
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioderived furanic compounds as replacements for BTX in chemical intermediate applications
Amir
Al Ghatta
 * and
Jason P.
Hallett
*
* and
Jason P.
Hallett
*
Department of Chemical Engineering, Imperial College, South Kensington Campus, London SW7 2AZ, Northern Ireland, UK. E-mail: a.al-ghatta16@imperial.ac.uk; j.hallett@imperial.ac.uk
First published on 13th April 2023
Abstract
The valorization of sugars and cellulose into high value-added compounds represents a promising alternative to petrochemical processes to produce biobased chemicals. This approach can increase sustainability and improve the environmental impact of the chemical industry. The petroleum fraction BTX, composed of benzene, toluene and the isomers of xylene, provides a large variety of bulk chemicals which are used for myriad applications, most notably for polymers and surfactants. The high demand for bioderived products which are carbon neutral has pushed research toward seeking alternative routes to replace BTX-derived compounds at large scale. Much research has been focused on lignin valorization due to its high aromatic content which can release BTX through hydrocracking, but the development of this process is limited by the low theoretical yield of useful aromatics. Cellulose and hemicellulose represent a valid alternative with the potential to synthesize a large variety of new furanic compounds which can replace aryl compounds derived from BTX. 5-HMF and furfural derived from the dehydration of sugars are the key platform chemicals for a large variety of reactions which lead to different intermediate molecules that can replace BTX with molecules that deliver the same performance or even the same molecule. 2,5-Furandicarboxylic acid (FDCA), caprolactam, phthalic anhydride, furan, maleic anhydride and alkylfurans can be derived from these molecules and can substitute petrochemical resins to obtain the resin polyethylene furoate (PEF), nylon 6,6, plasticizers and surfactants. Much research activities have been focused on the development of catalytic pathways for the efficient valorization of these molecules at high yield but few chemical routes are commercially viable. In this paper, the state of the art in obtaining valuable biobased chemical intermediates to replace BTX will be reviewed by analyzing the synthesis of 5-HMF and furfural and their further transformation. Emphasis is given to the process sustainability, chemical challenges, and prospects for future research.
Sustainability spotlightThe chemical industry has evolved in developing new chemical technologies to provide intermediate building blocks which can provide cost effective commodity products. These chemical intermediates can be classified into aromatic and linear building blocks. While the implementation of green processes has been partially successful in replacing linear building blocks with eco-friendly ones, the replacement of aromatic ones remains challenging and still need to face different scientific conceptual barriers to overcome the applicability on a large scale. Most of the research done so far has been focused in developing few aspects of the whole overall picture which could lead to the success of green technology in this field. New approaches that can unify multiple studies with critical considerations on process development have the potential to result in new efficient processes that can exploit waste and valorise it into new green products with low carbon impact, in line with the UN SDG 12. |
1 Introduction
The increasing policies on CO2 emissions and the depletion of petroleum reserves have pushed researchers into seeking alternative solutions to petroleum-derived products.1–3 In Europe specific targets and plans were established for biofuel incorporation,4 while directives for biobased chemicals are minor and legislation that regulates the entire region is not in place5 The reason can be seen in the small fraction covered by chemical products compared to total production from petroleum.6 Moreover replacement of common petroleum derivatives with renewable chemicals faces important challenges mainly due to the high cost and low state of the art of these technologies.7,8 Biofuel technologies such as lignocellulosic ethanol and biodiesel production are more established and different technologies are available and commercially viable with government subsidies.9,10 Regarding biobased chemicals, many routes are available to replace most of the existing chemicals but only a few of these routes are economically viable for process scale up and much research remains to be done to overcome theoretical and technical barriers.3,11,12 Currently only 5% of chemical production worldwide is biobased.13 However, interest in developing alternative pathways to biofuels is high and most biobased chemicals have higher added value compared with ethanol, leading to an increasing trend in the holistic valorization of lignocellulosic biomass.14–17 Moreover, transformations of biomass-derived chemicals are more favored thermodynamically compared with petroleum derivatives since these chemicals are already functionalized while petroleum based feedstocks consist of alkanes and aromatics that are unfunctionalized and require high energy and multiple steps to introduce chemical functional groups onto the hydrocarbon skeleton, which is associated with high energy inputs due to the complexities of C–H transformation.3,11 Energy consumption from the synthesis of petrochemicals represents about 20% of the total energy usage in this industry.18 Sugars are seen as one of the most suitable feedstocks to produce a wide range of green chemicals19,20 with the potential to reduce CO2 emissions since the feedstocks are renewable and the energy for the processing of these feedstocks is locked into their chemical structure.21 Moreover, the price of sugars is competitive with BTX, establishing the potential of a sustainable and economically competitive chemical process. For example, the price of p-xylene and benzene are between 1–1.1 $ per kg and 0.5–0.6 $ per kg, respectively, while sugar prices are between 0.4–0.6 $ per kg.22,23 However, the biobased chemicals sector experiences high investment in R&D but not high commercial activity. In the Final Report for the European Commission Directorate-General Energy made by a consortium led by E4tech, Record and Wageningen entitled “From sugar platform to biofuels and biochemicals”,24 94 green products were identified between chemicals, biofuels and polymers derived from sugars obtained from the pretreatment of sustainable biomass feedstock while the US department of energy had previously identified only 12 promising candidates to be converted into useful intermediates at large scale.25Sugars derived from cellulose are key feedstocks for many chemical reactions. The production of sugars from cellulose is established and many different chemicals beyond biofuels can potentially replace petrochemicals.26 In terms of commercial impact, lignocellulosic (LC) ethanol has seen major development in the last decade and has been considered the only sustainable feedstock for ethanol production (replacing starch).27 Large investment and subsidies have been used to increase the commercial market of this fuel because carbon neutrality and transportation costs can be minimized due to the ability of these crops to grow on marginal land.28 This has favored the development of different technologies for the pretreatment of biomass for the production of sugars at high yield that can be fermented into ethanol. So far, steam explosion has proven to be a viable technology for high yield sugar production from grassy biomass and a 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ton per year operating plant was built in 2008 in Crescentino (Italy).29 The development at large scale of LC ethanol has permitted the spread of green ethylene and ethylene glycol obtained from the dehydration of ethanol,4,24 which provides the basis for the production of a large variety of chemicals exploiting the well-known chemical processing of ethylene and the large scale utilization of ethylene glycol to form polyesters. Other biobased chemicals produced at large scale are 1,3 propanediol and succinic acid which, through the development of genetically modified microorganisms, can be produced from sugars, replacing the routes which use fossil derived chemicals.30,31
000 ton per year operating plant was built in 2008 in Crescentino (Italy).29 The development at large scale of LC ethanol has permitted the spread of green ethylene and ethylene glycol obtained from the dehydration of ethanol,4,24 which provides the basis for the production of a large variety of chemicals exploiting the well-known chemical processing of ethylene and the large scale utilization of ethylene glycol to form polyesters. Other biobased chemicals produced at large scale are 1,3 propanediol and succinic acid which, through the development of genetically modified microorganisms, can be produced from sugars, replacing the routes which use fossil derived chemicals.30,31
While ethylene and ethylene glycol can be used to produce green polymers and intermediate chemicals, the substitution of aromatic compounds is much more challenging. These are synthesized from the aromatized hydrocarbon mixture BTX (Fig. 1).
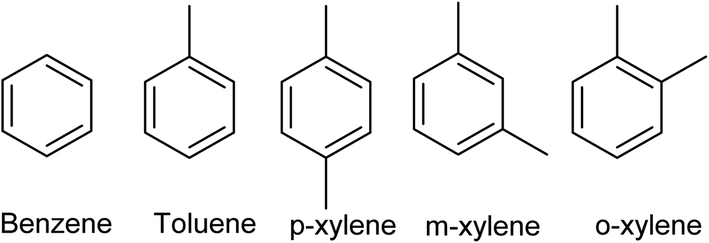 | ||
| Fig. 1 Main components in the BTX mixture. | ||
Coal was once the main source to produce this fraction through pyrolysis, but at present 96% of BTX is produced by catalytic reforming of the C5–C10 petroleum fraction since this approach is more selective and less energy intensive.32 Once produced, this mixture undergoes further separation to obtain the most valuable components, which are benzene, p-xylene and o-xylene while toluene and m-xylene are used as solvents or further converted to p-xylene or benzene.33,34 For many years, lignin has attracted interest as a potential source of BTX through hydrocracking. Despite intensive R&D efforts and many different approaches, scale-up of this concept has proven to be economically challenging and inefficient due mainly to the low theoretical yield of BTX that can be obtained from lignin.35 The highest yields reported to date are far too low (14%)36 and development has been slow over the last 30 years. For this reason, the combustion of lignin is preferred (at present) to supply energy to the biorefinery itself and sell the excess as electricity or use lignin directly as a feedstock in niche applications.37
Furan compounds derived from sugar dehydration are an intriguing option to replace many petrochemicals. More importantly they can replace the high energy process needed to produce and fractionate BTX, which involves a series of extractions, distillations and crystallizations.38,39 The large variety of sectors which BTX compounds supply includes fuel additives, paint, coatings, cleaning agents, foams and polymers and this have led to a constantly increasing demand for these bulk chemicals (Fig. 2). It is expected that by 2027 the market will grow by 6% reaching a total value of 275 billion $ per year.40 Due the high volumes and the high carbon footprint of these compounds an alternative green route would be highly beneficial for improving the sustainability of both the synthesis of the final products and its end-of-life usage.
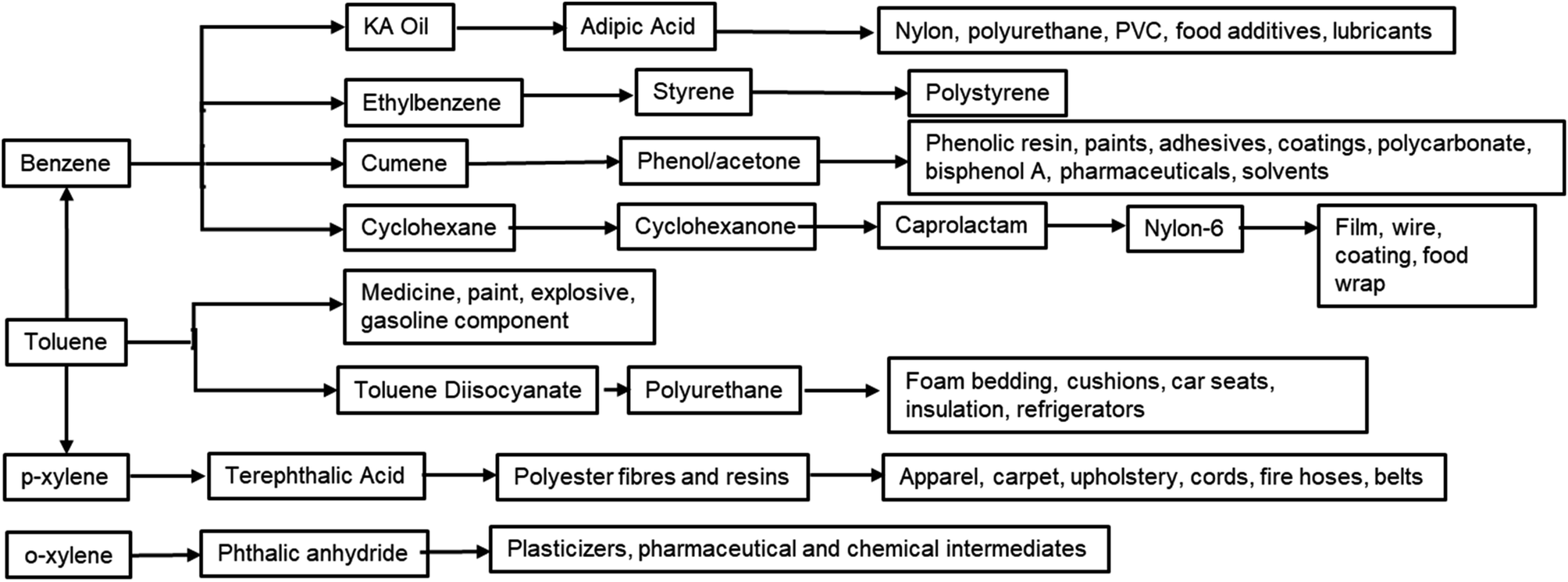 | ||
| Fig. 2 Chemicals that can be obtained from BTX. | ||
The substitution of the aromatic compounds with bioderived cellulose intermediates requires that the replacement of the aryl moiety with a furan leads to molecules with same properties and end function. It is widely recognized that the most common biobased furan sources are 5-hydroxymethylfurfural and furfural, obtained from the dehydration of C6 and C5 sugars from cellulose and hemicellulose. Their multifunctional chemical structures have attracted much attention since there is potential for these furanic compounds to undergo a wide variety of reactions that can produce both chemicals and fuels.41 A depiction of the large set of chemicals that can be obtained from 5-HMF and furfural is depicted in Fig. 3.42,43 Some of the compounds derived from furfural already find diverse applications and are produced in large scale because furfural can be derived directly from lignocellulosic biomass.44–46 Furfuryl alcohol is used as a monomer for the synthesis of polyfurfuryl alcohol, a thermoset resin used in many different applications.47 Furoic acid is used as a food preservative,48 while levulinic acid is used in personal care and cosmetic products.49 Furan, furfuryl amine and methylfuran are used as precursors to specialty chemicals and the solvent THF, used in different application at large scale (such as the polymerization of polyurethane).45 While the synthesis of biobased chemicals from furfural is established, multifunctional molecules derived from HMF are still undergoing extensive research but as yet no commercial activity has arisen due to challenges with HMF synthesis and stability.50 2,5-Diformylfuran (DFF) is widely present in the literature but the applications of this molecule are still unclear.51 2,5-Bishydroxymethylfuran (BHMF) can give rise to a different category of polymers but no commercial activity exists at present.52
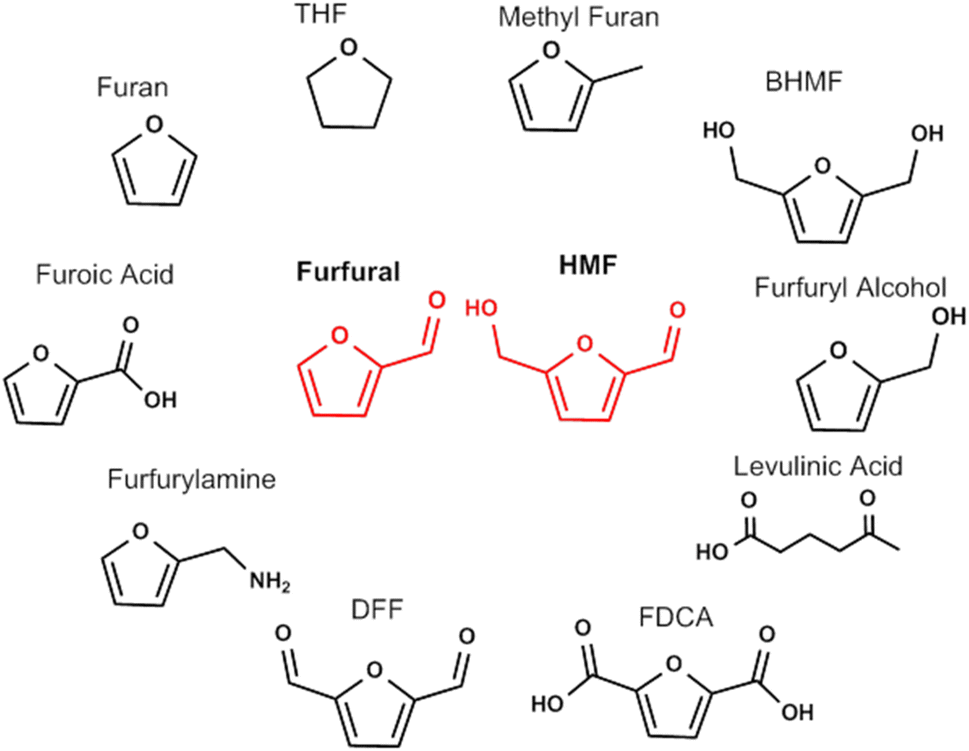 | ||
| Fig. 3 Chemicals that can be obtained from Furfural and HMF. | ||
Among these, the highest potential for direct application in the polymer sector belongs to 2,5-furandicarboxylic acid (FDCA).53
The diacid monomer 2,5-furandicarboxylic acid (FDCA) is considered one of the highest value molecules that can be derived from sugars and represents one of the premier candidates to substitute terephthalic acid (TA), derived from p-xylene, in the synthesis of polyesters. The market demand for FDCA is increasing due to the desire to obtain a completely green bioplastic that can replace polyethylene terephthalate (PET) within the food and packaging sector.54 However, an efficient techno-economical path from sugars to FDCA does not currently exist, mostly due difficulties in producing and isolating HMF, which makes FDCA cost prohibitive for applications in the polymer sector.
The substitution of FDCA for TA is a prime example where a furan ring can replace a benzene ring, producing a molecule with the same functionality. Other furan compounds that can potentially substitute BTX in different applications are reported in Fig. 4. Another important application in which furans can play a significant role is in the synthesis of surfactants. Linear alkyl benzene sulfonate (LAS) is used in many different products, especially for detergent and cleaning purposes, and is synthesized via alkylation of benzene. The high volumes in which these are commercialized renders the potential environmental impact very attractive in terms of reducing CO2 emissions. In addition, more favorable biodegradability is likely since furan rings are more biodegradable compared with aryl molecules.55,56 Different approaches have been developed for the alkylation of the furan ring to create a new category of biosurfactants derived from HMF and furfural and recently it has been demonstrated that these can actually exhibit higher performance compared with LAS, which currently needs to be heavily formulated to achieve optimum cleaning performance. Furan-derived surfactants can open new horizons in the detergent industry, where the need for and cost of the formulation is crucial.57,58 In other applications, furan can react by cycloaddition with maleic anhydride to form phthalic anhydride, a compound widely used as a plasticizer for PVC.59,60 Caprolactam is the main monomer for the synthesis of Nylon which is conventionally produced through multiple steps using benzene.61 Adipic acid has large utilization in polymers, food and medicine applications. It is produced from KA oil, a product derived from the oxidation of benzene.62
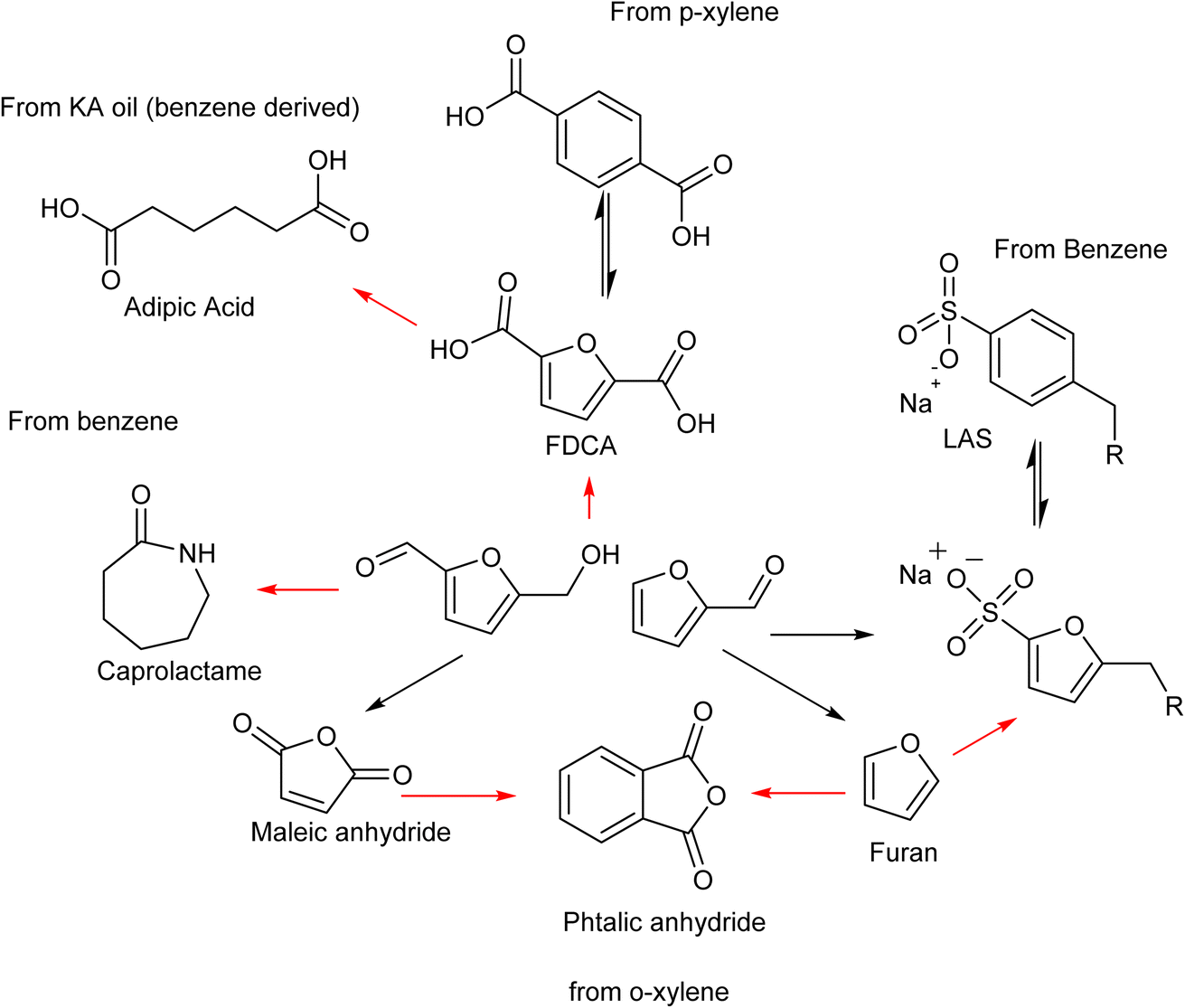 | ||
| Fig. 4 Chemical path to substitute some BTX derivatives with biomass derived 5-HMF and furfural. | ||
In this review, we will tour the state of the art of the production of most significant furanic molecules with potential for the substitution of BTX feedstocks and derivatives, their limitations, advantages and room for improvement. We will focus on the catalytic principles to obtain high selectivity and yield with further insights on the process separation and scalability of each product. We will critically assess the influence of performing the reaction in a defined environment which can favor separation and relate it to current techno-economic studies. We will not include caprolactam and adipic acid since the synthetic procedure from HMF and FDCA requires multiple steps for these we suggest other reviews published specifically for these compounds.63–65
2 5-Hydroxymethylfurfural
HMF is an intermediate chemical compound derived from C6 sugar dehydration.66 Its synthesis and application has been extensively studied for several years.67 HMF was first reported at the end of the 19th century where its presence was first observed in the heating of inulin and oxalic acid.68 Following the observation of this product, studies were done to demonstrate that the dehydration is catalyzed by acids and it was immediately demonstrated that solvent composition has a strong impact on the reaction behaviour.69,70The dehydration of sugars into HMF has been seen as the first fundamental step for the production of many biobased chemicals, but this platform faces multiple challenges due mainly to side products which can be categorized into self-condensation of HMF, condensation of HMF with the initial substrate and overhydration into levulinic and formic acid; any of these can lead to the formation of high molecular weight compounds classified as humins.71,72 A depictive scheme of the overall pathway is presented in Fig. 5.
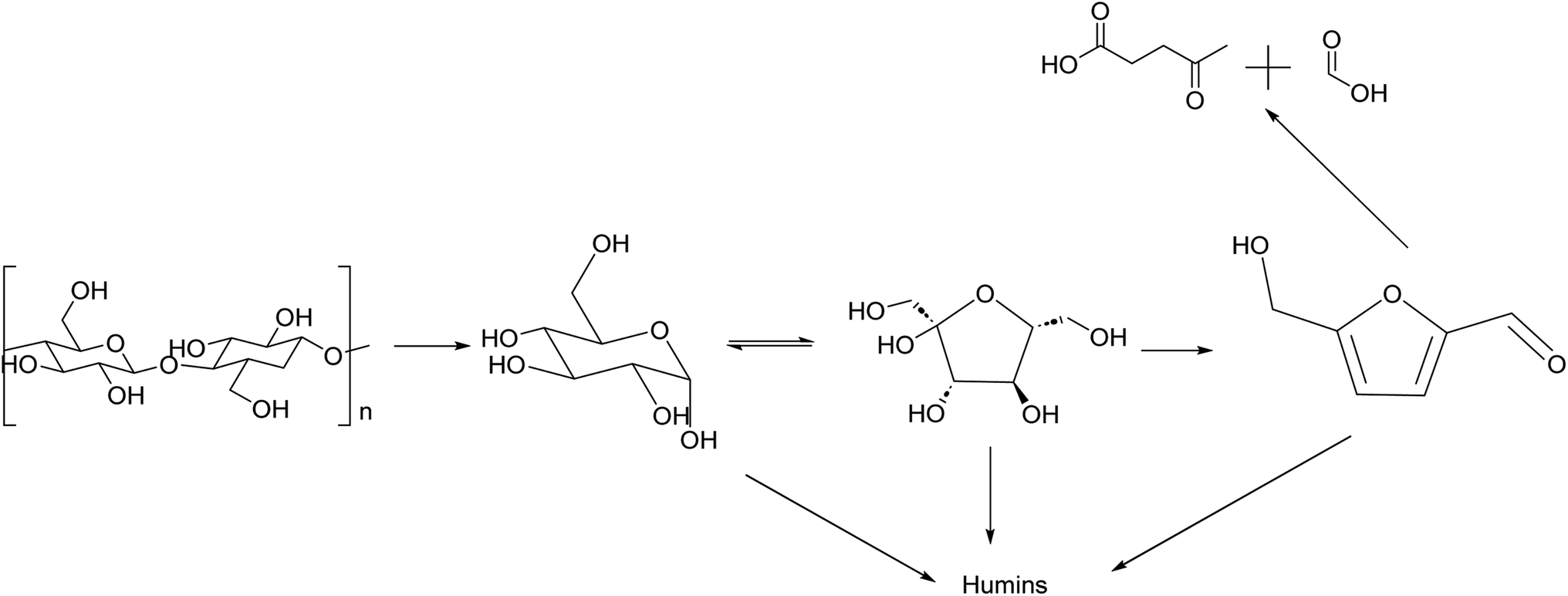 | ||
| Fig. 5 Reaction scheme of synthesis of HMF from cellulose with main side products produced along the reaction pathway. | ||
Many catalytic systems, both homogeneous and heterogeneous, have been developed for HMF production, resulting in more than 2000 publications in the last 20 years, including reviews covering different aspects of the synthesis.73–76 Most studies focus on developing catalytic systems for the selective conversion of different feedstocks into HMF by minimizing the side products. The main challenge lies in the fact that acid catalysts can promote both the formation of HMF and its degradation into humins and acids.77 Therefore a combination of catalyst design and process optimization needs to be carefully studied to minimize these side products. Solvents such as DMSO and ionic liquids can suppress the overdehydration into acids but the problem of humin formation still persists.77–79 Synthesis of HMF starting from cellulose or glucose is much more challenging compared with fructose; therefore to date a scalable system remains elusive.80 Fructose is a much more versatile feedstock for such transformations and many studies have reported high yields starting from this substrate.81 Although the cost of fructose is higher than glucose, its usage can still be competitive since today it can be produced efficiently through starch or cellulose hydrolysis followed by isomerization.82,83 Both of these steps are already applied in large scale to produce sweeteners chemoenzymatically through highly stable immobilized enzymes such as glucose isomerase.84,85 Multiple challenges exist for the development of an optimum catalytic system which can guarantee both high yield and good separation to produce HMF from fructose. Solvents have proven to have a strong effect in each of these aspects and different catalysts show different behavior in protic or aprotic environments.86 DMSO, ionic liquids and mixtures of water with organic solvents are examples of solvents which suppress some side reactions of 5-HMF.87 The application of each of these solvents has advantages and disadvantages concerning yield and separations. A perfect tradeoff which combines an optimum reaction-separation scheme still needs to be achieved due to problems related to the stability and physical properties of HMF, a key issue for large scale production.88,89
It is highly desirable that the solvent and catalyst are environmentally friendly and recyclable. In this section we review the synthesis of HMF focusing on the solvent effects, catalyst development and key issues which need to be faced to scale up this molecule.
2.1 Solvent effects
Solvents play a crucial role in determining the efficiency of the dehydration of sugars and cellulose into HMF as they strongly impact the choice of the catalyst, substrate versatility, reaction conditions and process sustainability. Water was the first investigated solvent for this purpose, however the acid dehydration of fructose into HMF proved to be inefficient in terms of both humins formation and overdehydration into formic and levulinic acids.90 Sulfuric acid, HCl and other organic acids have been tested and resulted generally in yields lower than 50%. Marginal improvements could be achieved under microwave radiation, reaching 53% yield with HCl and 64% using phosphate acids.91,92 The extensive research carried out so far on the development of an efficient catalytic system has demonstrated that a solvent which stabilizes HMF towards side reactions is needed to guarantee high selectivity towards this molecule.93 Different studies have focused on finding solutions which can remain environmentally sustainable and produce a satisfactory yield. DMSO, ionic liquids75,94 or water in combination with an organic solvent95 proved to hinder the formation of side products from HMF. More specifically, the stability of HMF needs to be ensured at the conditions under which the reaction is carried out, which is strongly influenced by the solvent used, since the activation energies of the main and side reactions are altered differently.96,97 Some studies relate this to the hydroxy group of HMF which leads to HMF degradation even at room temperature in 24 h. By modifying it with a protecting group, the stability is remarkably increased.89 In solution, the solvent can partially stabilize the molecule by interacting with the functional groups of HMF. A very useful understanding of the principles of the solvents can be done by observing the transformation in ionic liquids since the interactions can be easily triggered by changing the cation and anion. Ionic liquids can form hydrogen bonds with the OH group reducing the degradation at different temperatures79,98 however this stabilization is strongly dependent on the water content and the H-bonding strength of the anion. Bromide anions which have a mild H-bonding ability exhibit a higher degree of stabilization under a wide range of temperature compared with the two extremes Cl− and [OTf]−, suggesting that an optimum exists for maximum stability.79 In the same way, DMSO interacts with the hydroxy and carbonyl groups of HMF, protecting it from overdehydration under acidic conditions.99 DFT calculations have shown that these interactions increase the energy of the LUMO orbital, reducing its susceptibility to nucleophilic attack, which is an initial step in the formation of humins.78 Water content also plays an important role for both DMSO and ionic liquids. Indeed, in dry solvents HMF proved to be extremely unstable, forming consistently high amounts of humins under heating but this is suppressed through the addition of water or Lewis acids such as CrCl3, which in low amounts have a beneficial effect on the stability of the molecule.100–103 In [bmim][OTf], with only 3.5% of water content the degradation of HMF decrease from 40% conversion in dry solvents to 12% in wet ionic liquids at acidic conditions.104 However at high water content further side reactions are promoted since water can attack the furan ring, leading to ring opening followed by aldol condensation.105,106 Some degradation products derived from HMF degradation under acidic conditions are reported in Fig. 6 but it is outside the scope of this review to analyze in detail the mechanism of degradation since it has been estimated that over 100 active pathways exists for this reaction.107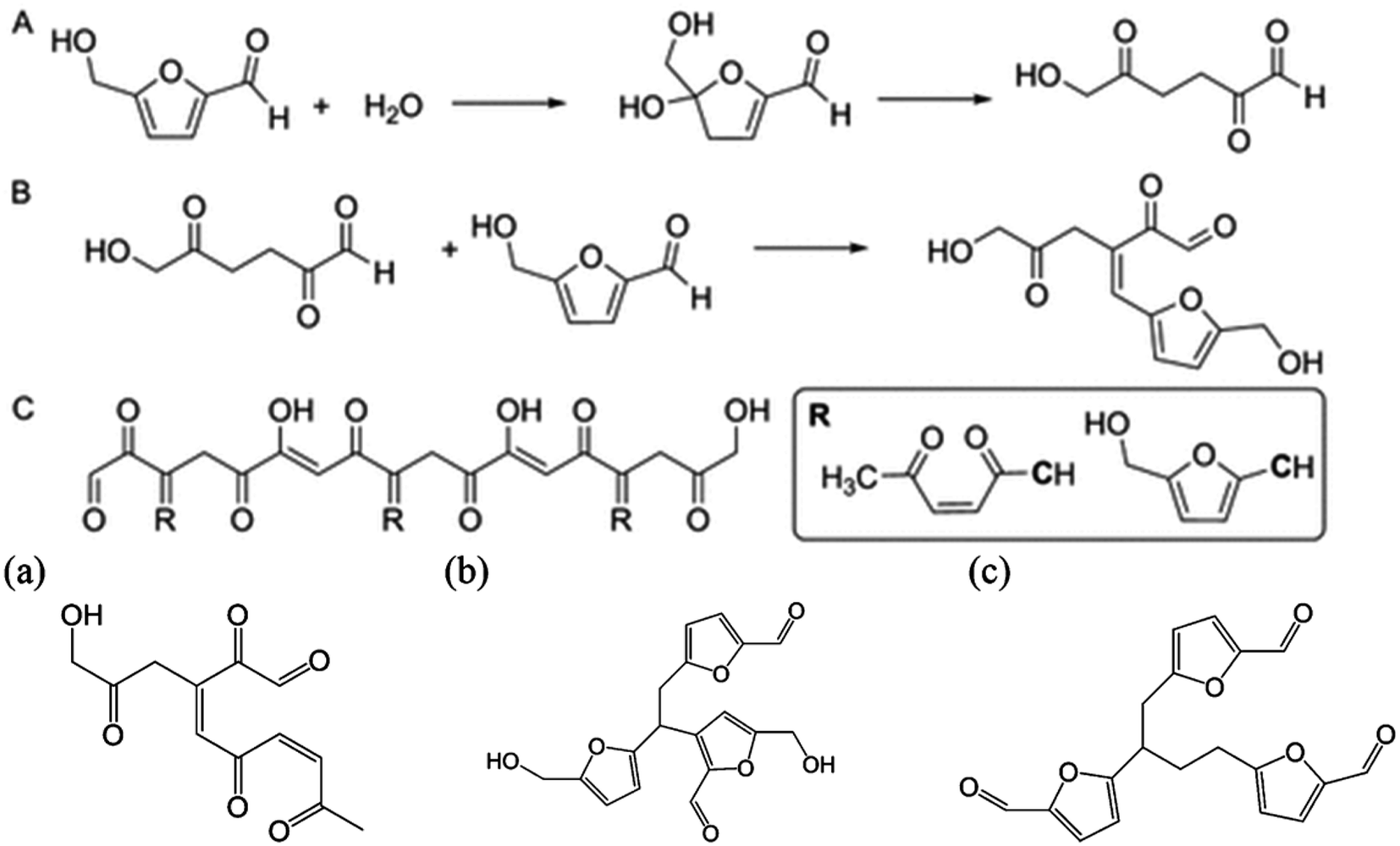 | ||
| Fig. 6 Proposed scheme and by-products of HMF degradation in water.78,105 | ||
Besides the stabilization effect, the solvent can behave as a catalytic promoter to favor sugar dehydration, by favoring the formation of certain fructose configurations (Fig. 7). Some studies have tried to relate the selectivity of the reaction towards the distribution of the fructose tautomers in solution. The high selectivity achieved in DMSO can be related to the β-fructofuranose which is recognized as the most favorable tautomer leading to HMF,108,109 while the fructopyranose, which is typically favored in water leads to oligomers.110 This is further supported by the fact that at higher temperature the β-fructofuranose configuration is favored, in line with the reaction in DMSO normally requiring temperatures higher than 100 °C.111 A detailed study performed recently by Fu and co-workers112 elucidated the conformation of fructose in different solvents and assigned the main product according the conformation. Different solvents favored different pathways of acid degradation leading to different types of oligomers, which was related to the fructose conformation in that environment. Specifically different low molecular weight compounds were observed for each conformation, α-fructofuranose and an open-chain lead mainly formic acid, α-fructopyranose leads to levulinic acid, β-fructopyranose to acetic acid and β-fructofuranose to HMF.112
 | ||
| Fig. 7 Fructose tautomers.112 | ||
The choice of the right solvent can allow more favorable reaction conditions and also operation with higher substrate loadings, improving process economics in terms of energy use and equipment costs, which ultimately will define the commercial success of the technology. Ionic liquids offer a remarkable combination of stabilization and catalyst enhancement. As an example, it has been reported that a Brønsted acid resin can achieve fructose dehydration in [bmim]Cl with a yield over 80% at fructose loadings of 40% at temperatures between 100–120 °C.113 The same results can be achieved in DMSO, but with much lower substrate loadings and higher temperatures.
Generally, for homogeneous catalysis, the interaction of the solvent with the catalyst is a key principle defining the efficiency of the reaction, especially when Lewis acids are used. For Brønsted acids the stabilization of the solvent towards HMF plays a more determinant role.99 The reason for this behavior is that Lewis acids undergo different complexation behavior with different solvents, which can reverse the selectivity of the catalyst.114–116 Meanwhile, Brønsted acidity can lead to HMF degradation and the solvent needs to stabilize HMF towards side reactions to prevent this.117,118 In the case of DMSO, different computational studies have revealed the role of DMSO in the dehydration mechanism. Ren and co-workers119 demonstrated that protons in DMSO tend to interact with the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the solvent rather than directly with the hydroxyl group of HMF. The complex [DMSOH]+ decreases the activation energy for the three dehydration steps compared with the acid behaving as a free proton (Fig. 8).119
O group of the solvent rather than directly with the hydroxyl group of HMF. The complex [DMSOH]+ decreases the activation energy for the three dehydration steps compared with the acid behaving as a free proton (Fig. 8).119
 | ||
| Fig. 8 Mechanism of fructose dehydration into HMF in DMSO.119 | ||
Lewis acids are the preferred catalysts when glucose is used as the feedstock for HMF production since they are more selective towards the glucose–fructose isomerization than Brønsted acids, but their efficiency is dictated by the solvent environment.114,120 CrCl3 has proven to be efficient for the conversion of glucose into HMF when the ionic liquid [bmim]Cl is used as solvent while longer reaction times and lower yields are achieved when the solvent is water or DMSO, due to different types of complexation which arise with chromium.121–123 When fructose is the substrate, both Brønsted and Lewis acids are suitable since no ring opening is involved in the mechanism but only dehydration of the hydroxyl groups.124
While DMSO and ionic liquids are able to stabilize HMF against side reactions, in water the main challenge is the overdehydration of HMF, which is promoted in a water rich environment since both reactions have similar activation energies.125 One solution is to remove the product from the acidic reaction mixture using a biphasic system through liquid–liquid extraction. Roman-Leshkov and co-workers reported that the combination of water with a hydrophobic organic solvent such as MIBK can reach selectivities of over 80% at high fructose loading using additives in both phases, such as dimethyl sulfoxide (DMSO), N-methylpyrrolidone (NMP) and polyvinylpyrrolidone (PVP). Specifically, the combination of PVP in the water phase and butanol in the organic phase led to the optimum yield for this system.126 The same authors reported that inorganic salts such as NaCl can have a beneficial effect by increasing the partition coefficient towards the organic phase due to the salting out effect, with the added advantage of using a cheap chemical compared with the more expensive DMSO, NMP and PVP, which also suffer from losses due to leaching into the organic phase.127,128 Moreover, the presence of the salts ensures immiscibility of the water and organic phase at high temperatures, which can eventually lead to a low partitioning due to the reduced amount of organic phase available for the extraction.129 The system has the main advantage that the separation of HMF can be easily achieved and the phases recovered and eventually recycled. This system has undergone different optimizations by changing the solvent and operating conditions. Improvements were achieved when the reaction was carried out in flow, reaching over 90% yield at short residence time (40 s).130 The system THF/water/NaCl proved to be one of the most versatile by many authors due to the low cost of THF compared with MIBK and the low boiling point which favors the separation and isolation of HMF.131 The role of sodium chloride in this case is twofold, creating an immiscibility region in the THF/water phase diagram at high temperature and improving the partition coefficient of HMF towards the organic solvent phase through the salting out effect. It was also observed that THF/water solvent system show better performances compared with ketones and alcohols when cellulose was used as substrate. This was explained by the formation of an emulsion (which improves the kinetics) and the formation of a thick water layer around the cellulose molecules which confers the hydrogen bonds needed in the hydrolysis.127,132 Other reports have shown that by modifying the THF phase with low amounts of butanol or performing the reaction under a CO2 environment one can further improve the extraction and the selectivity towards HMF.133–135
Depending on the solvent used, the water/organic solvent system may be highly tunable toward a high yield of HMF from fructose.136 Okano and co-workers reported a biphasic system of water/acetonitrile using the protic ionic liquid 1-methyl-3-(butyl-4-chlorosulfonyl) imidazolium chlorosulfate as catalyst and phase separator instead of inorganic salts. The authors reported yields over 80% with a fructose loading of 33% but the system suffers from catalyst leaching into the organic phase which compromises the recyclability.137 Another study showed that increasing the organic solvent phase using MIBK in a ratio of 4:1 to water increased the yield up to 70% at a fructose loading of 45%.138 A COSMO-RS simulation study has shown that o-propylphenol gave the highest partition coefficient amongst all organic solvents.139 In order to simplify the system, different studies focused on homogeneous mixtures such as water/acetone or water/GVL which also stabilize HMF.140,141
Ionic liquids have also demonstrated very interesting solvent effects on the reaction kinetics and selectivity toward HMF. Of particular note is their ability to dissolve cellulose at high loadings and mild conditions (10% in [bmim]Cl at 100 °C)142 compared to traditional solvent systems such as DMAC/LiCl, DMF/N2O2, DMSO/TBAF, NMMO, molten salt hydrates, aqueous solutions of alkali hydroxide and aqueous solutions of metal complexes. Ionic liquids represent a good tradeoff between toxicity, stability, flammability, dissolution capability and cost compared with other cellulose solvents.143 The use of aqueous biphasic systems or DMSO for cellulose processing has the main disadvantage of low cellulose solubility which makes the development of heterogeneous catalysts very tedious, as the systems require high temperatures and low substrate loadings. Beyond the solubility advantages, the ions in ionic liquids have shown favorable properties for HMF formation. Indeed, compared with reactions in DMSO and water, the use of ionic liquids can generally reduce the reaction time and temperature, even without the utilization of a catalyst. The solvent design strategy is normally dictated by the choice of the anion, which strongly impacts the selectivity and kinetics of the reaction. Under dry conditions, ionic liquids with bromide anions such as [bmim]Br can catalyze the dehydration of fructose at low temperatures (<100 °C) and high substrate loadings (30% by mass compared with the solvent) at near quantitative yield without the addition of any catalyst.144 The reaction can also be performed in the presence of low amounts of water, but by changing the reaction time and temperature. The presence of chloride anions has a beneficial effect but microwave heating or catalyst addition are needed. Mechanistic studies have demonstrated that high yields in fructose dehydration can be achieved in ionic liquids with an optimum combination of anion, cation and acid catalyst. Mass spectrometry, NMR and molecular dynamic studies have shown that the acid plays a key role in the beginning of the reaction mechanism while the anion and cation are involved in the second and third dehydration steps.145 A cation which is a H-bond donor has a beneficial effect on the reaction while the anion needs to be a good H-bond acceptor to coordinate with the hydrogen of the hydroxy groups on the substrate as depicted in Fig. 9.145,146
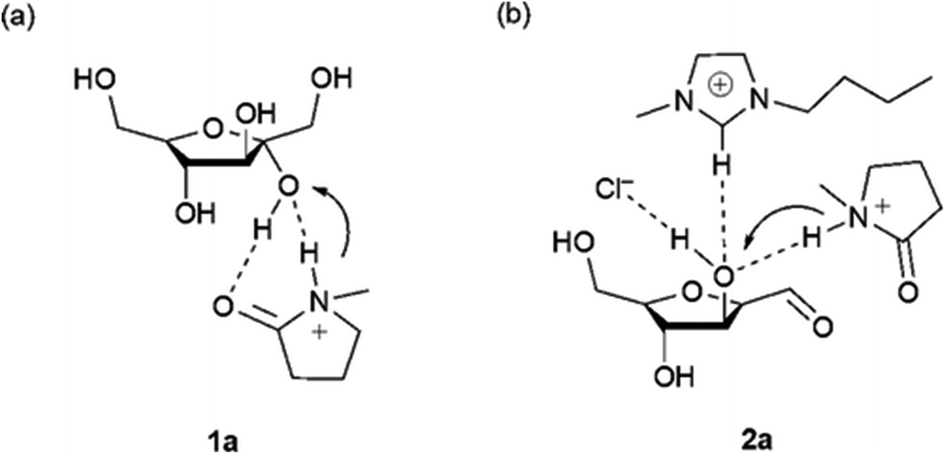 | ||
| Fig. 9 Coordination of the anion, cation and Brønsted acid catalyst with the hydroxyl group of fructose.145 | ||
The interaction energy between the ionic liquid and the hydroxyl group of fructose decreased by increasing the chain length of the imidazolium alkyl side chain, probably due increasing binary interactions of between anion and cation. As an example, the interaction energy increases by two-fold when moving from [bmim]Cl to [emim]Cl, which was accompanied by a drop in yield from over 90% to lower than 80%. This result can be related to the aggregation behavior of the ionic liquids with long alkyl chains, which can hinder proton transfer during catalysis by forming an anisotropic mixture.147 According to these findings, protic ionic liquids show outstanding behavior in carrying out this transformation since they can incorporate all required properties in one molecule and are also relatively cheap.148 Over 90% yield at high fructose loading can be achieved in [Hmim]Cl at short reaction time (30 min) and 90 °C.149 Another study used a room temperature ionic liquid by mixing [HNMP][CH3SO3] and [bmim]Cl, reaching over 90% yield when the solvent molar composition of [bmim]Cl is 86%.150,151 The dehydration proved to be less efficient when the proton source is the anion. The ionic liquid [bmim][HSO4] caused HMF degradation; however the yield could be improved by the addition of an organic solvent such as MIBK.152
The specific role of the cation was analyzed by Li and co-workers,144 who analyzed its influence in bromide based ionic liquids. Butylimidazolium chains demonstrated higher activity compared with [Hpy], [C12mim], [C8mim], [C6mim] and [Hmim].153 The authors observed through NMR and DFT calculations that the formation of hydrogen bonds between the ionic liquid and the fructose involves both anion and cation coordination with the proton and oxygen electron doublets, but reasons for the high performances of the bromide anion in carrying out a catalyst free dehydration is not fully understood. A study reported that the catalyst free dehydration of fructose using bromide ionic liquids can be boosted remarkably using the pyridinium ionic liquid [C10(Epy)2]2Br which can catalyze the conversion of fructose at very high loading (1:1 molar) with a yield of 89.7% at 100 °C.154
According these mechanistic studies of fructose dehydration into HMF, the H-bonding ability of the anion is a fundamental determinant of reaction efficiency. For this reason the transformation in ionic liquids which do not form strong H-bonds, such as the non-coordinating anions [PF6], [BF4], [OTf] and [NTf2], is slow and often leads to negligible yields even after long reaction times, high temperatures and at low substrate loadings.149,155 A triphasic system based on water and the hydrophobic ionic liquid [bmim][NTf2] led to 81% HMF yield using a vanadium phosphate catalyst.156 This represented a good compromise in separating HMF from the ionic liquid since the affinity of HMF for hydrophobic non-coordinating ionic liquids is low, therefore favoring the extraction even if [bmim][NTf2] suffers from leaching into water.157,158 The synthesis of HMF in these ionic liquids is highly desired since this can partially overcome the separation issues related to the Cl and Br or favor further HMF reaction to other valuable compounds as DFF.98,104,157,159 Good results in these type of systems were recently achieved in the ionic liquid [bmim][OTf].104 It was observed that low amounts of water could improve the stability of HMF at acidic conditions, which is low in environments where no H bonds are formed. By adjusting the water content to 3.5%, it was recently demonstrated that over 80% yield of HMF can be obtained in 10 min at 14% fructose loading at 80 °C. While in Cl− and Br− ionic liquids HCl and H2SO4 gave the same performance, with [bmim][OTf] only HCl was suitable, confirming that the Cl− anion is needed to coordinate with the hydroxyl group to catalyze the dehydration.145
Ions play an active role in catalyzing the transformation from glucose by coordinating metals to form active complexes. Zhao and co-workers100 established that high yields of HMF of over 70% can be obtained using glucose as a feedstock and CrCl2 as a catalyst at mild conditions in [bmim]Cl (100 °C, 10% loading). The reason for this high efficiency lies in the coordination of the Cl− anions with the Cr centers, forming [CrCl4]−, which is highly active towards the dehydration. In this case the interaction of the catalyst with the solvent is the key to an efficient system.
Other optimization studies were done by changing the cation and adding co-solvents. Switching the cation from imidazolium to ammonium salts has shown slightly improved results with CrCl3 catalysts for glucose dehydration, increasing the yield to 71.3% with tetrabutylammonium chloride (TBAC) at short reaction times (10 min) at 140 °C.160,161 Biphasic systems have been reported by Hu and co-workers which were able to increase the yield to 91% from 84% with a continuous extraction with ethyl acetate in a choline Cl/citric acid system.162 The advantage in using the ammonium ionic liquids lies in the low cost compared to the expensive imidazolium ones, but very high viscosities are known properties of these salts163 creating challenges for process operation.
Besides the anion, the cation of the ionic liquid has an impact on the reaction when starting from glucose. Wu and co-workers used a 1,8 diazabicyclo(5.4.0)undec-7-ene (DBU)-benzene sulfonate ionic liquid with CrCl3, obtaining yields of 85% from glucose under the same conditions as originally reported by Zhao and co-workers.100,164 The interesting behavior of this system is the high resilience of the yield towards an increase in glucose loading compared to other work reported for [bmim]Cl,103 with yields of 68.7% at 20% loading, further illustrating the capability of this ionic liquid to suppress side reactions.
While the impact of the anion on the catalysis is straightforward, the participation of the cation in glucose dehydration is not as clear since high activity has been often observed for both imidazolium and ammonium ionic liquids, suggesting no clear effect of hydrogen bonding or polarizability. DFT calculations analyzed the ion effects on the dehydration and found that higher acidity of the cation can help the activity of the dehydration reaction by functioning as a proton bridge.165 Another study unveiled a strong influence of the alkyl chain on the yield of the reaction, decreasing the yield by three fold when going from [bmim]Cl to [omim]Cl mirroring the decrease in acidity when increasing the alkyl chain length.166
Addition of co-solvents showed a positive influence on the reaction conditions as well. Some organic solvents and water proved to favor the dehydration of glucose to HMF probably due to partial stabilization of HMF.104 Bell and co-workers added acetonitrile up to a solvent composition of 33% with [bmim]Cl, achieving high yields of HMF from glucose using heteropolyacids based on molybdenum.167 Water proved to partially increase the yield of the [bmim]Cl/CrCl3 system up to a level of 15% solvent composition.103 However, the effect of the co-solvent is strictly related to the type of catalyst used and the ionic liquid chosen. Qi and co-workers needed to increase the water content to 50% to increase the yield of HMF from 7 to 53% when ZrO2 was used as catalyst in [C6mim]Cl; therefore a general conclusion on co-solvent effects in ionic liquids cannot be derived.168
Extensive studies with different substrates have also been performed using DMSO as solvent, which also proved to have favorable chemical properties for the reaction in addition to non-flammability and low viscosity. DMSO is one of the main compounds recognized as effective for high yields of HMF from sugars.67,169,170 Different theories have been elaborated on the mechanism of fructose dehydration in DMSO. Originally it was proposed that DMSO acts as both a base and Lewis acid which can favor the dehydration.171 But recently, more complex proposals have been elaborated due to experimental observations and DFT studies. However, concerns have been raised for the instability of DMSO under acidic and aerobic conditions, which leads to its decomposition into methane sulfonic acids, sulfuric acids and dimethyl sulfide. This causes difficulty in defining the efficiency of the reported catalysts since the acids that derive from the decompositions can catalyze the reaction.108 This topic has created much scientific debate in the literature. Recently, Svenningsen and co-workers have addressed this issue by showing that no activity towards the fructose dehydration was observed under acid free, anaerobic conditions, further supporting the theory of the key role of solvent decomposition.172 This would explain the high recyclability of Amberlyst-15 in this solvent compared to in DMF or ionic liquids, which are affected by leaching, since the acid formed can regenerate the catalyst.173,174 Another recent study contradicted these observations since no sulfuric acid was detected by BaCl2 testing and no residual acidity was observed through control experiments on DMSO stability.175 However, high selectivity can be achieved with the addition of a Brønsted acid even under anaerobic conditions through acid addition, highlighting the beneficial effect of DMSO for this reaction.
The dehydration of glucose also seems favored in DMSO to some extent but in this case water is needed to perform the reaction. A computational study has shown that water molecules act as bridges for hydrogen transfer during the isomerization of glucose to fructose which leads to lower transition state energies. This was experimentally confirmed by incorporation of deuterium in the C1 position where the ring opening arises.176 Due to the favorable properties of DMSO high yields have been reported using this solvent but generally the loading of fructose and glucose is low and harsher conditions are required compared with ionic liquids.
2.2 Catalyst development
The main challenge is to choose the right combination of catalyst and solvent which can guarantee stability of HMF while simultaneously performing the complex chemistry of glucose isomerization into fructose. For this reason, most reports have focused on heterogeneous catalysis since these are easier to functionalize. When DMSO and water are used as solvents the reaction of cellulose is limited by the solubility of the substrate; therefore higher temperatures are generally required.181,182
The solubility of the substrate is one of the key decisions between homogeneous or heterogeneous catalysts. Ionic liquids have exhibited the highest versatility in this choice since the ionic nature of these compounds guarantees high substrate solubilities and supports the activity of the Lewis acid salts in solution, while in water activity is limited due to solvation of the counterions of the metals.116 Yb and Nb have been reported as Lewis acid water compatible catalysts, but the yield achieved from glucose is less than 50%. For this reason, in water heterogenous catalysis is still preferred for cellulose and glucose processing.
On the other hand, the dehydration of fructose proved to be very efficient with Brønsted acids, and generally high yields can be achieved in short reaction times. Optimization of the catalyst in this case is limited since the major role in the dehydration is carried out by the proton and the solvent only needs to guarantee HMF stability.183 Other types of optimization were performed through studying Lewis acids. Zhao and co-workers reported that many different metal chloride salts based on Ru, Pt, Fe, Al and Cu can efficiently catalyze fructose dehydration in [emim]Cl at yields between 60 to 80% at low temperatures (80 °C) in 3 h.100 Other studies have focused on optimizing the reaction conditions for some of these catalysts. The addition of a ligand to CrCl2 proved to enhance the selectivity from fructose to HMF but at the cost of increased reaction times.184 By replacing the Cl− anion of the ionic liquid with [HSO4]−, CrCl3 proved to be very efficient in dehydrating fructose at high substrate loadings.151 Germanium(IV) chloride also showed good activity, reaching over 90% yield in [emim]Cl with 5% loading.185 Another study showed that with the same catalyst, the reaction can be performed at room temperature by DMSO addition.186 Many other metal catalysts have been studied, such as AlCl3 and LaCl3, which proved to give high yields in DMSO but at higher temperatures and lower fructose loadings.
Mittal and co-workers187 measured the Lewis acid strength of different metal chlorides using acetonitrile shifts with FT-IR in [bmim]Cl, showing that an optimum exists to give a high yield of HMF from fructose. NbCl5 proved to be more selective compared with stronger Lewis acids such as Ru, Cu and Fe chlorides and weaker ones such as SnCl2 and LiCl. However, high yields can be obtained by adjusting the reaction parameters or changing the ionic liquid anion as reported for FeCl3 or using a mixture of multiple catalysts such as Ir and Au.188,189
Generally, the use of homogeneous acid catalysts gives satisfactory results but it can compromise the work up of the system if further HMF conversions need to be carried out in the same solvent. In one of our reports, we demonstrated that the HCl used for the dehydration of fructose into HMF had an inhibitory effect on the subsequent oxidation of HMF into DFF.159 The development of a catalyst which can be separated from the reaction mixture is therefore highly desired. Liu and co-workers190 designed a system based on ChlCl with CO2 (40 bar) as a switchable solvent to generate carbonic acid in situ and decompose it by reducing the pressure, exploiting also the beneficial effect of CO2 in suppressing oligomer formation.134,135 The system proved to be very efficient, reaching 66% yield at a fructose loading of 1:1 in weight compared the solvent.190
Lewis acids exhibit different activity according to the substrate used. Cr and Sn salts proved to be the higher performing catalysts for glucose dehydration into HMF. Their activity changes according the solvent used and generally an inorganic salt can help boost their activity. The choice of the salt is also related to the solvent used. In aprotic solvents, CrCl3 showed that enhanced activity is reached when it is coordinated with a Cl− anion to form the complex [CrCl4]− while SnCl4 increases its activity when combined with bromide ions.122,191,192 In protic solvents such as water, this phenomenon is reversed as chromium has shown higher activity using KBr in combination with HCl and Sn with LiCl. However, here the complexation of the Lewis acids with the metals is unlikely due to water solvation of the anions, therefore the effect can be related to a combined synergy between the catalyst and the inorganic salt in HMF salting out, since these experiments were conducted in biphasic systems.192,193 However the difficulties in handling Sn salts due to their instability in air and high toxicity have make Cr a more ideal homogeneous catalyst for this reaction due to its low toxicity while in the +3 form.194 Therefore most of studies were reported to optimize Cr rather than Sn.
Further studies were done to increase the selectivity of the catalyst. The combination of chromium chloride with [bmim]Cl gave the highest performances in terms of reaction conditions, substrate loading and reaction time, though further work in improving the selectivity is highly desired. Despite the suppression of the overdehydration of HMF in ionic liquids, quantitative yields are not reached, indicating that other side reactions arise, mostly due to the reaction of HMF to form humins.100 The yield can be significantly improved by decreasing the concentration below 2 mg g−1, indicating that the origin of this side reaction is cross condensation of HMF with sugars. Further optimization studies showed that these can be partially suppressed by addition of water or Lewis acids.103,104 Studies also tried to improve the yield of HMF from glucose by using different ligands. These proved to have a strong effect on the reaction. Different studies have observed that carbenes can regulate the selectivity of the catalyst but strongly penalize the activity. Yong and co-workers184 found that NHC-ligand systems have a strong effect on the reaction when CrCl2 is used as catalyst and the size of the ligand regulates both the selectivity and conversion of the reaction in [bmim]Cl. Another study reported that the carbene behaves as a poison for the catalyst by showing that small excesses of ligand shut down the reaction.195 Indeed, the reaction proved to be much slower and harsher conditions were needed (120 °C, 6 h) compared with the [emim]Cl/CrCl3 system reported by Zhao and co-workers where full conversion was achieved in 3 h and 100 °C.196 The species involved in the catalysis have been discussed in several studies. It is generally agreed that the anion [CrCl4]− or some of its complexes are the active species which coordinates and stabilizes the open chain of glucose to HMF and that fructose is formed during the reaction.185 EXFAS and DFT studies have shown that the reason lies in the coordination of the catalyst with the substrate which is thermodynamically more favorable due to the planar configuration of CrCl3 and [CrCl4]−. Moreover, a kinetic study has shown that the reaction in [bmim]Cl chloride is first order with respect to CrCl3 and second order with respect to glucose, supporting the hypothesis that [CrCl4]− is actually the active form of the catalyst, coordinating two molecules of glucose with an activation energy of 134.9 kJ mol−1.197–199 Another study reported evidence that dimers such as [Cr3Cl4]− and [Cr2Cl4]2− can be formed and are capable of accepting hydrogen bonds from the hydroxyl groups of glucose, catalyzing the isomerization into the β-glucopyranose form that is considered the active species for further reaction. Such interactions are weak or not present for other metal salts based on Cu and Fe since they tend to assume a tetragonal conformation rather than planar.147,189,197,200 The low yield of HMF from glucose (<50%) in DMSO using the same catalyst is a further demonstration of the synergetic potential of the chromium salts with the chloride ions in the ionic liquid to perform the dehydration.201 This low efficiency is also explained when a large amount of water is present in the solvent since Cr undergoes complexation to an ‘aqua’ species which consists of free metal ions or mononuclear or multinuclear hydroxy complexes which are less effective in coordinating the open chain of fructose.202 Other Lewis acid-based catalysts proved less efficient than CrCl3 for this type of reaction. While the coordination of Cl− with the catalyst is valid for Cr, this was not the case for aluminum salts. Liu and co-workers, showed that while the metal salt AlCl3 was not effective in [emim]Cl, the trialkyl metal Al(Et)3 performed better even if still much less effective than the Cr salts (50% yield).203
Other types of Lewis acids proved to be less effective. Ge(IV)Cl4 and lanthanide chlorides have been tested by different researchers but the yield from glucose struggled to reach 50% and the substrate loading was generally lower compared to chromium salts.185,204 Another report showed that a combination of two Brønsted acids, 12-tungstophosphoric acid and boric acid, have a synergetic effect in increasing the yield of HMF.205,206
Heating methodology also strongly influences this reaction, with marked improvements observed when microwave heating is used, reaching over 90% isolated yield of HMF at 400 MW in 2 min in [bmim]Cl/CrCl3 system from glucose196 while under catalyst free conditions quantitative yields can be achieved in [bmim]Cl starting from fructose.207
For fructose dehydration, homogeneous Brønsted acids are a simpler system for this transformation and have proven very efficient in different solvents; however, the performance and optimum process conditions change according to the solvent used. For example, HCl in [bmim]Cl and [bmim]Br requires very short reaction times (<10 min) and quantitative yields have been reported at 10% fructose loading at 80 °C.155 The selectivity and yield remained high while increasing the loading to 30% but longer reaction times were needed (>30 min) due to the water generated in the system, which slowed the reaction. The reason for the higher performance of [bmim]Cl and [bmim]Br can be attributed in part to the favorable catalytic effect which the anion and cation impart to the reaction since (as mentioned in the previous section) these behave as solvents and catalysts at the same time. Roman-Leshkov126 and co-workers employed the system water/MIBK and reported a yield of 64% with a fructose loading of 30% (compared to water) in 4 h and 150 °C. Major improvements in terms of process conditions can be achieved using triazaheterocyclic compounds which achieved 70% yield at 90 °C in 2 h.208
Unsurprisingly, HMF production starting from cellulose has proven to be much more challenging. Cellulose requires hydrolysis, isomerization and dehydration which the current state of the art achieves in separate steps. The one pot transformation of this substrate represents a potential step-change improvement in operating and capital costs for a hypothetical plant.209 Besides the physical behavior, the dehydration of cellulose in ionic liquids is more thermodynamically favored compared with processes which use water or DMSO as solvent since the β-1,4-glycosidic linkages are easier to access in [emim]Cl due to the dissolution of cellulose breaking the fibril aggregates, making it easier to hydrolyze.210–212 Jian and co-workers213 performed an in situ13C-NMR study of the dehydration of cellulose with a combination of [bmim]Cl and the sulfonated ionic liquid [C4SO3Hmim]Cl. They observed that cellulose is converted into glucose at short reaction times while at longer reaction times HMF is observed even if at low yield (10%). The low yield is mainly due to the Brønsted acidity of the catalyst, which is not suitable for the dehydration of glucose into HMF, rather leading to side reactions of HMF with glucose or cellulose intermediates, leading to humins.211 The transformation into HMF is much more complex in this case since the breakdown of the ether bonds is required to convert cellulose into glucose, moreover the reaction is strongly influenced by moisture since stoichiometric amounts of water are needed for the reaction.214 Water content influences both the selectivity towards sugars and toward HMF with the highest selectivity observed at a molar ratio of water:cellulose of 10:1. Higher water content would increase the selectivity towards sugars. The study also analyzed the effect of the cation, showing that pyrrolidinium ionic liquids suppress the formation of HMF, favoring sugar formation.215 Further optimization of the water content was conducted by Qi and co-workers, who performed the dehydration using Amberlyst 15 as an acid catalyst with gradual water addition until reaching a water composition of 35%. This led to a glucose yield of 83%. After removing the catalyst by filtration, CrCl3 was added, obtaining a yield of 83% in 4 h.216 A report by Zhang and co-workers adopted a two-step approach to obtain high yields of HMF. By optimizing the water content (1 eq. compared with the theoretical glucose content) the authors achieved a high yield of sugars using HCl as catalyst and then CrCl2, obtaining 89% yield of HMF. The main disadvantage of these systems lies in the incompatibility of the two catalysts, which requires their subsequent separation and recycling.217
Other studies revealed that the addition of an organic solvent does not significantly improve the reaction yield218,219 because catalysts need further functionality to break down cellulose. Noble metals such as Ru and Pd salts did enhance the catalytic activity when used as co-catalysts for CrCl3.220,221 However, poorer results were generally obtained for high molecular weight cellulose such as cotton or filter paper, which have a degree of polymerization higher than 3000. For these, CrCl3 and AlCl3 proved to be more efficient.222 Microwave irradiation leads to the same yield using solely CrCl3 as catalyst, showing that the type of initial cellulose used has only a minor influence on the yield.196,223 Another study showed that slight improvements in the yield can be achieved by decreasing the cellulose loading to 5%.224
Other combinations of metal catalysts have been reported. Cr with Cu chloride salts are effective at 120 °C (55% yield).225 The formation of different furanic products during cellulose dehydration has been highlighted, including furylhydroxymethyl ketone (FHMK) and furfural formed as products alongside HMF. The authors reported that a combination of FeCl3 with CuCl2 can maximize the production of these three compounds (75.6% yield) but with low selectivity towards HMF.226
Other reports have studied combinations of Brønsted and Lewis acids.227–230 Acid functionalized ionic liquid cations including 1-(4-sulfonic acid) butyl-3-methylimidazolium perform better compared with inorganic acids or [HSO4]− ionic liquids when combined with a Lewis acid, increasing the yield by 20% with MnCl2.231 Based on this observation, a metallic bifunctional ionic liquid by substituting the hydrogen in the 1-(3-sulfonic acid) propane-3-methylimidazole hydrosulfate with Cr was reported.232,233 This catalyst in [bmim]Cl was able to achieve 53% yield at a relatively low temperature (120 °C) with a cellulose loading of 5%.233 The same catalytic cation proved to be efficient in [emim][OAc] in combination with CuCl2, achieving 70% yield of HMF but at 3.5% loading.234 A DFT computational study was done by Li and co-workers showed different advantages of this system, particularly the sulfonated group favors the formation of an 8 member ring which can decrease the transition state energy favoring both the isomerization of glucose to fructose and dehydration to HMF.165 Further functionalization of this cation by introducing aryl moieties at the C2 position of the imidazolium ring also enhanced the selectivity of the hydrolysis towards glucose.235
In Table 1 we reported a comparison of the main catalytic systems to perform this transformation.
| Solvent system | Type of catalyst | Substrate | Yields | Reference |
|---|---|---|---|---|
| Water/organic solvents | Mineral acids | Fructose (30%) | >70% | 126, 130, 131, 137 and 138 |
| [bmim] + Br or Cl | Mineral acids | Fructose (<40%) | >80% | 113, 145, 149–151, 160–162 |
| [bmim]HSO4 | CrCl3 | Fructose (10%) | >80% | 151 |
| [C10(Epy)2]2Br | — | Fructose (50%) | >80% | 154 |
| Chloride based ionic liquids + CO2 | — | Fructose (50%) | 66% | 190 |
| [bmim]Cl | Lewis acids | Fructose (<10%) | 80–90% | 100, 185 and 186 |
| [bmim] or DBU benzene sulfonate + Cl or Br | Chromium chlorides(II or III) or tin chlorides(II) | Glucose (<20%) | 70–85% | 100 and 164 |
| [bmim] Cl | Combination of metal chlorides | Cellulose (<5%) | 50–70% | 220–222, 225 and 226 |
| [Bmim]Cl/[OAc] | Combination of bronsted and lewis acid | Cellulose (<5%) | 50–70% | 231, 233 and 234 |
The solubility of the substrate and by-products has a strong influence on the catalytic activity, especially for heterogeneous catalysis since it can impact mass transfer and catalyst deactivation (Table 2). Ionic liquids are more suitable for this due to the high solubility of humins and all biomass derived compounds, while reactions performed in water have the drawback of insolubility of cellulose and humins which can lead to catalyst deactivation.240,241 Cellulose insolubility in water further reduces the rate of reaction, requiring high temperatures to compensate for the mass transfer limitations arising from solid–solid interactions, increasing the probability of forming unwanted humin side products.78,242,243 When water is used as the solvent for cellulose or sugar processing the catalyst needs to be regenerated at high temperatures to burn off all of the organic compounds on the catalyst surface, requiring a solid which doesn't undergo to decomposition at high temperature.243,244 It has been reported that to regenerate ZrP for further catalytic cycles, 600 °C is needed to remove all the humins from the catalyst surface.245 This make the use of carbon infeasible as a catalyst support in water since it cannot be regenerated at high temperature under oxidative conditions. Many catalysts which were designed to be active in water have been reported to suffer deactivation at the required temperatures of more than 150 °C, which leads to the deposition of side products onto the catalyst surface.140,246–249
| Solvent | Cellulose solubility | By-product solubility |
|---|---|---|
| [bmim]Cl | + | + |
| DMSO | − | + |
| Water | − | − |
In designing an optimum heterogeneous catalyst, different factors need to be considered. The catalyst preparation method strongly influences the final results since it can modify both the amount of Lewis/Brønsted acid sites and the morphology. Silicoaluminate zeolites are highly tunable, permitting regulation of the ratio of the acid sites by changing the Si/Al ratio.250 The commercial HB-zeolite with Si:Al ratios between 5 and 50 performs poorly for glucose dehydration, achieving low yield even with a long reaction time and high temperature, mostly due to the Brønsted acid characteristics which characterizes this catalyst.161,251 Calcination of the catalyst at 550 °C can improve the catalytic performance by increasing the ratio of Lewis over Brønsted acid sites from 0.32 to 1.72.252 It has been reported that silicoaluminates prepared by the sol–gel method with Si:Al equal to 5 can improve these results in the biphasic system water/MIBK at 10% loading giving a good yield of 63%.253 Another report showed that silicoaluminate prepared with phosphoric acid can increase the yield of HMF from glucose to over 70% in GVL/water or MIBK/water mixtures.141,253 We summarized the effect of the zeolites preparation methodology on the glucose dehydration in Table 3 (Fig. 10).
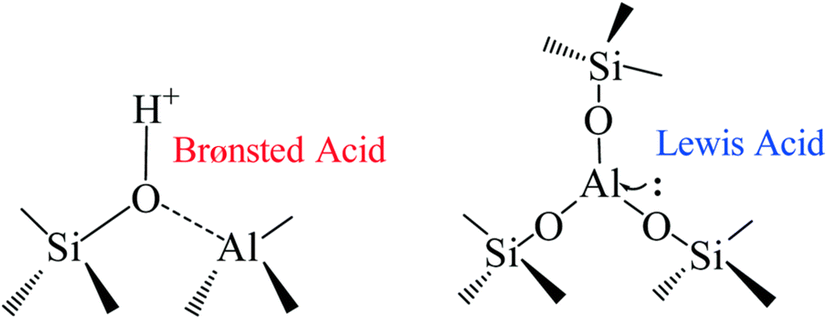 | ||
| Fig. 10 Brønsted and Lewis acid sites in silicoaluminate catalysts.250 | ||
The main advantage of heterogeneous catalysis lies in the possibility of conferring multifunctional character to the reaction, which brings a potential solution for the conversion of cellulose to HMF, since different functionalities are required for the three steps involved. Moreover, the advantages in separating heterogeneous catalysts from the reaction mixture simplifies the downstream process for further valorization of HMF. Most studies focus on tuning the chemical proprieties of the solid catalyst by finding the optimum ratio of Lewis and Brønsted acid sties which most suits the substrate used. The characterisation of these sites is often conducted by pyridine or ammonia adsorption and measurement through FT-IR or thermogravimetric analysis. However, it is difficult to universalize the strength of these sites since the measurements are strongly affected by the method used to evaluate the strength and the quantities, therefore any comparison is only valid within a single study. For cellulose processing, an equal distribution of Lewis and Brønsted acid sites has been reported as most efficient for this purpose, while for glucose the optimum ratio has been reported to be around 1.2, but these values may differ since the strength of these sites needs to be considered as well.243,248,254–256 The functionality of neutral Al2O3 can be triggered by treating the solid in an aqueous solution at different values of pH. Alumina treated with 0.05 M NaOH exhibited the highest selectivity (50%) which is attributed to the highest distribution of Lewis acid sites with low Brønsted acidity compared with alumina treated under acidic conditions, which is composed primarily of Brønsted acidic sites.257 Further improvements with alumina could be achieved in the metalloporphyrin form on Fe2O3 nanoparticles stabilized with chitosan.247
Zhang and co-workers studied different Cr, Sn and Sr oxides and the effect of using graphene oxide as a support. The authors found that while Sn and Cr oxides were ineffective in either oxide or supported form, amorphous SrO exhibits high activity, while calcination or deposition on graphene oxide has a negative effect on the yield. However heavy leaching was observed for Sr.258 Metal phosphates of Fe, Zr, Hf, Ta and Sn proved to be better performing. These are synthesized by treatment of the oxides with phosphoric acid. By regulating the amount of acid, different morphologies and functionalities can be obtained. Contrary to the oxide counterpart, the phosphates exhibit an amorphous morphology with small particle and pore sizes. The role of the phosphate counterion is to reduce the strength of the Lewis acidic sites which would lead to deactivation through strong adsorption of HMF onto the catalyst surface.259 FePO4 proved to be active towards cellulose dehydration in a water/NaCl/THF system. However the catalytic activity decreased over multiple runs due to partial solubility of the catalyst at high temperature, which during the cooldown phase of reaction undergoes a morphology change.260 High yields have been reported using these phosphate catalysts. Specifically, Zr proved to be more suitable for fructose dehydration while hafnium was superior for glucose and cellulose dehydration.248,261
Li and co-workers recently reported a recyclable sulfonated polyphenylene sulfide capable of 88% yield at 18% glucose loading. The authors showed that the degree of sulfonation has a high impact on the selectivity and yield of reaction at 26% mol sulfonic acid composition.262 Another study reports a Sn supported catalyst with a polymer formed by polymerization of p-toluene sulfonic acid with paraformaldehyde, which confers a Brønsted acid character to the catalyst, obtaining 60% yield from glucose.263 Zhang and co-workers designed a highly functionalized hydrophobic hybrid macroporous organic–inorganic catalyst polymer derived from divinylbenzene, oleic acid and ZrO2 which gave over 80% yield of HMF for the dehydration of fructose at dilute conditions with high recyclability.264
Many studies have adopted the approach of heterogenizing an established homogeneous catalyst analogue. To avoid the degradation of HMF in solution it is desirable that the reaction proceeds quickly, therefore high surface area and low particle size are needed and also a high pore accessibility for the substrate. A large variety of acidic resins can be used for fructose dehydration since macroporous ion exchange resins proved to have superior properties compared with gel resins for mass transfer.265 Specifically, the pore sizes need to be larger than the molecule involved, which in case of glucose is 0.9 nm while for cellulose larger than 30 nm.251,266 Small pore diameters will compromise the diffusion of HMF from the pore to the bulk, leading to side reactions.267 A study done by Abou-Yousef and co-workers268 showed that HMF can become trapped in the catalyst pores when zeolites are used, therefore the effective yield calculated by other authors may be biased by the different workup of the reaction mixture. In order to more accurately calculate the yield, the authors diluted the reaction mixture with water, filtered the zeolite catalyst, washed the catalyst with methanol and analyzed both the aqueous and methanol phases. The authors observed a consistent partitioning of HMF into both the aqueous and organic phases. In recent work, different vinyl polymers have been tested in a water/dioxane mixture, accentuating the importance of the pore size of the solid on the selectivity of the reaction. Specifically, the authors measured the accessibility of the Lewis acidic sites in the pore by titration of different Lewis bases with different sizes, showing that the solids with higher accessibility proved to be more effective for this reaction.267,269 Zhang and co-workers270 circumnavigated the problem by designing a heteropolyacid from choline chloride and H3PWO4 which at high temperature forms micelles of 10 nm, encapsulating the cellulose molecules and enhancing the catalytic activity (yield of 75%).
Other authors exploited different techniques to optimize the morphology of the catalyst. Ma and co-workers reported that by thermally treating zeolite L with ammonium nitrate, remarkable improvements of the catalyst morphology and chemical properties can be achieved through increases in the volume of micropores and the density of Brønsted and Lewis acid sites, obtaining near quantitative yields of HMF in [bmim]Br at 10% loading with recyclability up to 5 times.271 Other studies have focused on using carbon-based materials which are well known to have high surface areas and wide distributions of pores. The source of carbon and its preparation strongly influences its performance. Graphene synthetized by Tour's method proved to be the most effective in the dehydration compared with other type of graphene.272 While this approach was effective for the functionalization with Brønsted acidity, it has a negative effect when used as a support for Cr and Sn, while it enhanced the activity of Fe2O3 when [emim]Br is used as a solvent, reaching a yield of 83% in 4 h at 18% glucose loading.258 Amorphous carbon materials have also been widely studied for use in ionic liquids or DMSO.272 These are synthesized by thermal treatment of the feedstock followed by sulfonation to confer Brønsted acid character to the solid or impregnation of metal salts if Lewis acidity is needed.273,274 In order to facilitate the separation from the solution, treatment with iron salts has been reported, followed by calcination, forming a magnetic carbon material with incapsulated Fe2O3 in the carbon framework without altering its activity.275 The carbon obtained from biomass showed morphology formed by flakes or sphere-like particles when derived from sugars and cellulose, which grows by increasing the thermal treatment, reaching sizes larger than 1 μm, while lignin morphology is irregular.276–278 Chemical treatment with KOH increased the surface area, which is accompanied by an increase in selectivity and rate for the dehydration of fructose.279
Brønsted acidic resins have been extensively tested for the dehydration of fructose and the hydrolysis of cellulose into sugars with satisfactory results. Commercial ion exchange resins such as Amberlyst 15 and 70 have proven efficient in different solvents with yields over 90% in short reaction times (<30 min) at moderate temperatures (<120 °C).140,262,280 However, the catalyst leaches when ionic liquids are used by partially exchanging the cation with the acidic proton of the resin while in DMSO the catalyst proved to be highly recyclable but with lower substrate loadings (<1%).149 The reason for such high recyclability may lie in the acid decomposition of DMSO, which can regenerate the lost proton from the surface of the catalyst. If the ionic liquid is recycled this should not represent a problem since an equilibrium will be reached between the protons in solution and the cations on the catalyst surface. The high efficiency of the system using ionic liquids was confirmed in a continuous reactor which proved to be highly efficient.281,282 The challenge in developing such systems lies in the viscosity of ionic liquids which complicates the workup of the reaction during catalyst recovery and recycling. Some studies overcome this problem through the addition of a co-solvent in small amounts. Different studies have focused on the addition of an organic solvent or water to reduce the viscosity without penalizing the yield. Small amounts of acetone or glycerol carbonate can be used without excessive loss of selectivity.113,283 Other studies show that the wettability of the catalyst must be considered to enhance the yield. Different wettabilities can be achieved by regulating the amount of sulfonic acid groups on the surface of the resin and a minimum wettability of 110° is needed to activate the reaction.284,285 Another study showed that the addition of an alkali metals in ionic liquids can have huge influence on the selectivity of Brønsted acid resins for cellulose conversion, when LiCl was used a total furan yield of 82% was achieved while higher selectivity specifically towards HMF was achieved (70%) when KCl was used.286 A remarkable improvement was reported by Xiao and co-workers287 who studied the two heteropoly acid catalysts H3PW12O40 and H4SiW12O40, showing outstanding activity and selectivity from fructose at short reaction time (5 min) and low temperature (80 °C). By increasing the reaction time to 40 min the system could be charged with fructose up to a 170% loading, keeping the yield over 98%. Moreover, the catalyst could be recycled more than 10 times at the maximum substrate loading making this system very promising for scale up.245 These heteropolyacids proved to be efficient also in water. Lv and co-workers used a heteropolyacids (HSiW) supported by silica, obtaining 70% yield at 14% fructose loading and 160 °C.288 A silver based heteropolyacid was also developed which at 30% fructose loading gave 78% yield with the further advantage of reducing the temperature to 120 °C.246
Attempts to immobilize Lewis acids for the conversion of glucose and cellulose proved to be more challenging due to the leaching of the metals into solution. Different trials were done to exploit the high efficiency of chromium. Chromium nanoparticles stabilized with CO ligands can catalyze the dehydration of glucose at 120 °C, but at long reaction times (6 h) and low yields were obtained (<50%).289 Different catalysts were prepared by ion exchange and even if the catalysts performed well in the first cycle, leaching was observed when divinylbenzene resins, zeolites and silica were used as supports.290–294 The leaching from these supports can be partially improved by the further immobilization of ligands as a Schiff base or isophthalic anhydride, but the problem still persists.294,295
Hydroxyapatite prevented chromium leaching providing high recyclability and a potential solution for the design of an efficient, scalable catalyst.296 The catalyst performed well for fructose dehydration but major improvements are needed when glucose is used as a starting material since microwave heating is required.296,297 Further improvements were reached when Al2O3 was used as support for both the dehydration of fructose and glucose with yields over 70% in [bmim]Cl.256 Other studies have focused in the immobilization of tin which exhibits lower leaching compared with chromium. Tetrahedral Sn is preferred over octahedral since this is related to the inactive SnO2.298 By adding phosphate as a counterion, the catalyst remained highly insoluble and moderation of the Lewis acid sites improved the performance.299 Studies done with mesoporous silica and MnO2 as supports reported no leaching of Sn and the catalyst was recycled several times without loss of activity in Table 4 we summarized the different systems described.298,300
| Solvent System | Type of catalyst | Substrate | Yields | Reference |
|---|---|---|---|---|
| Halogenated ionic liquids | Zeolites | Fructose (30%) | <90% | 271 |
| Water/organic solvent | Zirconium or hafnium phosphate | Fructose, glucose, cellulose (2%) | 70–90% | 248 and 261 |
| [emim]Br | Sulfonated polyphenylene sulfide | Cellulose, glucose (18%) | 65–90% | 258 and 262 |
| Iron oxide supported on carbon | ||||
| DMSO/water organic solvent/ionic liquids | Ion exchange resin | Fructose (10–50%) | 80–100% | 140, 262 and 280 |
| DMSO | Supported Cr on hydroxyapatite or Al2O3 | Fructose and glucose (<4%) | 50–70% | 256 and 296 |
2.3 Issues related to separation and perspective
Despite the many different options available to perform the transformation of sugars into HMF, the production at large scale is still hindered due mostly to the stability of HMF and separation issues.88 Galkin and co-workers have highlighted that HMF undergoes decomposition at room temperature and that the route of synthesis impacts its stability.89,301 Specifically, HMF in the liquid phase synthesized by Brønsted acid dehydration leads to faster degradation due to increased self-diffusion and residual acidity present during the work up of the reaction. Storage of HMF at room temperature leads to gradual degradation due to oligomerization and etherification side reactions accompanied by a gradual change to a dark color.89,301 This adds significant costs to the process for refrigeration and due to the complexity of separation caused by the high boiling point and instability of HMF at high temperature. Moreover, the selectivity of the reaction plays an important role in defining the purification process. Simeonov and co-workers were able to crystallize HMF from ionic liquids by the addition of ethanol and ethyl acetate and observed that the purity of HMF varies between 70 to 90% according to the selectivity of the reaction since by-products can co-crystalize along with the HMF.281 This was observed also by Liu and co-workers when the dehydration of fructose was tested at different substrate loadings, observing a decreased purity of HMF when extracted with MIBK when high loadings of fructose were used, which was accompanied by lower reaction selectivity.190Ionic liquids give high selectivity with different catalysts but removal of HMF from ionic liquids has proven to be extremely challenging due to the involatility of these salts. A study by Wei and co-workers reported the distillation of HMF from an ionic liquid under high vacuum (300 Pa), high temperature (180 °C) and rapid gas bubbling of nitrogen or low boiling point organic compounds, achieving a purity of 90% with 90% recovery in 10 min (Fig. 11).101 The strong interactions of the chloride and bromide anions of the ionic liquids with the hydroxyl group on HMF reduced the partition coefficient with other organic solvents, making the extraction disfavored and necessitating large volumes of organic solvent to completely separate HMF.98,160,191,192 Performing the reaction in non-coordinated ionic liquids which do not form hydrogen bonds with the hydroxy group can partially solve this problem but the choice of the organic solvent is much more limited due to the higher miscibility of the organic solvents with ionic liquids with [OTf]− and [NTf2]− anions.
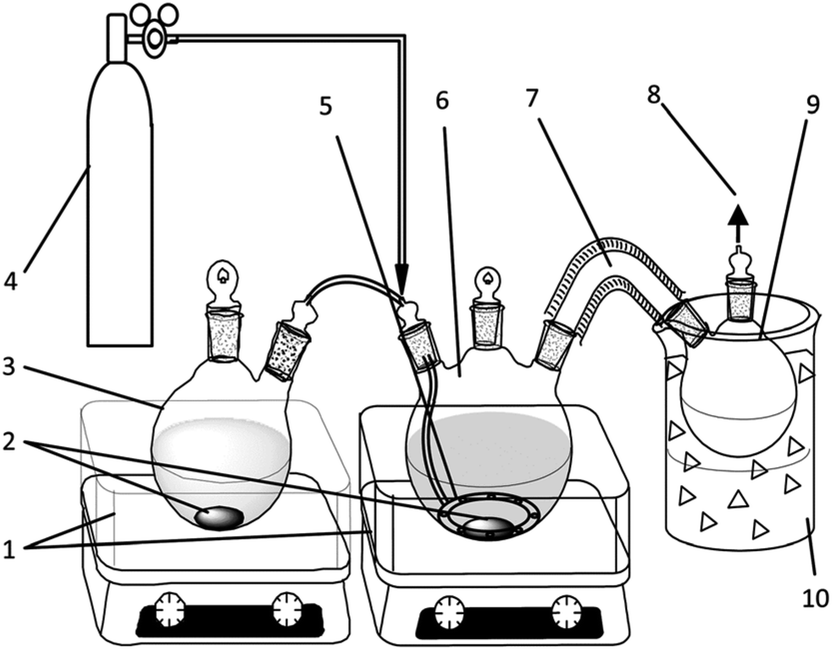 | ||
| Fig. 11 System proposed by Wei and co-workers. (1) oil bath heater, (2) magnetic stirrer, (3) two neck flask for vapor generator(organic solvent at its boiling point temperature), (4) nitrogen cylinder, (5) ring type gas distributor, (6) three neck flask (ionic liquid and HMF), (7) connector, (8) connection to vacuum oil pump, (9) product collection, (10) ice bath.101 | ||
Major improvements in the partition coefficient can be achieved by performing the reaction in a hydrophobic ionic liquid with an [NTf2]− anion and performing the extraction with water.156 However, these ionic liquids do not perform well for this type of reaction and they leach into in the water phase, which is highly undesirable due to the high cost and toxicity of these solvents.302
Other authors have proposed extraction of HMF from ionic liquids with supercritical CO2 (Fig. 12).303 In contrast to organic solvents, supercritical CO2 is considered a green solvent because of its non-toxicity and non-flammability; hence extraction with this solvent can establish an organic solvent-free process. Sun and co-workers have studied the partitioning of HMF between [bmim]Cl and CO2 at different pressures and temperatures, showing that lower temperature favours the extraction and a minimum of 20 bar pressure is necessary to achieve 70% extraction, which is still considered too low for a scalable process.304 Other studies used CO2 as a phase separator to increase the extraction with organic solvents. Following the observation that at relatively low CO2 pressure (7.5 bar) the mixture of [omim]Cl and acetone separate, Shi and co-workers303 designed a system where HMF is extracted continuously from [omim]Cl with CO2 and acetone reaching a yield of 84% from fructose. Major drawbacks of the system lie in the selectivity of extraction since catalysts and sugars are partially soluble in the CO2–acetone mixture.
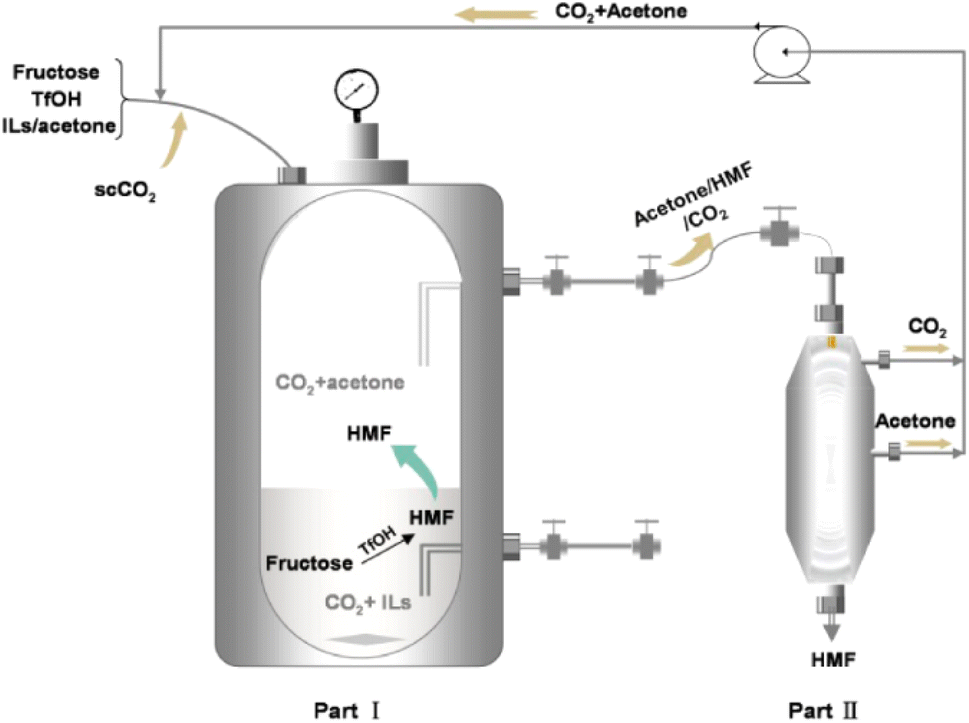 | ||
| Fig. 12 System of Shi and co-workers for the extraction of HMF from ionic liquids with a mixture of acetone and CO2 with continuous concentration.303 | ||
Since the main interaction is H-bonding with the ionic liquids, we proposed in one of our studies to convert the HMF in situ to compounds which are easier to separate.157 We identified DFF as the most suitable option since the ionic liquids show low interaction with aldehydes, allowing an easy separation. In this case DFF could be separated by sublimation at high purity.159 However, further research is needed to improve the transformation of HMF into DFF in ionic liquids in terms of catalyst design and recyclability.
The isolation of HMF is also challenging when DMSO is used as solvent. Some authors have exploited the high affinity of DMSO with water and used an organic solvent to extract HMF from a water/DMSO phase, however leaching of DMSO into the organic solvent arises and excessive amounts of water are needed achieve a favorable partitioning of HMF, compromising the regeneration of the solvent for successive cycles.102,305 Other studies have shown that HMF can be extracted efficiently from DMSO using some commercial carbon resins but the recovery was not discussed.306
The water/NaCl/organic system has so far proven the most efficient process to isolate HMF from a reaction mixture due to the higher partition coefficient and the salting out effect. However, major challenges need to be considered for this type of system since the cost of solvent regeneration can compromise the process economics and CO2 emissions. The price of HMF is currently estimated at more than 1.5 $ per kg, with considerable CO2 emissions even under optimized conditions, making HMF too expensive for most bulk and intermediate chemical products.177,179 The main inefficiency is that no heat is generated during production since the enthalpy of reaction for HMF formation is close to zero, requiring high utility costs and to satisfy the energy demand and achieve a purity over 99%.177
3 Furfural
Furfural is the main building block which is derived from hemicellulose.307,308 Contrary to HMF, it is produced at large scale with an annual production of 400000 tons per year with China as the main producer.309,310 This compound has been seen as one of the main valuable biochemicals that can be obtained from biomass waste, specifically pentosan rich biomass which is not easily digested into ethanol, giving furfural the appellative of ‘gold from garbage’.311,312 Furfural is one of the few examples in which a biobased building block is actually more economical competitive than petrochemical routes.44,313 Currently, most furfural plants use a modified version of the historical Quaker Oats technology developed in 1920, with the main focus on reducing hazardous waste generation from the process and optimizing the recovery of side products to improve the economic and environmental sustainability of the plant.312,314 The wide availability and low cost of the feedstock are the driving forces which makes these processes feasible and allow large added value. Biomass feedstocks include a large variety of waste products rich in pentosan such as corncob, oat hulls and cottonseed hulls.315–317 However, the total annual production of furfural doesn't reflect the typical numbers of a commodity chemical but rather a specialty chemical for niche applications. It is mostly used to produce polyfurfuryl alcohol, specialty solvents and intermediates for food preservatives.47,318 The price of furfural moreover has experienced high volatility over the last 20 years, fluctuating between 0.4 to over 2 $ per kg due to changes in demand, legislation on waste disposal and feedstock availability, making it difficult to identify a spot price for furfural.319 A technoeconomic analysis reported that capital and operating costs associated to a furfural plant are relatively low and profitability of a plant with a capacity of 10000 tons per year can be between 1 to 2 million $ per year. This value, however, does not reflect that all the furfural plants are not optimized to valorize all biomass fractions, and as such cellulose and lignin are treated as byproducts and are burned (in some cases without energy recovery) or disposed with the wastewater. This further highlights the potential of achieving more economic gains from a completely integrated biorefinery by co-production of ethanol.320–322
In this section we analyze the state of the art of furfural production since we believe it can be a valuable future building block to replace linear alkyl benzene sulfonate (LAS) in surfactant applications. We will analyze the different factors which govern the furfural synthesis (solvent and catalyst) to highlight where major efforts are needed to establish a more efficient and sustainable process at large scale.
The main advantage in furfural production is the relative ease of removal from the reaction mixture for furfural compared with HMF due to its lower boiling point (161.7 °C at 1 atm) and formation of a minimum boiling azeotrope with water at 98 °C and 35% furfural content.323,324 The removal of furfural from solution suppresses undesired side reactions with the substrate or its intermediate (resinification), reactions with sugars or fragmentation.316,325 Most of the current furfural production plants operate through vapor stripping of furfural at high pressure (6–15 bar), obtaining a vapor stream at a furfural concentration between 3 to 10 weight % which is then sent to a distillation column which concentrates it to a point where two liquid phases form. The furfural rich fraction (purity between 80–90%) undergoes further refining to reach a purity above 98% while the diluted water fraction (10–15%) is recycled in the distillation column. It has been reported that the presence of glucose derived from the hydrolysis of cellulose in the reaction mixture is particularly detrimental, making the optimization from biomass a crucial factor to achieve high yield.326,327 This was the basis for the development of the SupraYield process where the water mixture with biomass is depressurized at a specific gradient at high temperature, allowing a gradual removal of furfural during formation, reaching yields up to 80% with the further advantage that is versatile for different feedstocks.328–330
Furfural tends to undergo autoxidation when stored in presence of oxygen and light causing an undesired dark coloring, However, furfural is considered stable since the decrease in purity is generally less than 1% over 6 months.331 Similarly to HMF, the yield is strictly related to the choice of the solvent and catalyst which directly impacts the kinetics of the reaction and the stability of the final product.
In this section we will focus mostly on those studies which uses raw biomass as feedstock, since this route is more economically viable compared with the use of isolated xylose, arabinose, pentosans or hemicellulose.332 Indeed, the price of isolated xylose is estimated to be above 4 $ per kg which would render further conversion to furfural economically irrelevant, even if some studies have shown that a two-step hydrolysis and dehydration would lead to higher yield compared to a one step conversion.333 However, studies which use isolated sugars or hemicellulose are useful to understanding the limiting step of the reaction, allowing a better re-optimization of the process.334 The overall objective of the field is to focus on the cost reduction for separation, which requires a robust system which can work at high substrate loading with a simultaneous improvement of the reaction yield.335–337
3.1 Solvent effect
Currently, furfural is produced by acid hydrolysis of biomass in water. In this process, the biomass chips are treated in a dilute solution of sulfuric acid (<1%) at high temperature (170–200 °C) during which a mixture of furfural and water vapor is separated and distilled to obtain furfural at 98% purity. The know-how involved with the separation of the furfural generally differentiates one process from another. Several studies have focused on improving the production of furfural through improvements in separation techniques and exploration of new solvents which can suppress the side reactions. Often these systems are studied in combination with an extracting solvent (such as MIBK or toluene) typically at a ratio of 2:1 to increase the yield to over 70%.338–341
As with fructose, the yield can be increased easily to over 70% when a biphasic system with a hydrophobic solvent is used.342–345 However, general principles which were valid for glucose and fructose are not valid in this case. Raines and co-workers have shown that typical solvent systems which are efficient for glucose and fructose are affected by much lower yields when xylose is used as the substrate.346 The role of different solvents on the selectivity of the reaction does not obey the same trends for pentose sugars as for hexose sugars. Solvents such as ionic liquids, DMSO, DMAc and low boiling point organic solvents (ethanol, methanol) do not show any particular advantage in favoring the yield compared to water, rather the combination of catalyst and solvent plays a major role.347,348 Studies done in DMAc showed that the addition of ionic liquids such as [bmim]Cl and [bmim]Br proved beneficial for the reaction when glucose and fructose were used as substrates while this was not the case when xylose was used.346 When water is used as a solvent, the addition of salts with small cation (such as Li) and large anion (such as I) can increase the selectivity of the reaction by enhancing the formation and stability of the intermediate 1,2-enediol and improving the separation through a salting out effect when a biphasic system is used.349–351 Generally, it has been shown that there is a relationship between the basicity of the anion and the effectiveness of reaction. Iodide and bromide ions proved more efficient compared with chloride during Brønsted and Lewis acid catalysis.346,352 Such behavior has been explained by a stabilization effect of the anion towards the intermediate enol and the lower solvability of iodide compared with chloride anions. Vigier and co-workers have further analyzed the effect of the cation, showing that ionic liquids with hydroxyl functionality (such as choline) would improve the selectivity by introducing a different reaction pathway through a xyloside intermediate which increased the yield compared to pure water, allowing conversion at much higher concentrations of xylose (33%).353
Organic solvent/water systems have been established to give high yields of furfural from biomass. The organic solvents suppress the aldol condensation of furfural with lignin, allowing the design of a potential process for the optimum valorization of all components of biomass. Water/acetone (3:7) and water/THF (1:3) are potential systems to exploit this.354–356 GVL boosts the turnover of the acid catalyst, allowing a decrease in the reaction temperature and improvements in the xylose loading up to 10% without affecting the yield. This has been related to the stabilization of a protonated intermediate which decreases the activation energy of the overall reaction by about twofold when GVL is used as solvent.357–360 It was observed that beside favoring the dehydration step of xylose, GVL is a good solvent also for the production of sugars when small amount of acid is added (0.1%),361,362 an open system was reported where furfural was boiled from the reaction mixture at high temperature (140 °C) achieving an isolated yield of 79% at a feedstock loading of 6.6% (from corn cob).363 In this case the optimization of the water content is an important factor from both a thermodynamic aspect (since it is involved in the reaction mechanism) and for the removal of furfural by lowering the boiling point of the mixture.360
Further studies with GVL have shown that this solvent can further dissolve and depolymerize lignin by producing a high quality pulp (over 85% in purity), making this solvent a good candidate for an optimum biorefinery.364–366
Ionic liquids have been also investigated for furfural production with the aim of exploiting the higher solubility of biomass potentially reducing mass transfer limitations. [bmim]Cl proved to stabilize furfural under acidic conditions with the further advantage that vacuum can be applied to the reaction mixture to remove furfural more efficiently as it is formed.367,368 However, this aspect has not been extensively studied in the literature with much fewer examples compared to HMF.369 A published review summarizes well the different examples in this area.367 The use of [bmim]HSO4 and [bmim]Cl proved to be inefficient when xylose is used as substrate with yields lower than 50% resulting in large amount of humins.227,370–372 Applying microwave heating at low substrate concentrations improved the efficiency up to 90% from hemicellulose but yields from biomass still remained poor (<15%).373 Another study reported the usage of [emim]Br and SnCl4 which showed a yield of 54% but the system suffered of low loading and high toxicity of the catalyst used.374 The use of these solvents starting from pure biomass did not show any particular advantage in terms of yield and process efficiency.375 One of the main problems may lie in the regulation of the water content in these mixtures and its role on the xylose dehydration mechanism.376,377 This represents an aspect which requires further studies for this particular reaction.
3.2 Catalyst development
The formation of furfural from biomass requires a catalyst which is capable of performing multiple reactions steps, including hemicellulose hydrolysis into xylose and its subsequent dehydration into furfural. Overall this reaction can be performed hydrothermally at temperatures above 200 °C.378,379 The addition of an acid can decrease the temperature down to 140 °C and generally improve the yield up to 80% even if the nature of the acid used influences the selectivity and the mechanism of formation of the side products.348,380,381 Different studies using different Brønsted acids and ionic liquids for the dehydration of xylose have suggested that a pKa below 0 is needed to perform efficiently the reaction and an optimum value of Hammett acidity has been identified to be 2.08 (sulfuric acid is 1.67).339,357 The high efficiency in producing furfural with a Brønsted acid resembles the fructose dehydration step following a subsequent elimination of hydroxyl group from the ring. However, this is different when using Lewis acids. Some experiments have shown that catalysts inefficient for glucose and fructose dehydration such as FeCl3 actually proved to perform well for furfural using biomass as feedstock reaching yields up to 79% when GVL is used as solvent, indicating a different coordinating sphere for xylose compared with hexose sugars.382 Generally, FeCl3, AlCl3 and CrCl3 show poor selectivity in water but it has been shown that this can be improved in combination with a Brønsted acid by optimizing the pH.383 This synergy has been an object of many studies and experimental testing which have suggested a combined effect on the hydrolysis between the metal and the coordination of the metal ions with the intermediate enediol.350,384–386 This was also evident when FeCl3 and SnCl2 were immobilized on acid exchange resins, achieving yields over 90%, further highlighting the importance of both Brønsted and Lewis acids.387,388 Durán and co-workers showed that metal oxides such as ZrO2, ZnO and TiO2 have the same effect in combination with a homogeneous Brønsted acid catalyst using rice hull as a feedstock.333,389 To further explain this, Huber and co-workers have evaluated the Brønsted and Lewis acid sites of different acid catalysts and concluded that Zr–P has the highest selectivity over silicoaluminate, γ-alumina and HY zeolite, identifying the optimum ratio of Brønsted/Lewis acid sites at about 30. Further experiments on the activity and adsorption/desorption of furfural in HY zeolite showed that microstructured pores lead to furfural degradation since they retain furfural on the catalyst surface, inducing polymerization.390 Another study has shown that the activity of the metal chlorides can be improved through the addition of chloride anions at ratio 1:2, suggesting that the complexation of the metal centre has an impact on the selectivity.391,392
Different computational and experimental studies have attempted to explain the different selectivities of catalysts towards furfural. Indeed, evidence has shown that there are different mechanisms of reaction involved for different catalysts used.393 Labelling experiments conducted in D2O have shown that homogenous Brønsted acids proceed through a ring rearrangement which resembles the fructose mechanism394 while Lewis acids proceed through a ring opening. For the latter case, the observation of large xylulose intermediate with no incorporation of D2O when using Lewis acids such as Cr and Sn suggests a hydride shift mechanism from xylose which is favored by the coordination of the Lewis acid with the hydroxyl and carbonyl group in the open chain (Fig. 13a).346,388,395
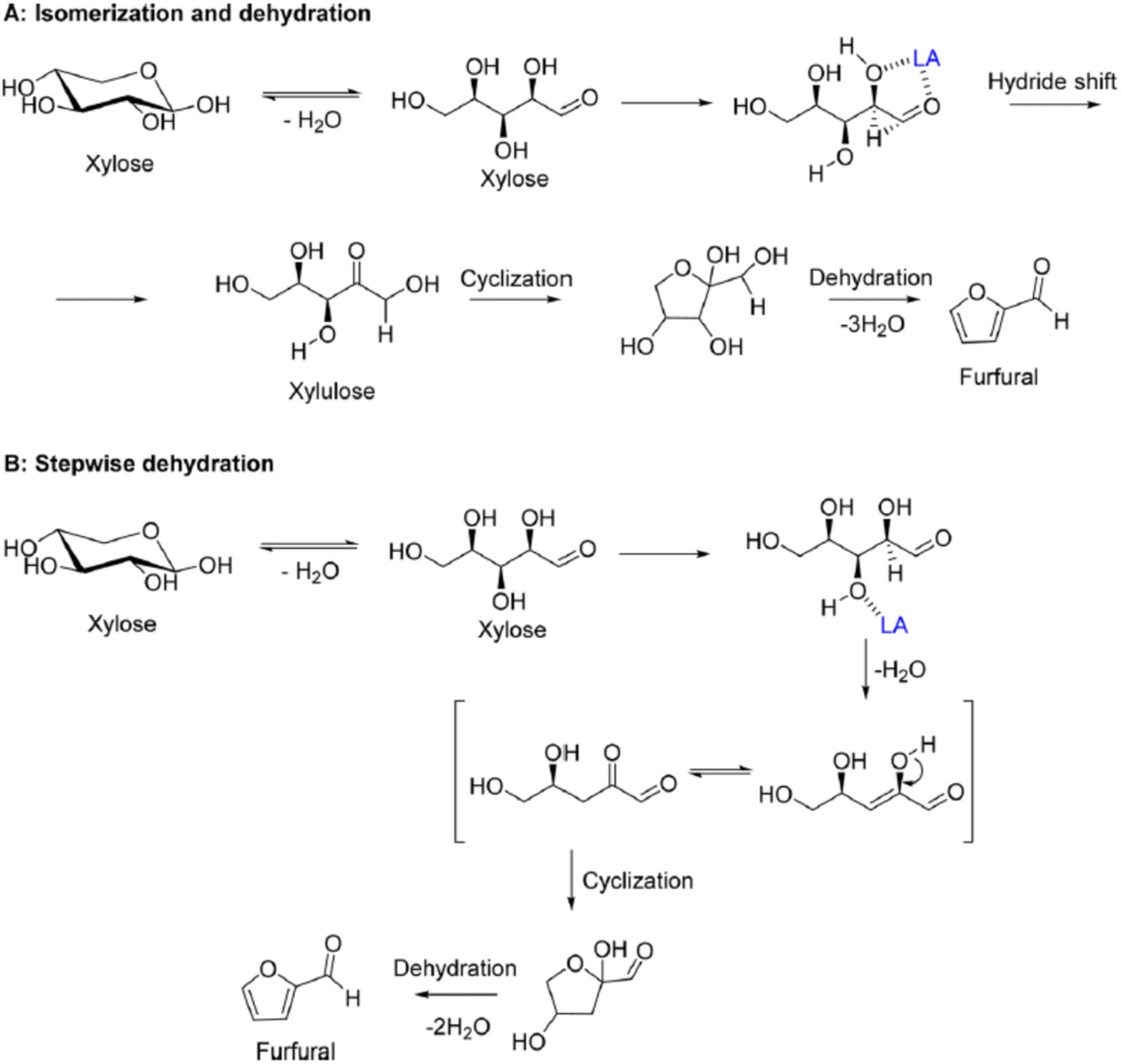 | ||
| Fig. 13 Reaction mechanisms proposed by Gupta and co-workers396 for the synthesis of furfural using homogeneous (A) and heterogeneous Lewis acids (B). | ||
Other experiments using heterogeneous Lewis acids have demonstrated different coordination with xylose can happen, leading to different reaction mechanisms with higher yields. Gupta and co-workers396 neutralized the Brønsted acids of Nb2O5 through ion exchange with sodium and observed a different mechanism in D2O which involved an incorporation of the deuterium atom in the final molecule, indicating that the Lewis acid type can further influence the mechanism which can lead to a higher selectivity. The authors proposed a stepwise dehydration step for the heterogeneous Nb2O5 where the deuterium atom is incorporated through the formation of a di-ketone (Fig. 13b).
According to these mechanisms, the higher selectivity observed in a combined Lewis and Brønsted acid mechanism lies on the implementation of two steps. The isomerization of xylose into xylulose is favored by the Lewis acid in an open chain form and the dehydration of the xylulose into furfural is kinetically more favored compared with xylose under Brønsted acid conditions.397
Generally, the utilization of heterogeneous catalysts is limited by the multiphasic nature of the system due to the insolubility of the lignin and cellulose in most of the solvents, which would compromise the recovery of the catalyst and may result in carbon deposition.359,398,399 Even if Brønsted acid resins demonstrated good performance, the direct use of biomass and the presence of K and Na impurities will cause a gradual decrease of the activity due to ion exchange with the protons.359,360,400 A study using silicoaluminate phosphates (SAPO) showed that the recyclability of the catalyst is dependent on the biomass content (3.3%) confirming this aspect.401,402 Performing the biomass pretreatment in two subsequent steps can improve the overall yield. This can be achieved with hot water pretreatment under acidic conditions to selectively extract the hemicellulose so that it can be converted with a heterogeneous catalyst.398,403–405 It is outside the scope of this review to analyze pretreatment technologies for the separation of hemicellulose.
3.3 Furfural perspective
Within a biorefinery concept, the production of furfural will be integrated with the valorization of other main components such as cellulose336 and lignin which can be produced alongside furfural. The integration of furfural production with biomass fraction can improve the biorefinery value by providing an economically competitive aromatic building block. The challenge lies with maximizing furfural production in combination with removing lignin form the cellulosic pulp to improve its saccharification and its applicability as a biofuel. This should be done by reducing the amount of energy and steam required to achieve the separation and purification of furfural.406 While lignin is generally burned to provide the energy for the plant, cellulose represents a valuable feedstock for a large variety of applications including second generation bioethanol production.321,407 An integrated biorefinery approach has the potential to strongly favor the quality of the final pulp since hemicellulosic side products can negatively impact the cellulose saccharification and enzyme activity. Furfural removal can overcome this since it limits the degradation as demonstrated by Mao and co-workers at pilot scale. Corn-cob was treated at high temperature (190 °C) recovering furfural through stripping with acetic acid vapors followed by steam explosion in a second step, showing that the delignification of the pulp can be improved.408The production of furfural has proven to be very advantageous using Brønsted acids in water, which can be further optimized by implementing separation technologies to decrease the side reaction and increase the yield. This has led to the scale up of different processes which focus mostly on the implementation of efficient separation techniques instead of an improvement in catalysis. The use of Lewis acids did not lead to enormous advantages and their addition would complicate the system and its recyclability rather than provide a true economic benefit. Moreover, the addition of a Lewis acid can lead to the degradation of the cellulose, decreasing the overall added value. One of the main challenges in terms of optimization lies in the identification of a favorable reaction pathway which would lead to an improvement in the process. The solvent effect proved to be less deterministic compared to when glucose or fructose are used. The necessity to utilize raw biomass for economic feasibility compromises the design of heterogenous catalysts, making Brønsted acids the most realistic option. GVL proved to be the most efficient solvent to carry out this reaction but its utilization in a combined production of high-quality cellulose still needs to be established in addition of lignin recovery and solvent regeneration. Ionic liquids proved to be efficient solvents for biomass pre-treatment but the combined production of furfural is still a topic at proof-of-concept level.
4 2,5-Furandicarboxylic acid
The synthesis and production of terephthalic acid (TA) from p-xylene at high yield once represented a breakthrough in the chemical industry and established PET in the market.409 Today, few improvements are achievable for the production of TA and many plants operating at large scale have been built.410–414 The success of TA derives from the Amoco-Mid Century process which uses acetic acid as solvent and a mixture of Mn/Co/Br as catalyst.415 The main advantage of this system is the separation of the final product through precipitation and recycling of the solvent with the dissolved catalyst at high efficiency, reducing the cost of the whole system and delivering all the efficiencies of homogeneous catalysis.416 Moreover, this reaction is characterized by fast rates which enables high productivity thanks to the formation of radicals which catalyze the reaction at high yield, further enhanced by the stability of the final product. Major challenges have been faced in obtaining TA at the desired purity (>99.6%) since the main side products, which consist of incompletely oxidized intermediates, act as chain terminators during the subsequent polymerization, hindering the formation of high molecular weight PET resins.417 Different technologies have been developed based on enhancing the conversion or purifying the raw TA through multiple crystallization steps.417,418 The efficiency of TA synthesis enabled scale-up of PET production to large volumes by providing a cheap starting material to produce the polymer. This makes the substitution of TA very challenging. Recently it has been announced that a TA plant facility at Corpus Christi (US) will be built with a capacity of 1.2 million ton per year with an integrated PET line of 1 million ton per year.419The diacid monomer 2,5-furandicarboxylic acid (FDCA) is considered one of the most valuable molecules that can be chemically obtained from sugars.20,53 Market demand for FDCA is increasing due to the desire to obtain a completely green bioplastic that can replace PET in the food and packaging sector.54 However, viable technologies which give an efficient techno-economical pathway from sugars to FDCA do not currently exist. This is due to the challenges in isolating the HMF intermediate at low cost and further issues related to HMF stability, which limits its applicability in catalysis.420–422 A plant for the production of FDCA at moderate scale was announced by Avantium which uses furanic ether derivatives of HMF and oxidizes these to FDCA using the Amoco-Mid Century catalytic system.423–425 However the process so far is experiencing challenges due to the formation of humins in all reaction steps which limits the recyclability of the catalyst and solvent.
Homogeneous catalytic oxidation using the classic Amoco-Mid Century system gives yields over 80% starting from HMF although this reaction is affected by side reactions due to the intrinsic instability of HMF, which is also more susceptible to overoxidation.426 The side reactions lead to the formation of humins, maleic anhydride, furoic acid and CO2.427 The reaction is usually performed at high temperatures over 180 °C, necessitating short reaction times to limit the formation of these side products. While the catalyst has proven to be very active, a substantial impact is caused by mass transfer limitations due to slow diffusion of oxygen into the liquid phase. A semi batch reactor with continuous bubbling was reported to improve the selectivity compared with a batch pressurized process.428 Similar to terephthalic acid, FDCA can be easily separated from the reaction mixture by crystallization, allowing the recycling of the solvent.429,430 However, this system has proven poorly recyclable due to the accumulation of side products in the reaction mixture, which cannot easily be removed from acetic acid, leading to catalyst deactivation and the need to flare most of the spent solvent. Therefore, much research is focused on finding different catalytic routes for the efficient production of FDCA in a green and sustainable way. For this reason, the transformation of HMF into FDCA it is one of the main examples of a green reaction for catalyst design, and many publications have been reported and are continuously released on different catalysts which can achieve high yield and high recyclability, summarized in several reviews.20,53,431,432
Heterogeneous catalysis has thus far proven to be the most efficient option for FDCA production, with noble metals showing the highest catalytic activity, further enhanced by the addition of a homogeneous base. Noble metals complex the hydroxy group on HMF and catalyze a β-hydride shift elimination.433,434 Molecular oxygen regenerates the catalyst surface by forming water or peroxides and does not directly participate in the transformation of HMF into FDCA. The introduction of the atomic oxygen into FDCA derives from the formation of geminal diols with water, which are the active species that adsorb onto the catalyst surface.435–437 This mechanism in water (Fig. 14) is a recognized concept in organic chemistry from alcohol and aldehyde oxidation and can be found in any organic chemistry textbook.435,438,439
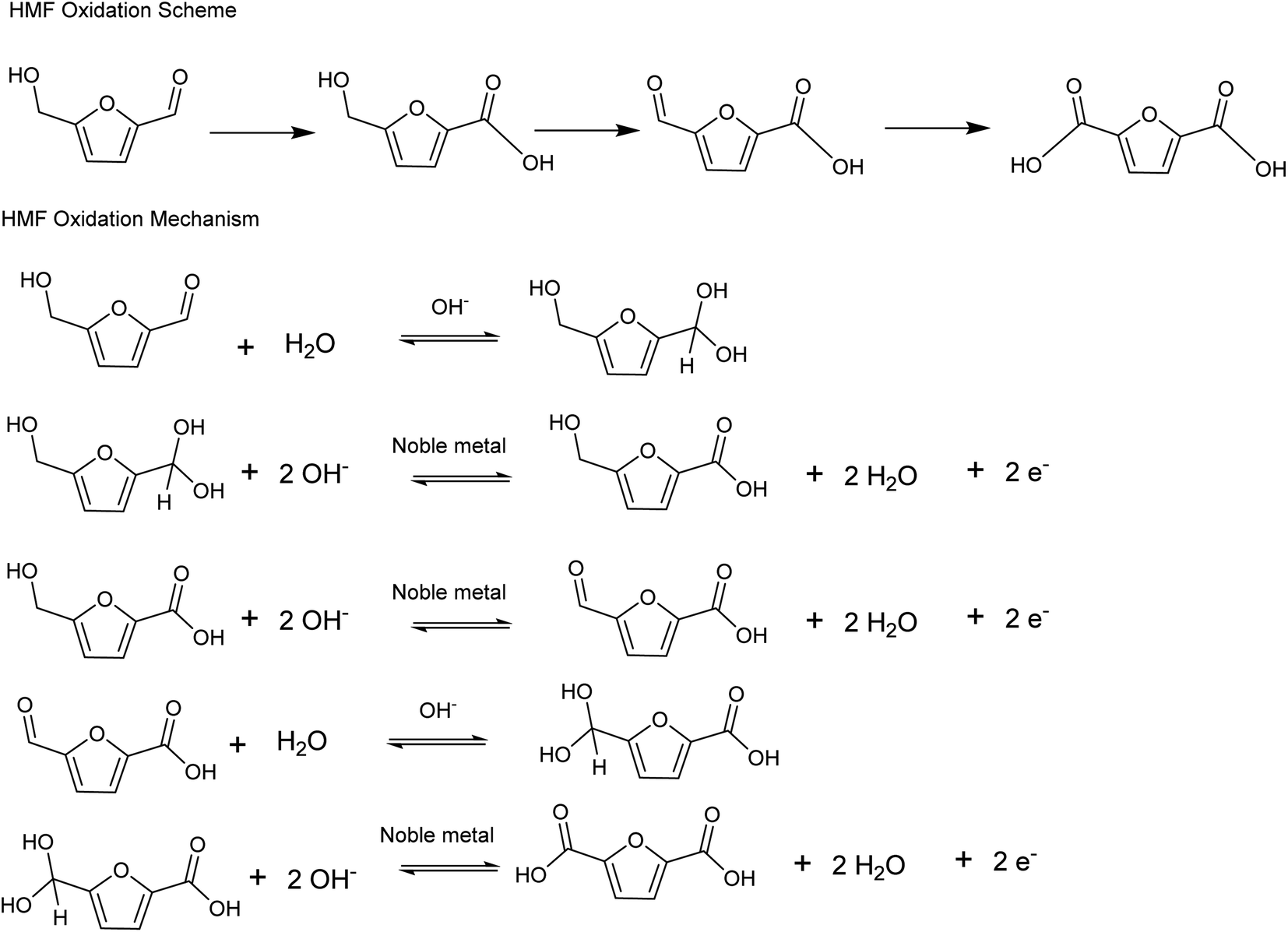 | ||
| Fig. 14 Reaction scheme proposed by Davis and co-workers for the oxidation of HMF into FDCA passing through geminal diols.435 | ||
The addition of a homogeneous base can increase the population of geminal diols, enhancing the rate of the reaction at milder conditions, but disadvantages lies in the higher costs related to the use of a chemical which is not recyclable.440 Moreover, base addition can lead to the formation of other side products through the Cannizzaro reaction or HMF degradation into humins which, even in low amounts, strongly deactivates the catalyst.441 The addition of a base also compromises the separation of FDCA from water since it drastically increases its solubility (via deprotonation) and twice stoichiometric amounts of acid need to be added to precipitate the product, creating twice stoichiometric amounts of salt waste.442
The selectivity towards FDCA is greatly affected by the solvent used.443 Water is the preferred solvent since the oxidation of aldehydes is thermodynamically favored through the formation of geminal diols, which serve as the intermediate for the formation of the carboxylic acids.444 By switching to a solvent other than water, other intermediate products become favored. For example, in aprotic organic solvents the dialdehyde DFF is mostly formed, while in alcohols the related ester is preferred.445–447
When water is used as the solvent, the reaction is limited mostly by the oxidation of the intermediate FFCA, due to the low equilibrium constant of this compound towards the geminal diol, slowing the rate of oxidation. This is an extremely important consideration when developing a catalytic system as FFCA acts as a chain terminator during the polymerization of FDCA (similar to 4-carboxybenzaldehyde in the analogous terephthalic acid route). Therefore, a water rich environment is necessary to increase the population of geminal diols and a highly active catalyst needs to ensure that FFCA is completely converted.448
The use of water has several advantages, as water is cheap, non-toxic and can provide simple separation of FDCA through acidification. However, limitations arise due to the low solubility of FDCA in this solvent which can clog the catalyst surface while forming when heterogeneous catalysts are employed. Therefore, the reaction is usually carried out under highly dilute conditions with concentrations below 0.1 M, adding the disadvantage that the water phase cannot be recycled since the reaction is negatively affected by the pH and the presence of chloride salts, which decreases the selectivity of the reaction.449,450
Several catalytic pathways have been studied to produce FDCA at high yield in a cost-effective way. Most of the literature studies involve heterogeneous catalysis based on carbonaceous and metal-based materials which are good candidates for production at large scale because of high tunability which can improve the reaction conditions. Other studies have focused on chemoenzymatic routes based on isolated enzymes and microorganisms which can give high selectivity under milder reaction conditions. Electrocatalysis has also been extensively considered by many research groups, highlighting the advantages of hydrogen co-production.451,452
A few studies have reported the conversion of furfural into FDCA. These have used carboxylation with CO2 using carbonate salts to activate the C–H bond in the furfural ring in a non-catalytic system (Fig. 15). In order to favor the deprotonation of the C–H bond, Kanan and co-workers used K or Cs carbonates in equimolar amounts at high temperature to achieve a molten salt under 8 bar of static CO2, achieving an isolated yield of 89% at 200 °C with CsCO3.453 Ultimately the authors optimized the system in a continuous flow of CO2 with a mixture of K:Cs carbonate salt (4:1) at 275 °C which allowed water removal and improved the mass transfer of CO2 in the highly viscous melt, allowing production at the 100 gram scale in a fixed-bed flow reactor.454
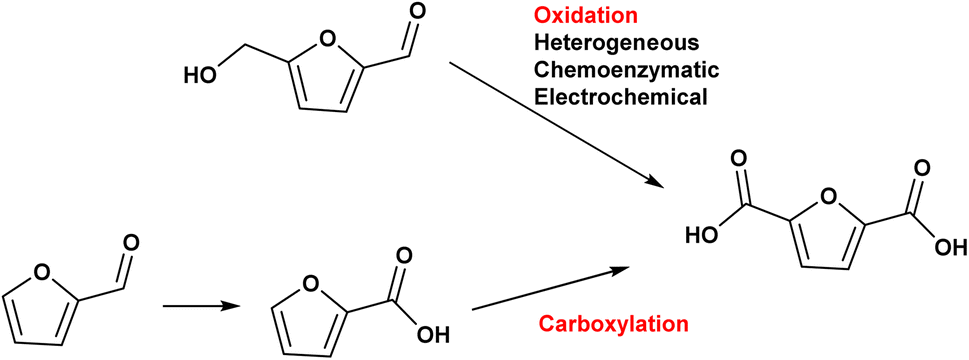 | ||
| Fig. 15 Reaction scheme to obtain FDCA from HMF and furfural. | ||
In the next sections, the different routes to produce FDCA will be summarized, highlighting the individual challenges.
4.1 Catalyst design
Noble metals (Au, Pt, Pd, Ru) represent ideal active sites to perform the oxidation reaction since they give high selectivity through interaction with the alcohol groups and can be easily regenerated using molecular oxygen or other stoichiometric oxidants.455–457 Due their high costs, the catalysts need to be recyclable without appreciable loss in activity, which is often challenging because of catalyst poisoning, agglomeration or leaching of the active sites which reduces the selectivity of the reaction.439,458 Their versatility is due to their multiple accessible oxidation states even if lower oxidation states are less selective in the oxidation and enhance leaching of metal from the support. However overoxidation of the noble metal can arise when high oxygen pressures are used and this is strongly dependent on the support, but the catalytic activity can often be restored through treatment with H2.459–461 For example, overoxidation arises for ruthenium when deposited on hydroxyapatite while this was not observed for supports based on Co.460,462The formation of side products such as humins or other oligomers also causes catalyst deactivation due to irreversible adsorption or clogging of the active sites, necessitating regeneration.463 This is usually performed by calcination of the catalyst at high temperature or through DMSO washing which would burn or dissolve the impurities. Thermal treatments are usually incompatible with these catalysts since most of them are based on nanodispersed noble metals which would agglomerate upon heating, decreasing their catalytic activity considerably.464,465 These steps are also economically prohibitive at large scale because of energy or chemical costs. To avoid this limitation, catalyst design needs to focus on maximizing the selectivity to avoid undesired side product formation, operation at low oxygen pressures and catalyst stability to avoid leaching of the noble metal and agglomeration of the active sites.
The activity depends on the type of support and deposition technique.466–468 Atomic layer deposition is more efficient for the catalyst activity compared with the more widely used deposition techniques such as impregnation and co-precipitation.469 It has been observed that the dispersion of the noble metal on the catalyst surface is a more relevant parameter than the surface area of the support and a remarkable enhancement in catalytic performance can be achieved with deposition techniques that give a uniform distribution with small particle sizes.469,470 It is important that this dispersion remains stable, avoiding agglomeration over multiple catalytic cycles. Wide varieties of supports have been tested and reported to perform differently according to the metal used since the interaction of the noble metal and the support contributes to the overall activity of the catalyst.471 Cerium and titanium oxides gave the higher yields of FDCA when Au is used as the catalyst even if the particle sizes of the noble metals are similar when activated carbon or Fe2O3 are used as support. Meanwhile, ZrO2 proved to be more efficient as support for Pt than Pd, further supporting that the interaction between noble metal and support plays an important role in determining the selectivity of the reaction.459,466,472,473 However, these mono-metal oxides are normally not sufficiently stable and more functional supports need to be used. Improvements in the performance of zirconia can be achieved by adding MgO which enhances the mobility of oxygen anions by introducing vacancies in the tetragonal structure of zirconia.474
Pichler and co-workers have studied the effect of the preparation techniques of ZrO2 support when Ru is used as catalyst. Surface casting methods using SBA-15 as a template were more efficient compared with other methodologies. This was due to a kinetic effect originating in the high surface area (256 m2 g−1) since all other ZrO2 supports actually showed the same carbon balance, indicating that the overall selectivity is not affected.475 A recent study by Megías-Sayago and co-workers have shown that the role of the zirconium oxide is to create Brønsted acidic groups in the vicinity of the noble metal site to form the O− species under a basic solvent environment and deprotonate the alcohol when coordinated with the noble metal (Fig. 16).476
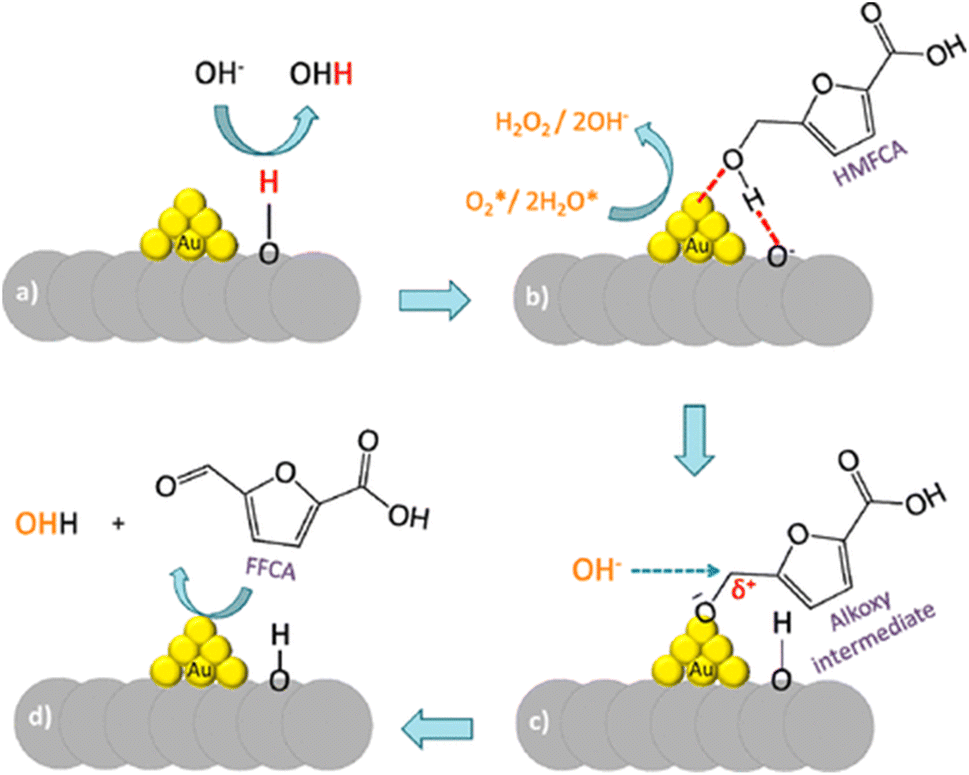 | ||
| Fig. 16 Role of the Brønsted acid groups on the catalyst support proposed by Megias-Sayago and co-workers.476 | ||
The use of a coordinating polymer derived from the polymerization of benzenetricarboxylic acid with the deposition of cerium and Pt can further enhance the activity compared with pure CeO2, preventing leaching of the noble metal and improving recyclability.477 A support-free catalyst was developed by optimizing the synthesis of platinum nanoparticles which can achieve high yields in short reaction times, but much higher metal loadings were needed compared with the supported catalysts. There are also issues regarding instability of nanoparticles in solution and the hazards presented by using nano powders.478–480
It is important that particle sizes of metals deposited on the support are as small as possible with a high degree of dispersion, usually measured by TEM.481 The role of the support is to enhance the stability of the noble metal particles, avoiding migration which reduces the dispersion and leads to leaching.482 Different techniques of catalyst preparation can be used to stabilize the noble metal particles. Additives such as PVP can be used during impregnation to coat the metal particles, avoiding agglomeration.483 Doping with Ce or using mesostructured supports can also improve the dispersion of nanoparticles on the surface and minimize the agglomeration.470,484 However, the addition of the polymer might not be sufficient to avoid the agglomeration since this is highly dependent on the type of support. Moreover, the PVP can leach and degrade during the reaction, reducing the noble metal stability after extensive use.
Pd and Au deposited in Al2O3 exhibited higher agglomeration after the first cycle, forming clusters of 20 nm when ZrO2/La2O3 and TiO2 was used.466,484 HY and NaY zeolites could support particle sizes as low as 1 nm for Au because of the cage structure which can trap the noble metals, however these supports proved to be affected by agglomeration when ruthenium is used and leaching when Au was used.485–487 Other studies have shown that the functionality and amorphousness of carbon materials can enhance the formation of small particles by acting as nucleation sites and interacting with metal centers through H-bond interactions. The particle size can be decreased by increasing the oxygen content in the support and this leads to better performance than non-functionalized carbons such as graphene.488,489 Activated carbon can give high dispersion through sol–gel deposition methods and leaching is avoided under basic conditions, however when a base is not added consistent leaching was observed.490–492 While the loss of noble metals is minimized under basic conditions, the type of base used influences the particle growth over different catalytic cycles. For example, NaHCO3 enhanced the particle growth more than NaOH for Au/SiO2.470 Partial improvements can be achieved by doping the carbon surfaces with nitrogen, which enhances the defects and disorder of the support, but the problem still persist after several cycles.493 A recent study done by Donoeva and co-workers494 analyzed the effect of functionalized graphene, observing that acid functionalized groups on the carbon surface have a negative effect on the selectivity, to the contrary of basic groups derived from doping with nitrogen. The authors related this effect to the surface charge of the support measured by the point of zero charge (PZV) and the pH of the solution. Under basic conditions the acidic sites of the support form negative charges on the surface which would repel hydroxy groups from the catalyst surface.
Other studies have reported magnetic catalysts formed by microspheres of Fe2O3 coated by carbonaceous material derived from glucose pyrolysis which prevented leaching of Pt but a uniform distribution of nanoparticles was not achieved, with particles of 10 nm on the catalyst surface leading to low yield.495,496 Further improvements were achieved when graphene was used to coat the iron oxide particles, achieving a uniform distribution with particle sizes on the order of 4.2 nm but the catalyst suffered from leaching.497
Mixtures of different metals can enhance catalyst stability and activity. Combinations of metals such as AuCu, PdPt, AuAg and AuPd perform better than single metals, achieving quantitative yields in short reaction times.488,498–501 Specifically, the enhancement of the Au particles in combination with a noble metal was seen in the formation of an alloy cluster with an electron transfer from Pd to the Au atoms, forming negatively charged gold atoms on the surface which are more active towards this reaction.502,503 The same effect was observed for Pd/Pt alloys.500 Bismuth also proved to enhance further the catalytic activity of noble metals by increasing resistance to poisoning and favoring the coordination of the noble metal with the substrate by interacting with the furan ring.459,504,505
The desire to remove the homogeneous base has led to research studying multifunctional heterogeneous catalysts with basic character. Hydrotalcites are a good choice due to alkaline character arising from HCO3 groups on the surface, leading to good performance when Au or a mixture of Pd/Pt is deposited, but partial decomposition results in the release of Mg ions into the aqueous solution.500,506–508 The supports also do not prevent leaching of the noble metals when Au is used.509 Another type of alkaline support was prepared by carbonization with MgO which gave high yields, but the catalyst still suffered from leaching and aggregation.510 A polyionic liquid was tested as a support, chosen because the hydrophilicity of the support and anionic character should enhance the dispersion of the noble metals and favor the adsorption and desorption of the reactants.511,512 Another support based on zinchydroxycarbonate proved to stabilize the noble metal particles at very small sizes (1.6 nm) and prevent leaching.513 A study done on commercial Ru/C showed that the role of the base is only to avoid the precipitation of FDCA onto the catalyst surface by increasing its solubility, but this problem could be circumnavigated by increasing the temperature from 90 to 120 °C.491,514 However, the main challenge in such systems is the large decrease in pH which enhances the leaching, therefore the support needs strong interactions so that this is avoided. Reaction set-up and reaction conditions also impact the yield. Liguori and co-workers performed the reaction in continuous flow with a residence time of 303 s and oxygen pressure of 20 bar using Pt/C. This set-up was 100% selective with quantitative yield and reduced the reaction time which in typical batch mode is more than 2 h.515
In order to find a suitable solution to the leaching problem and retain high performance with recyclability, new supports are needed. A mixture of different metal oxides based on manganese, cobalt and cerium have been reported to give high performance. Without the deposition of noble metals, a mixture of these metal oxides is still active but a homogeneous base is required, along with high temperature and pressure. Han and co-workers reported that cerium oxide can enhance the catalytic activity for the aerobic oxidation when combined with manganese oxide, but high oxygen pressures and temperatures are required. Preparation methods should focus on enhancing the population of the Ce3+, which provides oxygen vacancies in the lattice, favoring the regeneration of the manganese active sites which work as a mediator for electron transfer to the ruthenium centers.516 Other reports have demonstrated that cobalt oxide has the same effect as Ce when used with manganese when prepared by co-precipitation or grinding.517–519 In particular, a mixture of manganese and cobalt oxides prepared by grinding provides small particle sizes of 6 nm with a highly triggerable Lewis base functionality at different Co–Mn ratios.520 These compounds enhance the catalytic activity when noble metals are deposited; Ru more than other noble metals Au, Pt and Pd, as strong interactions with the oxide surface give stability and high catalytic activity.521 Mishra and co-workers reported that a mixture of manganese and cobalt in tetrahedral form synthesized by coprecipitation can stabilize ruthenium nanoparticles (3.2 nm), avoiding agglomeration and leaching, achieving quantitative yield over multiple runs.462 Another report522 obtained the same results using a mixture of manganese and cerium oxides, which stabilized the Ru nanoparticles over multiple catalytic cycles with quantitative yield, but harsher conditions were required compared with the work of Mishra and co-workers.462
Noble metals have proven successful in the synthesis of FDCA at high yield but performance is determined by the quality of the HMF used since side products derived from HMF decomposition deactivate the catalyst.430,523 Yi and co-workers reported that the impurities formed during HMF dehydration using isopropanol as solvent can be removed by dissolution in water followed by filtration of the side products. The final HMF product would be suitable for the oxidation using Au/HT (Fig. 17).524
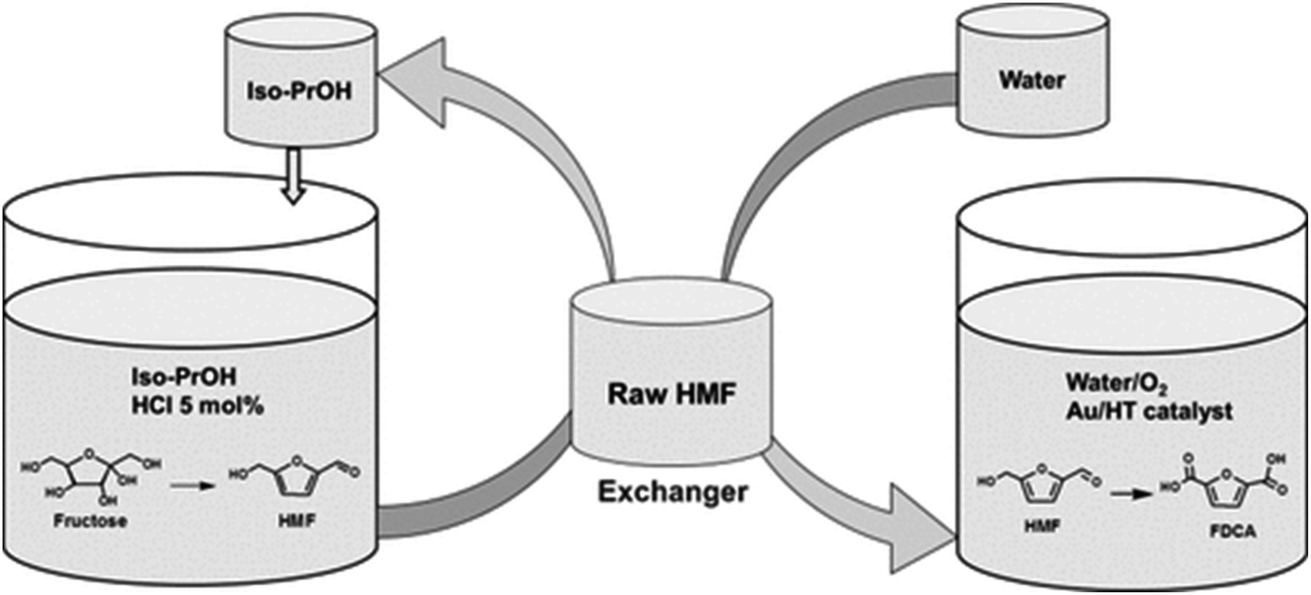 | ||
| Fig. 17 Two-step process reported by Yi and co-workers for the synthesis of FDCA from fructose.524 | ||
In order to establish more versatility towards the production of high purity HMF, noble metal free catalysts are highly sought after. These could reduce the risk of deactivation and avoid the expense of regeneration or replacement of noble metals. Non-noble metal based catalysts have been reported, mostly based on manganese, cobalt and nitrogen doped carbon materials.525,526 The surface area plays a determinant role, therefore catalyst design needs to be directed toward high porosity.527 Mn/Fe oxide mixtures and cobalt complexes are highly active towards the oxidation but a stoichiometric oxidant must be used.528–530 Other studies have focused on using carbon materials, in particular nitrogen doped graphene synthesized from PVP and methylimidazole as a nitrogen source, calcinated at 900 °C with Zn(NO3) to produce an active metal free catalyst for the oxidation.531 Unfortunately, the catalyst performed poorly, requiring long reaction times and suffering from deactivation over multiple cycles. Verma and co-workers showed that calcinated chitosan at 300 °C has very good recyclability but the reaction times are long.532 Further improvements could be achieved by deposition of Co or Mn onto the surface of the carbon material. Liu and co-workers developed a nanodispersed CoO2 (25 nm) on mesoporous carbon which is highly selective towards the oxidation but long reaction times are needed.533 The deposition of Co and Mn on lignin doped with nitrogen proved to be more efficient. In particular, nitrogen proved to activate cobalt towards the oxidation and the addition of manganese further enhances its activity and stability compared to the unsupported catalyst or other supports such as silica.436,518,534 Another study reported activated MnO2 as a recyclable catalyst in combination with sodium bicarbonate to obtain high yields of FDCA (91%) through the formation of the peroxide species MnOOH which can be regenerated with molecular oxygen.535 Bao and co-workers studied the effect of calcinating manganese MOFs at different temperatures and found that by preparing the catalyst at temperatures over 400 °C, highly porous nanoflakes of Mn2O3 were formed which exhibited high activity for three catalytic cycles.536 Hayashi and co-workers have studied the effect of the crystalline structure of manganese oxide on the catalytic performances and relate their catalytic activities with the energy of formation of oxygen vacancies in the lattice calculated by DFT. β and λ MnO2 proved to have the lower energy which corresponded to the higher activity towards FDCA. The authors further elaborated a protocol to increase the surface area of β-MnO2 from 14 to 82 m2 g−1 to further enhance its catalytic activity.537
Ventura and co-workers recently demonstrated that a mixture of CeO2, CuO and MnO2 prepared by milling can achieve synergy without the deposition of Ru, achieving yields of 98.6% with no formation of humins or leaching observed. The catalyst did deactivate through the formation of metallic copper during the catalytic cycle which could be regenerated through calcination.538
In order to increase the selectivity and perform the oxidation at room temperature, different enzymes have been studied. Dijkman and co-workers reported an oxidoreductase enzyme HMFO with FAD as prosthetic group which can oxidase HMF at high yield (95%), but the enzyme proved to deactivate through leaching of the FAD group which needed to be reintegrated.539,540 Another study reported a cascade reaction which involves two different enzymes to oxidize specifically the alcohol and aldehyde group with galactose oxidase M3-5 and PaoABC. The GoaseM3-5 has been reported as wild and a mutant type which can achieve high yield of DFF in shorter reaction times with the aid of horseradish peroxidase (HRP).541,542 The PoaABC could be added subsequently, achieving yields close to 100%.541 Both steps require the addition of catalase to decompose the H2O2 which acts as a deactivating agent for the enzymes.
Another cascade reaction was reported using laccase in combination with TEMPO to selectively produce FFCA which was subsequently coupled with a P. putida KT2440 to obtain FDCA at 100% yield or performed in a single step by immobilizing Laccase on an iron/silica support.543–545 However, these enzymes proved to have extremely low rates (>10 h) requiring further research to boost the activity.
We summarized different studies regarding HMF oxidation in Table 5.
| Catalyst | Support | Base requirement | Recyclability | Reference |
|---|---|---|---|---|
| Au | CeO2 or TiO2 | Yes | Requires reactivation | 470 |
| Pd | ZrO2 | Yes | Requires reactivation | 467 |
| Au | Hydrotalcite | No | 5 times | 524 |
| Pt | CeO2 with polymer | Yes | 5 times | 477 |
| Mixture noble metals | Activated carbon | Yes | 5 times | 488, 498–501 and 509 |
| Ru | C | Yes | 5 times, slight decrease in activity. Leaching observed | 491 and 514 |
| Ru | MnCo or MnCe | No | 5 times, no leaching observed | 516 and 520 |
| Mn/Co on carbon material | Yes | 3 times with slight decrease in activity | 533 | |
| CeO, CuO, MnO2 | Yes | 5 times but requires regeneration | 538 | |
| MnO2 | Yes | 5 times. Turnover number not defined | 535 |
4.2 Solvent effects and impact on catalysis
While the synthesis of FDCA has been successful using water as a solvent, the oxidation is more challenging when other solvents are used. The use of ionic liquids or DMSO is desired since high yields of HMF can be achieved from sugars and cellulose (though separation is difficult), raising the possibility of a one-pot reaction which can lead to FDCA which can then be separated by water addition. Moreover these solvents can solubilize (protonated) FDCA at much higher concentrations than water, permitting process intensification and reducing catalyst deactivation.102,157 When performing the reaction in [bmim]Cl, an iron based catalyst proved to be highly recyclable because the impurities are soluble in the solvent, avoiding clogging of the catalyst active sites.546 Halide-based ionic liquids are preferred since they can solubilize both FDCA and humins. Cl− and Br− based ionic liquids are also cheaper since fewer reaction steps are required for their synthesis, whereas [OTf]− and [NTf2]− ionic liquids require a metathesis step (usually from a halide precursor) with expensive fluorinated salts. In addition, Cl− and Br− produce ionic liquids that are capable of dissolving cellulose and glucose, creating the opportunity to directly use a cheaper feedstock. On the other hand, the solubility of oxygen in these ILs is very low compared to those containing [OTf]− or [NTf2]− anions, which already show lower oxygen solubility than organic solvents or water.547 However the development of an efficient catalyst in these solvents is highly challenging because of the negative impact of the coordinating anions on the selectivity when noble metals are used. Seddon and Stark showed that the activity of a Pd-based catalyst is switched from oxidation to etherification in presence of Cl− ions.548 Other systems have been reported for the oxidation of alcohols in ionic liquids which mostly consist of homogeneous catalysts, and for the selective oxidation of alcohols to aldehydes, while only a few examples report oxidation to carboxylic acids. Moreover these catalysts do not possess a wide substrate scope and are rarely efficient towards furan compounds.549,550 One example is the synthesis of carboxylic acids with Ni(acac)2, which is an efficient catalyst for the oxidation of different benzyl aldehydes in non-coordinating ionic liquids with [BF4]− and [PF6]− anions.551 This is likely due to the formation of benzoperoxides due to radical formation of benzyl aldehydes under the exposure to light, which leads to the capture of molecular oxygen, forming a benzoperoxy radical which undergoes disproportionation, forming the carboxylic acid.552 Since these radicals are not formed for furan compounds, this catalytic system is not suitable for the oxidation of HMF.The oxidation of 5-HMF in coordinating ionic liquids has only recently been studied and only a few publications exist. Stahlberg and co-workers studied the oxidation of 5-HMF with RuOH on different supports.553 All catalysts demonstrated poor selectivity toward DFF and FDCA in most of the imidazolium ionic liquids studied. The best selectivity was achieved with [emim][OAc] under very harsh conditions (140 °C, 20 bar O2 pressure) obtaining 40% yield at full conversion after 24 h. This study demonstrated that while Ru proved to be an outstanding catalyst to oxidize HMF to both DFF and FDCA at high yields in organic solvents or water,445,491 this was not the case when ionic liquids were used, indicating a strong influence of the ions on the mechanism of the reaction. Moreover, it is well known that dialkylimidazolium acetate ionic liquids form carbenes above 60 °C which in this case could be the main species responsible for the oxidation.554 It has already been demonstrated in many reports that carbenes are very good catalysts for aldehyde oxidation,555,556 therefore the role of Ru in the system may be marginal. Leaching of the catalyst, separation of FDCA, low yield, harsh conditions, low HMF loadings and poor solvent stability are the main issues in this catalytic system, which represented the first reported example of HMF oxidation in ionic liquids. However, the work of Stahlberg553 and co-workers shows interesting insight regarding the catalytic oxidation in ionic liquids compared to conventional oxidation systems. While HMF is not stable at high temperatures in chloride-based ionic liquids, this degradation seems to be accelerated when the catalyst is added, indicating that a different reaction pathway is undertaken when the hydroxyl group is engaged in H-bonding with an anion.
Yan and co-workers designed a noble metal-free catalyst for the aerobic oxidation in ionic liquids of 5-HMF based on Zr and Fe oxides. The reaction was still restricted to modest yields (60%), low substrate loadings and harsh conditions (160 °C, 20 bar O2).546 The authors demonstrated that the Lewis acidity of the support does not have a significant impact on the reaction while the Lewis basicity impacts both the selectivity and reaction rate. A control study done by the authors showed that the rate limiting step is the oxidation of the alcohol moiety of HMF while the aldehydes are oxidized more rapidly. This suggests that a different mechanism is involved in non-aqueous environments at high temperatures. Likely, the low strength of the acid sites due to the absence of the noble metals indicates that the hydroxyl group of HMF is not adsorbed efficiently onto the catalyst surface while the strong basicity allows the adsorption of the aldehyde group (which is more electrophilic) without requiring a geminal diol. Another study performed by these authors revealed that, for this type of catalyst, Ce doping did not bring any additional benefit compared with other type of catalysts,557 indicating that oxygen mobility on the catalyst surface is not a limiting step as shown in other studies carried out in water.516 The authors developed a two-step reaction to produce FDCA directly from sugars. In the first step, fructose was converted to HMF with Amberlyst-15 as the catalyst, then the oxidation was performed in a second step with the Fe/Zr catalyst, achieving 46% yield overall. This represents an innovative chemical system where both reactions are performed with recyclable heterogeneous catalysts. The main drawback lies in the low concentration of HMF (0.1 M) which makes separation challenging. Moreover, the prospect of working at high loadings is limited by the harsh conditions used (160 °C) and the long reaction time (24 h) due to the pronounced instability of HMF under these conditions.
Significant improvements in catalyst performance in [bmim]Cl have been developed recently by Chen and co-workers, who used heteropoly acids based on Mo and V to achieve 89% yield of FDCA from HMF and 44% yield from glucose at 140 °C and 10 bar O2 in 6 h. The authors demonstrated a synergistic effect between the interaction of the cation and anion of the ionic liquid with the catalytic activity of the molybdenum centre.558 Though the catalytic activity is high, the catalyst suffers from leaching, losing activity after the third run. Moreover, the system still suffers from difficult product separation due to the low substrate concentration used (10 mg g−1).
All of the results reported in ionic liquids are HPLC yields, and product isolation was not attempted due to the low concentrations of FDCA achieved, making the separation infeasible. Recycling was demonstrated only for the catalyst and not the ionic liquid. Chen and co-workers558 proposed an in situ polymerization with ethylene glycol to form PEF directly. This option would be extremely beneficial since the resin is completely insoluble in water and therefore addition of only a small amount would lead to precipitation. However, polycondensation does not seem a feasible option since PEF requires high temperatures (>200 °C) whereas, in the case of ionic liquids, carbenes are often formed under these conditions, favoring depolymerisation.559 Recently, we reported a new system using [bmim]Cl with MnO2 which leads to high yield of FDCA starting from HMF. The main advantage lies on the formation of the imidazolium salt of FDCA which proved easy to separate by precipitation through addition of ethanol as antisolvent to be subsequently acidified into FDCA. This system allowed the implementation of a two-step process where first the sugars are converted into HMF and then converted into FDCA salt through addition of MnO2. Although high yields were achieved (over 85%) from glucose with efficient separation at low loading (<2%), the process suffers from catalyst leaching and deactivation further compromised by water removal in the intermittent steps which necessitates an expensive, high energy step to dry the ionic liquid.560,561
Other studies have exploited hydrophilic organic solvents for the dehydration of fructose and oxidation of HMF by regulating the water content. Dumesic and co-workers have designed a two-step, one-pot process using a mixture of water and GVL (50:50) as the solvent to perform both the dehydration of fructose and oxidation of HMF. The authors performed the dehydration by using FDCA as an acid catalyst and performed the oxidation using a commercial Pt/C at 40 bar O2 prior to removal of humins by activated charcoal. The product could be then recovered by cooling, precipitating most of the FDCA at high purity and recycling the solvent to perform a further cycle.562 Rathod and co-workers used a bifunctional Pd supported to sulfonated graphene to confer the acid functionality required to perform the dehydration followed by the addition of base and oxygen bubbling, obtaining 64% isolated yield but, compared with previous work, FDCA needed to undergo to extensive purification and highly dilute conditions were used.563
Other authors have focused on using DMSO instead of GVL since DMSO is more efficient for the dehydration of sugars. The use of dry DMSO for the oxidation step proved inefficient under aerobic conditions and a stoichiometric oxidant such as tertbutyl peroxide was needed to improve the yield using FeO/CoO as catalyst. However this approach, besides having the disadvantages of requiring stoichiometric oxidants, gave low yields of below 60%.564 A study by Chen and co-workers reported that Pt/C can be used to obtain high yields in a mixture of water/DMSO (3:1 in mass) with the further addition of K2CO3, showing good recyclability upon reduction with H2.565 A Ru catalyst supported by a Zn and Fe oxide spinel was reported as another active catalyst which can avoid the addition of base in the oxidation step. In this study it was shown that an optimum solvent composition of water/DMSO exists which can obtain high yields at short reaction times.566 In pure water, the catalyst displayed low activity while in dry DMSO the reaction was highly selective towards DFF. An optimum ratio of 2:1 gave the highest yield of FDCA, indicating that the presence of DMSO can favor the reaction depending on the catalyst used. Further studies need to be done to validate this concept and the role DMSO plays in the activity of the catalyst. Another work by Liu and co-workers reported that Ru/C can be used in combination with NaHCO3 in DMSO and water (2:1 in mass) to perform the oxidation of HMF at 20% loading with 65% yield.567 In this case higher oxygen pressure was used (40 bar).
4.3 Challenges in FDCA scale-up and feasibility
The synthesis of FDCA has been subject of extensive research, mostly focused on improving the catalyst design. In the last 3 years catalysts based on Ru, Co and Mn have proven to be the best performing, with neglectable metal leaching, high stability, high yield and high recyclability when water is used as the solvent. Enzymes also give high selectivity and yield but issues with recyclability arise due to the homogeneous nature of the catalyst and the separation of the final product by acidification which would denature the enzyme, deactivating it for further cycles. In addition, the kinetics are extremely slow (>10 h) reducing productivity. Immobilization of these enzymes has been successful but the enzymes still lose their activity after multiple cycles.541 Microorganisms could be a potential option since they can be engineered to resist substrate deactivation by expressing the active enzymes in the cells, but the productivity and rate need to be improved due to the long lag time (30 h) required due to the cytotoxic effects of HMF.568 Due to the high cost of the enzymes, much more research and development is needed to improve the recyclability of these catalysts.Most of the literature has focused on performing the oxidation in water starting from HMF, however this approach is limited by the necessity to isolate HMF, which has proven to be economically prohibitive.96 Dumesic and co-workers estimated that isolated HMF would have a minimum selling price of 1.4–1.7 $ per kg which makes it non-ideal for the synthesis of FDCA since the terephthalic acid price lies below 1.2 $ per kg.96 In order to meet the high purity of HMF required to avoid catalyst deactivation, low concentrations of fructose are used, increasing the cost for solvent evaporation.524 This also increases energy use and the CO2 emissions associated with the overall process. In one of our studies, we demonstrated that even at a fructose loading of 30% by weight and quantitative yield of HMF, the energy produced in the oxidation step would not be sufficient to satisfy the energy requirements for the solvent recovery, although a substantial reduction in selling price can be achieved.177 This problem is magnified when furfural is used as the feedstock since the carboxylation step doesn't release heat (reducing the potential for energy integration) in addition to the high temperatures required (>250 °C). Moreover, the Cs salt used during the reaction must be regenerated electrochemically, which represents another expensive and energy intensive unit. A detailed energy analysis is needed to better estimate the cost and CO2 emissions associated with the whole process.453
Further improvements in the production of FDCA can be achieved by converting HMF in situ in a two-step one-pot reaction, avoiding the expensive HMF isolation. Substantial reductions in both CO2 emissions and FDCA selling price can be achieved when using solvents in which high yields of HMF can be obtained, such as DMSO and ionic liquids. The main challenge lies in achieving sufficient oxidation activity in a non-aqueous environment since the catalysts studied for aqueous oxidation struggle in other solvents. Sugar dehydration is much more favored in non-volatile polar solvents such as DMSO and ionic liquids, while the oxidation is accelerated in a basic water environment. Moreover, the dehydration of sugars produces most of the side products which would deactivate the catalyst. Therefore, the optimal solvent system should permit purification without excessive loss of HMF. This remains a challenge; for example, Yi and co-workers showed that purification with activated carbon leads to excessive loss of HMF when using a mixture of GVL/water.524,562
DMSO has also been used to perform the dehydration and oxidation reactions but most often the reaction proved to be selective towards DFF rather than FDCA. Although DFF represents a valuable chemical which can subsequently converted into FDCA, its separation will determine the feasibility of this process, which is dependent on the solvent used and the cost to regenerate that solvent. Different reports have designed catalysts which are capable of synthesizing DFF in one step starting from fructose using solvents such as DMSO and DMAC.569,570 Once DFF is isolated, it can then be converted into FDCA in water or though the Amoco Mid Century process.571 Molybdenum oxide based catalysts are very efficient for the direct conversion of fructose into DFF and most studies focus on enhancing the stability of the molybdenum oxide since it suffers heavy leaching during the oxidation.572–574 These reactions are typically convened in high boiling point solvents such as DMSO, and water has a negative impact on the selectivity since it leads to overoxidation to FFCA, necessitating dilute conditions (<10% weight) which increases the solvent regeneration energy, already intensive due to their high hydrophilicity. The problem of this separation can be bypassed by performing the reaction in ionic liquids since DFF can be easily separated by sublimation at low temperatures (<80 °C) and one can therefore achieve a highly pure product at low cost.157 A system based on TEMPO-CuCl in non-coordinating ionic liquids [bmim][OTf] and [bmim][NTf2] demonstrated good selectivity from HMF and fructose and good isolated yield, but with the drawback of catalyst deactivation after the first cycle. Further studies need to be address the stability of the catalyst to enable recycling of the solvent, which is the key point for the production of DFF in an economical and sustainable manner.159
Regulating the water content can switch the selectivity towards FDCA when DMSO is used but generally the kinetics proved to be slower and conversion limited to FFCA since the equilibrium constant for the geminal diol for this compound is low. Moreover, the separation of the final product is tedious mostly due to the high solvation ability of DMSO. Gajula and co-workers proposed water as antisolvent for the separation of FDCA from DMSO, however large amounts of water are needed requiring a highly concentrated solution of FDCA to achieve a recovery over 90%.102 To date these high concentrations of FDCA are unachievable in DMSO since low fructose concentrations are needed (often less than 2%) to limit the formation of humins and the degradation of HMF at high temperatures (which is enhanced at high substrate concentrations).565,566 Liu and co-workers were able to achieve a high yield of FDCA from fructose (65%) starting with a fructose concentration of 19% but separation was still not possible since their system requires a base which substantially increases the solubility of FDCA in water.567
Oxidation in ionic liquids is still at a proof-of-concept level because of the challenges encountered in the catalysis and the inability to use noble metals due to the negative influence of the anions on the selectivity plus the high solvation ability of the ionic liquids towards metals which causes leaching. Trials in [bmim]Cl using polyoxomethylates and Fe/Zr oxide catalysts were done at low substrate loading (<12 mg g−1), making product recovery infeasible.157
4.4 Biobased adipic acid
Various research have been done for the production of adipic acid through hydrogenation of FDCA followed by ring opening (Fig. 18).575 Baek and co-workers576 studied a ruthenium catalyzed reaction over different supports using water as solvent. The metal support has been selected to maximize the yield of THFDCA and reduce the leaching of ruthenium, while the ring opening is performed in a second step. The THFDCA is isolated through solvent extraction and crystallization followed by ring opening in a second pot. Upon the observation that using HI as catalyst, higher selectivity are observed towards ring opening compared other mineral acids, a sulfonated ionic liquids with iodide anion has been designed reaching yields over 99%.576,577 While this approach can guarantee high overall yield with high recyclability of both catalyst and ionic liquid, the implementation of a one-step process will be highly beneficial to avoid complex separation techniques which will strongly hinder the scalability of the process. Another approach have implemented a one pot process designing a multifunctional catalyst based on Pt supported on NbO2 obtaining 40% yield.578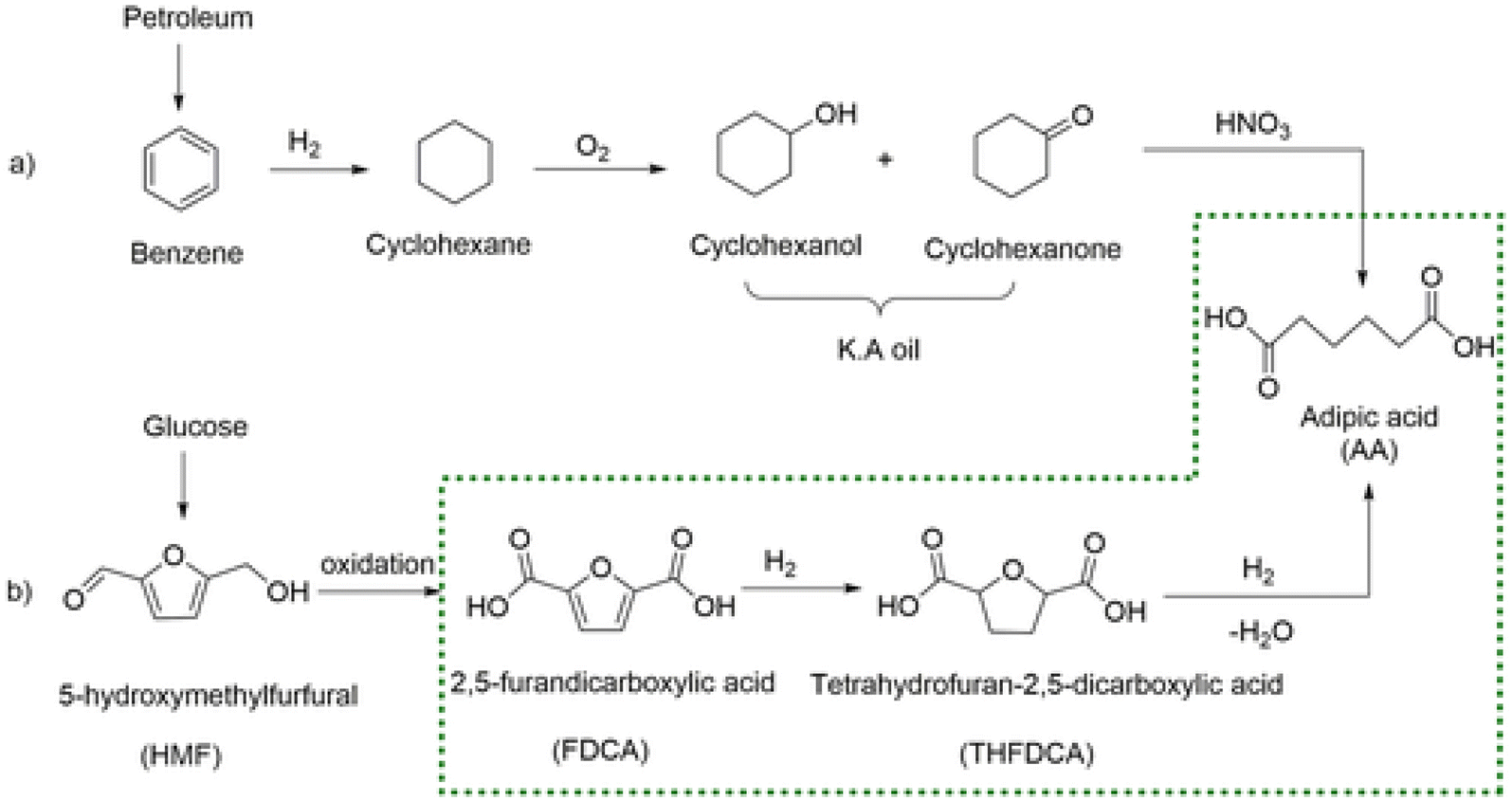 | ||
| Fig. 18 Catalytic path to produce adipic acid using FDCA as substrate. Reported by Baek and co-workers.576 | ||
The hydrogenation of FDCA to produce adipic acid has been a topic of few studies in the last 5 years and further improvements need to be implemented in understanding the main factors involved in the selectivity of this transformations. Moreover, such reaction path is strongly penalized by the isolation of HMF and the oxidation into FDCA which makes this option far from implementation.
5 Furan based surfactants as replacements for LAS
Surfactants are one of the most used commodity chemicals, commercialized mostly in cleaning products which account for approximately 60% of worldwide surfactant production.579 The remainder find large-scale application in many chemical sectors such as oil & gas recovery, paint, food, emulsifiers for polymerization and niche applications in pharmaceuticals.580,581 The design of a surfactant is directly tied to the application, which defines the structure of the hydrophobic and hydrophilic groups. The most commercialized surfactants today are the alkylbenzene sulfonates used mostly in cleaning products, oil & gas recovery and paint. These are produced through the Friedel–Crafts alkylation of benzene with primary or secondary alkenes.582,583 The choice of the alkyl chain has been a frequent topic of research and development since it strongly influences the environmental impact of the surfactant. Originally these were produced with a branched alkyl chain (BAS) which proved to be more efficient in cleaning, but due to their low biodegradability they have been substituted with linear alkyl chains (LAS) which are more biodegradable and can achieve the same performance as BAS. Their wide use is attributable to their low price and high performance. This is compounded by the wide R&D done in developing formulations for LAS to boost performance in a defined environment. Indeed, these surfactants exhibit limitations in their pure form due to low solubility in water, high Kraft Point (minimum micellization temperature) and low resistance to hard water. Different compounds have been developed to overcome these issues, including hydrotropes, builders and chelants which keep the surfactant in the liquid phase and guarantee good cleaning ability in a mineral rich environment.57The petrochemical nature of LAS has pushed researchers into finding alternative solutions which can use biobased alternatives to this molecule. Most commercial solutions today are based on the formulation of cleaning products which use the biobased sodium dodecyl sulfate (SDS) as a surfactant which has more benign environmental credential in terms of biodegradability, toxicity and environmental impact. However, the performance of these greener products does not match LAS formulations and results in higher costs due to the higher price of SDS. This limits the expansion of this market which is mostly driven by low price and high performance.
One of the main reasons for the low performance of SDS is the lack of an aromatic moiety which generally gives lower critical micelle concentrations, lower surface tension between water and oil and higher affinity to hydrophobic substances. The substitution of benzene with furan rings represents a valid alternative for this purpose which can deliver better green credentials and fill the gap of performance between SDS and LAS. The surfactants need to be stable and provide a synthetic approach that can be scalable and avoids complex purification techniques and high operating costs. The synthesis of a sulfonated surfactant proceeds through two main steps: (1) synthesis of the hydrophobe and (2) sulfonation. The first step generally requires a catalytic approach while the sulfonation step is done using a sulfonating agent. At large scale the sulfonation is carried out in a falling film reactor using gaseous SO3 in sulfonating plant. This method has proven to be the standard for the manufacture of all sulfonated surfactants and results in lower operating costs since SO3 is a cheap sulfonating agent and can be easily removed from the final mixture, reducing the amount of residual salts which are unwanted in a formulation.584 Further aspects such as regulation of the final colour and salt content make this technique the most desired one for this application.585 The compatibility of the hydrophobe with this technique is crucial for the commercial success of a new sulfonated surfactant since all other sulfonating agents are more expensive, requiring complex separation and purification techniques which undermines the feasibility of the process. However, this aspect is not covered in the literature since it requires large amounts of hydrophobe and a specific expensive set-up to generate SO3; therefore most new surfactant syntheses reported use other sulfonating agent such as NaHSO3, chlorosulfonic acid, oleum or a pyridine SO3 complex, which are not representative of a true process. Therefore, this aspect will be neglected in this review, which will mainly focus on the synthesis of the hydrophobe.
Recently a review on furan based surfactants has been published which well summarizes the different paths to produce anionic, cationic and amphoteric surfactants.586 In this section we will focus exclusively on those that can replace LAS which is a BTX derivative.
5.1 Properties and environmental impact of furan-based surfactants
The introduction of a new surfactant requires two distinct sets of evaluation based on the performance and the environmental impact.The utilization of furan-based surfactants has shown different advantages concerning the basic properties even if a large impact has been observed according to the type of linker used. Dauenhauer and co-workers have shown that the LAS analogue linear alkyl furan sulfonate exhibits similar CMC values as LAS and superior performance in terms of resistance to hard water, not exhibiting any precipitation at high calcium concentrations.57 Moreover, a Kraft Point of 30 °C was measured, indicating a better suitability of this surfactant for cold water applications. By substituting the linker with a ketone group the Kraft Point could be further improved, reaching values below 0 °C but the CMC increased to over 10000 ppm which makes it unsuitable for cleaning applications. A good compromise could be obtained by using an ester linkage. In one of our recent publications, we reported that sulfonated alkyl furoates (SAF) exhibit Kraft points lower than 0 °C in combination with low CMC and high resistance to hard water, making this surfactant ideal for cleaning at low temperatures and in hard water with the prospective of delivering the same performance as LAS in cleaning applications. This can lead to enormous advantages in the final formulation since it would eliminate the need for chelants and builders. Ethylenediaminetetraacetate (EDTA), sodium tripolyphosphate (STPP) and sodium nitrilotriacetate, which are typical additives to chelate calcium, exhibits toxicity to human and aquatic life.587–591 However, the pH range of these surfactants is limited due to the risk of hydrolysis. Other attempts have been made to synthesise the ether linker analogue but these proved to be unsuccessful due to the instability of the hydrophobe during sulfonation which leads to complete degradation of the product.
Another approach reported the use of furfural to produce the building block 5-hydroxyfuranone through oxygenation with UV light followed by etherification. The final surfactant exhibited CMCs similar to LAS with the further advantage of being more biodegradable.592
Not many studies have considered the environmental impact of these surfactants but a careful analysis needs to be carried out concerning their biodegradability and the produced metabolites. It has already been demonstrated that these surfactants are biodegradable, reaching about 70% biodegradation under aerobic conditions in 28 days for a chain length of 12 carbons, which is compared with about 60% for LAS.593 However from a waste-water treatment point of view it is highly desirable that most of the organic compounds are anaerobically digested, since biogas is the main added value vector of WWT. LAS not only are incompatible with anaerobic digestion but have the potential to exhibit an inhibitory effect on the anaerobic bacterial community.594
In one of our recent publication, we showed that furan surfactants exhibit limited toxicity towards E. Coli and protozoa, about 50% less than LAS, representing a potentially more environmentally benign product.595 One of the main advantages of the furan-based surfactants is the biodegradability of the furan head group when compared with benzene.
Although it has been largely demonstrated that both LAS and SDS are readily biodegradable by aerobic bacteria,596 different considerations have to be taken according to the surfactant resistance to the hard water since additives are designed to not precipitate the surfactant during its application nor when is diluted in a rich, diversified environment. This can cause the surfactant to precipitate before entering the digestors with a higher chance to be dispersed in the environment. This was reported by studies of raw sewage which reported very high concentrations (up to 35%) of LAS in primary settling tanks where insoluble materials are filtered before entering the digestors.597,598 Moreover, since LAS are not bioderived they have a positive net CO2 emission after aerobic digestion. One factor that can affect the biodegradability of the surfactant is the linker. Ester cleavage can be achieved by anaerobic digestion and fatty alcohol digestion has been well demonstrated, providing advantages for sulfonated alkyl furoate over LAS.599,600 Moreover, furfural has been demonstrated as a highly compatible feedstock in anaerobic digestion, indicating that the furan ring can be digested in an efficient way, contrary to the benzene ring characteristic of LAS.55,56 The anaerobic degradation of furan surfactants with ester linkage is therefore likely superior to LAS.
5.2 Synthesis of furan based surfactants
Optimization of the production of furan surfactants has been the topic of few studies. The synthesis of different furan based surfactants have been reported, passing through the intermediate dodecanoyl furan.57,601 Currently three different pathways have been reported for the synthesis of this intermediate, as reported in Fig. 19.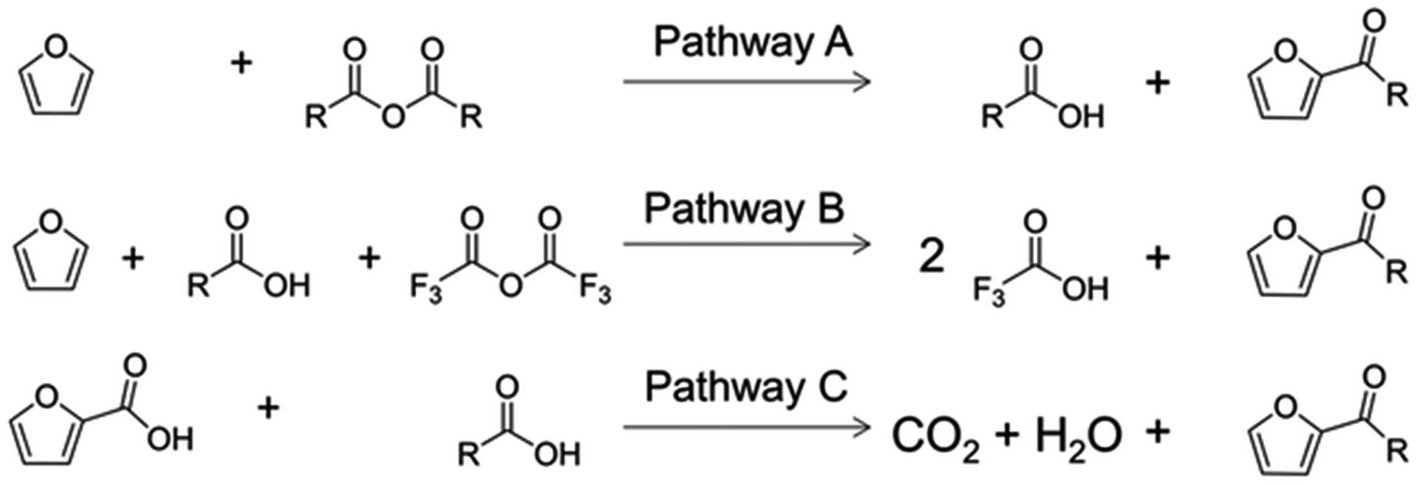 | ||
| Fig. 19 Three different pathways to produce dodecanoyl furan reported in the literature. | ||
Pathways A and B each proved to be very efficient with yields over 90%, carrying out the first reaction with a solid acid catalyst (such as a zeolite) and the second with trifluoroacetic anhydride. Pathway A is preferred due to its more efficient atom economy but it requires high temperatures (180 °C), while Pathway B can be performed at room temperature. Recently the use of a metal catalyst based on Fe3O4 has been studied (Pathway C) but the reaction requires high temperatures (>200 °C) and suffers from low yields (<40%).602
Other research reported the utilization of HMF as aromatic building block. The alcohol group can be etherified directly from fructose by using an ionic liquid as acid catalyst and an excess of fatty alcohol (20:1 in mol) to form the hydrophobic part of the surfactant while the aldehyde can be sulfonated with NaHSO3.603,604 However, these surfactants proved to have low solubility with very high Kraft Point (over 70 °C) with strong instability of the sulfonated aldehyde head.603,604
A study from Hoffmann and co-workers592 has developed a new synthetic route to produce a highly biodegradable surfactant using furfural as building block. The process involves the conversion of furfural through UV assisted oxidation into hydroxyfuranone which is subsequently alkylated using a fatty alcohol forming the hydrophobic part of the molecule. This is then sulfonated with NaHSO3. The final molecule proved to have similar CMC of LAS and high biodegradability. However, these routes have the main drawback that UV technologies are not scalable and the sulfonation on the aldehyde group of HMF leads to instability of the final product.
Recently we reported the synthesis of the furoate surfactants which proved to be highly scalable. In our work we used furoic acid and a fatty alcohol as feedstock which can be esterified at 150 °C at yields over 95% with the utilization of low concentrations of sulfuric acid (1%). The main advantage of this approach is the lack of purification steps. A technoeconomic assessment of the proposed process indicated that the operating costs associated with the synthesis of the hydrophobic part of the molecule are competitive for producing a low cost surfactant but this will be strictly dependent on the production cost of furoic acid which large scale production is not in place yet.595,605
Other studies have been reported to produce biobased LAB through cycloaddition of furan with dodecene through but further studies are needed to improve the selectivity and recyclability of the catalyst, in addition to design an efficient process for the decarboxylation of furan.606
5.3 Perspective of furan surfactants
The implementation of furan-based surfactants still requires intensive R&D regarding application development and process design. Ultimately, the success of these molecules in the market is strongly dependent on the technoeconomic feasibility in producing the furan head group. Therefore the design of efficient processes to produce furfural and HMF from biomass is crucial to minimize the operating costs and improve the competitiveness against LAS.607 In the short term, reaction paths which utilize furfural have better prospective to be economically feasible since the production of this intermediate is already implemented in the supply chain efficiently. However, the conversion into intermediate chemicals, such as furoic acid and furan, still need to be optimized and important aspects in catalyst recyclability and waste generation need to be addressed.Characterization of the basic proprieties (such as CMC and Kraft Point) of furan-based surfactants have been carried out extensively in literature, but application development has still much room of improvement in the field of formulation design. Indeed, the compatibility of furan surfactants within a formulation (cleaning, agricultural, lubricant, etc.) still need to be validated with further assessment on the physical proprieties and phase diagrams. Further evaluation on the environmental credentials and biodegradability is required to complete the sustainability profile of these molecules.
6 Maleic anhydride
Maleic anhydride (MA) is an important chemical intermediate in the production of many commodity chemicals and materials with a total annual production of 500000 tons per year. Half of the MA produced worldwide is used for unsaturated polyester resins which find application as reinforcement materials for automobile, marine craft, buildings etc. The second most used application (about 10%) is for the synthesis of agrochemicals used as pesticides, plant growth regulators and herbicides. Other minor applications are use as flavoring agents (fumaric acid) and lubricant additives.608,609 It is commercially produced today mainly through the oxidation of benzene or butane at high temperature (over 450 °C) over vanadium based catalysts. The use of benzene leads to higher selectivity but has less favourable atom economy.610,611 Butane has been implemented as a cheaper and more atom economical option compared with benzene, but this reaction still struggles to achieve yields, in excess of 60%.611,612 Both technologies have been optimized over the years to improve the recovery of the heat of reaction which requires specific reactor designs due to high exothermicity and product purification, which requires the separation and recovery of several different side products.613–615
In contact with water, maleic anhydride undergoes equilibrium conversion to maleic acid. In literature some authors consider the combined synthesis of the two forms a useful path of optimization, however maleic acid has a very niche market while maleic anhydride is a building block for much different (and larger scale) applications.
Biobased maleic anhydride can be produced from both linear or aromatic feedstock which can be functionalized or unfunctionalized.616 The utilization of furan based chemicals can lead to multiple chemical routes to produce maleic anhydride. Most commonly furan, HMF and furfural are used to lead to maleic anhydride, however each proceeds through different approaches which are based on gas or liquid phase oxidation.617 Furfural is the preferred substrate since it is the cheapest furan building block that can be obtained from biomass and has a more favourable atom economy compared with the petrochemical-derived analogue benzene.
The production of biobased maleic anhydride opens the possibility of producing its derivative phthalic anhydride through Diels–Alder condensation with furan, but the feasibility of this process is dependent on the cost of furan which is derived from decarboxylation of furfural.
Studies regarding the synthesis of maleic anhydride are quite limited but they proved to be efficient by readopting the conventional system used for the oxidation of benzene and butane.618
6.1 Phase influence
Technoeconomic assessments have indicated that the processes which use solvents limit economic feasibility while the gas phase reaction is both more viable and more competitive, capable of delivering a price close to the petrochemical route.619 When HMF is used, only liquid phase oxidation can be done due to the high boiling point of this compound, while furan and furfural can be applied in a gas phase reaction, replicating the conventional benzene oxidation. The influence of the solvent on the liquid phase oxidation has proven to have a determinant role in the selectivity, particularly the water content which controls the hydration of the maleic anhydride to maleic acid, a reversible reaction that can be corrected in the downstream process. Studies have demonstrated that the total maleic content can be increased by about 15% if it this equilibrium reaction is allowed to happen (suggesting instability of the anhydride form), however this would lead to further costs of purification, making the selective conversion to the anhydride more attractive.620 The optimum solvent to carry out such reactions is difficult to assess and is related to the type of catalyst used and the interactions between the catalyst and solvent. However due to thermodynamic reasons, these types of oxidations performed in water would lead to maleic acid, while a water-free condition would lead to the anhydride.621 Oxidations performed with VO(acac)2 showed a huge solvent effect going from 60% when acetonitrile was used to 3% in DMSO, which may be related to a hypothetical radical mechanism.622 Acetic acid was also analysed as solvent and co-solvent with acetonitrile for different catalytic systems. In combination with acetonitrile it has the effect of hydrolysing the maleic anhydride to the acid form which would increase the total combined yield. However, these systems would necessitate expensive separation techniques which undermines the feasibility of the approach. The gas phase oxidation therefore provides some advantages even if high temperatures are required (generally over 300 °C). Under these conditions, the reaction proceeds stepwise through decarboxylation to furan and oxidation to furanone, resembling the oxidation of butane which passes through the furan intermediate as well (Fig. 20).623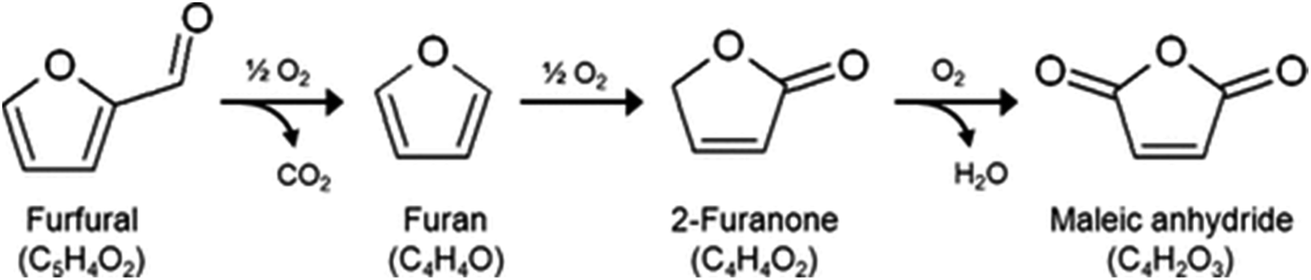 | ||
| Fig. 20 Reaction scheme of the gas phase oxidation of furfural to maleic anhydride. | ||
6.2 Catalyst development
Catalyst development conducted to date has mostly followed the same approach as the benzene oxidation processes. Here vanadium oxides are the active species for the oxidation, while molybdenum acts as a moderator of the active sites, regulating the amounts of V5+ and V4+.624 Specifically, the catalyst should favour the C–C cleavage rather than the selective oxidation of the alcohol and aldehyde. This is highly dependent on the solvent used; indeed vanadium based heteropolyacids favour the oxidation of HMF into FDCA when ionic liquids are used but towards maleic anhydride when in acetonitrile.620 Pure V2O5 or supported (with SiO2 or Al2O3) performs well for both the liquid (in acetic acid) and gas phase oxidation in terms of yield (over 70%) and recyclability with minimal impurities for both HMF and furfural.622,625 Other forms of vanadium such as molybdenum vanadate are highly active with an optimum ratio between molybdenum and vanadium III–IV but are poorly recyclable.626 Work aimed at boosting the activity of the catalyst by supporting the vanadium oxide on functionalized supports such as graphene oxide with Schiff base results in near quantitative yields on the first cycle starting from HMF, but the yield dropped quickly over recycles. Xu and co-workers have shown that the formation of maleic anhydride is favoured at high pressure and an optimum temperature of 90 °C to maximise the yield against the parallel alcohol oxidation to diformylfuran (DFF).627 The formation of MA at such low temperatures indicates that a different mechanism is involved in this case since C–C cleavage requires more energy compared the selective oxidation of an alcohol group, further suggested by the formation of formic acid as a side product. A kinetic study done by Yin and co-workers suggested that the mechanism undergoes first to a the disproportionation reaction where the hydroxyl group of HMF is converted to formic acid, then the decarbonization of the aldehyde leads to CO.6207 Diesel–Alder reactions
7.1 Phthalic anhydride
Phthalic anhydride (PA) it is an important building block for the production of plasticizers for PVC, and it is has been defined as the ‘most important product for the large scale organic synthesis’ with volumes that reached 3 million tons per year.60 Originally produced from naphthalene, this compound is generally produced by obeying the same principles as the benzene oxidation to MA. Most processes today use the BTX derivative o-xylene as feedstock due to its lower price and better atom economy.628 The most used commercial catalyst for this purpose is V2O5/TiO2 at temperatures between 280–360 °C.629–631The bio-route to obtain phthalic anhydride differs from the conventional petrochemical approach since it is strongly dependent on the economic feasibility of the production of MA.632 A green pathway to produce PA was reported by Mahmoud and co-workers who studied the cycloaddition of MA with furan to form an oxanorbornene intermediate followed by a dehydration to obtain PA (Fig. 21).633 In this reaction path the authors were able to carry out both steps at low temperature under solvent free conditions, producing the intermediate at high yield (81%) followed by the addition of mixed sulfonic-carboxylic anhydride as catalyst to obtain PA at a total yield of 80%.
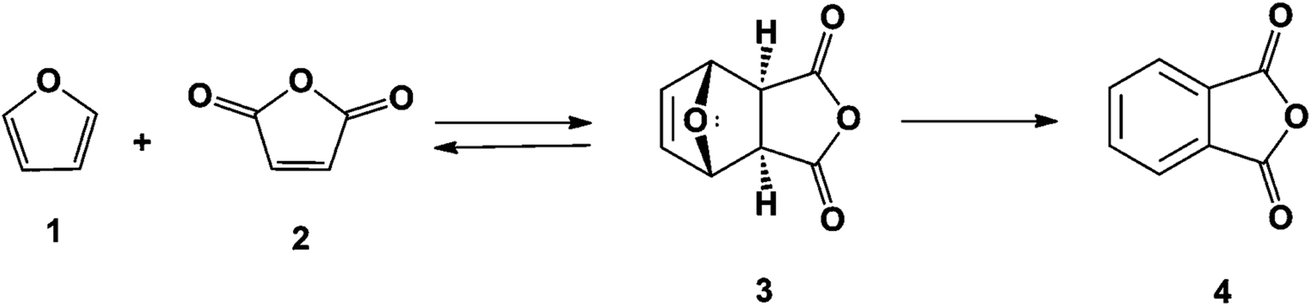 | ||
| Fig. 21 Reaction path to produce biobased phthalic anhydride as elaborated by Mahmoud and co-workers.633 | ||
Other studies have shown that these products can be obtained through a one step process starting from HMF. Jia and co-workers have developed a catalytic system based on MoO3 and Cu(NO3)2 achieving about 63% yield using K2S2O8 as stoichiometric oxidant.634,635
The production of biobased PA still needs much research as very few publications are reported for an efficient and competitive chemical route to produce this chemical at large scale. The path reported by Mahmoud and co-workers633 has the potential to produce high yields of PA but the process is strictly limited by the cost of production of maleic anhydride and furan and the separation, which still needs to be validated. The production of biobased furan can be done efficiently by decarboxylation of furfural but generally its production cost compared with the petrochemical routes needs to be evaluated.
A one step process can strongly favour the complexity of the process but currently the reaction has limited substrate scope (HMF). More environmentally friendly paths need to be developed starting from furfural using molecular oxygen as a more benign oxidant.
7.2 Biobased terephthalic acid
The implementation of furan-based products in the supply chain involves the implementation of different approaches in process design and performance evaluation of the final product. The production of building blocks that have the same structures compared the oil-based ones will allow an easier implementation within the current chemical processes without the need to establish new technologies which can result in high capital cost. For this reason, the establishment of a process which can produce the same molecules as the ones conventionally used, can simplify the large-scale production of biobased chemicals. Different reports have studied the synthesis of biobased p-xylene from furan compounds through Diesel–Alder reaction with the final aim to produce biobased terephthalic acid for PET production.636 The production of p-xylene will allow the utilization of the current Amoco Mid Century process to produce the final desired molecule (Fig. 22a).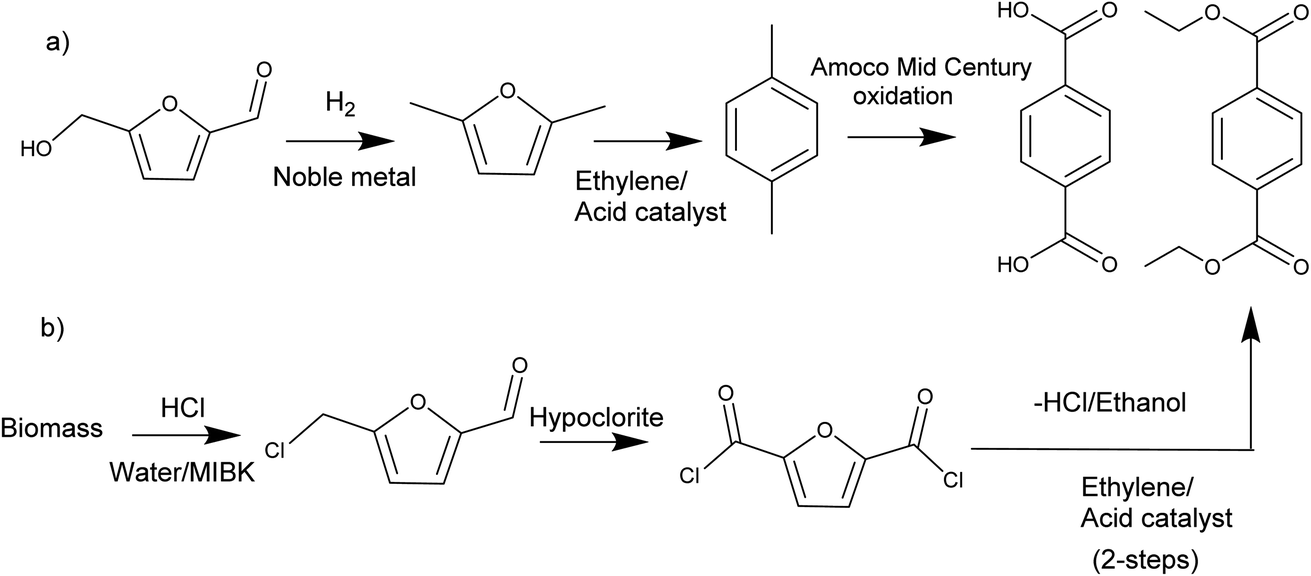 | ||
| Fig. 22 Two different reaction scheme to produce biobased terephthalic acid. | ||
In order to convert furan rings into aryl ones, electron donating functional groups attached to the furan ring are needed are thermodynamically preferred, therefor HMF and furfural are not suitable substrate for this scope.637,638 Much of the literature use instead dimethyl furan (DMF) as reactant with ethylene, developing a catalytic/solvent system to maximise the selectivity.638 The role of the catalyst is to perform efficiently the dehydration of the intermediate oxa-norbornene, therefor an acidic character is needed to carry the reaction at high yield.639,640 Different catalysts with Lewis and Brønsted acid characters proved to be suitable for this purpose guaranteeing high yield of reaction at a temperature range between 200 to 300 °C.641,642
While this transformation proved to be extremely efficient reaching yields over 80% at different conditions,643,644 different aspects need to be taken into account to assess the environmental sustainability and process economics. Indeed, these processes are strictly dependent on the production of DMF, which feasibility is still related to the isolation of HMF notwithstanding both hydrogenation and cycloaddition reaction can be carried out efficiently. This aspect needs further consideration since hydroconversion processes proved to be strongly affected by traces of solvents used for the sugar dehydration (such as DMSO), which makes the purification step more complex and expensive.645 Moreover, since p-xylene is used for the terephthalic acid production, the overall process will require a reduction followed by an oxidation with the Amoco-Mid Century process which can result in an energy inefficient process. A technoeconomic analysis with an assessment of the carbon impact is required to validate the environmental credentials.
A different chemical path was reported by Mascal and co-workers (Fig. 22b) using 5-chloromethyl furfural (CMF) as building block. The main advantage of this molecule is that it can be produced directly from biomass by simple treatment with HCl in a biphasic system.646,647 CMF can be isolated from the organic phase and isolated efficiently thanks its stability at high temperature.648 This opens the prospective towards new oxidation methodology which can avoid the reduction step. The oxidation of CMF into 2,5-furandicarbonyl chloride can be carried out efficiently using hypochlorite649 allowing to perform efficiently the Diesel–Alder reaction650 prior treatment in ethanol, ultimately producing terephthalate ester without passing through p-xylene. The main advantage of this route is that avoids defunctionalisation of the functional groups which results in an overall more energetically favourable process.
7.3 Ethyl benzene
Different studies have been done to explore the replacement of ethyl benzene through the usage of furan-based building blocks. Difficulties lies on the alkylation of the furan ring using ethylene which doesn't obey the same mechanistic principles of aryl ones. For this reason, one of the approaches is the conversion of furan into benzene with further alkylation with biobased ethylene through conventional Friedel–Crafts process.651 This inevitably implies the decarboxylation of furfural into furan which can be carried out using metal oxides in the vapor phase.652 Each reaction step has been studied separately in literature and an overall implementation of the process seems achievable. However, the conversion of furan into benzene using ethylene didn't prove to be efficient due to the lack of substituents, therefore low activation of both substrates led to poor selectivity for this reaction.653 A study reported a one step process to produce an alkyl furan using ethanol and furan as reactants. Specifically, the usage of ethanol instead of ethylene at an optimum ratio 10:1 compared furan, reduces the formation of benzofuran, which is the main side product of the reaction. The reaction is carried in the gas phase over an optimized zeolite catalyst reaching a selectivity over 60%.654 The advantages in carrying such reaction path is the lower costs in the feedstock, since the dehydration of ethanol is performed in situ, and it avoids the dependence on the Friedel–Crafts alkylation (Fig. 23).
 | ||
| Fig. 23 Reaction paths to produce ethyl benzene reported by Tsang and co-workers.654 | ||
No further studies are reported to produce ethyl benzene using furan-based building blocks. Improvements in this field relies in the optimization of each reaction step to maximize the yield and the identification of the main limiting steps of the whole process in terms of yields and separations.
8 Conclusions
The utilization of furan-based compounds for the substitution of the petroleum based BTX fraction has been a popular topic of R&D over the last 30 years. Studies have proven that substitution of the aryl moiety of some commodity chemical products such as PET and LAS with furan-based alternatives can lead to final products that have superior performance compared with the petrochemical analogues, giving the prospect of establishing a product that is both greener and better, reducing CO2 emissions associated with the end-life-usage. In particular, FDCA has been extensively studied but the research approaches have been based solely on catalyst development rather on the optimization of the whole process in terms of separation. The adoption of the aerobic oxidation in water can be feasible if backintegration of HMF production from sugars is in place, decreasing the costs of isolation and purification of HMF. However, side products produced during the sugar dehydration needs to be taken into account since humins are a poison for most of the catalysts developed. The production of HMF from sugars and cellulose has been widely studied reporting a strong effect of the solvent on the selectivity of the process. Also in this case, very few studies consider the difficulties in separating HMF in the reaction mixture, focusing mostly on the yield of reaction and catalyst/solvent design. The separation in DMSO and ionic liquids still remains a question not completely answered while the only viable route is with water/organic solvents even if this suffers from low versatility in terms of substrate scope, since is inefficient when glucose or cellulose are used.The production of the main furan feedstock is the key to produce all the downstream processes. The production of furfural in large scale has been in place for a century but only a few significant developments have been achieved. The processes consist in stripping the furfural out with steam at high pressure. The remaining residue is used for heat and power production. Current furfural plants have a maximum capacity of 50000 tons per year but the production struggles to expand due to low demand of this compound which currently finds application in low volume polymers and solvents. Current research currently is increasing the application of furfural derivatives in surfactants and maleic anhydride. The latter proved to be compatible with the vanadium based catalysts used for the benzene and butane oxidation, but more energy is required to vaporize the feed due to the higher boiling point of furfural.
The efficient production of maleic anhydride from furfural has opened the possibly to scale-up this molecule with the further integration of phthalic anhydride, which can be synthetized at low temperature. However the feasibility of production of furan from furfural and its environmental impact needs further examination since it can be a limitation for the whole process.
Different studies have been reported for the production of furan based surfactants. The feasibility of the production of furfural derivatives such as furan and furoic acids also needs to be studied. The introduction of furan surfactants in the market can lead to different advantages in terms of green credentials and performance but currently this route is limited by the economical production of the building blocks. Research has focused extensively on the development of catalyst and reaction conditions that can achieve high yield, but further studies needs to be addressed on the isolation of the final product, an aspect which is going to determine the success of any furan technology.
Conflicts of interest
There are no conflicts to declare.References
- J. Rogelj, A. Popp, K. V Calvin, G. Luderer, J. Emmerling, D. Gernaat, S. Fujimori, J. Strefler, T. Hasegawa, G. Marangoni, V. Krey, E. Kriegler, K. Riahi, D. P. Van Vuuren, J. Doelman, L. Drouet, J. Edmonds, O. Fricko, M. Harmsen, P. Havlík, F. Humpenöder, E. Stehfest and M. Tavoni, Nat. Clim. Change., 2018, 8, 329–333 CrossRef.
- P. Erickson, M. Lazarus and G. Piggot, Nat. Clim. Change., 2018, 8, 1037–1043 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- S. L. Stattman, A. Gupta, L. Partzsch and P. Oosterveer, Sustain, 2018, 10, 1–17 CrossRef.
- S. N. Jogdand, Current Status of Bio-Based Chemicals, 2014 Search PubMed.
- B. Kamm and M. Kamm, Appl. Microbiol. Biotechnol., 2004, 64, 137–145 CrossRef CAS PubMed.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1427–1445 CrossRef CAS.
- J. P. M. Sanders, J. H. Clark, G. J. Harmsen, H. J. Heeres, J. J. Heijnen, S. R. A. Kersten, W. P. M. van Swaaij and J. A. Moulijn, Chem. Eng. Process.: Process Intensif., 2012, 51, 117–136 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- D. R. Doods, R. A. Gross, D. R. Dodds and R. A. Gross, Science, 2007, 318, 1250–1251 CrossRef PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- F. Cherubini, Energy Conv. Manag., 2010, 51, 1412–1421 CrossRef CAS.
- F. Cherubini and A. H. Strómman, Energy Fuels, 2010, 24, 2657–2666 CrossRef CAS.
- L. Shen, J. Haufe and M. Patel, Market Study on Succinic, Itaconic Acid and FDCA, Final Rep., 2011, 1–173 Search PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- A. L. Marshall and P. J. Alaimo, Chem.–Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed.
- S. Sarkar, A. Kumar and A. Sultana, Energy, 2011, 36, 6251–6262 CrossRef CAS.
- Ó. Ögmundarson, M. J. Herrgård, J. Forster, M. Z. Hauschild and P. Fantke, Nat. Sustain., 2020, 3, 167–174 CrossRef.
- Z. Fang and R. L. Smith, Production of Platform Chemicals from Sustainable Resources, 2017 Search PubMed.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- H. H. Khoo, W. L. Ee and V. Isoni, Green Chem., 2016, 18, 1912–1922 RSC.
- Asian aromatics, derivative trades moderate after crude-led spike, ICIS, https://www.icis.com/explore/resources/news/2016/03/11/9978050/asian-aromatics-derivative-trades-moderate-after-crude-led-spike/, accessed 20 April 2020 Search PubMed.
- Sugar – Monthly Price – Commodity Prices – Price Charts, Data, and News – IndexMundi, https://www.indexmundi.com/commodities/?commodity=sugar%26months=120, accessed 20 April 2020 Search PubMed.
- E4tech, Re-Cord and Wur, From the Sugar Platform to Biofuels and Biochemicals, 2015 Search PubMed.
- Top Value Added Chemicals from Biomass: Volume I-Results of Screening for Potential Candidates from Sugars and Synthetic Gas, ed. T. Werpy and G. Petersen, National Renewable Energy Laboratory (NREL), Pacific Northwest National Laboratory (PNNL), 2004 Search PubMed.
- S. Takkellapati, T. Li and M. A. Gonzalez, Clean Technol. Environ. Policy, 2018, 20, 1615–1630 CrossRef CAS PubMed.
- M. Padella, A. O’Connell and M. Prussi, Appl. Sci., 2019, 9(21), 4523–4536 CrossRef CAS.
- S. Parsons, M. C. McManus and C. M. Taylor, in Greenhouse Gas Balances of Bioenergy Systems, Elsevier Inc., 2018, pp. 193–206 Search PubMed.
- A. V. Gusakov, Biofuels, 2013, 4, 567–569 CrossRef CAS.
- C. S. Lee, M. K. Aroua, W. M. A. W. Daud, P. Cognet, Y. Pérès-Lucchese, P. L. Fabre, O. Reynes and L. Latapie, Renewable Sustainable Energy Rev., 2015, 42, 235–244 CrossRef.
- A. Mazière, P. Prinsen, A. García, R. Luque and C. Len, Biofuel Bioprod. Biorefin., 2017, 11, 908–931 CrossRef.
- J. A. Moulijn, M. Makee and A. Van Diepen, Chemical Process Technology, Wiley-VCH, 2013 Search PubMed.
- S. Al-Khattaf, Chem. Eng. Process.: Process Intensif., 2007, 46, 964–974 CrossRef CAS.
- M. T. Ashraf, R. Chebbi and N. A. Darwish, Ind. Eng. Chem. Res., 2013, 52, 13730–13737 CrossRef CAS.
- H. Wang, Y. Pu, A. Ragauskas and B. Yang, Bioresour. Technol., 2019, 271, 449–461 CrossRef CAS PubMed.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- T.-Q. Yuan, F. Xu and R.-C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 346–352 CrossRef CAS.
- Y. H. Kim, Chem. Eng. Technol., 2016, 39, 2312–2322 CrossRef CAS.
- J. C. Lee, Y. K. Yeo, K. H. Song and I. W. Kim, Korean J. Chem. Eng., 2001, 18, 428–431 CrossRef CAS.
- BTX (Benzene, Toluene & Xylene) Market Analysis, 2019-2027, https://www.reportsanddata.com/report-detail/btx-benzene-toluene-and-xylene-market, accessed 18 April 2020 Search PubMed.
- K. Wilson and A. F. Lee, in Advances in Biorefineries: Biomass and Waste Supply Chain Exploitation, Elsevier Ltd, 2014, pp. 624–658 Search PubMed.
- A. Mittal, H. M. Pilath and D. K. Johnson, Energy Fuels, 2020, 34(3), 3284–3293 CrossRef CAS.
- M. Kabbour and R. Luque, in Biomass, Biofuels, Biochemicals, Elsevier, 2020, pp. 283–297 Search PubMed.
- A. E. Eseyin and P. H. Steele, Int. J. Adv. Chem., 2015, 3, 42 CrossRef.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- C. E. Carraher, J. Chem. Educ., 1978, 54, 269–270 CrossRef.
- G. G. Millán and H. Sixta, Catal, 2020, 10, 1101 CrossRef.
- M. Kabbour and R. Luque, Biomass, Biofuels, Biochem. Recent Adv. Dev. Platf. Chem., 2020, pp. 283–297 Search PubMed.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- M. Dashtban, A. Gilbert and P. Fatehi, RSC Adv., 2014, 4, 2037–2050 RSC.
- J. Dai, Green Energy Environ., 2021, 6, 22–32 CrossRef CAS.
- L. Hu, Y. Jiang, X. Wang, A. He, J. Xu and Z. Wu, Biomass Convers. Biorefin., 2020, 2020, 1–16 Search PubMed.
- E. de Jong, M. A. Dam, L. Sipos and G.-J. M. Gruter, ACS Symp. Ser. Am. Chem. Soc., 2012, 1105, 1–13 CAS.
- Polyethylene Furanoate Market Size | PEF Industry Report, 2016-2022, https://www.grandviewresearch.com/industry-analysis/polyethylene-furanoate-pef-market, accessed 13 January 2020 Search PubMed.
- C. Akobi, H. Hafez and G. Nakhla, Bioresour. Technol., 2016, 221, 598–606 CrossRef CAS PubMed.
- Z. Wang, Z. Liu, R. S. Noor, Q. Cheng, X. Chu, B. Qu, F. Zhen and Y. Sun, Sci. Total Environ., 2019, 674, 49–57 CrossRef CAS PubMed.
- D. S. Park, K. E. Joseph, M. Koehle, C. Krumm, L. Ren, J. N. Damen, M. H. Shete, H. S. Lee, X. Zuo, B. Lee, W. Fan, D. G. Vlachos, R. F. Lobo, M. Tsapatsis and P. J. Dauenhauer, ACS Cent. Sci., 2016, 2, 820–824 CrossRef CAS PubMed.
- T. Yu, X. Wang and C. Li, J. Environ. Eng., 2014, 140(9) DOI:10.1061/(ASCE)EE.1943-7870.0000736.
- K. I. Galkin and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22, 22 Search PubMed.
- V. Nikolov, D. Klissurski and A. Anastasov, Catal. Rev., 2006, 33, 319–374 CrossRef.
- R. Rulkens and C. Koning, Polym. Sci. A Compr. Ref. 10 Vol. Set, 2012, vol. 5, pp. 431–467 Search PubMed.
- A. Castellan, J. C. J. Bart and S. Cavallaro, Catal. Today, 1991, 9, 237–254 CrossRef CAS.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- T. Polen, M. Spelberg and M. Bott, J. Biotechnol., 2013, 167, 75–84 CrossRef CAS PubMed.
- B. L. Scallet and J. H. Gardner, J. Am. Chem. Soc., 1945, 67, 1934–1935 CrossRef CAS.
- B. F. M. Kuster, Starch – Starke, 1990, 42, 314–321 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- T. G. Bonner, E. J. Bourne and S. Mcnally, J. Chem. Soc., 1960, 2929 RSC.
- C. J. Moye and Z. S. Krzeminski, Aust. J. Chem., 1963, 16, 258–269 CrossRef CAS.
- J. Lewkowski, Synthesis, chemistry and applications of 5-hydroxymethylfurfural and its derivatives, Arkivoc, 2001 DOI:10.1002/chin.200302269.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem. Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- M. E. Zakrzewska, E. Bogel-łukasik and R. Bogel-łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- T. Ståhlberg, W. Fu, J. M. Woodley and A. Riisager, ChemSusChem, 2011, 4, 451–458 CrossRef PubMed.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- Z. Cheng, D. G. Vlachos, J. L. Everhart, G. Tsilomelekis, V. Nikolakis and B. Saha, Green Chem., 2018, 20, 997–1006 RSC.
- A. Al Ghatta, X. Zhou, G. Casarano, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustain. Chem. Eng., 2021, 9(5), 2212–2223 CrossRef CAS.
- Z. Xue, M. G. Ma, Z. Li and T. Mu, RSC Adv., 2016, 6, 98874–98892 RSC.
- Y. Qiao, N. Theyssen and Z. Hou, Recycl. Catal., 2015, 2(1) DOI:10.1515/recat-2015-0006.
- J. M. Johnson and F. D. Conforti, Fructose, Encyclopedia of Food Sciences and Nutrition, 2nd edn, 2003 Search PubMed.
- W. D. Crabb and J. K. Shetty, Curr. Opin. Microbiol., 1999, 2, 252–256 CrossRef CAS PubMed.
- P. C. Badger, Trends new Crop. new uses, 2002, vol. 1, pp. 17–21 Search PubMed.
- A. Boisen, T. B. Christensen, W. Fu, Y. Y. Gorbanev, T. S. Hansen, J. S. Jensen, S. K. Klitgaard, S. Pedersen, A. Riisager, T. Ståhlberg and J. M. Woodley, Chem. Eng. Res. Des., 2009, 87, 1318–1327 CrossRef CAS.
- A. Teimouri, M. Mazaheri, A. N. Chermahini, H. Salavati, F. Momenbeik and M. Fazel-Najafabadi, J. Taiwan Inst. Chem. Eng., 2015, 49, 40–50 CrossRef CAS.
- L. Chen, Y. Xiong, H. Qin, Z. Qi, L. Chen, Y. Xiong, Z. Qi and H. Qin, ChemSusChem, 2022, 15(13) DOI:10.1002/CSSC.202102635.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS PubMed.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem. Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- T. Thananatthanachon and T. B. Rauchfuss, ChemSusChem, 2010, 3, 1139–1141 CrossRef CAS PubMed.
- T. S. Hansen, J. M. Woodley and A. Riisager, Carbohydr. Res., 2009, 344, 2568–2572 CrossRef CAS PubMed.
- J. Lu, Y. Yan, Y. Zhang and Y. Tang, RSC Adv., 2012, 2, 7652–7655 RSC.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- A. Chinnappan, C. Baskar and H. Kim, RSC Adv., 2016, 6, 63991–64002 RSC.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- T. W. Walker, A. K. Chew, H. Li, B. Demir, Z. C. Zhang, G. W. Huber, R. C. Van Lehn and J. A. Dumesic, Energy Environ. Sci., 2018, 11, 617–628 RSC.
- A. Chuntanapum, T. L. K. Yong, S. Miyake and Y. Matsumura, Ind. Eng. Chem. Res., 2008, 47, 2956–2962 CrossRef CAS.
- H. Wang, S. Liu, Y. Zhao, H. Zhang and J. Wang, ACS Sustain. Chem. Eng., 2016, 4, 6712–6721 CrossRef CAS.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- Z. Wei, Y. Liu, D. Thushara and Q. Ren, Green Chem., 2012, 14, 1220–1226 RSC.
- S. Gajula, K. Inthumathi, S. R. Arumugam and K. Srinivasan, ACS Sustain. Chem. Eng., 2017, 5, 5373–5381 CrossRef CAS.
- S. Eminov, A. Brandt, J. D. E. T. Wilton-Ely and J. P. Hallett, PLoS One, 2016, 11, 1–15 CrossRef PubMed.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, ChemSusChem, 2019, 12, 4452–4460 CrossRef PubMed.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitz, Appl. Catal., B, 2016, 181, 874–887 CrossRef CAS.
- V. Nikolakis, S. H. Mushrif, B. Herbert, K. S. Booksh and D. G. Vlachos, J. Phys. Chem. B, 2012, 116, 11274–11283 CrossRef CAS PubMed.
- M. H. Tucker, R. Alamillo, A. J. Crisci, G. M. Gonzalez, S. L. Scott and J. A. Dumesic, ACS Sustain. Chem. Eng., 2013, 1, 554–560 CrossRef CAS.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2013, 117, 2102–2113 CrossRef CAS PubMed.
- X. Fu, Y. Hu, Y. Zhang, Y. Zhang, D. Tang, L. Zhu and C. Hu, ChemSusChem, 2020, 13, 501–512 CrossRef CAS PubMed.
- M. Benoit, Y. Brissonnet, E. Guélou, K. De Oliveira Vigier, J. Barrault and F. Jérôme, ChemSusChem, 2010, 3, 1304–1309 CrossRef CAS PubMed.
- X. Li, K. Peng, X. Liu, Q. Xia and Y. Wang, ChemCatChem, 2017, 9, 2739–2746 CrossRef CAS.
- T. Wang, J. A. Glasper and B. H. Shanks, Appl. Catal., A, 2015, 498, 214–221 CrossRef CAS.
- A. Døssing, Coord. Chem. Rev., 2014, 280, 38–53 CrossRef.
- P. Körner, D. Jung and A. Kruse, ChemistryOpen, 2019, 8, 1121–1132 CrossRef PubMed.
- D. Jung, P. Körner and A. Kruse, Biomass Convers. Biorefin., 2019, 1–16 Search PubMed.
- L.-K. K. Ren, L.-F. F. Zhu, T. Qi, J.-Q. Q. Tang, H.-Q. Q. Yang and C.-W. W. Hu, ACS Catal., 2017, 7, 2199–2212 CrossRef CAS.
- S. Jia, K. Liu, Z. Xu, P. Yan, W. Xu, X. Liu and Z. C. Zhang, Catal. Today, 2014, 234, 83–90 CrossRef CAS.
- S. Jia, Z. Xu and Z. C. Zhang, Chem. Eng. J., 2014, 254, 333–339 CrossRef CAS.
- S. Bali, M. A. Tofanelli, R. D. Ernst and E. M. Eyring, Biomass Bioenergy, 2012, 42, 224–227 CrossRef CAS.
- T. Wang, Y. J. Pagán-Torres, E. J. Combs, J. A. Dumesic and B. H. Shanks, Top. Catal., 2012, 55, 657–662 CrossRef CAS.
- S. Dutta, S. De and B. Saha, Biomass Bioenergy, 2013, 55, 355–369 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- Y. Roman-Leshkov, J. N. Chheda and J. a Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
- S. Mohammad, G. Grundl, R. Müller, W. Kunz, G. Sadowski and C. Held, Fluid Phase Equilib., 2016, 428, 102–111 CrossRef CAS.
- S. Mohammad, C. Held, E. Altuntepe, T. Köse, T. Gerlach, I. Smirnova and G. Sadowski, Fluid Phase Equilib., 2016, 416, 83–93 CrossRef CAS.
- J. Lueckgen, L. Vanoye, R. Philippe, M. Eternot, P. Fongarland, C. de Bellefon and A. Favre-Réguillon, J. Flow Chem., 2018, 8, 3–9 CrossRef CAS.
- J. Li, W. Zhang, S. Xu and C. Hu, Front. Chem., 2020, 8, 70 CrossRef CAS PubMed.
- N. Shi, Q. Liu, Q. Zhang, T. Wang and L. Ma, Green Chem., 2013, 15, 1967–1974 RSC.
- Z. Tan, S. Sivoththaman and F. Liu, Sustainable Environ. Res., 2014, 24, 149–157 Search PubMed.
- X. Fu, J. Dai, X. Guo, J. Tang, L. Zhu and C. Hu, Green Chem., 2017, 19, 3334–3343 RSC.
- R. Lee, J. Harris, P. Champagne and P. G. Jessop, Green Chem., 2016, 18, 6305–6310 RSC.
- I. K. M. Yu, D. C. W. Tsang, S. S. Chen, L. Wang, A. J. Hunt, J. Sherwood, K. De Oliveira Vigier, F. Jérôme, Y. S. Ok and C. S. Poon, Bioresour. Technol., 2017, 245, 456–462 CrossRef CAS PubMed.
- T. Okano, K. Qiao, Q. Bao, D. Tomida, H. Hagiwara and C. Yokoyama, Appl. Catal., A, 2013, 451, 1–5 CrossRef CAS.
- H. Ma, F. Wang, Y. Yu, L. Wang and X. Li, Ind. Eng. Chem. Res., 2015, 54(10), 2657–2666 CrossRef CAS.
- L. C. Blumenthal, C. M. Jens, J. Ulbrich, F. Schwering, V. Langrehr, T. Turek, U. Kunz, K. Leonhard and R. Palkovits, ACS Sustain. Chem. Eng., 2016, 4, 228–235 CrossRef CAS.
- F. Huang, Y. Su, Y. Tao, W. Sun and W. Wang, Fuel, 2018, 226, 417–422 CrossRef CAS.
- L. Zhang, G. Xi, Z. Chen, Z. Qi and X. Wang, Chem. Eng. J., 2017, 307, 877–883 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC.
- Y. N. Li, J. Q. Wang, L. N. He, Z. Z. Yang, A. H. Liu, B. Yu and C. R. Luan, Green Chem., 2012, 14, 2752–2758 RSC.
- J. Zhang, X. Yu, F. Zou, Y. Zhong, N. Du and X. Huang, ACS Sustain. Chem. Eng., 2015, 3, 3338–3345 CrossRef CAS.
- L. Lai and Y. Zhang, ChemSusChem, 2010, 3, 1257–1259 CrossRef CAS PubMed.
- C. Shi, Y. Zhao, J. Xin, J. Wang, X. Lu, X. Zhang and S. Zhang, Chem. Commun., 2012, 48, 4103–4105 RSC.
- F. J. V. Gschwend, A. Brandt-Talbot, C. L. Chambon and J. P. Hallett, ACS Symp. Ser., 2017, 1250, 209–223 CrossRef CAS.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4, 517–520 CrossRef CAS.
- Y. Xiao and X. Huang, RSC Adv., 2018, 8, 18784–18791 RSC.
- S. Eminov, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustain. Chem. Eng., 2014, 2, 978–981 CrossRef CAS.
- F. Tao, H. Song and L. Chou, RSC Adv., 2011, 1, 672–676 RSC.
- J. Ryu, J. W. Choi, D. J. Suh, D. J. Ahn and Y. W. Suh, Catal. Commun., 2012, 24, 11–15 CrossRef CAS.
- A. Chinnappan, A. H. Jadhav, H. Kim and W. J. Chung, Chem. Eng. J., 2014, 237, 95–100 CrossRef CAS.
- C. Li, Z. K. Zhao, A. Wang, M. Zheng and T. Zhang, Carbohydr. Res., 2010, 345, 1846–1850 CrossRef CAS PubMed.
- C. Tian, X. Zhu, S. H. Chai, Z. Wu, A. Binder, S. Brown, L. Li, H. Luo, Y. Guo and S. Dai, ChemSusChem, 2014, 7, 1703–1709 CrossRef CAS PubMed.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustain. Chem. Eng., 2019, 7, 16483–16492 CrossRef.
- C. J. Clarke, L. Bui-Le, P. J. Corbett and J. P. Hallett, Ind. Eng. Chem. Res., 2019, 54(10), 12536–12544 Search PubMed.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustain. Chem. Eng., 2020, 8, 2462–2471 CrossRef CAS.
- L. Hu, Y. Sun and L. Lin, Ind. Eng. Chem. Res., 2012, 51, 1099–1104 CrossRef CAS.
- H. Jadhav, E. Taarning, C. M. Pedersen and M. Bols, Tetrahedron Lett., 2012, 53, 983–985 CrossRef CAS.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- A. Bhattacharjee, A. Luís, J. H. Santos, J. A. Lopes-da-Silva, M. G. Freire, P. J. Carvalho and J. A. P. Coutinho, Fluid Phase Equilib., 2014, 381, 36–45 CrossRef CAS PubMed.
- L. Wu, J. Song, B. Zhang, B. Zhou, H. Zhou, H. Fan, Y. Yang and B. Han, Green Chem., 2014, 16, 3935–3941 RSC.
- J. J. Li, J. J. Li, D. Zhang and C. Liu, J. Phys. Chem. B, 2015, 119, 13398–13406 CrossRef CAS PubMed.
- Q. Cao, X. Guo, S. Yao, J. Guan, X. Wang, X. Mu and D. Zhang, Carbohydr. Res., 2011, 346, 956–959 CrossRef CAS PubMed.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253–1262 RSC.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Bioresour. Technol., 2012, 109, 224–228 CrossRef CAS PubMed.
- R. M. Musau and R. M. Munavu, Biomass, 1987, 13, 67–74 CrossRef CAS.
- Z. Hu, B. Liu, Z. Zhang and L. Chen, Ind. Crops Prod., 2013, 50, 264–269 CrossRef CAS.
- A. S. Amarasekara, L. T. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS PubMed.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591–5600 CrossRef CAS.
- G. Sampath and S. Kannan, Catal. Commun., 2013, 37, 41–44 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Ind. Eng. Chem. Res., 2008, 47, 9234–9239 CrossRef CAS.
- M. R. Whitaker, A. Parulkar, P. Ranadive, R. Joshi and N. A. Brunelli, ChemSusChem, 2019, 12, 2211–2219 CrossRef CAS PubMed.
- F. Zhou, X. Sun, D. Wu, Y. Zhang and H. Su, ChemCatChem, 2017, 9, 2784–2789 CrossRef CAS.
- A. Al Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, Green Chem., 2021, 23, 1716–1733 RSC.
- P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318–346 CrossRef CAS.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- K. Enomoto, T. Hosoya and H. Miyafuji, Cellulose, 2018, 25, 2249–2257 CrossRef CAS.
- A. Xu, L. Cao, B. Wang and J. Ma, Adv. Mater. Sci. Eng., 2015 DOI:10.1155/2015/406470.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- P. Körner, D. Jung and A. Kruse, Green Chem., 2018, 20, 2231–2241 RSC.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345–9348 CrossRef CAS PubMed.
- Z. Zhang, Q. Wang, H. Xie, W. Liu and Z. K. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS PubMed.
- Z. Zhang, B. Liu and Z. Zhao, Carbohydr. Polym., 2012, 88, 891–895 CrossRef CAS.
- N. Mittal, G. M. Nisola and W. J. Chung, Tetrahedron Lett., 2012, 53, 3149–3155 CrossRef CAS.
- X. Zhou, Z. Zhang, B. Liu, Q. Zhou, S. Wang and K. Deng, J. Ind. Eng. Chem., 2014, 20, 644–649 CrossRef CAS.
- X. Tong, M. Li, N. Yan, Y. Ma, P. J. Dyson and Y. Li, Catal. Today, 2011, 175, 524–527 CrossRef CAS.
- F. Liu, J. Barrault, K. De Oliveira Vigier and F. Jérôme, ChemSusChem, 2012, 5, 1223–1226 CrossRef CAS PubMed.
- Q. Hou, W. Li, M. Zhen, L. Liu, Y. Chen, Q. Yang, F. Huang, S. Zhang and M. Ju, RSC Adv., 2017, 7, 47288–47296 RSC.
- G. Tian, X. Tong, Y. Cheng and S. Xue, Carbohydr. Res., 2013, 370, 33–37 CrossRef CAS PubMed.
- P. Wrigstedt, J. Keskiväli, M. Leskelä and T. Repo, ChemCatChem, 2015, 7, 501–507 CrossRef CAS.
- B. J. Stoecker, J. Trace Elem. Exp. Med., 1999, 12, 163–169 CrossRef CAS.
- E. F. Dunn, D. Liu and E. Y. X. Chen, Appl. Catal., A, 2013, 460–461, 1–7 CrossRef CAS.
- C. Li, Z. Zhang and Z. K. Zhao, Tetrahedron Lett., 2009, 50, 5403–5405 CrossRef CAS.
- E. A. Pidko, V. Degirmenci and E. J. M. Hensen, ChemCatChem, 2012, 4, 1263–1271 CrossRef CAS.
- J. Zhang, Y. Cao, H. Li and X. Ma, Chem. Eng. J., 2014, 237, 55–61 CrossRef CAS.
- E. A. Pidko, V. Degirmenci, R. A. Van Santen and E. J. M. Hensen, Inorg. Chem., 2010, 49, 10081–10091 CrossRef CAS PubMed.
- Y. Zhang, E. A. Pidko and E. J. M. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS PubMed.
- C. B. Rasrendra, J. N. M. Soetedjo, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, Top. Catal., 2012, 55, 543–549 CrossRef CAS.
- G. Cetin, S. Kocaoba, W. H. Höll and G. Akcin, Solvent Extr. Ion Exch., 2012, 30, 88–100 CrossRef CAS.
- D. Liu and E. Y. X. Chen, Appl. Catal., A, 2012, 435–436, 78–85 CAS.
- T. Ståhlberg, M. G. Sørensen and A. Riisager, Green Chem., 2010, 12, 321–325 RSC.
- L. Hu, Y. Sun, L. Lin and S. Liu, Biomass Bioenergy, 2014, 47, 289–294 CrossRef.
- T. Ståhlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem.–Eur. J., 2011, 17, 1456–1464 CrossRef PubMed.
- C. Li, Z. K. Zhao, H. Cai, A. Wang and T. Zhang, Biomass Bioenergy, 2011, 35, 2013–2017 CrossRef CAS.
- Z. Huang, Y. Pan, Y. Chao, W. Shen, C. Wang and H. Xu, RSC Adv., 2014, 4, 13434–13437 RSC.
- L. Dessbesell, S. Souzanchi, K. T. Venkateswara Rao, A. A. Carrillo, D. Bekker, K. A. Hall, K. M. Lawrence, C. L. J. Tait and C. Xu, Biofuel Bioprod. Biorefin., 2019, 13(5), 1234–1245 CrossRef CAS.
- R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schüth, ChemSusChem, 2010, 3, 266–276 CrossRef CAS PubMed.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS PubMed.
- L. Vanoye, M. Fanselow, J. D. Holbrey, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390–396 RSC.
- F. Jiang, Q. Zhu, D. Ma, X. Liu and X. Han, J. Mol. Catal. A: Chem., 2011, 1(2), 8–12 CrossRef.
- S. Eminov, P. Filippousi, A. Brandt, J. Wilton-Ely and J. Hallett, Inorganics, 2016, 4, 1–15 CrossRef.
- W. H. Hsu, Y. Y. Lee, W. H. Peng and K. C. W. Wu, Catal. Today, 2011, 174, 65–69 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Cellulose, 2011, 18, 1327–1333 CrossRef CAS.
- Y. Zhang, H. Du, X. Qian and E. Y. X. Chen, Energy Fuels, 2010, 24, 2410–2417 CrossRef CAS.
- S. Kassaye, C. Pagar, K. K. Pant, S. Jain and R. Gupta, Bioresour. Technol., 2016, 220, 394–400 CrossRef CAS PubMed.
- H. Zhang, Z. Wang and H. Gao, BioResources, 2018, 13(1), 1189–1201 CAS.
- B. Kim, J. Jeong, D. Lee, S. Kim, H.-J. Yoon, Y.-S. Lee and J. K. Cho, Green Chem., 2011, 13, 1503 RSC.
- Y. Su, M. H. Brown, G. Li, X. Zhou, J. E. Amonette, L. J. Fulton, D. M. Camaioni and Z. C. Zhang, Appl. Catal., A, 2011, 391, 436–442 CrossRef CAS.
- E. Leng, M. Mao, Y. Peng, X. Li, X. Gong and Y. Zhang, ChemistrySelect, 2019, 4, 181–189 CrossRef CAS.
- Z. Zhang and Z. K. Zhao, Bioresour. Technol., 2010, 101, 1111–1114 CrossRef CAS PubMed.
- L. Zhou, Y. He, Z. Ma, R. Liang, T. Wu and Y. Wu, Carbohydr. Polym., 2015, 117, 694–700 CrossRef CAS PubMed.
- Y. Su, M. H. Brown, X. Huang, X. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 2, 117–122 CrossRef.
- H. Abou-Yousef, H. El Barbary and P. Steele, J. Fuel Chem. Technol., 2013, 41, 214–222 CrossRef CAS.
- S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93–99 CrossRef CAS.
- F. Tao, H. Song and L. Chou, J. Mol. Catal. A: Chem., 2012, 357, 11–18 CrossRef CAS.
- Y. B. Yi, J. L. Lee, Y. H. Choi, S. M. Park and C. H. Chung, Environ. Chem. Lett., 2012, 10, 13–19 CrossRef CAS.
- Y. Shen, Y. Zhang, Y. Chen, Y. Yan, J. Pan, M. Liu and W. Shi, Energy Technol., 2016, 4, 600–609 CrossRef CAS.
- F. Tao, H. Song, J. Yang and L. Chou, Carbohydr. Polym., 2011, 85, 363–368 CrossRef CAS.
- Y. Zhang, J. Pan, M. Gan, H. Ou, Y. Yan, W. Shi and L. Yu, RSC Adv., 2014, 4, 11664–11672 RSC.
- L. Zhou, R. Liang, Z. Ma, T. Wu and Y. Wu, Bioresour. Technol., 2013, 129, 450–455 CrossRef CAS PubMed.
- Z. D. Ding, J. C. Shi, J. J. Xiao, W. X. Gu, C. G. Zheng and H. J. Wang, Carbohydr. Polym., 2012, 90, 792–798 CrossRef CAS PubMed.
- K. Zhuo, Q. Du, G. Bai, C. Wang, Y. Chen and J. Wang, Carbohydr. Polym., 2015, 115, 49–53 CrossRef CAS PubMed.
- X. Li, L. Zhang, S. Wang and Y. Wu, Front. Chem., 2020, 7, 948 CrossRef PubMed.
- P. Sudarsanam, R. Zhong, S. Van Den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- Q. Sun, S. Wang, B. Aguila, X. Meng, S. Ma and F. S. Xiao, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- M. E. Zakrzewska, E. Bogel-Yukasik and R. Bogel-Yukasik, Energy Fuels, 2010, 24, 737–745 CrossRef CAS.
- A. Al Ghatta, X. Zhou, G. Casarano, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustain. Chem. Eng., 2021, 9, 2212–2223 CrossRef CAS.
- Y. Yu and H. Wu, Energy Fuels, 2010, 24, 1963–1971 CrossRef CAS.
- R. Otomo, T. Yokoi, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2014, 470, 318–326 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2015, 18, 165–175 RSC.
- A. Jain, A. M. Shore, S. C. Jonnalagadda, K. V. Ramanujachary and A. Mugweru, Appl. Catal., A, 2015, 489, 72–76 CrossRef CAS.
- C. Fan, H. Guan, H. Zhang, J. Wang, S. Wang and X. Wang, Biomass Bioenergy, 2011, 35, 2659–2665 CrossRef CAS.
- B. Yuan, J. Guan, J. Peng, G. zhou Zhu and J. hong Jiang, Chem. Eng. J., 2017, 330, 109–119 CrossRef CAS.
- Z. Cao, Z. Fan, Y. Chen, M. Li, T. Shen, C. Zhu and H. Ying, Appl. Catal., B, 2019, 244, 170–177 CrossRef CAS.
- K. Y. Nandiwale, N. D. Galande, P. Thakur, S. D. Sawant, V. P. Zambre and V. V. Bokade, ACS Sustainable Chem. Eng. Am. Chem. Soc., 2014, 2, 1928–1932 CrossRef CAS.
- X. Li, Q. Xia, V. C. Nguyen, K. Peng, X. Liu, N. Essayem and Y. Wang, Catal. Sci. Technol., 2016, 6, 7586–7596 RSC.
- L. Hu, Z. Wu, J. Xu, Y. Sun, L. Lin and S. Liu, Chem. Eng. J., 2014, 244, 137–144 CrossRef CAS.
- Y. Feng, M. Zuo, T. Wang, W. Jia, X. Zhao, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, J. Taiwan Inst. Chem. Eng., 2019, 96, 431–438 CrossRef CAS.
- I. Jiménez-Morales, M. Moreno-Recio, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2015, 164, 70–76 CrossRef.
- X. Li, K. Peng, Q. Xia, X. Liu and Y. Wang, Chem. Eng. J., 2018, 332, 528–536 CrossRef CAS.
- J. J. Wiesfeld, R. Gaquere and E. J. M. Hensen, ACS Sustain. Chem. Eng., 2019, 7, 7552–7562 CrossRef CAS.
- C. Song, H. Liu, Y. Li, S. Ge, H. Wang, W. Zhu, Y. Chang, C. Han and H. Li, Chin. J. Chem., 2014, 32, 434–442 CrossRef CAS.
- Q. Hou, M. Zhen, W. Li, L. Liu, J. Liu, S. Zhang, Y. Nie, C. Bai, X. Bai and M. Ju, Appl. Catal., B, 2019, 253, 1–10 CrossRef CAS.
- M. Zhang, K. Su, H. Song, Z. Li and B. Cheng, Catal. Commun., 2015, 69, 76–80 CrossRef CAS.
- I. Jiménez-Morales, A. Teckchandani-Ortiz, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 144, 22–28 CrossRef.
- H. Xia, S. Xu, X. Yan and S. Zuo, Fuel Process. Technol., 2016, 152, 140–146 CrossRef CAS.
- H. Xu, Z. Miao, H. Zhao, J. Yang, J. Zhao, H. Song, N. Liang and L. Chou, Fuel, 2015, 145, 234–240 CrossRef CAS.
- Z. Li, K. Su, J. Ren, D. Yang, B. Cheng, C. K. Kim and X. Yao, Green Chem., 2018, 20, 863–872 RSC.
- T. Zhang, W. Fan, W. Li, Z. Xu, H. Xin, M. Su, Y. Lu and L. Ma, Energy Technol., 2017, 5, 747–755 CrossRef CAS.
- Y. Zhang, Y. Shen, Y. Chen, Y. Yan, J. Pan, Q. Xiong, W. Shi and L. Yu, Chem. Eng. J., 2016, 294, 222–235 CrossRef CAS.
- Y. Li, H. Liu, C. Song, X. Gu, H. Li, W. Zhu, S. Yin and C. Han, Bioresour. Technol., 2013, 133, 347–353 CrossRef CAS PubMed.
- W. H. Peng, Y. Y. Lee, C. Wu and K. C. W. Wu, J. Mater. Chem., 2012, 22, 23181–23185 RSC.
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS PubMed.
- H. Abou-Yousef and E. B. Hassan, J. Ind. Eng. Chem., 2014, 20, 1952–1957 CrossRef CAS.
- M. Du, A. Agrawal, S. Chakraborty, S. J. Garibay, R. Limvorapitux, B. Choi, S. T. Madrahimov and S. T. Nguyen, ACS Sustain. Chem. Eng., 2019, 7, 8126–8135 CrossRef CAS.
- X. Zhang, D. Zhang, Z. Sun, L. Xue, X. Wang and Z. Jiang, Appl. Catal., B, 2016, 196, 50–56 CrossRef CAS.
- Z. Ma, H. Hu, Z. Sun, W. Fang, J. Zhang, L. Yang, Y. Zhang and L. Wang, ChemSusChem, 2017, 10, 1669–1674 CrossRef CAS PubMed.
- Q. Hou, W. Li, M. Ju, L. Liu, Y. Chen and Q. Yang, RSC Adv., 2016, 6, 104016–104024 RSC.
- U. Tyagi, N. Anand and D. Kumar, Bioresour. Technol., 2018, 267, 326–332 CrossRef CAS PubMed.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
- L. Hu, X. Tang, Z. Wu, L. Lin, J. Xu, N. Xu and B. Dai, Chem. Eng. J., 2015, 263, 299–308 CrossRef CAS.
- F. Guo, Z. Fang and T. J. Zhou, Bioresour. Technol., 2012, 112, 313–318 CrossRef CAS PubMed.
- X. Qi, H. Guo, L. Li and R. L. Smith, ChemSusChem, 2012, 5, 2215–2220 CrossRef CAS PubMed.
- J. Zhao, C. Zhou, C. He, Y. Dai, X. Jia and Y. Yang, Catal. Today, 2016, 264, 123–130 CrossRef CAS.
- L. Hu, G. Zhao, X. Tang, Z. Wu, J. Xu, L. Lin and S. Liu, Bioresour. Technol., 2013, 148, 501–507 CrossRef CAS PubMed.
- Y. Dou, S. Zhou, C. Oldani, W. Fang and Q. Cao, Fuel, 2018, 214, 45–54 CrossRef CAS.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2012, 5, 1388–1391 CrossRef CAS PubMed.
- S. P. Simeonov and C. A. M. Afonso, J. Chem. Educ., 2013, 90(10), 1373–1375 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. M. Aida and R. L. L. Smith, ChemSusChem, 2009, 2, 944–946 CrossRef CAS PubMed.
- H. Gao, Y. Peng, J. Pan, J. Zeng, C. Song, Y. Zhang, Y. Yan and W. Shi, RSC Adv., 2014, 4, 43029–43038 RSC.
- Y. Zhang, J. Pan, Y. Shen, W. Shi, C. Liu and L. Yu, ACS Sustain. Chem. Eng., 2015, 3, 871–879 CrossRef CAS.
- H. Cai, C. Li, A. Wang, G. Xu and T. Zhang, Appl. Catal., B, 2012, 123–124, 333–338 CrossRef CAS.
- Y. Xiao and Y. F. Song, Appl. Catal., A, 2014, 484, 74–78 CrossRef CAS.
- G. Lv, L. Deng, B. Lu, J. Li, X. Hou and Y. Yang, J. Cleaner Prod., 2017, 142, 2244–2251 CrossRef CAS.
- J. He, Y. Zhang and E. Y. X. Chen, ChemSusChem, 2013, 6, 61–64 CrossRef CAS PubMed.
- H. Liu, H. Wang, Y. Li, W. Yang, C. Song, H. Li, W. Zhu and W. Jiang, RSC Adv., 2015, 5, 9290–9297 RSC.
- A. R. Aylak, S. Akmaz and S. N. Koc, Part. Sci. Technol., 2017, 35, 490–493 CrossRef CAS.
- Y. Y. Lee and K. C. W. Wu, Phys. Chem. Chem. Phys., 2012, 14, 13914–13917 RSC.
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef CAS.
- D. Chen, F. Liang, D. Feng, M. Xian, H. Zhang, H. Liu and F. Du, Chem. Eng. J., 2016, 300, 177–184 CrossRef CAS.
- Y. Wang, Z. Gu, W. Liu, Y. Yao, H. Wang, X. F. Xia and W. Li, RSC Adv., 2015, 5, 60736–60744 RSC.
- J. Lan and Z. Zhang, J. Ind. Eng. Chem., 2015, 23, 200–205 CrossRef CAS.
- Z. Zhang and Z. K. Zhao, Bioresour. Technol., 2011, 102, 3970–3972 CrossRef CAS PubMed.
- K. Li, M. Du and P. Ji, ACS Sustain. Chem. Eng., 2018, 6, 5636–5644 CrossRef CAS.
- Q. Hou, M. Zhen, L. Liu, Y. Chen, F. Huang, S. Zhang, W. Li and M. Ju, Appl. Catal., B, 2018, 224, 183–193 CrossRef CAS.
- Q. Xu, Z. Zhu, Y. Tian, J. Den, J. Shi and Y. Fu, BioResources, 2014, 9, 303–315 CAS.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 185–189 CrossRef CAS PubMed.
- C. J. Clarke, L. Bui-Le, P. J. Corbett and J. P. Hallett, Ind. Eng. Chem. Res., 2020, 59, 12536–12544 CrossRef CAS.
- C. Shi, J. Xin, X. Liu, X. Lu and S. Zhang, ACS Sustain. Chem. Eng., 2016, 4, 557–563 CrossRef CAS.
- X. Sun, Z. Liu, Z. Xue, Y. Zhang and T. Mu, Green Chem., 2015, 17, 2719–2722 RSC.
- J. Wang, J. Ren, X. Liu, G. Lu and Y. Wang, AlChE J., 2013, 26, 2558–2566 CrossRef.
- N. Rajabbeigi, R. Ranjan and M. Tsapatsis, Microporous Mesoporous Mater., 2012, 158, 253–256 CrossRef CAS.
- Y. Luo, Z. Li, X. Li, X. Liu, J. Fan, J. H. Clark and C. Hu, Catal. Today, 2019, 319, 14–24 CrossRef CAS.
- S. Dutta, S. De, B. Saha and M. Imteyaz Alam, Catal. Sci. Technol., 2012, 2, 2025–2036 RSC.
- K. Dalvand, J. Rubin, S. Gunukula, M. Clayton Wheeler and G. Hunt, Biomass Bioenergy, 2018, 115, 56–63 CrossRef.
- M. Kabbour and R. Luque, Biomass, Biofuels, Biochem. Recent Adv. Dev. Platf. Chem., 2020, pp. 283–297 Search PubMed.
- H. J. Brownlee and E. S. Carl Miner, Ind. Eng. Chem., 1948, 40(2), 201–204 CrossRef CAS.
- D. T. Win, Aust. J. Technol., 2005, 8, 185–190 Search PubMed.
- T. Douzou, K. Cheenkachorn, M. Sriariyanun and M. Rattanaporn, Appl. Sci. Eng., 2020, 13, 3–10 Search PubMed.
- H. Brownlee and C. S. Miner, Ind. Eng. Chem., 1948, 40, 201–204 CrossRef CAS.
- G. H. Mains and F. B. Laforge, Ind. Eng. Chem., 1924, 356–359 CrossRef CAS.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many By-Products, 2000 Search PubMed.
- C. Di Blasi, C. Branca and A. Galgano, Ind. Eng. Chem. Res., 2010, 49, 2658–2671 CrossRef CAS.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- Furfural Market Global Forecast to 2024 | MarketsandMarkets, https://www.marketsandmarkets.com/Market-Reports/furfural-market-101056456.html, accessed 15 September 2021 Search PubMed.
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193–2205 RSC.
- R. N. Ntimbani, S. Farzad and J. F. Görgens, Ind. Crops Prod., 2021, 162, 113272 CrossRef CAS.
- A. Giuliano, D. Barletta, I. De Bari and M. Poletto, Comput. Aided Chem. Eng., 2018, 43, 585–590 CAS.
- O. Ershova, J. P. Pokki, A. Zaitseva, V. Alopaeus and H. Sixta, Chem. Eng. Sci., 2018, 176, 19–34 CrossRef CAS.
- P. S. Metkar, E. J. Till, D. R. Corbin, C. J. Pereira, K. W. Hutchenson and S. K. Sengupta, Green Chem., 2015, 17, 1453–1466 RSC.
- B. Danon, L. Van Der Aa and W. De Jong, Carbohydr. Res., 2013, 375, 145–152 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria, J. Requies, M. B. Güemez and P. L. Arias, Green Chem., 2012, 14, 3132–3140 RSC.
- J. E. Romo, K. C. Miller, T. L. Sundsted, A. L. Job, K. A. Hoo and S. G. Wettstein, ChemCatChem, 2019, 11(19), 4715–4719 CrossRef CAS.
- S. R. Schay, The production of furfural from sunflower husks using the s-suprayield process, Doctoral dissertation, 2010.
- D. R. Arnold and J. L. Buzzard, Proc. South African Chem. Eng. Congr., 2003, 3–5 Search PubMed.
- N. Hidayat, A. N. Hidayat and M. Gozan, AIP Conf. Proc., 2019, 2062, 020048 CrossRef.
- K. J. Zeitsch, Sugar Ser., 2000, 13, 28–33 Search PubMed.
- D. Steinbach, A. Kruse and J. Sauer, Biomass Convers. Biorefin., 2017, 7, 247–274 CrossRef CAS.
- H. D. Mansilla, J. Baeza, S. Urzúa, G. Maturana, J. Villaseñor and N. Durán, Bioresour. Technol., 1998, 66, 189–193 CrossRef CAS.
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Y. Murzin, Chem. Rev., 2011, 111, 5638–5666 CrossRef PubMed.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Güemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS PubMed.
- C. B. T. L. Lee and T. Y. Wu, Renewable Sustainable Energy Rev., 2021, 137, 110172 CrossRef CAS.
- Q. Wang, W. Qi, W. Wang, Y. Zhang, N. Leksawasdi, X. Zhuang, Q. Yu and Z. Yuan, Renewable Energy, 2019, 144, 139–146 CrossRef CAS.
- A. Mittal, S. K. Black, T. B. Vinzant, M. O'Brien, M. P. Tucker and D. K. Johnson, ACS Sustain. Chem. Eng., 2017, 5, 5694–5701 CrossRef CAS.
- B. M. Matsagar and P. L. Dhepe, New J. Chem., 2017, 41, 6137–6144 RSC.
- I. Agirrezabal-Telleria, J. Requies, M. B. Güemez and P. L. Arias, Appl. Catal., B, 2014, 145, 34–42 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- J. H. Kim, S. M. Cho, J. H. Choi, H. Jeong, S. M. Lee, B. Koo and I. G. Choi, Appl. Sci., 2021, 11, 1–12 Search PubMed.
- A. Cañada-Barcala, D. Rodríguez-Llorente, L. López, P. Navarro, E. Hernández, V. I. Águeda, S. Álvarez-Torrellas, J. C. Parajó, S. Rivas and M. Larriba, ACS Sustain. Chem. Eng., 2021, 9, 10266–10275 CrossRef.
- B. M. Matsagar, S. A. Hossain, T. Islam, H. R. Alamri, Z. A. Alothman, Y. Yamauchi, P. L. Dhepe and K. C.-W. Wu, Sci. Rep., 2017, 7, 1–7 CrossRef CAS PubMed.
- B. M. Matsagar, M. K. Munshi, A. A. Kelkar and P. L. Dhepe, Catal. Sci. Technol., 2015, 5, 5086–5090 RSC.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS PubMed.
- R. J. H. Grisel, J. C. Van Der Waal, E. De Jong and W. J. J. Huijgen, Catal. Today, 2014, 223, 3–10 CrossRef CAS.
- W. Riansa-Ngawong and P. Prasertsan, Carbohydr. Res., 2011, 346, 103–110 CrossRef CAS PubMed.
- K. R. Enslow and A. T. Bell, ChemCatChem, 2015, 7, 479–489 CrossRef CAS.
- T. vom Stein, P. M. Grande, W. Leitner and P. Domínguez de María, ChemSusChem, 2011, 4, 1592–1594 CrossRef CAS PubMed.
- G. Marcotullio and W. D. Jong, Green Chem., 2010, 12, 1739–1746 RSC.
- G. Marcotullio and W. De Jong, Carbohydr. Res., 2011, 346, 1291–1293 CrossRef CAS PubMed.
- S. Jiang, C. Verrier, M. Ahmar, J. Lai, C. Ma, E. Muller, Y. Queneau, M. Pera-Titus, F. Jérôme and K. De Oliveira Vigier, Green Chem., 2018, 20, 5104–5110 RSC.
- Q. Wang, X. Zhuang, W. Wang, X. Tan, Q. Yu, W. Qi and Z. Yuan, Chem. Eng. J., 2018, 334, 698–706 CrossRef CAS.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, Green Chem., 2013, 15, 3140–3145 RSC.
- L. Jiang, D. Ding, L. Xu, Q. Guo and Y. Fu, RSC Adv., 2014, 4, 14985–14992 RSC.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53(44), 11872–11875 CrossRef CAS PubMed.
- C. Sener, A. H. Motagamwala, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2018, 11, 2321–2331 CrossRef CAS PubMed.
- H. Li, A. Deng, J. Ren, C. Liu, Q. Lu, L. Zhong, F. Peng and R. Sun, Bioresour. Technol., 2014, 158, 313–320 CrossRef CAS PubMed.
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed.
- A. H. Motagamwala, W. Won, C. T. Maravelias and J. A. Dumesic, Green Chem., 2016, 18, 5756–5763 RSC.
- H. Lin, J. Chen, Y. Zhao and S. Wang, Energy Fuels, 2017, 31, 3929–3934 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2012, 6, 76–80 RSC.
- W. Fang and H. Sixta, ChemSusChem, 2015, 8, 73–76 CrossRef CAS PubMed.
- Z. Xue, X. Zhao, R. Sun and T. Mu, ACS Sustain. Chem. Eng., 2016, 4, 3864–3870 CrossRef CAS.
- Y. Luo, Z. Li, Y. Zuo, Z. Su and C. Hu, ACS Sustain. Chem. Eng., 2017, 5, 8137–8147 CrossRef CAS.
- S. Peleteiro, S. Rivas, J. L. Alonso, V. Santos and J. C. Parajó, Bioresour. Technol., 2016, 202, 181–191 CrossRef CAS PubMed.
- C. Sievers, I. Musin, T. Marzialetti, M. Valenzuela, B. Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2(7), 665–671 CrossRef CAS PubMed.
- S. Lima, M. M. Antunes, M. Pillinger and A. A. Valente, ChemCatChem, 2011, 3, 1686–1706 CrossRef CAS.
- S. Peleteiro, A. M. da C. Lopes, G. Garrote, J. C. Parajó and R. Bogel-Łukasik, Ind. Eng. Chem. Res., 2015, 54, 8368–8373 CrossRef CAS.
- C. Sievers, I. Musin, T. Marzialetti, M. B. V. Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2, 665–671 CrossRef CAS PubMed.
- S. Peleteiro, A. M. da Costa Lopes, G. Garrote, R. Bogel-Łukasik and J. C. Parajó, Ind. Crops Prod., 2015, 77, 163–166 CrossRef CAS.
- L. Zhang, H. Yu and P. Wang, Bioresour. Technol., 2013, 136, 515–521 CrossRef CAS PubMed.
- Y. Nie, Q. Hou, W. Li, C. Bai, X. Bai and M. Ju, Molecules, 2019, 24(3), 594 CrossRef PubMed.
- S. Peleteiro, V. Santos, G. Garrote and J. C. Parajó, Carbohydr. Polym., 2016, 146, 20–25 CrossRef CAS PubMed.
- A. V. Carvalho, A. M. da C. Lopes and R. Bogel-Łukasik, RSC Adv., 2015, 5, 47153–47164 RSC.
- H. Ren, M.-H. Zong, H. Wu and N. Li, Ind. Eng. Chem. Res., 2016, 55, 1788–1795 CrossRef CAS.
- A. R. C. Morais and M. D. D. J. Matuchaki, Jürgen Andreaus and Rafal Bogel-Lukasik, Green Chem., 2016, 18, 2985–2994 RSC.
- Q. JING and X. LÜ, Chin. J. Chem. Eng., 2007, 15, 666–669 CrossRef CAS.
- T. H. Kim, H. J. Ryu and K. K. Oh, Bioresour. Technol., 2016, 218, 367–372 CrossRef CAS PubMed.
- G. Marcotullio, M. A. T. Cardoso, W. De Jong and A. H. M. Verkooijen, Int. J. Chem. React. Eng., 2019, 7(1) DOI:10.2202/1542-6580.1980.
- L. Zhang, H. Yu, P. Wang and Y. Li, Bioresour. Technol., 2014, 151, 355–360 CrossRef CAS PubMed.
- Efficient Production of Furfural from Corncob by an Integrated Mineral-Organic-Lewis Acid Catalytic Process | Zhang | BioResources, https://ojs.cnr.ncsu.edu/index.php/BioRes/article/view/BioRes_12_2_2965_Zhang_Efficient_Production_Furfural_Corncob, accessed 12 September 2021 Search PubMed.
- O. Ershova, K. Nieminen and H. Sixta, ChemCatChem, 2017, 9, 3031–3040 CrossRef CAS.
- M. Lopes, K. Dussan and J. J. Leahy, Chem. Eng. J., 2017, 323, 278–286 CrossRef CAS.
- O. Yemiş and G. Mazza, Waste Biomass Valorization, 2017, 10, 1343–1353 CrossRef.
- K. Sun, Y. Shao, P. Liu, L. Zhang, G. Gao, D. Dong, S. Zhang, G. Hu, L. Xu and X. Hu, Fuel, 2021, 300, 120990 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 CrossRef CAS.
- R. K. Mishra, V. B. Kumar, A. Victor, I. N. Pulidindi and A. Gedanken, Ultrason. Sonochem., 2019, 56, 55–62 CrossRef CAS PubMed.
- R. Weingarten, G. A. Tompsett, W. C. Conner and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS.
- Y. Zhao, H. Xu, K. Lu, Y. Qu, L. Zhu and S. Wang, Energy Sci. Eng., 2019, 7, 2237–2246 CrossRef CAS.
- Y. Yang, C.-W. Hu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 405–410 CrossRef CAS PubMed.
- M. J. Antal, T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71–85 CrossRef CAS.
- N. R. Vinueza, E. S. Kim, V. A. Gallardo, N. S. Mosier, M. M. Abu-Omar, N. C. Carpita and H. I. Kenttämaa, Biomass Bioenergy, 2015, 74, 1–5 CrossRef CAS.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Catal., 2017, 7, 2430–2436 CrossRef CAS.
- V. Choudhary, S. Caratzoulas and D. G. Vlachos, Carbohydr. Res., 2013, 368, 89–95 CrossRef CAS PubMed.
- H. Gao, H. Liu, B. Pang, G. Yu, J. Du, Y. Zhang, H. Wang and X. Mu, Bioresour. Technol., 2014, 172, 453–456 CrossRef CAS PubMed.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer, J. H. Yeap, H. C. Wong and J. A. Dumesic, Top. Catal., 2013, 56, 1775–1781 CrossRef CAS.
- R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751–761 CrossRef CAS PubMed.
- P. Prasenjit Bhaumik and, Laxmikant Dhepe, RSC Adv., 2014, 4, 26215–26221 RSC.
- P. Bhaumik and P. L. Dhepe, ACS Catal., 2013, 3, 2299–2303 CrossRef CAS.
- A. Deng, J. Ren, H. Li, F. Peng and R. Sun, RSC Adv., 2015, 5, 60264–60272 RSC.
- T. H. Kim, Y. J. Jeon, K. K. Oh and T. H. Kim, Korean J. Chem. Eng., 2013, 30, 1339–1346 CrossRef CAS.
- F. López, M. T. García, M. J. Feria, J. C. García, C. M. de Diego, M. A. M. Zamudio and M. J. Díaz, Chem. Eng. J., 2014, 240, 195–201 CrossRef.
- T. Shen, Y. Hu, R. Hu, W. Zhuang, M. Li, H. Niu, H. Xu, C. Zhu and H. Ying, Ind. Crops Prod., 2020, 153, 112565 CrossRef CAS.
- M. S. Hossain, C. Theodoropoulos and A. Yousuf, Biochem. Eng. J., 2019, 144, 89–103 CrossRef CAS.
- L. Mao, L. Zhang, N. Gao and A. Li, Green Chem., 2013, 15, 727–737 RSC.
- R. A. Sheldon, Stud. Surf. Sci. Catal., 1991, 66, 573–594 CrossRef CAS.
- M. Li, T. Ruddy, D. Fahey, D. H. Busch and B. Subramaniam, ACS Sustain. Chem. Eng., 2014, 2, 823–835 CrossRef CAS.
- S. Fakirov and M. Evstatiev, Polym. J., 1990, 31, 431–434 CAS.
- K. Ravindkanath and R. A. Mashelkar, Polym. Eng. Sci., 1984, 24, 30–41 CrossRef.
- M. Z. G. Holmvik and F. Zeng, Analysis: New purified terephthalic acid plants may shoulder China's paraxylene imports amid wave of new PX plants, https://www.spglobal.com/platts/en/market-insights/latest-news/petrochemicals/101218-analysis-new-purified-terephthalic-acid-plants-may-shoulder-chinas-paraxylene-imports-amid-wave-of-new-px-plants Search PubMed.
- Reportlinker, Global Polyethylene Terephthalate (PET) Industry Outlook to 2022 – Capacity and Capital Expenditure Forecasts with Details of All Active and Planned Plants, https://www.prnewswire.com/news-releases/global-polyethylene-terephthalate-pet-industry-outlook-to-2022---capacity-and-capital-expenditure-forecasts-with-details-of-all-active-and-planned-plants-300606699.html%0D Search PubMed.
- R. A. F. Tomás, J. C. M. Bordado and J. F. P. Gomes, Chem. Rev., 2013, 113, 7421–7469 CrossRef PubMed.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10(2), 91–105 CrossRef CAS.
- M. Li, F. Niu, X. Zuo, P. D. Metelski, D. H. Busch and B. Subramaniam, Chem. Eng. Sci., 2013, 104, 93–102 CrossRef CAS.
- J. H. Zhou, G. Z. Shen, J. Zhu and W. K. Yuan, in Studies in Surface Science and Catalysis, Elsevier Inc., 2006, vol. 159, pp. 293–296 Search PubMed.
- M&G Chemicals Corpus Christi Facility, http://www.mgcorpuschristi.com/en/corpus-christi/the-projects.
- J. Konnerth and P. Solt, ChemSusChem, 2020, 13(14), 3544–3564 CrossRef PubMed.
- Purified Terephthalic Acid (PTA) Prices and Pricing Information | ICIS, https://www.icis.com/explore/resources/news/2007/11/06/9076460/purified-terephthalic-acid-pta-prices-and-pricing-information/, accessed 7 January 2020 Search PubMed.
- S. r. o. Weastra, Market Study on Succinic Acid, Itaconic Acid and FDCA, 2011 Search PubMed.
- J. Liu, Y. Tang, K. Wu, C. Bi and Q. Cui, Carbohydr. Res., 2012, 350, 20–24 CrossRef CAS PubMed.
- G. J. Gruter and F. Dautzenberg, Method for the synthesis of 5-alkoxymethylfurfural ethers and their use, Chem. plant raw Mater, EP1834950A1, 2012.
- K. J. P. Schouten, J. C. V. D. Waal, M. Varini and G. J. M. Gruter, European Patent, application no. EP3297995B1, 2019.
- X. Zuo, P. Venkitasubramanian, D. H. Busch and B. Subramaniam, ACS Sustain. Chem. Eng., 2016, 4, 3659–3668 CrossRef CAS.
- S. Chen, Y. Cheng, H. Ban, Y. Zhang, L. Zheng, L. Wang and X. Li, Ind. Eng. Chem. Res., 2021, 24(47), 16887–16898 CrossRef.
- X. Zuo, A. S. Chaudhari, K. Snavely, F. Niu, H. Zhu, K. J. Martin and B. Subramaniam, AIChE J., 2017, 63, 162–171 CrossRef CAS.
- H. Ban, T. Pan, Y. Cheng, L. Wang and X. Li, J. Chem. Eng. Data, 2018, 63, 1987–1993 CrossRef CAS.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- H. Zhou, H. Xu and Y. Liu, Appl. Catal., B, 2019, 244, 965–973 CrossRef CAS.
- S. E. Davis, A. D. Benavidez, R. W. Gosselink, J. H. Bitter, K. P. De Jong, A. K. Datye and R. J. Davis, J. Mol. Catal. A: Chem., 2014, 388–389, 123–132 CrossRef CAS.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford, 2nd edn, 2001 Search PubMed.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- J. H. J. Kluytmans, A. P. Markusse, B. F. M. Kuster, G. B. Marin and J. C. Schouten, Catal. Today, 2000, 57, 143–155 CrossRef CAS.
- S. Subbiah, S. P. Simeonov, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Green Chem., 2013, 15, 2849–2853 RSC.
- Y. Zhang, X. Guo, P. Tang and J. Xu, J. Chem. Eng. Data, 2018, 63, 1316–1324 CrossRef CAS.
- F. Wang, Z. Yuan, B. Liu, S. Chen and Z. Zhang, J. Ind. Eng. Chem., 2016, 38, 181–185 CrossRef CAS.
- Oxidation of Aldehydes to Carboxylic Acids – Chemgapedia, http://www.chemgapedia.de/vsengine/vlu/vsc/en/ch/2/vlu/oxidation_reduktion/oxi_carbs_ald.vlu/Page/vsc/en/ch/2/oc/reaktionen/formale_systematik/oxidation_reduktion/oxidation/ersatz_h_durch_o_n/ox_aldehyde_zu_carbonsaeuren/mechanismus.vscml.html, accessed 13 January 2020 Search PubMed.
- C. A. Antonyraj, J. Jeong, B. Kim, S. Shin, S. Kim, K. Y. Lee and J. K. Cho, J. Ind. Eng. Chem., 2013, 19, 1056–1059 CrossRef CAS.
- B. Liu, Z. Zhang, K. Lv, K. Deng and H. Duan, Appl. Catal., A, 2014, 472, 64–71 CrossRef CAS.
- K. Ghosh, R. A. Molla, M. A. Iqubal, S. S. Islam and S. M. Islam, Appl. Catal., A, 2016, 520, 44–52 CrossRef CAS.
- F. Neaţu, R. S. Marin, M. Florea, N. Petrea, O. D. Pavel and V. I. Pârvulescu, Appl. Catal., B, 2016, 180, 751–757 CrossRef.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- V. R. Gangwal, J. Van Der Schaaf, B. F. M. Kuster and J. C. Schouten, J. Catal., 2005, 229, 389–403 CrossRef CAS.
- Y. Zhao, M. Cai, J. Xian, Y. Sun and G. Li, J. Mater. Chem. A, 2021, 9, 20164–20183 RSC.
- M. Guo, X. Lu, J. Xiong, R. Zhang, X. Li, Y. Qiao, N. Ji and Z. Yu, ChemSusChem, 2022, 15, e202201074 CAS.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966–2972 RSC.
- C.-T. Chen, C. Van Nguyen, Z.-Y. Wang, Y. Bando, Y. Yamauchi, M. T. S. Bazziz, A. Fatehmulla, W. A. Farooq, T. Yoshikawa, T. Masuda and K. C.-W. Wu, ChemCatChem, 2018, 10, 361–365 CrossRef CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- P. Vinke, W. van der Poel and H. van Bekkum, Stud. Surf. Sci. Catal., 1991, 59, 385–394 CrossRef CAS.
- J. Xie, P. Duan, N. Kaylor, K. Yin, B. Huang, K. Schmidt-Rohr and R. J. Davis, ACS Catal., 2017, 7, 6745–6756 CrossRef CAS.
- H. Ait Rass, N. Essayem and M. Besson, ChemSusChem, 2015, 8, 1206–1217 CrossRef CAS PubMed.
- T. Gao, Y. Yin, W. Fang and Q. Cao, Mol. Catal., 2018, 450, 55–64 CrossRef CAS.
- J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef CAS PubMed.
- D. K. Mishra, H. J. Lee, J. Kim, H. S. Lee, J. K. Cho, Y. W. Suh, Y. Yi and Y. J. Kim, Green Chem., 2017, 19, 1619–1623 RSC.
- B. Sang, J. Li, X. Tian, F. Yuan and Y. Zhu, Mol. Catal., 2019, 470, 67–74 CrossRef.
- O. Schade, P. Dolcet, A. Nefedov, X. Huang, E. Saraçi, C. Wöll and J.-D. Grunwaldt, Catalysts, 2020, 10, 342 CrossRef CAS.
- A. Lolli, R. Amadori, C. Lucarelli, M. G. Cutrufello, E. Rombi, F. Cavani and S. Albonetti, Microporous Mesoporous Mater., 2016, 226, 466–475 CrossRef CAS.
- B. Siyo, M. Schneider, J. Radnik, M. M. Pohl, P. Langer and N. Steinfeldt, Appl. Catal., A, 2014, 478, 107–116 CrossRef CAS.
- Y. Y. Gorbanev, S. Kegnæs and A. Riisager, Top. Catal., 2011, 54, 1318–1324 CrossRef CAS.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J. D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- H. Chen, J. Shen, K. Chen, Y. Qin, X. Lu, P. Ouyang and J. Fu, Appl. Catal., A, 2018, 555, 98–107 CrossRef CAS.
- N. Masoud, B. Donoeva and P. E. de Jongh, Appl. Catal., A, 2018, 561, 150–157 CrossRef CAS.
- S. Albonetti, A. Lolli, V. Morandi, A. Migliori, C. Lucarelli and F. Cavani, Appl. Catal., B, 2015, 163, 520–530 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- A. Lolli, R. Amadori, C. Lucarelli, M. G. Cutrufello, E. Rombi, F. Cavani and S. Albonetti, Microporous Mesoporous Mater., 2016, 226, 466–475 CrossRef CAS.
- A. I. M. Rabee, S. D. Le, K. Higashimine and S. Nishimura, ACS Sustain. Chem. Eng., 2020, 8, 7150–7161 CrossRef CAS.
- C. M. Pichler, M. G. Al-Shaal, D. Gu, H. Joshi, W. Ciptonugroho and F. Schüth, ChemSusChem, 2018, 11, 2083–2090 CrossRef CAS PubMed.
- C. Megías-Sayago, K. Chakarova, A. Penkova, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, ACS Catal., 2018, 8, 11154–11164 CrossRef.
- W. Gong, K. Zheng and P. Ji, RSC Adv., 2017, 7, 34776–34782 RSC.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- B. Siyo, M. Schneider, M. M. Pohl, P. Langer and N. Steinfelt, Catal. Lett., 2014, 144, 498–506 CrossRef CAS.
- N. Yan, Y. Yuan and P. J. Dyson, Dalton Trans., 2013, 42, 13294–13304 RSC.
- C. Megías-Sayago, A. Lolli, D. Bonincontro, A. Penkova, S. Albonetti, F. Cavani, J. A. Odriozola and S. Ivanova, ChemCatChem, 2020, 12, 1177–1183 CrossRef.
- A. Villa, M. Schiavoni and L. Prati, Catal. Sci. Technol., 2012, 2, 673–682 RSC.
- M. H. Hussain, N. F. Abu Bakar, A. N. Mustapa, K. F. Low, N. H. Othman and F. Adam, Nanoscale Res. Lett., 2020, 15, 1–10 CrossRef CAS PubMed.
- C. Megías-Sayago, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, Catal. Today, 2019, 333, 169–175 CrossRef.
- J. Cai, H. Ma, J. Zhang, Q. Song, Z. Du, Y. Huang and J. Xu, Chem. Eur. J., 2013, 19, 14215–14223 CrossRef CAS PubMed.
- W. Zhuang, X. Liu, L. Chen, P. Liu, H. Wen, Y. Zhou and J. Wang, Green Chem., 2020, 22(13), 4199–4209 RSC.
- P. Kandasamy, P. Gogoi, A. T. Venugopalan and T. Raja, Catal. Today, 2021, 375, 145–154 CrossRef CAS.
- A. Villa, M. Schiavoni, S. Campisi, G. M. Veith and L. Prati, ChemSusChem, 2013, 6, 609–612 CrossRef CAS PubMed.
- N. Mei, B. Liu, J. Zheng, K. Lv, D. Tang and Z. Zhang, Catal. Sci. Technol., 2015, 5, 3194–3202 RSC.
- S. E. Davis, L. R. Houk, E. C. Tamargo, A. K. Datye and R. J. Davis, Catal. Today, 2011, 160, 55–60 CrossRef CAS.
- G. Yi, S. P. Teong and Y. Zhang, Green Chem., 2016, 18, 979–983 RSC.
- C. A. Antonyraj, T. T. Huynh, K. W. Lee, Y. J. Kim, S. Shin, J. Shik and J. Ku Cho, Chem. Sci. J., 2039, 130, 156 CrossRef.
- X. Han, C. Li, Y. Guo, X. Liu, Y. Zhang and Y. Wang, Appl. Catal., A, 2016, 526, 1–8 CrossRef CAS.
- B. Donoeva, N. Masoud and P. E. De Jongh, ACS Catal., 2017, 7, 4581–4591 CrossRef CAS PubMed.
- B. Liu, Y. Ren and Z. Zhang, Green Chem., 2015, 17, 1610–1617 RSC.
- Y. Zhang, Z. Xue, J. Wang, X. Zhao, Y. Deng, W. Zhao and T. Mu, RSC Adv., 2016, 6, 51229–51237 RSC.
- C. Zhou, W. Shi, X. Wan, Y. Meng, Y. Yao, Z. Guo, Y. Dai, C. Wang and Y. Yang, Catal. Today, 2019, 330, 92–100 CrossRef CAS.
- S. Albonetti, T. Pasini, A. Lolli, M. Blosi, M. Piccinini, N. Dimitratos, J. a. Lopez-Sanchez, D. J. Morgan, A. F. Carley, G. J. Hutchings and F. Cavani, Catal. Today, 2012, 195, 120–126 CrossRef CAS.
- A. Lolli, S. Albonetti, L. Utili, R. Amadori, F. Ospitali, C. Lucarelli and F. Cavani, Appl. Catal., A, 2015, 504, 408–419 CrossRef CAS.
- H. Choudhary and K. Ebitani, Chem. Lett., 2016, 45, 613–615 CrossRef CAS.
- D. Bonincontro, A. Lolli, A. Villa, L. Prati, N. Dimitratos, G. M. Veith, L. E. Chinchilla, G. A. Botton, F. Cavani and S. Albonetti, Green Chem., 2019, 21, 4090–4099 RSC.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef PubMed.
- C. A. Antonyraj, N. T. T. Huynh, S. K. Park, S. Shin, Y. J. Kim, S. Kim, K. Y. Lee and J. K. Cho, Appl. Catal., A, 2017, 547, 230–236 CrossRef CAS.
- H. Ait Rass, N. Essayem and M. Besson, Green Chem., 2013, 15(8), 2240–2251 RSC.
- F. Li, X. L. Li, C. Li, J. Shi and Y. Fu, Green Chem., 2018, 20, 3050–3058 RSC.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824 RSC.
- L. Ardemani, G. Cibin, A. J. Dent, M. a. Isaacs, G. Kyriakou, A. F. Lee, C. M. a. Parlett, S. a. Parry and K. Wilson, Chem. Sci., 2015, 6, 4940–4945 RSC.
- T. Gao, T. Gao, W. Fang and Q. Cao, Mol. Catal., 2017, 439, 171–179 CrossRef CAS.
- H. Xia, J. An, M. Hong, S. Xu, L. Zhang and S. Zuo, Catal. Today, 2019, 319, 113–120 CrossRef CAS.
- X. Han, L. Geng, Y. Guo, R. Jia, X. Liu, Y. Zhang and Y. Wang, Green Chem., 2015, 18, 1597–1604 RSC.
- Q. Wang, W. Hou, S. Li, J. Xie, J. Li, Y. Zhou and J. Wang, Green Chem., 2017, 19, 3820–3830 RSC.
- S. Siankevich, S. Mozzettini, F. Bobbink, S. Ding, Z. Fei, N. Yan and P. J. Dyson, Chempluschem, 2018, 83, 19–23 CrossRef CAS PubMed.
- Z. Gui, W. Cao, S. Saravanamurugan, A. Riisager, L. Chen and Z. Qi, ChemCatChem, 2016, 8, 3636–3643 CrossRef CAS.
- F. Kerdi, H. Ait Rass, C. Pinel, M. Besson, G. Peru, B. Leger, S. Rio, E. Monflier and A. Ponchel, Appl. Catal., A, 2015, 506, 206–219 CrossRef CAS.
- F. Liguori, P. Barbaro and N. Calisi, ChemSusChem, 2019, 12, 2558–2563 CrossRef CAS PubMed.
- X. Han, C. Li, X. Liu, Q. Xia and Y. Wang, Green Chem., 2017, 19, 996–1004 RSC.
- A. Jain, S. C. Jonnalagadda, K. V. Ramanujachary and A. Mugweru, Catal. Commun., 2015, 58, 179–182 CrossRef CAS.
- S. Zhang, X. Sun, Z. Zheng and L. Zhang, Catal. Commun., 2018, 113, 19–22 CrossRef CAS.
- T. Gao, Y. Yin, G. Zhu, Q. Cao and W. Fang, Catal. Today, 2019, 355, 252–262 CrossRef.
- K. T. V. Rao, J. L. Rogers, S. Souzanchi, L. Dessbesell, M. B. Ray and C. C. Xu, ChemSusChem, 2018, 11, 3323–3334 CrossRef CAS PubMed.
- T. Gao, H. Zhang, C. Hu, F. Jing and W. Fang, Ind. Eng. Chem. Res., 2020, 50(39), 17200–17209 CrossRef.
- T. Gao, J. Chen, W. Fang, Q. Cao, W. Su and F. Dumeignil, J. Catal., 2018, 368, 53–68 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- G. Yi, S. P. Teong, X. Li and Y. Zhang, ChemSusChem, 2014, 7, 2131–2135 CrossRef CAS PubMed.
- K. Yu, Y. Liu, D. Lei, Y. Jiang, Y. Wang, Y. Feng, L. L. Lou, S. Liu and W. Zhou, Catal. Sci. Technol., 2018, 8, 2299–2303 RSC.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS PubMed.
- X. hai Zhou, K. he Song, Z. huan Li, W. min Kang, H. ru Ren, K. mei Su, M. liang Zhang and B. wen Cheng, Ceram. Int., 2019, 45, 2330–2337 CrossRef.
- L. Gao, K. Deng, J. Zheng, B. Liu and Z. Zhang, Chem. Eng. J., 2015, 270, 444–449 CrossRef CAS.
- A. B. Gawade, A. V. Nakhate and G. D. Yadav, Catal. Today, 2018, 309, 119–125 CrossRef CAS.
- D. X. Martínez-Vargas, J. Rivera De La Rosa, L. Sandoval-Rangel, J. L. Guzmán-Mar, M. A. Garza-Navarro, C. J. Lucio-Ortiz and D. A. De Haro-Del Río, Appl. Catal., A, 2017, 547, 132–145 CrossRef.
- C. V Nguyen, Y.-T. Liao, T.-C. Kang, J. E. Chen, T. Yoshikawa, Y. Nakasaka, T. Masuda and K. C.-W. Wu, Green Chem., 2016, 18, 5957–5961 RSC.
- S. Verma, M. N. Nadagouda and R. S. Varma, Sci. Rep., 2017, 7, 1–6 CrossRef PubMed.
- X. Liu, M. Zhang and Z. Li, ACS Sustain. Chem. Eng., 2020, 8, 4801–4808 CrossRef CAS.
- F. Yang, Y. Ding, J. Tang, S. Zhou, B. Wang and Y. Kong, Mol. Catal., 2017, 435, 144–155 CrossRef.
- E. Hayashi, T. Komanoya, K. Kamata and M. Hara, ChemSusChem, 2017, 10, 654–658 CrossRef CAS PubMed.
- L. Bao, F. Sun, G. Zhang and T. Hu, ChemSusChem, 2020, 13, 548–555 CrossRef CAS PubMed.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 899–900 Search PubMed.
- M. Ventura, F. Nocito, E. De Giglio, S. Cometa, A. Altomare and A. Dibenedetto, Green Chem., 2018, 20, 3921–3926 RSC.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem. Int. Ed., 2014, 53, 6515–6518 CrossRef CAS PubMed.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS.
- S. M. McKenna, P. Mines, P. Law, K. Kovacs-Schreiner, W. R. Birmingham, N. J. Turner, S. Leimkühler and A. J. Carnell, Green Chem., 2017, 19, 4660–4665 RSC.
- Y. Z. Qin, Y. M. Li, M. H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- L. Zou, Z. Zheng, H. Tan, Q. Xu and J. Ouyang, RSC Adv., 2020, 10, 21781–21788 RSC.
- C. Zhang, X. Chang, L. Zhu, Q. Xing, S. You, W. Qi, R. Su and Z. He, Int. J. Biol. Macromol., 2019, 128, 132–139 CrossRef CAS PubMed.
- K.-F. Wang, C. Liu, K. Sui, C. Guo and C.-Z. Liu, ChemBioChem, 2018, 19, 654–659 CrossRef CAS PubMed.
- D. Yan, J. Xin, Q. Zhao, K. Gao, X. Lu, G. Wang and S. Zhang, Catal. Sci. Technol., 2018, 8, 164–175 RSC.
- Z. Lei, C. Dai and B. Chen, Chem. Rev., 2014, 114, 1289–1326 CrossRef CAS PubMed.
- K. R. Seddon and A. Stark, Green Chem., 2002, 4, 119–123 RSC.
- I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507–1509 CrossRef CAS PubMed.
- L. Lin, J. Liuyan and W. Yunyang, Catal. Commun., 2008, 9, 1379–1382 CrossRef CAS.
- N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990–2010 CrossRef CAS.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- T. Stahlberg, E. Eyjolfsdottir, Y. Y. Gorbanev, I. Sdaba and A. Riisager, Catal. Lett., 2012, 142, 1089–1097 CrossRef CAS.
- D. Liu, Y. Zhang and E. Y.-X. Chen, Green Chem., 2012, 14, 2738 RSC.
- A. K. Khatana, V. Singh, M. K. Gupta and B. Tiwari, Synth, 2018, 50, 4290–4294 CrossRef CAS.
- O. Bortolini, C. Chiappe, M. Fogagnolo, P. P. Giovannini, A. Massi, C. S. Pomelli and D. Ragno, Chem. Commun., 2014, 50, 2008–2011 RSC.
- D. Yan, J. Xin, C. Shi, X. Lu, L. Ni, G. Wang and S. Zhang, Chem. Eng. J., 2017, 323, 473–482 CrossRef CAS.
- R. Chen, J. Xin, D. Yan, H. Dong, X. Lu and S. Zhang, ChemSusChem, 2019, 12, 2715–2724 CrossRef CAS PubMed.
- P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333–1347 CrossRef CAS.
- A. Al Ghatta and J. P. Hallett, Green Chem., 2022, 24, 3309–3313 RSC.
- A. Rafat, A. Al Ghatta, P. Verdia, M. S. Koo and J. P. Hallett, ACS Sustain. Chem. Eng., 2021, 9, 10524–10536 CrossRef.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, 1–8 Search PubMed.
- P. V. Rathod and V. H. Jadhav, ACS Sustain. Chem. Eng., 2018, 6, 5766–5771 CrossRef CAS.
- S. Wang, Z. Zhang and B. Liu, ACS Sustain. Chem. Eng., 2015, 3, 406–412 CrossRef CAS.
- G. Chen, L. Wu, H. Fan and B. G. Li, Ind. Eng. Chem. Res., 2018, 57, 16172–16181 CrossRef CAS.
- Z. Yang, W. Qi, R. Su and Z. He, Energy Fuels, 2016, 31, 533–541 CrossRef.
- H. Liu, X. Cao, T. Wang, J. Wei, X. Tang, X. Zeng, Y. Sun, T. Lei, S. Liu and L. Lin, J. Ind. Eng. Chem., 2019, 77, 209–214 CrossRef CAS.
- H. Yuan, J. Li, H. dong Shin, G. Du, J. Chen, Z. Shi and L. Liu, Bioresour. Technol., 2018, 247, 1184–1188 CrossRef CAS PubMed.
- Q. Wang, W. Hou, T. Meng, Q. Hou, Y. Zhou and J. Wang, Catal. Today, 2019, 319, 57–65 CrossRef CAS.
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustain. Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS.
- F. Xia, J. Ma, X. Jia, M. Guo, X. Liu, H. Ma, J. Gao and J. Xu, Chem. Asian J., 2019, 14, 3329–3334 CrossRef CAS PubMed.
- J. Zhao, A. Jayakumar, Z. T. Hu, Y. Yan, Y. Yang and J. M. Lee, ACS Sustain. Chem. Eng., 2018, 6, 284–291 CrossRef CAS.
- C. Zhou, J. Zhao, H. Sun, Y. Song, X. Wan, H. Lin and Y. Yang, ACS Sustain. Chem. Eng., 2019, 7, 315–323 CrossRef CAS.
- J. Zhao, A. Jayakumar and J. M. Lee, ACS Sustain. Chem. Eng., 2018, 6, 2976–2982 CrossRef CAS.
- M. Lang and H. Li, ChemSusChem, 2022, 15, e202101531 CrossRef CAS PubMed.
- A. Vy Tran, S. K. Park, H. Jin Lee, T. Yong Kim, Y. Kim, Y. W. Suh, K. Y. Lee, Y. Jin Kim and J. Baek, ChemSusChem, 2022, 15, e202200375 CAS.
- M. J. Gilkey, A. V. Mironenko, D. G. Vlachos and B. Xu, ACS Catal., 2017, 7, 6619–6634 CrossRef CAS.
- L. Wei, J. Zhang, W. Deng, S. Xie, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 8013–8016 RSC.
- S. K. Suri, M. S. Thakur and S. Bhardwaj, J. Am. Oil Chem. Soc., 1993, 70, 59–64 CrossRef CAS.
- Surfactants Market Size Growth & Share | Global Industry Report, 2022, https://www.grandviewresearch.com/industry-analysis/surfactants-market, accessed 4 November 2019 Search PubMed.
- L. Elsgaard, S. O. Petersen and K. Debosz, Environ. Toxicol. Chem., 2001, 20, 1656–1663 CrossRef CAS PubMed.
- J. L. G. de Almeida, M. Dufaux, Y. Ben Taarit and C. Naccache, J. Am. Oil Chem. Soc., 1994, 71, 675–694 CrossRef CAS.
- J. A. Kocal, B. V. Vora and T. Imai, Appl. Catal., A, 2001, 221, 295–301 CrossRef CAS.
- I. Dolganova, I. Dolganov, E. Ivanchina and E. Ivashkina, J. Surfactants Deterg., 2018, 21, 175–184 CrossRef CAS.
- D. W. Roberts, Org. Process Res. Dev., 2003, 7, 172–184 CrossRef CAS.
- X. Yue and Y. Queneau, ChemSusChem, 2022, 2022, e202102660 Search PubMed.
- G. K. Morse, R. Perry and J. N. Lester, Sci. Total Environ., 1995, 166(1), 179–192 CrossRef CAS.
- R. S. Lanigan and T. A. Yamarik, Int. J. Toxicol., 2002, 21, 95–142 CrossRef CAS PubMed.
- Y. Liu and J. Chen, Encyclopedia of Ecology, Elsevier, 2nd edn, 2014 Search PubMed.
- L. G. Scharpf, I. D. Hill, P. L. Wright, J. B. Plank, M. L. Keplinger and J. C. Calandra, Nature, 1972, 239, 231–234 CrossRef CAS PubMed.
- Y. YU, J. Zhao and A. E. Bayly, Chin. J. Chem. Eng., 2008, 16, 517–527 CrossRef CAS.
- A. Gassama, C. Ernenwein, A. Youssef, M. Agach, E. Riguet, S. Marinković, B. Estrine and N. Hoffmann, Green Chem., 2013, 15, 1558–1566 RSC.
- D. S. van Es, S. Marinkovic, X. Oduber and B. Estrine, J. Surfactants Deterg., 2013, 16, 147–154 CrossRef.
- H. N. Gavala and B. K. Ahring, Biodegradation, 2002, 13, 201–209 CrossRef CAS PubMed.
- A. Al Ghatta, R. C. Aravenas, Y. Wu, J. M. Perry, J. Lemus and J. P. Hallett, ACS Sustain. Chem. Eng., 2022, 10, 8846–8855 CrossRef CAS PubMed.
- V. M. Leon, A. Gomez-Parra and E. Gonzalez-Mazo, Environ. Sci. Technol., 2004, 38(8), 2359–2367 CrossRef CAS PubMed.
- M. J. Scott and M. N. Jones, Biochim. Biophys. Acta – Biomembr., 2000, 1(2), 235–251 CrossRef PubMed.
- J. L. Berna, A. Moreno and J. Ferrer, J. Chem. Technol. Biotechnol., 2007, 50, 387–398 CrossRef.
- R. L. VanEtten, G. A. Clowes, J. F. Sebastian and M. L. Bender, J. Am. Chem. Soc., 1967, 89, 3253–3262 CrossRef CAS.
- L. Florencio, J. A. Field and G. Lettinga, Braz. J. Chem. Eng., 1997, 14, 409–416 CrossRef CAS.
- A. V. Naik, K. E. Joseph, M. Shetty, M. A. Ardagh and P. J. Dauenhauer, ACS Sustain. Chem. Eng., 2020, 8, 18616–18625 CrossRef CAS.
- H. Nguyen, Y. Wang, D. Moglia, J. Fu, W. Zheng, M. Orazov and D. G. Vlachos, Catal. Sci. Technol., 2021, 11, 2762–2769 RSC.
- G. A. Kraus and J. J. Lee, J. Surfactants Deterg., 2013, 16, 317–320 CrossRef CAS.
- G. A. Kraus and T. Guney, Green Chem., 2012, 14, 1593–1596 RSC.
- A. Al Ghatta, J. M. Perry, H. Maeng, J. Lemus and J. P. Hallett, RSC Sustainability, 2023, 1, 303–309 RSC.
- W. Zuo and H. W. Wong, Green Chem. Lett. Rev., 2017, 10, 393–403 CrossRef CAS.
- A. Al Ghatta, P. Y. S. Nakasu and J. P. Hallett, Curr. Opin. Green Sustain. Chem., 2023, 41, 100792 CrossRef CAS.
- O. M. Musa, Handbook of Maleic Anhydride Based Materials: Syntheses, Properties and Applications, Springer, 2016 Search PubMed.
- T. R. Felthouse, J. C. Burnett, B. Horrell, M. J. Mummey and Y.-J. Kuo, Kirk-Othmer Encycl. Chem. Technol., 2000 DOI:10.1002/0471238961.1301120506051220.A01.PUB2.
- G. Centi, F. Trifiro, J. Ebner and V. Franchetti, Chem. Rev., 1988, 88, 55–80 CrossRef CAS.
- P. V. Mangili, P. G. Junqueira, L. S. Santos and D. M. Prata, Clean Technol. Environ. Policy, 2019, 21, 1073–1090 CrossRef CAS.
- F. Trifirò and R. Grasselli, Top. Catal., 2014, 54, 1188–1195 CrossRef.
- B. C. Trivedi and B. M. Culbertson, Maleic Anhydride, Springer, 1982 Search PubMed.
- R. M. Contractor, D. I. Garnett, H. S. Horowitz, H. E. Bergna, G. S. Patience, J. T. Schwartz and G. M. Sisler, Stud. Surf. Sci. Catal., 1994, 82, 233–242 CrossRef CAS.
- J. Gascón, C. Téllez, J. Herguido and M. Menéndez*, Ind. Eng. Chem. Res., 2005, 44, 8945–8951 CrossRef.
- G. Pavarelli, J. Velasquez Ochoa, A. Caldarelli, F. Puzzo, F. Cavani and J.-L. Dubois, ChemSusChem, 2015, 8, 2250–2259 CrossRef CAS PubMed.
- D. R. Kreile, V. A. Slavinskaya, M. V. Shimanskaya and E. Y. Lukevits, Chem. Heterocycl. Compd., 1972, 5(4), 429–430 CrossRef.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V Gonçalves, Sustain. Chem. Process., 2015,(3), 1–11 CAS.
- I. Agirre, I. Gandarias, M. L. Granados and P. L. Arias, Biomass Convers. Biorefin., 2019, 10, 1021–1033 CrossRef.
- J. Lan, J. Lin, Z. Chen and G. Yin, ACS Catal., 2015, 5, 2035–2041 CrossRef CAS.
- S. Shi, H. Guo and G. Yin, Catal. Commun., 2011, 12, 731–733 CrossRef CAS.
- X. Li and Y. Zhang, Green Chem., 2016, 18, 643–647 RSC.
- E. R. Nielsen, Ind. Eng. Chem., 1942, 41, 365–368 CrossRef.
- G. Lv, C. Chen, B. Lu, J. Li, Y. Yang, C. Chen, T. Deng, Y. Zhu and X. Hou, RSC Adv., 2016, 6, 101277–101282 RSC.
- N. Alonso-Fagúndez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef PubMed.
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC.
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- Phthalic Anhydride (PA) Production and Manufacturing Process | ICIS, https://www.icis.com/explore/resources/news/2007/11/06/9076143/phthalic-anhydride-pa-production-and-manufacturing-process/, accessed 1 October 2021.
- C. R. Dias, M. F. Portela and G. C. Bond, Rev. Catal., 1997, 39, 169–207 CrossRef CAS.
- G. C. Bond, J. Catal., 1989, 116, 531–539 CrossRef CAS.
- C. R. Dias, M. F. Portela and G. C. Bond, J. Catal., 1995, 157, 344–352 CrossRef CAS.
- S. Giarola, C. Romain, C. K. Williams, J. P. Hallett and N. Shah, Chem. Eng. Res. Des., 2016, 107, 181–194 CrossRef CAS.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2013, 16, 167–175 RSC.
- W. Jia, Y. Sun, M. Zuo, Y. Feng, X. Tang, X. Zeng and L. Lin, ChemSusChem, 2020, 13, 640–646 CrossRef CAS PubMed.
- W. Jia, W. Li, X. Zhao, Y. Feng, M. Zuo, Y. Sun, X. Tang, X. Zeng and L. Lu, Catal. Sci. Technol., 2021, 11, 5656–5662 RSC.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- I. van Scodeller, K. De Oliveira Vigier, E. Muller, C. Ma, F. Guégan, R. Wischert and F. Jérôme, ChemSusChem, 2021, 14, 313–323 CrossRef CAS PubMed.
- S. Dutta and N. S. Bhat, Biomass Convers. Biorefin., 2020, 13, 541–554 CrossRef.
- T. Salavati-Fard, S. Caratzoulas and D. J. Doren, J. Phys. Chem. A, 2015, 119, 9834–9843 CrossRef CAS PubMed.
- Z. Cui, Y. Fang and T. Tan, Catal. Lett., 2020, 150, 794–801 CrossRef CAS.
- H. J. Cho, L. Ren, V. Vattipalli, Y. H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef CAS.
- C. L. Williams, K. P. Vinter, C. C. Chang, R. Xiong, S. K. Green, S. I. Sandler, D. G. Vlachos, W. Fan and P. J. Dauenhauer, Catal. Sci. Technol., 2015, 6, 178–187 RSC.
- C. C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2013, 16, 585–588 RSC.
- Y. P. Wijaya, H. P. Winoto, Y. K. Park, D. J. Suh, H. Lee, J. M. Ha and J. Jae, Catal. Today, 2017, 293–294, 167–175 CrossRef CAS.
- A. Turkin, S. Eyley, G. Preegel, W. Thielemans, E. Makshina and B. F. Sels, ACS Catal., 2021, 11, 9204–9209 CrossRef CAS.
- M. Mascal, ACS Sustainable Chem. Eng., 2019, 7(6), 5588–5601 CrossRef CAS.
- M. Mascal, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS PubMed.
- S. Dutta, L. Wu and M. Mascal, Green Chem., 2015, 17, 3737–3739 RSC.
- J. K. Ogunjobi, T. J. Farmer, C. R. McElroy, S. W. Breeden, D. J. MacQuarrie, D. Thornthwaite and J. H. Clark, ACS Sustain. Chem. Eng., 2019, 7, 8183–8194 CrossRef CAS.
- P. A. Gushchin, I. M. Kolesnikov, V. A. Vinokurov, E. V. Ivanov, V. A. Lyubimenko and V. N. Borshch, J. Catal., 2017, 352, 75–82 CrossRef CAS.
- V. Vorotnikov, G. Mpourmpakis and D. G. Vlachos, ACS Catal., 2012, 2, 2496–2504 CrossRef CAS.
- Z. Li, Y. Jiang, Y. Li, H. Zhang, H. Li and S. Yang, Catal. Sci. Technol., 2022, 12, 1902–1921 RSC.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, ACS Catal., 2018, 8, 1843–1850 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |