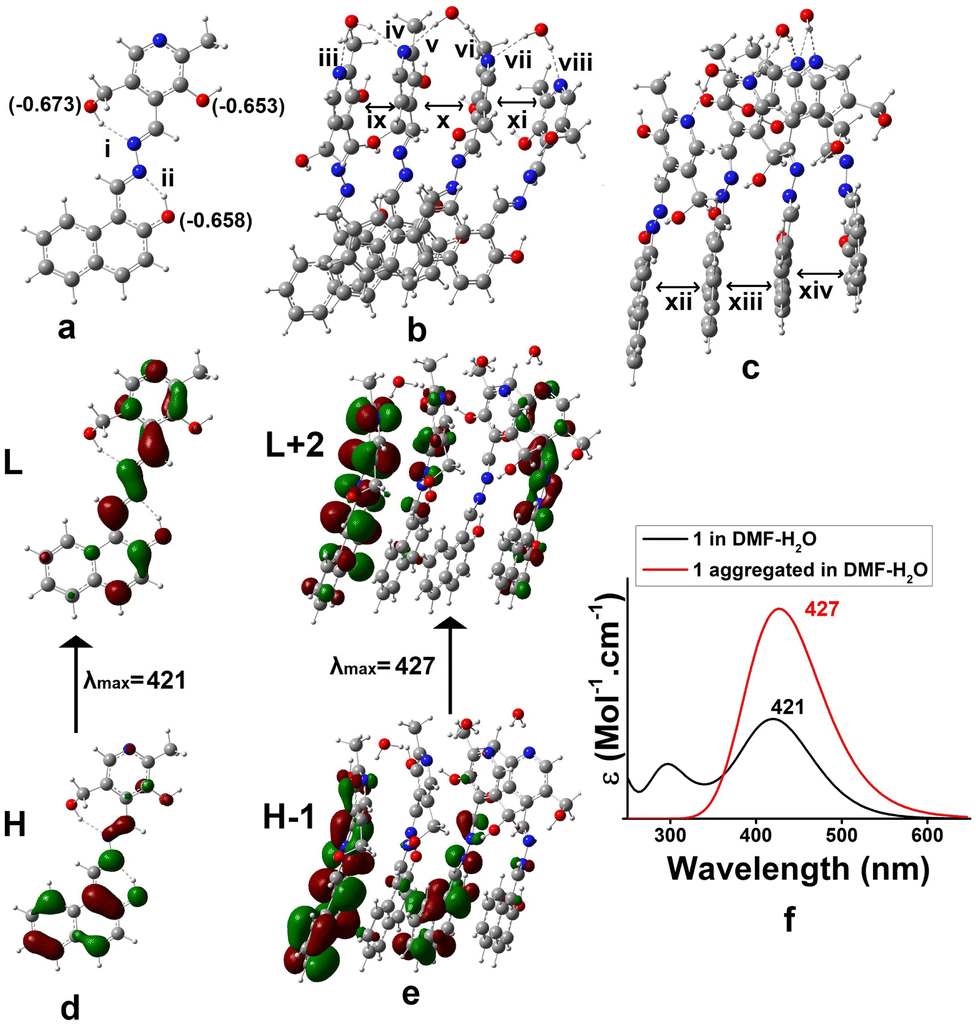 | ||
| Fig. 4 (a) Optimized geometry of 1 in DMF (Mulliken charges of the O-atom are given in parentheses), (b) and (c) optimized geometries of the aggregated form in DMF–H2O from different angles, (d) electronic transition of 1 and (e) aggregated form of 1 in DMF–water, and (f) UV-vis spectra of 1 in DMF and aggregation of 1 in DMF–water (i = 1.93, ii = 1.64, iii = 1.94, iv = 2.10, v = 2.16, vi = 1.99, vii = 2.21, viii = 1.93, ix = 3.61, x = 3.40, xi = 3.54, xii = 3.44, xiii = 3.41, xiv = 3.66 all distances are in Å unit). | ||
The oscillatory rheological experiment on DMF–H2O (10:1, v/v) gel of 1 was performed to determine its viscoelastic properties. In this study, the amplitude sweep and frequency sweep at 25 °C were ascertained at the cgc of the gel. In the amplitude sweep study at a constant frequency of 1 Hz, the curve for storage modulus (G′) runs over the curve for loss modulus (G′′) which indicated the elastic semisolid nature of the gel (Fig. 5a). The gel started to flow at 5.46% of strain. On the other hand, in the frequency sweep experiment at the constant strain of 0.1%, the curves for G′ and G′′ run parallel showing a frequency independent change. The magnitude of the average value of G′ to G′′ was determined to be 3.39 (Fig. 5b).
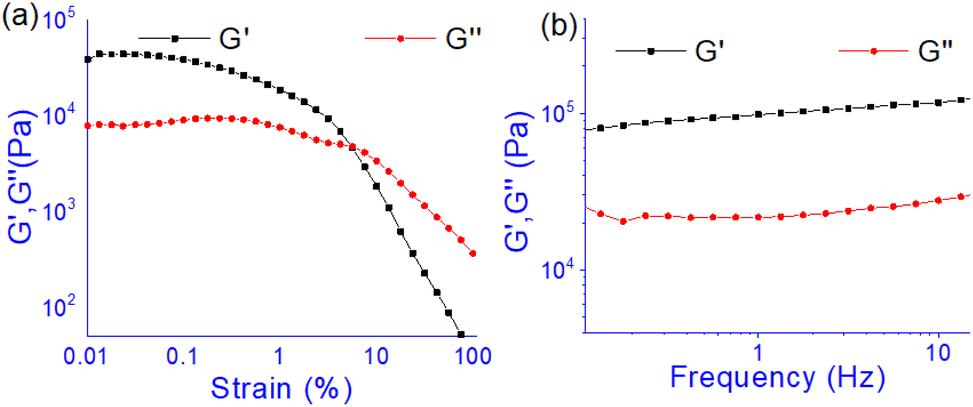 | ||
| Fig. 5 Rheological study: (a) amplitude sweep (at constant frequency of 1 Hz) and (b) frequency sweep (at constant 0.1% strain) experiments. Gel was prepared at the cgc in DMF/H2O (10:1, v/v). | ||
The summary of the rheological data is shown in Table 1. The study reveals the good viscoelastic response and true gel nature of the aggregated form.
| Gel | Critical strain (%) | Cross-over (% strain) |
|
|
|
|
|---|---|---|---|---|---|---|
| 1 | 0.1 | 5.46 | 87234 |
25719 |
3.39 | 0.294 |

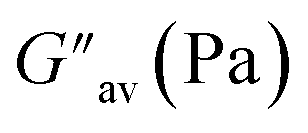
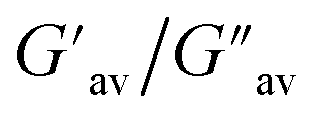
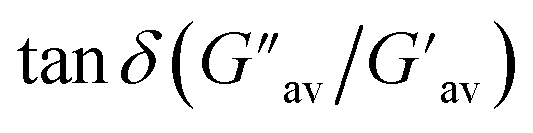
Anion and pH sensitivities of the gel
Prior to studying the interaction of 1 towards various anions, the solvatochromic effect of 1 in UV-vis was studied in different solvents (Fig. 6). In DMSO, compound 1 showed an intense band at 520 nm and exhibited a different spectral behaviour from other solvents. This is explained to be due to the involvement of phenolic –OH groups in hydrogen bonding and subsequent deprotonation in DMSO. This is similar to our earlier report.36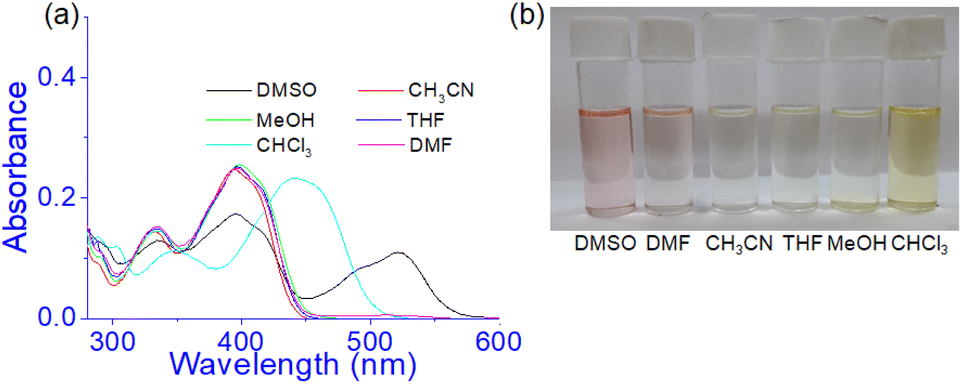 | ||
| Fig. 6 (a) Absorption spectra of 1 (c = 2.5 × 10−5 M) in different solvents and (b) change in colour of the solution in visible light. | ||
However, in CH3CN, THF, DMF, and CH3OH no distinct change in absorbance was noted at 395 nm except in CHCl3. In CHCl3, the absorbance at ∼400 nm underwent a red shift to 440 nm and the solution became light brown. The strong pink color of the solution in DMSO was due to the existence of some amounts of deprotonated species which were relatively less in DMF for which the DMF solution showed a very faint pink color (Fig. 6b). Thus the study intimated that compound 1 may be usable to study the anion interaction in the mentioned solvents except DMSO and accordingly, we took DMF as the working solvent. Moreover, to maintain the uniformity in condition with the gel state, DMF:H2O (10:1, v/v) was considered throughout the study in both solution and gel states.
First, to study the interaction of anions with the gel state, the DMF:H2O (10:1, v/v) gel of 1 which was prepared at the minimum gelation concentration [7 mg dissolved in 1 mL DMF:H2O (10:1, v/v); c = 2.08 × 10−2 M] was treated with 1 equivalent amount of different anions such as CN−, F−, AcO−, H2PO4−, HSO4−, NO3−, Cl−, Br−, and I− (all were used as tetrabutylammonium (TBA) salts). Conversely, aqueous solutions of the same anions in 1 equivalent amount were added to the slightly warmed DMF solution of 1 (Fig. 7a). In this event, compound 1 (7 mg) was dissolved in DMF (0.91 mL) and then 0.09 mL of water which contains 1 equivalent amount of different anions with respect to gelator 1 was added to prepare the gel (c = 2.08 × 10−2 M). In either case, the gel was not observed in the CN− containing vial. Either the gel was disintegrated or the gel was not formed upon contact with the CN− ion. The broken gel changed to deep red from yellow within 30 minutes. Importantly, phase change of the gel was not noted with other anions in the study. Only in contact with F− and AcO− ions, the gel became red from yellow without rupturing (Fig. 7a). F−-treated reddish gel was changed to the yellow gel in the presence of calcium perchlorate (Fig. 7b). Under identical conditions, AcO−-treated gel did not show a sharp change and it was thus well distinguished from F−. The F−-induced red gel showed fibrous morphology as confirmed by the confocal image. While F− was added sequentially from 0.25 equivalent to 1 equivalent amount, the fibres became gradually thin and distinct (Fig. 8). The rheological study revealed the slight weakness of the gel compared to the pristine gel. The crossover point dropped from 3.39 to 2.89 upon contact with F− (Fig. 4S and Table 3S†). Thus the gel of 1 was an effective matrix to detect and distinguish CN−, F− and AcO− from each other.
 | ||
| Fig. 7 Phase changes of (a) 1 (c = 2.08 × 10−2 M) in the presence of 1 equiv. amount of different anions (anions were taken as TBA salts), and (b) distinction between the AcO− and F− ions containing gels using Ca(ClO4)2. | ||
 | ||
| Fig. 8 Confocal microscopic images of gel 1 (in DMF:H2O (10:1, v/v; c = 2.08 × 10−2 M)) when treated with 0.25, 0.5 and 1 equivalent amounts of F− ions (λex = 405 nm). | ||
To check the gel phase sensitivity of 1 for CN−, initially CN− was added in 0.5 equiv. amount to the gel of 1 in DMF:H2O (10:1, v/v). However, under this condition, the gel sustained for a long time. When the amount of CN− was increased to 1 equivalent, the gel collapsed (Fig. 5S†). The solution became deep red in colour. Thus in the present investigation, 1 equivalent amount of CN− was the minimum amount for its recognition through sol–gel methodology.
Due to the phenolic –OH and imine functionalities in 1, the stability of the gel was checked at different pHs. The gels were stable in the pH window 2–13, but the gel did not survive below pH 2 likely due to the protonation of the pyridine ring nitrogen (Fig. 6S†).
Anion interaction in solution
The anion interaction was investigated in the solution phase through UV-vis and fluorescence titration experiments (Fig. 9) of 1 in DMF:H2O (10:1, v/v). A solution of 1 (c = 2.5 × 10−5 M) was prepared in DMF:H2O (10:1, v/v). Anions were taken as TBA-salts and dissolved in DMF:H2O (10:1, v/v) (c = 1.0 × 10−3 M). In the study, addition of CN− to the solution of 1 produced a ratiometric change in the absorption spectra. A decrease in absorption at 397 nm was noted with the development of a new intense band at 500 nm (Fig. 9a). Similar changes were observed with the F− ion but to lesser extents as evidenced from Fig. 9b. The remaining anions showed only a minor change in intensity at ∼397 nm without exhibiting any strong peak at ∼500 nm (Fig. 9b). The absorption at 500 nm for the basic anions F− and CN− ions is attributed to the loss of the phenolic –OH protons in 1. Owing to the small amount of water in the solvent, such deprotonation occurs in both cases. However, addition of water suppressed this peak for F− while that of CN− was intact with decreased intensity (Fig. 7S†). Due to the greater hydration of F− than CN−, the deprotonation of 1 is suppressed with F− and it is easily distinguished from CN−.
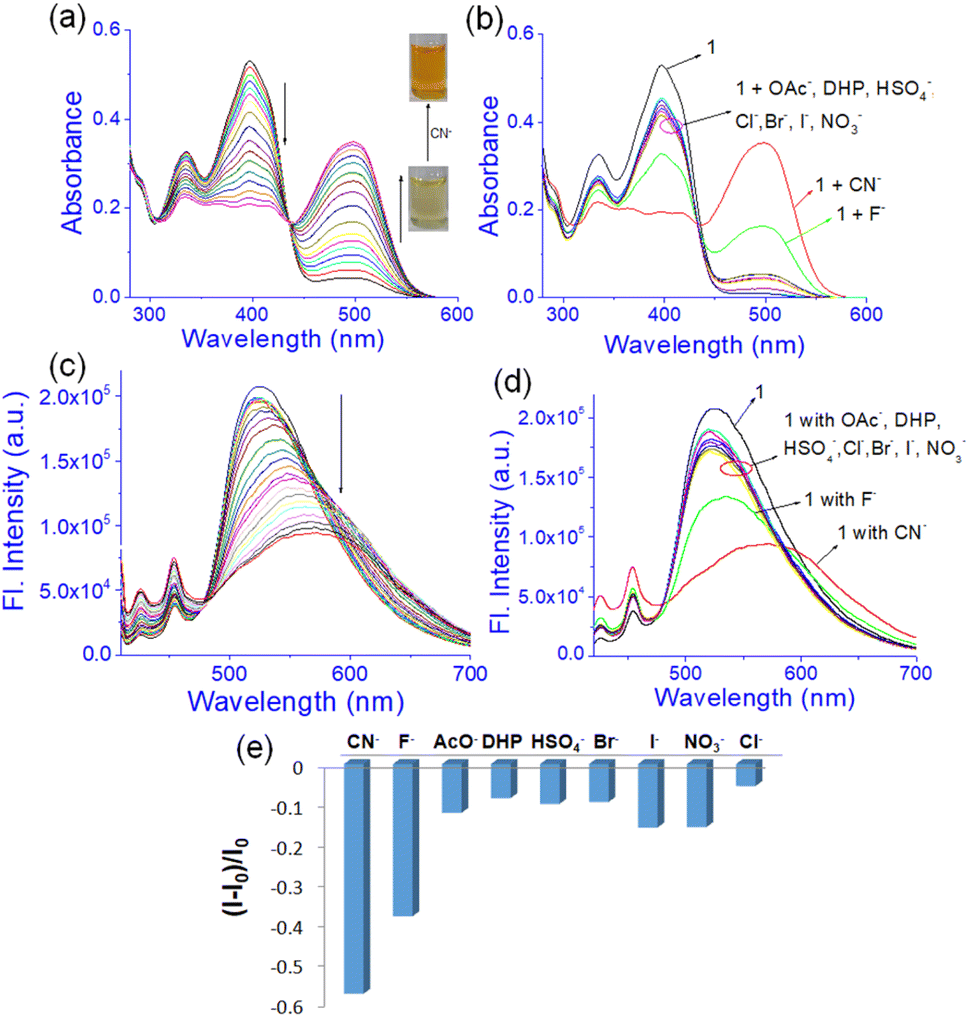 | ||
| Fig. 9 Photograph showing (a) change in the absorbance of compound 1 upon titration with 10 equiv. amounts of CN− and (b) different anions; (c) change in emission (λex = 400 nm) of 1 upon titration with 30 equiv. amounts of CN−; (d) change in emission (λex = 400 nm) of compound 1 upon addition of 30 equiv. amounts of different anions and (e) emission intensity plot with the addition of different anions in DMF–H2O (10:1, v/v) [c of compound 1 is 2.5 × 10−5 M and the anion concentration is 1 × 10−3 M]. | ||
In fluorescence, emission was observed at 525 nm in DMF:H2O (10:1, v/v) while compound 1 was excited at 400 nm. This emission was changed differently with the anions. Upon interaction with CN−, the emission at 525 nm was gradually shifted to 570 nm with decreasing intensity (Fig. 9c). A similar change was recorded upon addition of F− but to a lesser extent (Fig. 9d and e). Other anions brought minor changes in the emission spectra (Fig. 9d and e). The selectivity of 1 for CN− was examined in the presence of other anions as shown in Fig. 8S.† No anion except F− and AcO− interfered in the event.
To examine the effect of pH, solutions of 1 were prepared in DMF–H2O (10:1, v/v) at different pH's (2, 4, 6, 8, 10, 12, and 13) without using any buffer at a concentration of 2.5 × 10−5 M. The colour of solution was changed from yellow (pH ≤ 8) to light red (pH = 10) to deep red (pH ≥ 12) (Fig. 9S†). In UV-vis spectroscopy, the absorption at 417 nm underwent small positional movement up to pH 8. Above pH 8, a new peak at 498 nm for deprotonation of the phenolic –OH was observed (Fig. 9S†). In fluorescence spectroscopy, the emission of 1 at 520 nm was slightly increased with an increase in pH and became considerable at pHs 12–13 with a shift of 25 nm to the longer wavelength (Fig. 9S†). This is contrary to the interaction of F−/CN− ions which gave a reverse change in emission. This is presumably due to the aggregation of the phenolates in the presence of released water during the pH change of the medium.
The interaction of 1 towards CN− was 1:1 as realized from the Benesi–Hildebrand plot (Fig. 10S†).37 It was similar to F− interaction (Fig. 10S†). The detection limits38 for CN− and F− were ascertained to be 8.34 × 10−5 M and 2.62 × 10−4 M, respectively.
To check the interaction mechanism, the 1H NMR spectrum of 1 was gathered in the presence of CN− and F− in d6-DMSO. Upon addition of tetrabutylammonium fluoride (TBAF), deprotonation of –OH (phenolic Ha, Hb) occurred and there was upfield chemical shift of the imine (Hc and Hd) and other aromatic ring protons (Fig. 10a). The addition of tetrabutylammonium cyanide (TBACN) brought different observations. It deprotonated the phenolic –OHs and attacked the imine groups for which the spectra became complex to analyse (Fig. 10b). The new signals at 4.97 ppm and 4.27 ppm were indicative of the nucleophilic attack of CN− at the imine groups.39 The mechanism of interaction is highlighted in Fig. 10c where F− deprotonates to give 1b and CN− results in both deprotonation and nucleophilic attack to produce complicated mixtures of 1c–1e.
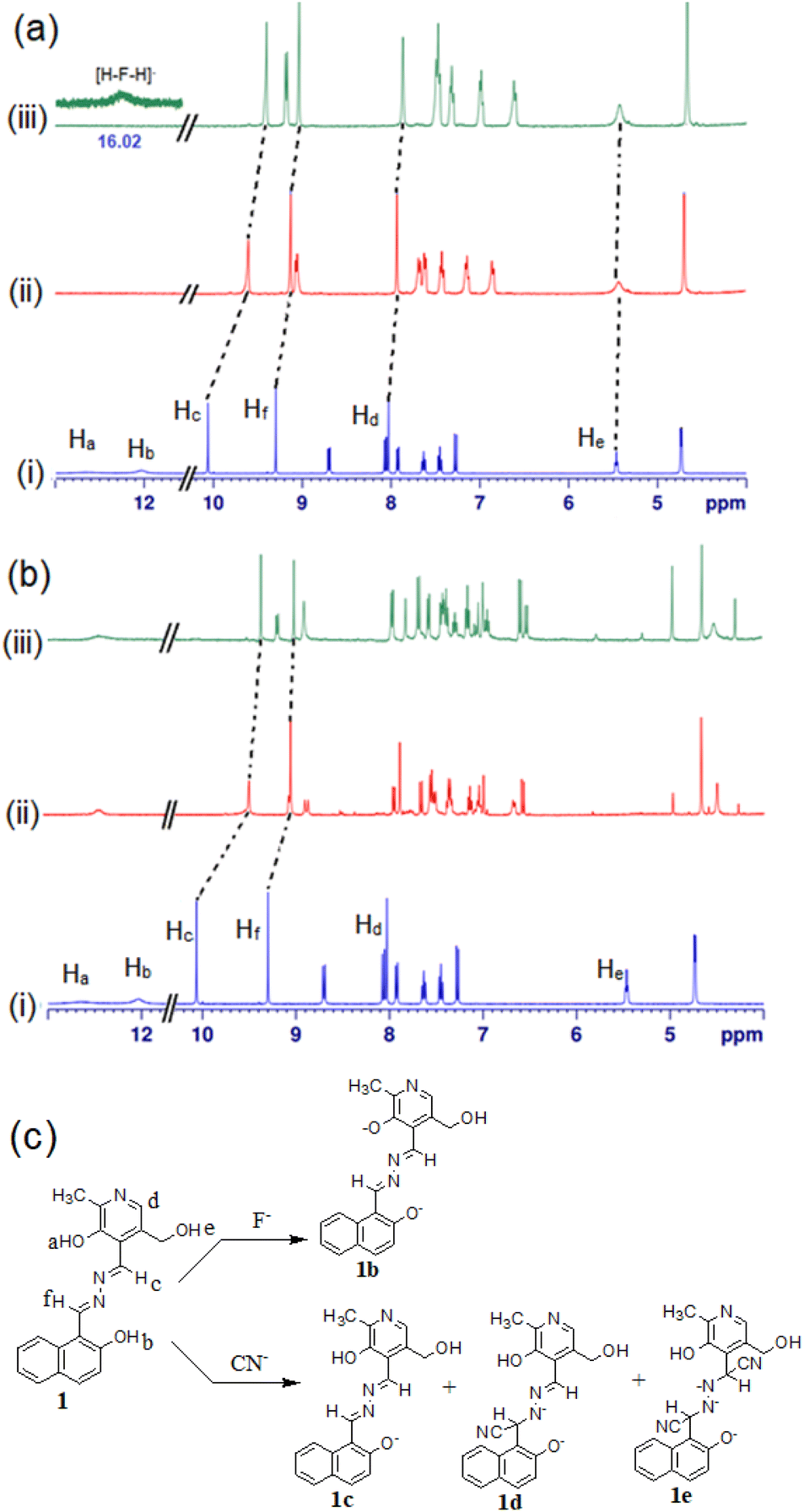 | ||
| Fig. 10 Partial 1H NMR (400 MHz, d6-DMSO) of (a) (i) compound 1 and with (ii) 1 equiv. and (iii) 2 equiv. amounts of TBAF; (b) (i) compound 1 and with (ii) 1 equiv. and (iii) 2 equiv. amounts of TBACN; (c) mode of interaction of compound 1 with F− and CN−. | ||
To have much insight about the anion interaction in solution, a theoretical study was also performed. TDDFT calculations on the forms 1b (dianionic form via F− interaction) and 1c (monoanionic form via CN− interaction) introduced UV-vis spectra which coincided with the experimental findings (Fig. 11). The electronic transitions from H → L for di-anionic and mono-anionic forms, shown in Fig. 11d and e, gave λmax at 490 nm and 498 nm respectively followed by disappearance of the peak at 406 nm. The information matches properly with the experimental results and confirmed our suggested mode of interactions.
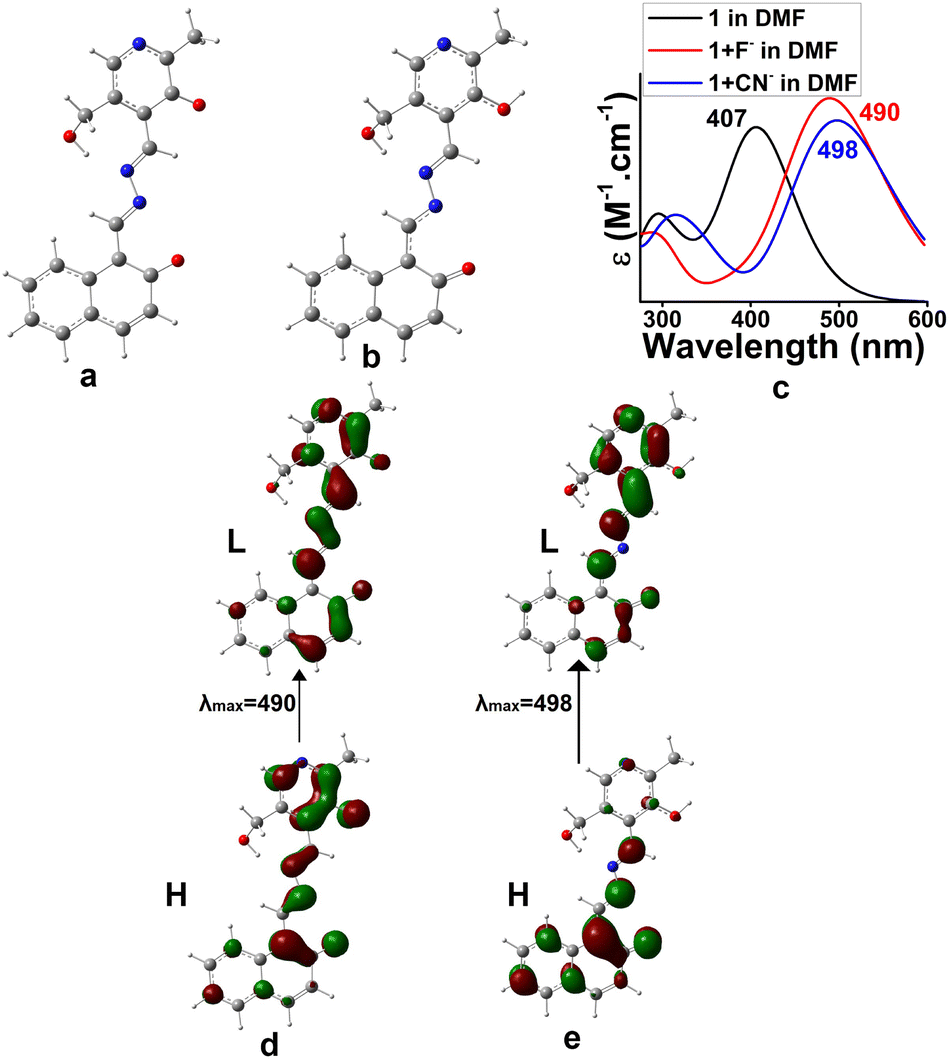 | ||
| Fig. 11 Optimized geometries of (a) di-anionic form 1b for F− binding, (b) mono-anionic form 1c for CN− binding in DMF, (c) UV-vis spectra of 1b, 1c and 1 in DMF, (d) electronic transition for (a) and (e) electronic transition for (b). | ||
Anion interaction in the solid state
The present study reveals that compound 1 is sensitive to CN− and F− compared to other anions in both solution and gel states through different observations. For the scope of attachment of the structure 1 onto the solid phase, we attached it on Merrifield resin (Fig. 11S†). The light yellow bead 1a, in this context, was employed in the study. On treatment of the beads with F− and CN− in DMF, the color of the beads changed from light yellow to light red. In the event, a minimum concentration of 10 mM of the anions brought the change and the change was sharp with CN− (Fig. 12). However, the addition of Ca(ClO4)2 changed the color of the beads to their original colour and it was much pronounced in the case of F−-treated beads. Cyanide treated beads were unable to return to its original colour. Importantly, in the case of F−, Ca2+-treated beads were further employed in the event and similar results were obtained. In this way, three cycles were possible for the detection of F−. After three cycles, the beads lose their efficiency. In the event, change of the beads with F− is associated purely with deprotonation and reprotonation phenomena. In the case of CN−, the attack of CN− to the imine bond as discussed in Fig. 10 hindered the colour change. | ||
| Fig. 12 Solid state recognition: (a) sensitivity of 1a towards different concentrations of F− and CN− in DMF; (b) change in of the beads of 1a after treatment with F− and CN− (c = 10 mM) followed by Ca(ClO4)2 in DMF. | ||
Conclusion
In conclusion, a new pyridoxal-based Schiff base 1 has been synthesized. The compound with several hydrogen bond functionalities forms yellow gels in different solvent combinations such as DMF:H2O (10:1, v/v) and DMSO:H2O (10:1, v/v). The gel derived from DMF:H2O (10:1, v/v) is undertaken in the study. The gel is thermoreversible and pH stable and exhibits fibrous morphology. The viscoelastic nature is confirmed from the higher values of storage modulus (G′) over the loss modulus (G′′) in the rheological study. The aggregation of the molecules in DMF is aggravated by adding a small amount of water which assists the packing of molecules through hydrogen bonding. No gelation occurs without water. A DFT study is carried out to explain the observations.
The DMF:H2O (10:1, v/v) gel is anion responsive. It displays CN−-induced gel-to-sol transformation with a colour change. In contrast, other basic anions F− and AcO− bring a gel-to-gel colour change without rupturing. In the case of the F−-gel, red colour is rapidly changed to its original yellow colour in the presence of Ca(ClO4)2 which scavenges F− ions from the medium. Such observation is distinct from the AcO−-gel which responds weakly under identical conditions. Thus the DMF:H2O (10:1, v/v) gel acts as an excellent matrix for recognition of CN− and F− with distinguishable features. The distinction of these ions is noticeable in solution also. Importantly, compound 1 exhibits a marked change with CN− and F− ions in both UV-vis and fluorescence. The spectral changes are pronounced with CN− addition to the solution of 1 in DMF:H2O (10:1, v/v). In UV-vis, the peak at ∼500 nm due to the addition of CN− and F− is tunable. Addition of water abolished this peak for F− while that of CN− remained visible with a decreased intensity. It is established that compound 1 interacts differently with these basic anions. While F− shows observable changes through deprotonation of the phenolic –OH, under similar conditions CN− is involved in both deprotonation and nucleophilic attack to the imine groups of 1 to bring the changes. A DFT study has been done to validate the different observations.
For practical application, compound 1 is hooked to the Merrifield resin bead which shows a colour change upon contact with CN− and F− in DMF. While F− ion treated red beads exhibit reversibility in colour change upon treatment with Ca2+ ions, CN− impregnated beads are silent in the event. The present investigation is a new addition to the reported pyridoxal-based molecular gelators explored in anion sensing by us only.6,7
Experimental
Materials and methods
All the reagents and chemicals were purchased from Spectrochem, India and Sigma-Aldrich. Tetrabutylammonium salts were purchased from Sigma-Aldrich. Dried and purified solvents wherever necessary were used. Solvents for NMR experiments were obtained from Aldrich. Thin layer chromatography was done on Merck precoated silica gel 60-F254 plates. 1H and 13C NMR spectra were recorded using a Bruker 400 MHz instrument using TMS as the internal standard. High resolution mass data were acquired using a Bruker Daltonics MicroTOF-Q-II Mass Spectrometer and Q-T of ESI-MS instrument (model HAB 273). FTIR measurements were done using a PerkinElmer L120-00A spectrometer (νmax in cm−1) using KBr cells and KBr pellets, respectively. Scanning electron microscopy (SEM) images were obtained on an EVO LS-10 ZEISS instrument. Fluorescence and UV-vis studies were performed using a Horiba Fluoromax 4C spectrofluorimeter and Shimadzu UV-2450 spectrophotometer, respectively. Confocal microscopy images were obtained using Carl Zeiss LSM 800 instrument.Synthesis
Gelation test and SEM imaging
Compound 1 (7 mg) was taken in DMF (0.91 mL) and warmed slightly. After cooling the solution, H2O (0.09 mL) was added to form a gel. The same was followed for preparing the gel in DMSO:H2O (10:1, v/v). No flow of the mass upon inversion of the vial confirmed the gel formation. A thin film of the gel was dried under vacuum for SEM imaging and then coated with a thin layer of gold metal.
General procedures for fluorescence and UV-vis titrations
Compound 1 was dissolved in DMF:H2O (10:1, v/v) to make a stock solution at a concentration of 2.50 × 10−5 M. Stock solutions of the anions were prepared in the same solvent maintaining a concentration of 1.0 × 10−3 M. A 2 mL solution of 1 was taken in a cuvette and different anionic guests were added individually in different amounts to this solution. On addition, the emission as well as absorbance was recorded.
Calculation of the detection limit38
To determine the detection limit, the absorbance of the compound was measured 5 times to evaluate the standard deviation in blank measurement. The plot of absorbance values against concentrations of the analyte gave a slope. Then the use of the values in the equation: detection limit = 3σ/k (σ = standard deviation of blank measurement, and k = slope) gave the detection limit.Theoretical study
A theoretical study was performed using the Gaussian 09 program. Geometry optimization of 1 was done by employing density functional theory. All calculations were performed with the help of the B3LYP hybrid functional and with basis set 6-31G(d). For the calculation of the aggregation process dispersion corrected hybrid functional B3LYP-D3 was applied.40 Structures of the optimized geometries were confirmed by frequency calculations to be true minima. To understand the implicit solvent effects (DMF–H2O) the Polarizable Continuum Model (PCM)41 was applied in time-dependent density functional theory (TD-DFT) calculations considering 30 singlet excitations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KG thanks SERB, New Delhi, India for financial support [CRG/2021/005928/OC Dated 20.12.2021]. SG thanks CSIR, New Delhi, India for a fellowship.References
- A. Mbarek, G. Moussa and J. Leblond Chain, Molecules, 2019, 24, 1803 CrossRef CAS PubMed.
- K. Ghosh, T. Sen and R. Fröhlich, Tetrahedron Lett., 2007, 48, 7022–7026 CrossRef CAS.
- S. Patel, D. Bariya, R. Mishra and S. Mishra, Steroids, 2022, 179, 108981 CrossRef CAS PubMed.
- M. Strianese, S. Milione, V. Bertolasi and C. Pellecchia, Inorg. Chem., 2013, 52, 11778–11786 CrossRef CAS PubMed.
- S. K. Sahoo, New J. Chem., 2021, 45, 8874–8897 RSC.
- C. Pati and K. Ghosh, New J. Chem., 2019, 43, 2718–2725 RSC.
- K. Ghosh and C. Pati, Tetrahedron Lett., 2016, 57, 5469–5474 CrossRef CAS.
- S. G. Roy, A. Kumar, N. Misra and K. Ghosh, Mater. Adv., 2022, 3, 5836–5844 RSC.
- R. G. Weiss and P. Terech, Materials with Self-Assembled Fibrillar Networks, 2006 Search PubMed.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC.
- A. Panja, R. Raza and K. Ghosh, ChemistrySelect, 2020, 5, 11874–11881 CrossRef CAS.
- S. Ghosh, P. Jana and K. Ghosh, Anal. Methods, 2021, 13, 695–702 RSC.
- L. Li, R. Sun, R. Zheng and Y. Huang, Mater. Des., 2021, 205, 109759 CrossRef CAS.
- H. Maeda, Chem. – Eur. J., 2008, 14, 11274–11282 CrossRef CAS PubMed.
- R. R. Dash, A. Gaur and C. Balomajumder, J. Hazard. Mater., 2009, 163, 1–11 CrossRef CAS PubMed.
- Y. B. Patil and K. M. Paknikar, Process Biochem., 2000, 35, 139–1151 CrossRef.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- H.-H. Yang, P.-P. Liu, J.-P. Hu, H. Fang, Q. Lin, Y. Hong, Y.-M. Zhang, W.-J. Qu and T.-B. Wei, Soft Matter, 2020, 16, 9876–9881 RSC.
- B. R. Jali, A. K. Barick, P. Mohapatra and S. K. Sahoo, J. Fluorine Chem., 2021, 244, 109744 CrossRef CAS.
- J. Han, J. Zhang, M. Gao, H. Hao and X. Xu, Dyes Pigm., 2019, 162, 412–439 CrossRef CAS.
- X. Cao, A. Gao, J.-t. Hou and T. Yi, Coord. Chem. Rev., 2021, 434, 213792 CrossRef CAS.
- P. B. Pati, Sens. Actuators, B, 2016, 222, 374–390 CrossRef CAS.
- R. S. Fernandes and N. Dey, J. Mol. Struct., 2022, 1262, 132968 CrossRef CAS.
- D. Udhayakumari, Spectrochim. Acta, Part A, 2020, 228, 117817 CrossRef CAS PubMed.
- S. Panja, A. Panja and K. Ghosh, Mater. Chem. Front., 2021, 5, 584–602 RSC.
- R. Raza, N. Baildya and K. Ghosh, ChemPlusChem, 2022, 87, e202200270 CrossRef CAS PubMed.
- A. Ghosh, S. Ta, M. Ghosh, S. Karmakar, A. Banik, T. K. Dangar, S. K. Mukhopadhyay and D. Das, Dalton Trans., 2016, 45, 599–606 RSC.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- M. J. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, Revision D. 1, Gaussian Inc.Wallingford CT, 2009, vol. 27, p. 34 Search PubMed.
- S. Ghosh, S. Ghosh, N. Baildya and K. Ghosh, New J. Chem., 2022, 46, 8817–8826 RSC.
- P.-T. Chou, G.-R. Wu, C.-Y. Wei, C.-C. Cheng, C.-P. Chang and F.-T. Hung, J. Phys. Chem. B, 2000, 104, 7818–7829 CrossRef CAS.
- B. Zhu, X. Zhang, Y. Li, P. Wang, H. Zhang and X. Zhuang, Chem. Commun., 2010, 46, 5710–5712 RSC.
- A. Raina, Y. Singh, K. K. Yadav and T. Ghosh, J. Chem. Sci., 2020, 132, 128 CrossRef CAS.
- P. E. Hartnett, C. M. Mauck, M. A. Harris, R. M. Young, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 749–756 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Figures showing the changes in IR data, rheological study, emission and absorption spectra, binding curves, detection limit, and copies of 1H, 13C NMR and HRMS spectra. See DOI: https://doi.org/10.1039/d2su00135g |
| This journal is © The Royal Society of Chemistry 2023 |