
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEco-friendly synthesis of cardanol-based AB monomer for formaldehyde-free phenolic thermosets†
Benoit
Briou
 *a,
Lucas
Jégo
a,
Thomas
De Dios Miguel
b,
Nicolas
Duguet
*b and
Sylvain
Caillol
*c
*a,
Lucas
Jégo
a,
Thomas
De Dios Miguel
b,
Nicolas
Duguet
*b and
Sylvain
Caillol
*c
aOrpia Innovation, CNRS, Bâtiment Chimie Balard, 1919 Route de Mendes, 34000 Montpellier, France. E-mail: b.briou@orpiainnovation.com
bUniv Lyon, Université Claude Bernard Lyon1, CNRS, INSA-Lyon, CPE-Lyon, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires, ICBMS, UMR 5246, Equipe CAtalyse, SYnthèse et ENvironnement (CASYEN), 1 rue Victor Grignard, 69100, Villeurbanne, France. E-mail: nicolas.duguet@univ-lyon1.fr
cICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: sylvain.caillol@enscm.fr
First published on 16th May 2023
Abstract
Phenolic thermosets are mainly synthesized from two very toxic and petroleum-based molecules, phenol and formaldehyde. This study aims at replacing these two species with a unique bio-based phenolic molecule bearing an aldehyde function. For this purpose, cardanol, a phenolic component extracted from cashew nut shells, has been chemically modified to give 8-(3-hydroxyphenyl)octanal. Two synthetic routes were investigated, each one consisting in three steps and sharing the same key intermediate. The first route consists in the epoxidation of cardanol, followed by hydrolysis to form diols, and finally an oxidative cleavage of these diols using NaIO4. A second route has been considered consisting in an epoxidation, followed by the formation of β-hydroxy hydroperoxides with H2O2, and finally a thermal cleavage. The green potential of these two methods was compared and discussed with other routes described in the literature. The resulting aldehyde was subsequently used as an AB monomer to obtain phenolic network by direct step growth homopolymerization, enabling the substitution of both phenol and formaldehyde at the same time. The resin formed thereby shows a low Tg due to the C8 alkyl chain of 8-(3-hydroxyphenyl)octanal as well as a good thermal stability.
Sustainability spotlightPhenolic thermosets have found numerous applications thanks to their high thermal and chemical resistance and their rigid mechanical behavior. However, most of them are prepared from phenol and formaldehyde, that are fossil-based reagents classified as CMR (Carcinogenic Mutagenic Reprotoxic). To address these concerns, we have prepared a unique bio-based platform molecule, incorporating both a phenol and an aldehyde moiety, which was synthesized from cardanol, a phenolic compound extracted from cashew nut shells. Several routes were investigated and discussed based on their safety score and green metrics. The resulting monomer was used to prepare phenolic networks with improved sustainability profile (bio-based origin, no CMR compounds). |
Introduction
Since their discovery in 1907 by L. Baekeland,1 phenolic thermosets have been used in a broad range of applications thanks to their high thermal and chemical resistance and their rigid mechanical behaviour. Phenolic thermosets are commonly used in foams, molding compounds, adhesives, electric laminates, acid-resistant coatings and fiber-reinforced composites among others.2,3 There are two different ways to obtain phenolic thermosets. Both consist in the reaction between a phenol derivative with reactive sites on aromatic ring and a molecule containing an aldehyde moiety. When the reaction is performed with an alkali catalyst, it leads to resol networks in only one step.4 Whereas with an acid catalyst, a second step is needed to add a crosslinker – mainly formaldehyde – to obtain novolac thermosets.5 Generally, and historically, the most used reactants are phenol and formaldehyde due to their high reactivity and low price (Fig. 1A). However, they are both very toxic substances classified as CMR (Carcinogenic Mutagenic Reprotoxic) by the ECHA (European Chemicals Agency) and are also from fossil origin. In the current context in favor to non-toxic and bio-based resources for social and ecological reasons, many studies were performed to find less harmful and bio-based substituents. In the literature, formaldehyde was substituted by less volatile aldehydes, with higher molar masses. However, these aldehydes turned out to be less reactive toward phenol.2 Other aldehydes such as lignin-based aldehydes,6,7 benzaldehyde,8,9 furfural10,11 and nonanal12 were recently used as alternatives to formaldehyde in phenolic networks. Concerning the partial or total replacement of phenol, bio-based phenols present in bio-oil13 or natural phenols like eugenol14 and cardanol12,15,16 were also investigated (Fig. 1B). Cardanol, produced by vacuum distillation of cashew nutshell liquid (CNSL), is a promising non-edible and non-toxic renewable resource used in a wide range of applications.16–19 In fact, cardanol is a mixture of 3-pentadecenyl phenols with saturated (5–8%) or unsaturated alkyl chains, with monoene (48–49%), diene (16–17%) and triene (29–30%) as illustrated in Fig. 2. Such a mixture increases the difficulty to analyze and predict the behavior and properties of material obtained thereof and could lead to various side-reactions. Moreover, its long alkyl chain (C15) is well known for bringing plasticizing effect.12 When desired, softness and low glass temperature (Tg) induced from this intern plasticizer could be valuable. However, this behavior is very limiting for many thermosets and reduces their range of applications. Other natural phenols such as eugenol or lignin-based phenols have shorter alkyl chains (C3), thus favoring the synthesis of very hard materials. In this context, finding a phenolic compound leading to intermediate properties between long and short dangling alkyl chains would be of high interest. Therefore, it is relevant to find a way to synthesize a phenolic compound with an intermediate alkyl chain length, preferentially between six and nine carbons.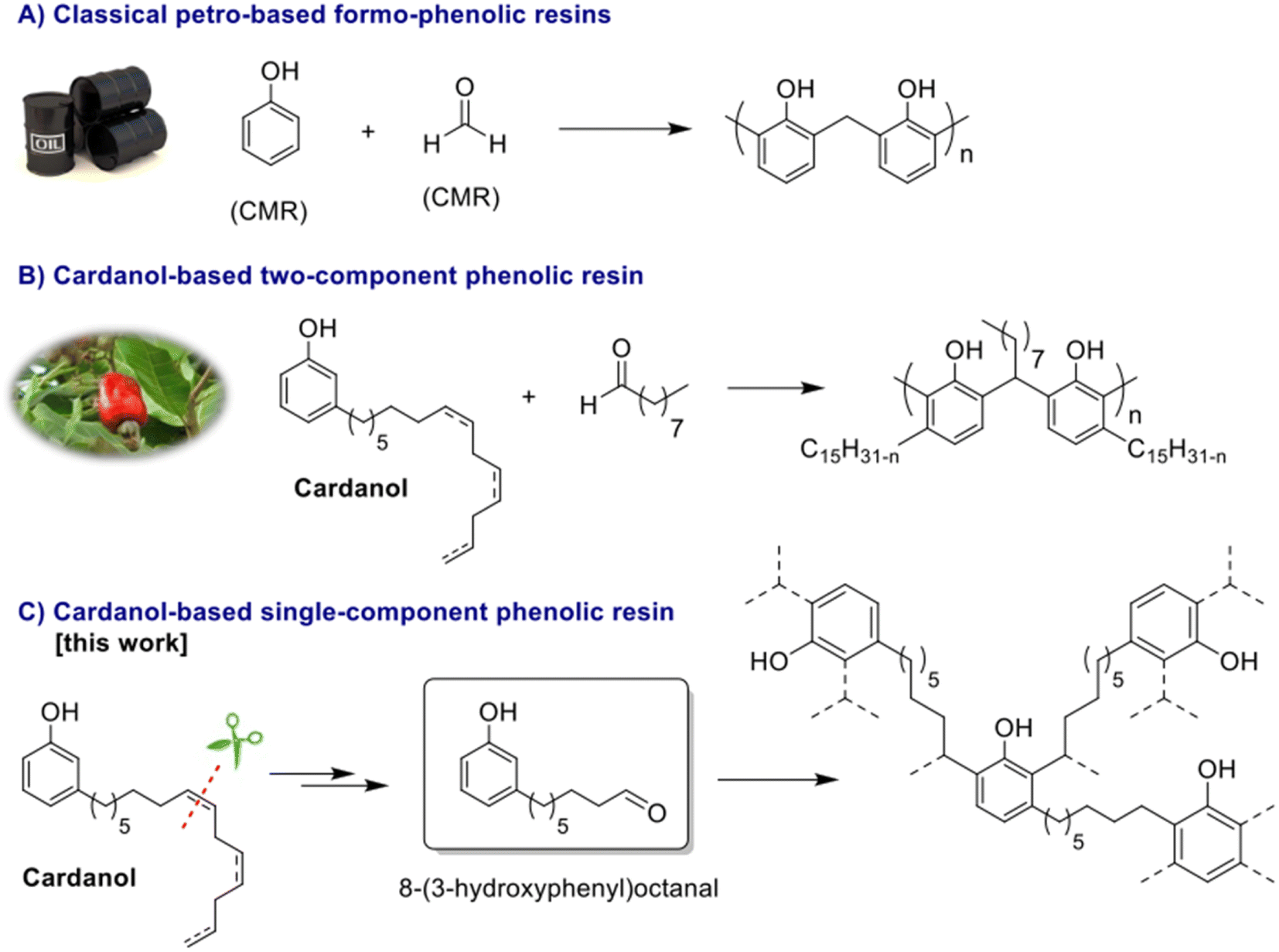 | ||
| Fig. 1 Routes to phenolic resins. | ||
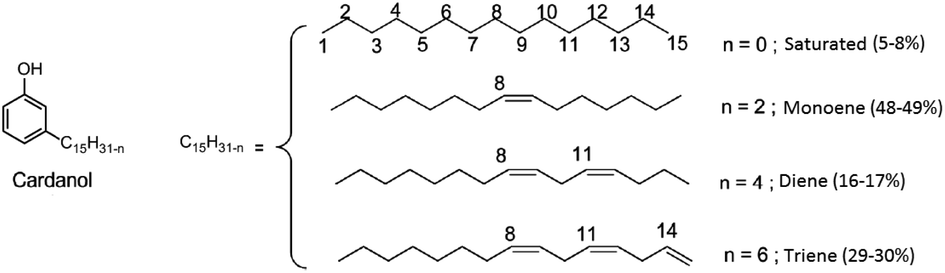 | ||
| Fig. 2 The global chemical structure of cardanol with its various fatty chains. | ||
It is acknowledged in organic chemistry that a double bond of a molecule could be cleaved, shortening the alkyl chain involved.20 These reactions can also be applied to cardanol. In fact, cleavage of the cardanol double bonds was already reported by Graham et al.21 in 2002 via reductive ozonolysis and permitted to obtain 8-(3-hydroxyphenyl)octanal with high yield (96%). This molecule has a C8 alkyl chain and the advantage to bear both a phenol and an aldehyde moiety. The presence of these two functional groups on the same molecule would allow a step growth homopolymerization without using formaldehyde or other aldehydes. However, ozonization is a dangerous method due to the intrinsic toxicity of ozone and the explosive nature of ozonide intermediates. Another synthetic pathway was described through an oxidative cleavage of diols but it led to a low yield.21
In this context, it would be highly desirable to find alternative methods to produce 8-(3-hydroxyphenyl)octanal. Given the structural similarities of cardanol and unsaturated vegetable oils, it might be possible to transpose some selective (towards the formation of aldehydes) methods reported on vegetable oil derivatives to cardanol. For instance, it was reported that fatty diols can be cleaved into aldehydes using NaIO4.22 Recently, it was also demonstrated that fatty epoxides can be cleaved into aldehydes, through the formation of β-hydroxy hydroperoxides and their subsequent thermal cleavage in flow.23 To the best of our knowledge, such methods of forming aldehydes have never been reported on cardanol or CNSL derivatives.
In this paper, we report a greener and safer synthesis of 8-(3-hydroxyphenyl)octanal from cardanol epoxides, through the formation of either diols or β-hydroxy hydroperoxides.24 The different methods to obtain 8-(3-hydroxyphenyl)octanal are compared and discussed based on green chemistry metrics. The 8-(3-hydroxyphenyl)octanal homopolycondensation and the thermal properties of the resulting phenolic thermosets were explored (Fig. 1C).
Experimental
Materials
Cardanol (NX 2026) was provided by Cardolite. Sulfuric acid (H2SO4), phosphorous acid, phosphotungstic acid, formic acid, sodium periodate (NaIO4), para-toluene sulfonic acid (p-TSA), t-amyl alcohol, magnesium sulfate, silica and 30%/50%-hydrogen peroxide were purchased from Sigma-Aldrich. Cyclohexane, acetonitrile (MeCN), ethanol and ethyl acetate were purchased from Analytic Lab. All these reagents were used as received.Characterizations and measurements
All new compounds were characterized by spectroscopic data. Reactions were monitored by TLC using aluminium silica gel (60F254). They were carried out on a plate of 0.20 mm silica gel. For revelations, UV (λ = 254 nm) light was provided (Universal UV lamp CAMAC). A phosphomolybdic acid solution or KMnO4 solution was used to reveal the TLC plate if necessary. Purification by flash chromatography was performed using silica gel 60H (40–63μ). Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker DRX 300 or Bruker ALS 300 (1H: 300 MHz, 13C: 75 MHz). Chemical shifts are given with reference to residual DMSO or CHCl3 central peaks: 2.50 and 7.26 ppm for proton, 39.52 and 77.16 ppm for carbon, respectively. J values are given in Hertz (Hz). Abbreviations are defined as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quadruplet, qt = quintet, hex = hexuplet, hept = heptuplet, m = multiplet, br = broad. Gas chromatography (GC) analyses were performed using a Shimadzu GC (GC-2025) apparatus equipped with a ZB-5-MS capillary column (30 m length, 0.25 mm i.d., 0.25 μm film thickness). The carrier gas was N2, at a total flow of 113.2 mL min−1, a column flow at 1.09 mL min−1 and the injection mode is split (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100). The column temperature was initially at 100 °C for 3 min, and then was gradually increased to 315 °C (35 °C min−1) and the temperature was kept during 6 min. The injector and FID temperature were respectively set at 250 °C and 315 °C. Thermogravimetric analysis (TGA) (under air) of the polymers (ca. 10 mg) was carried out using a TA Instruments TGA 51 apparatus. The polymer samples were heated from room temperature to 500 °C at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) analyses, under a N2 atmosphere, of the purified polymers (ca. 10 mg) were carried out using a TA Instruments Q100 instrument equipped with the DSC 200 F3 software. The instrument was calibrated with noble metals and checked before analysis with an indium sample (Tm = 156 °C). The heating or cooling range was from −50 °C to 150 °C at a scanning rate of 20 °C min−1.
:100). The column temperature was initially at 100 °C for 3 min, and then was gradually increased to 315 °C (35 °C min−1) and the temperature was kept during 6 min. The injector and FID temperature were respectively set at 250 °C and 315 °C. Thermogravimetric analysis (TGA) (under air) of the polymers (ca. 10 mg) was carried out using a TA Instruments TGA 51 apparatus. The polymer samples were heated from room temperature to 500 °C at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) analyses, under a N2 atmosphere, of the purified polymers (ca. 10 mg) were carried out using a TA Instruments Q100 instrument equipped with the DSC 200 F3 software. The instrument was calibrated with noble metals and checked before analysis with an indium sample (Tm = 156 °C). The heating or cooling range was from −50 °C to 150 °C at a scanning rate of 20 °C min−1.
Epoxidation of cardanol to CxEp
Cardanol (20 g, 66.45 mmol, 1 equiv.), formic acid (9.169 g, 199.35 mmol, 3 equiv.) and H2SO4 (0.065 g, 0.66 mmol, 0.01 equiv., 1 mol%) were mixed and heated at 50 °C. 30%-H2O2 (132.9 mmol/double bond, 1.5 equiv./double bond) was added dropwise to the mixture which was stirred at 65 °C for 3 h. After the completion of the reaction, there was two phases, one aqueous, one organic. The organic phase was diluted with ethyl acetate (25 mL). Water was removed by liquid/liquid extraction. The organic phase was then washed three times with water (25 mL) and dried over MgSO4. After filtration, the filtrate was concentrated under vacuum.Hydrolysis of epoxidized cardanol to CxDOH
In a 100 mL round-bottom flask, CxEp (16.8 g, 48.55 mmol, 1 equiv.) was mixed with phosphorous acid (79.63 mg, 0.97 mmol, 0.02 equiv., 2 mol%), ethyl acetate (15 mL) and water (15 mL) at 90 °C for 4 h under mechanical stirring at reflux. Water was removed from the mixture by liquid–liquid extraction then the mixture was diluted in ethyl acetate (10 mL), washed three times with water (10 mL) and dried over MgSO4. After filtration, the solvent of the organic layer was removed by vacuum.Oxidative cleavage of cardanol diol to CAl
To a stirred solution of CxDOH (17.2 g, 43.32 mmol, 1 equiv.) in ethanol (15 mL)–H2O (15 mL) at 25 °C, NaIO4 was added portion wise (13.9 g, 65 mmol 1.5 equiv.). After stirring for 2 h, the reaction mixture was filtered, the filtrate concentrated in vacuo and the residue extracted with ethyl acetate (25 mL), washed three times with water (25 mL). After drying with MgSO4, the organic extract was concentrated, and the crude product was purified by chromatography on a silica column (cyclohexane/ethyl acetate 9:1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2.47 (t, J = 7.6 Hz, 2H, CH2-Ph), 6.44–6.66 (m, 3H, aromatic CH), 7.04 (td, J = 7.3, 1.4 Hz, 1H, aromatic CH), 9.20 (s, OH), 9.66 (s, 1H, CHO). 13C NMR (75 MHz, d6-DMSO), δC (ppm) 22.0 (CH2), 28.96 (CH2), 28.97 (CH2), 29.1 (CH2), 31.3 (CH2), 35.6
O), 2.47 (t, J = 7.6 Hz, 2H, CH2-Ph), 6.44–6.66 (m, 3H, aromatic CH), 7.04 (td, J = 7.3, 1.4 Hz, 1H, aromatic CH), 9.20 (s, OH), 9.66 (s, 1H, CHO). 13C NMR (75 MHz, d6-DMSO), δC (ppm) 22.0 (CH2), 28.96 (CH2), 28.97 (CH2), 29.1 (CH2), 31.3 (CH2), 35.6 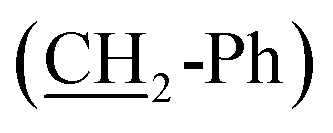 , 43.4
, 43.4 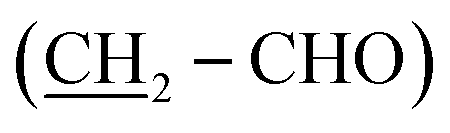 , 113.0 (CHAr), 115.6 (CHAr), 119.4 (CHAr), 129.5 (CHAr), 144.1 (Cq), 157.7 (Cq-OH), 204.0 (Cqaldehyde).
, 113.0 (CHAr), 115.6 (CHAr), 119.4 (CHAr), 129.5 (CHAr), 144.1 (Cq), 157.7 (Cq-OH), 204.0 (Cqaldehyde).
Ring-opening of cardanol mono-epoxides to cardanol β-hydroxy hydroperoxides (C1HP)
In a 100 mL double jacketed reactor, a mixture of cardanol mono-epoxides C1Ep (400 mg, 1.2 mmol) was suspended/dissolved in t-amyl alcohol (5 mL) used as a solvent. Phosphotungstic acid (PTA, 4 mg, 0.1 mol%) was added followed by dropwise addition of H2O2 (50 wt% in water, 100 μL, 1.3 mmol, 1.1 equiv.). The reactor was closed, purged with an argon atmosphere and stirred at 10 °C for 16 h (the reaction was monitored by GC and TLC). The reaction mixture was filtered over a Celite pad and washed with ethyl acetate (10 mL). The filtrate was concentrated under reduced pressure to give the crude product. The residue was purified by column chromatography (cyclohexane/ethyl acetate 90:10 to 70:30).
:38 mixture of isomers) = 11.22 (s, 1H, OOH), 9.18 (s, 1H, Ar-OH), 7.03 (d, J = 1.4, 1H, HAr), 6.71–6.46 (m, 3H, HAr), 4.48 (d, J = 4.8, 1H, OHminor isomer), 4.37 (d, J = 5.2, 1H, OHmajor isomer), 3.90–3.55 (m, 2H, 2 CH), 2.46 (t, J = 7.2, 2H, CH2), 1.69–1.12 (m, 22H, 11 CH2), 0.86 (t, J = 6.9, 3H, CH3). 13C NMR (75 MHz, d6-DMSO) δ (ppm). δc = 157.21 (CAr–OH), 143.69 (CAr), 129.04 (CAr), 118.88 (CAr), 115.10 (CAr), 112.52 (CAr), 87.36 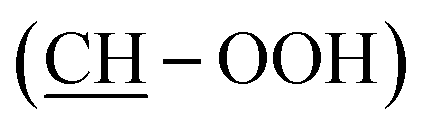 , 69.23
, 69.23 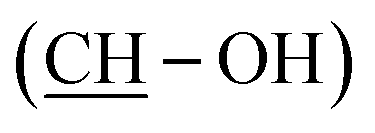 , 35.16 (CH2), 31.36 (CH2), 31.22 (CH2), 31.08 (CH2), 30.88 (CH2), 29.22 (CH2), 28.81 (CH2), 28.68 (CH2), 25.89 (CH2), 25.75 (CH2), 22.10 (CH2), 22.06 (CH2), 13.95 (CH3).
, 35.16 (CH2), 31.36 (CH2), 31.22 (CH2), 31.08 (CH2), 30.88 (CH2), 29.22 (CH2), 28.81 (CH2), 28.68 (CH2), 25.89 (CH2), 25.75 (CH2), 22.10 (CH2), 22.06 (CH2), 13.95 (CH3).
Thermal cleavage of cardanol β-hydroxy hydroperoxides to CAl
A solution of cardanol β-hydroxy-hydroperoxide C1HP (0.150 g, 0.4 mmol) in CH3CN (15 mL) was prepared with a concentration of 10 g L−1. The solution was pumped through an oven heated at 300 °C with a flow rate between 1 and 2 mL min−1. Then, the solvent, heptanal and other volatile aldehydes were evaporated under reduced pressure to give 8-(3-hydroxyphenyl)octanal (CAl) (30 mg, 32% yield) as a colorless oil.Synthesis of the homo-8-(3-hydroxyphenyl)octanal resin (HCAlR)
In an aluminum pan, 8-(3-hydroxyphenyl)octanal CAl (200 mg, 0.90 mmol, 1 equiv.) alone or mixed with APTS (7.8 mg, 0.045 mmol, 0.05 equiv., 5 mol%) was heated at 120 °C for 30 min. A black solid was obtained, HCAlR.Calculation of the green metrics used
Atom Economy (AE) is calculated following the eqn (1):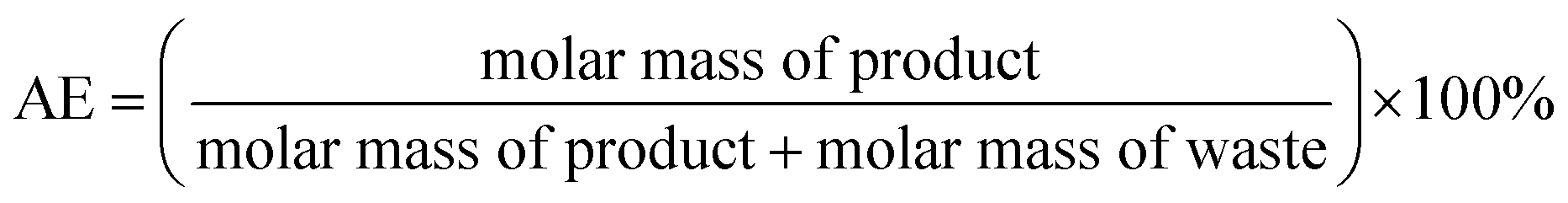 | (1) |
E-factor is calculated following the eqn (2):
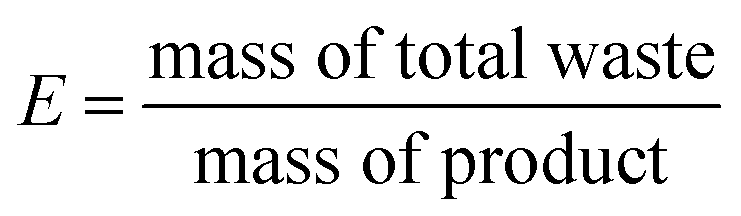 | (2) |
The atom efficacy is calculated following the eqn (3):
| Atom efficacy = yield × AE | (3) |
Evaluation of the safety score (SC) was established using the safety data sheets (SDS) of the products involved in the synthesis of the molecule of interest, whether reagents, catalysts or solvent, during each step: reaction, extraction, purification. A score is assigned for each category of hazard considering the number of sub-categories of this hazard. The maximum hazard category score is the category number +1. The minimum score is therefore +1 if it is the lowest category, 0 if there is no such danger. In our view, the following hazards: corrosive, irritant and STOT, present a less significant hazard than the other hazards, their rating is therefore halved. In contrast, because of the increased danger for humans and the process, an additional note is assigned to the following products: +2 for flammable gas, +3 for radioactive and/or explosive. The SC of a product is then the sum of the notes of each category of danger present on its SDS.
Results and discussion
Synthesis of 8-(3-hydroxyphenyl)octanal
The 8-(3-hydroxyphenyl)octanal synthesis was previously reported by reductive ozonolysis of cardanol (Scheme 1, route a).21 In their article, Graham et al.21 proceed the ozonization at −78 °C in ethyl acetate. The mixture was treated with ozonized air for 3 h, left overnight at low temperature and then, ozonization was resumed for 9 h. A step of hydrogenation was carried out in the presence of catalyst Pd–C, which resulted in an uptake of hydrogen. Undesired volatile aldehydes were removed by evaporation in vacuo. 8-(3-Hydroxyphenyl)octanal was successfully isolated with a yield of 84.7–103.6% after hydrogenation. The authors explained this high value of 103.6% by the presence of traces of retained solvent and unozonized saturated cardanol. However, because of its instability, O3 is a dangerous product. It is toxic, explosive, pollutant25–27 and needs to be produced close to the place it is used which could be an important limitation for an industrial production in emerging countries and even more at a laboratory scale. To avoid the use of ozone in the preparation of 8-(3-hydroxyphenyl)octanal, a safer 3-step synthetic route was investigated (Scheme 1, route b). The strategy consists in modifying the double bonds of cardanol into diol moieties and proceeding to an oxidative cleavage to obtain the desired 8-(3-hydroxyphenyl)octanal. The following 3-step sequence involves an epoxidation, a hydrolysis and an oxidative cleavage.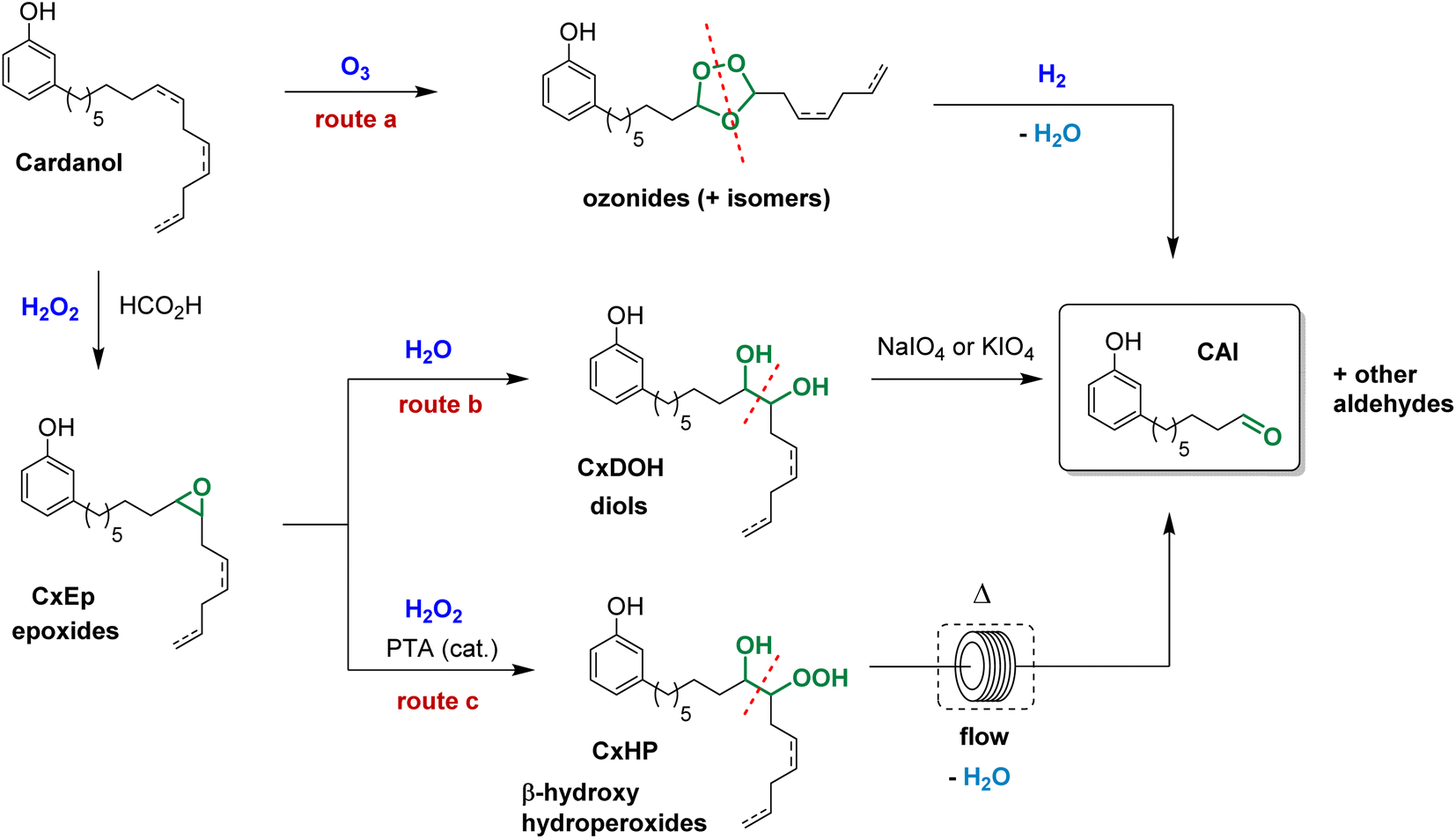 | ||
| Scheme 1 The three different pathways to obtain 8-(3-hydroxyphenyl)octanal from cardanol. | ||
First, cardanol was epoxidized using 30%-H2O2 (1.5 equiv./alkene) in the presence of formic acid. Under these conditions, the overall epoxidation of double bonds reached 82%. In details, the conversion of internal double bonds was almost complete while the epoxidation of the terminal alkene only reached 55%. This clearly demonstrates that internal double bonds are more reactive than terminal ones, as previously reported28 (Fig. 3). The uncomplete epoxidation of the terminal double bond does not represent a problem considering that the aim of this approach is to cleave the cardanol's alkyl chain at the first internal double bond (position 8). In fact, this uncomplete epoxidation limits the quantity of side products that could be produced during the oxidative cleavage sequence. In the following step, the mixture of cardanol epoxides (CxEp) was hydrolyzed to CxDOH in the presence of phosphorous acid to give the corresponding cardanol diols with 98% yield. The presence of hydroxyl moieties has been confirmed by NMR. On the 1H NMR spectrum of the cardanol diol (CxDOH) (Fig. 3), the signals j,k,l (3.0–3.2 ppm) of the epoxides were shifted to the o,m,n signals (3.4–4.2 ppm), which is consistent with a diol chemical shift.
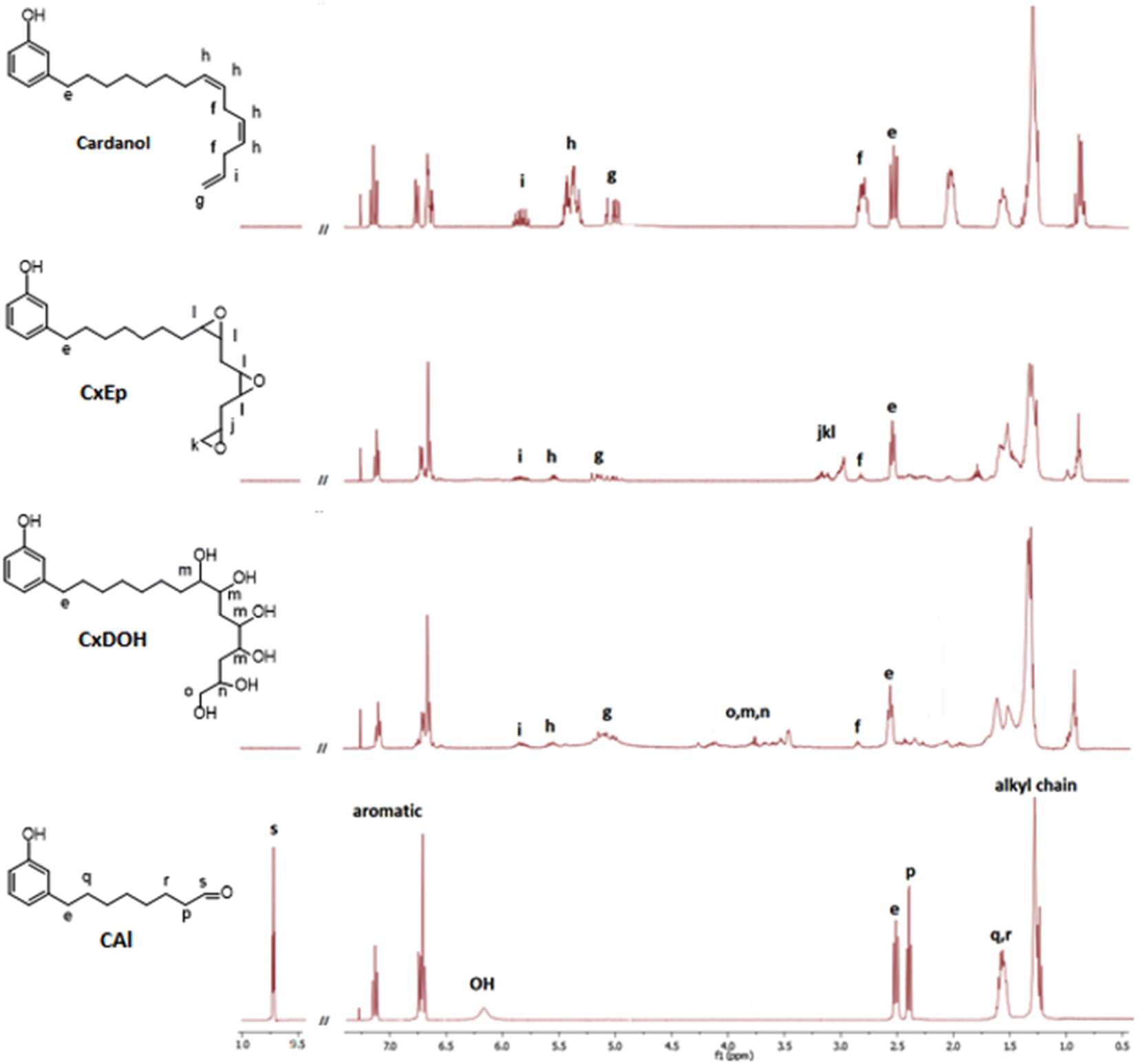 | ||
| Fig. 3 1H NMR (300 MHz, CDCl3) spectra of cardanol, CxEp, CxDOH and CAl. | ||
In addition, it was clearly noticeable in the 13C NMR spectrum that the signal of epoxy carbon shift from 45–60 ppm to the region of carbon bringing hydroxy function at 65–75 ppm, confirming the previous inference. Finally, the cleavage of alkyl chain by oxidation of the diol function was performed using NaIO4 (1 equiv./diol function). The calculation of the quantity of NaIO4 involved all the internal diols able to react such as about 1.5 moiety by molecule. The cleavage yield was 95% but the crude product included 8-(3-hydroxyphenyl)octanal and a mixture of other aldehydes. After purification by column chromatography, 8-(3-hydroxyphenyl)octanal was obtained with a yield of 52%. The loss in yield could be explained by the difficulty to separate each aldehyde from the other and some 8-(3-hydroxyphenyl)octanal has been sacrificed to obtain pure molecules. A purification by distillation could avoid such loss, as previously demonstrated by Graham et al.21 The structure of 8-(3-hydroxyphenyl)octanal was confirmed by 1H NMR spectrum (Fig. 3). Aromatic signals between 6.5 and 7.5 ppm and the hydroxy signal stated that the compound brings a phenol moiety. Alkyl chain signals between 1.2 and 3.0 ppm was still noticeable but their number has decreased. The ending CH3 signal at 0.8–0.95 ppm from CxDOH has disappeared. A new signal at 9.7 ppm validates the presence of an aldehyde function. These changes of signal were corroborating with the cleavage of the cardanol alkyl chain, replacing the CH3 by an aldehyde moiety at the end of shorter alkyl chain (C8 instead of C15). No traces of epoxy, diol or double bonds were detected. This 3-step synthesis gave 8-(3-hydroxyphenyl) octanal with a total yield of 74% (42% after purification by column chromatography).
In this work, 8-(3-hydroxyphenyl)octanal was prepared from cardanol in 3 steps following an epoxidation/hydrolysis/oxidative cleavage sequence (Scheme 1, route b). Despite that good yields can be achieved through this route, the key step requires the use of NaIO4 as an oxidant (1 equiv./diol function), thus resulting in the production of large quantities of waste. Moreover, NaIO4 is a harmful reactant and is, at some concentration, toxic for both humans and the environment. In this context, we have also investigated an alternative and potentially greener route (Scheme 1, route c).
We have previously reported that epoxidized fatty derivatives can be cleaved into the corresponding aldehydes through the formation of β-hydroxy hydroperoxides.23 These species were formed by ring-opening of epoxides with H2O2 and were next thermally cleaved in flow conditions to give the desired aldehydes.23 Based on these results, we envisioned that a similar strategy could be applied to produce 8-(3-hydroxyphenyl)octanal from cardanol, that is a much more complex substrate (Scheme 2). The first step of epoxidation was performed in similar conditions that was previously described in the route b. Then, the mixture of cardanol epoxides CxEp in t-amyl alcohol was treated at 20 °C for 16 h with aqueous 50%-H2O2 (1.1 equiv./epoxide function) in the presence of phosphotungstic acid (PTA, 0.1 mol%/epoxide function) to give a mixture of cardanol β-hydroxy hydroperoxides CxHP. The formation of hydroperoxide functions can be observed in 1H NMR spectrum by the presence of a broad peak around 11.5 ppm. The broad peak indicates the formation of many regioisomers and diastereoisomers as could be expected for cardanol derivatives. Finally, the mixture of β-hydroxy hydroperoxides was thermally cleaved at 300 °C under flow conditions (residence time of about 1 min). Satisfyingly, the formation of the desired aldehyde CAl was detected in GC along with other cleavage products. Formaldehyde was not detected under these reaction conditions. However, its formation could not be excluded considering that the terminal double bond of cardanol was partially converted to the epoxyde in the first step.
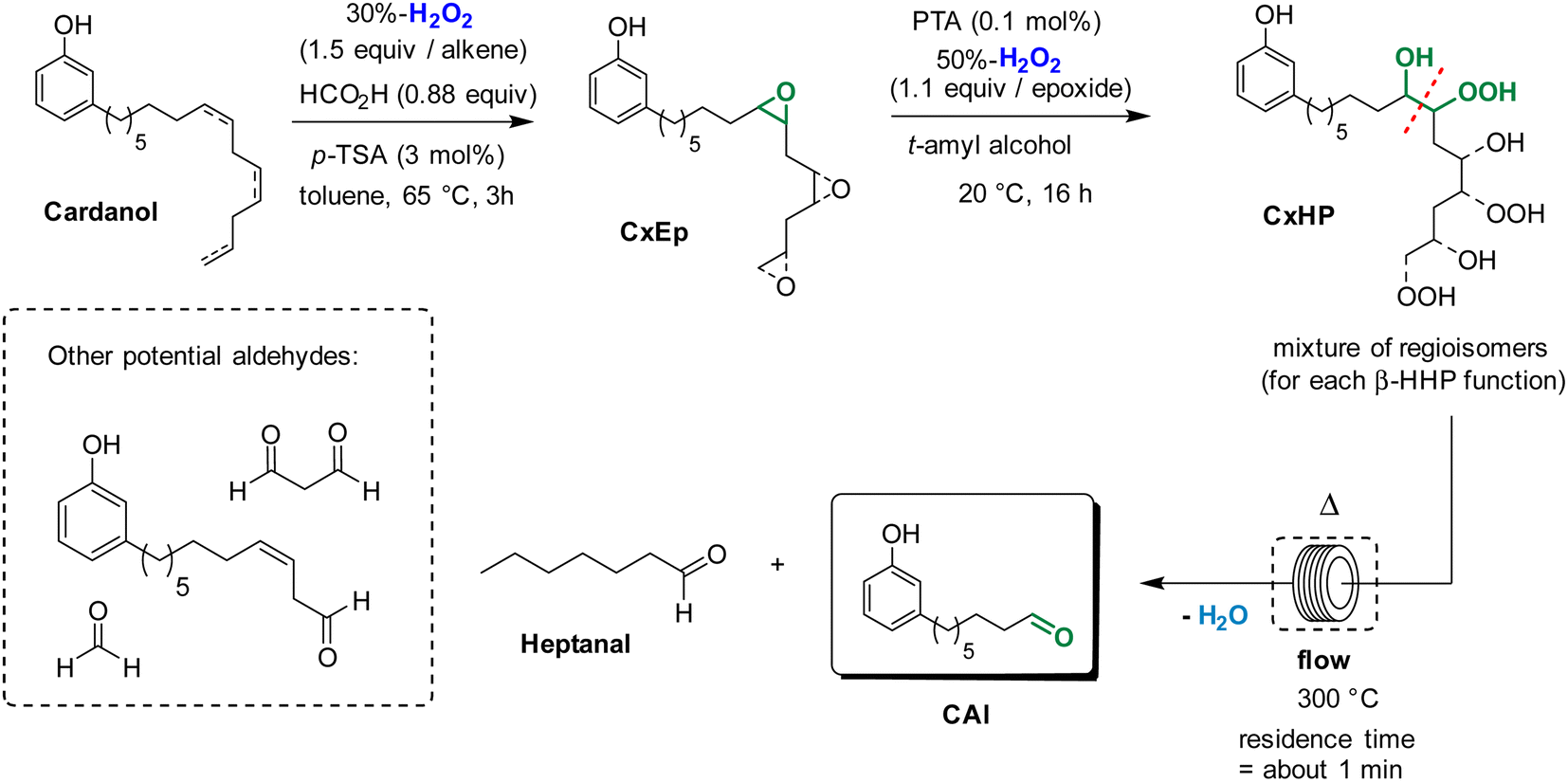 | ||
| Scheme 2 Preparation of 8-(3-hydroxyphenyl)octanal from cardanol through thermal cleavage of β-hydroxy hydroperoxides. | ||
The purification of the mixture by column chromatography afforded 8-(3-hydroxyphenyl)octanal with 23% yield. This relatively low yield can be explained by the fact that the ring opening of epoxides with aqueous 50%-H2O2 is not completely selective. Indeed, the competitive ring opening of epoxides with H2O (from aqueous H2O2) leads to the formation of the corresponding diols, that can not be cleaved under the thermal conditions.
As observed with vegetable oil derivatives, the selectivity towards β-hydroxy hydroperoxides is higher than 90% when the fatty chain bears only one double bond (i.e. using methyl oleate derivatives). However, this selectivity drops to about 50% when the fatty chain bears multiple double bonds (i.e. using methyl linoleate derivatives).23 Considering that cardanol is actually a mixture of compounds with a fatty chain bearing none C0 (5–8%), one C1 (48–49%), two C2 (16–17%) or three double bonds C3 (29–30%), a lot of diols were produced in that case. This could explain why only a small amount of the desired aldehyde was produced. To support this hypothesis, the mixture of epoxidized cardanol CxEp was purified by column chromatography (see ESI† for details) (Scheme 3). A fraction of cardanol mono-epoxides was isolated with 19% yield. From 1H NMR, it was found that this fraction of mono-epoxides contains 87% of saturated chain (C1Ep:0), 3% of one double bond (C1Ep:1) and 10% of two double bonds (C1Ep:2) (Fig. 4, top). The formation of these 3 cardanol mono-epoxides was also validated by HRMS (see ESI†). The mixture of mono-epoxides was treated with 50%-H2O2 under the same conditions to give the corresponding β-hydroxy hydroperoxide C1HP with 43% yield after purification by column chromatography. In that case, the formation of the β-hydroxy hydroperoxide function was assessed by 1H NMR with the presence of a sharp peak at 11.2 ppm (Fig. 4, middle). The GC analysis of compound C1HP shows that it can be readily cleaved at 300 °C (temperature of the GC injector) to give the desired aldehyde CAl and heptanal (Fig. 5).
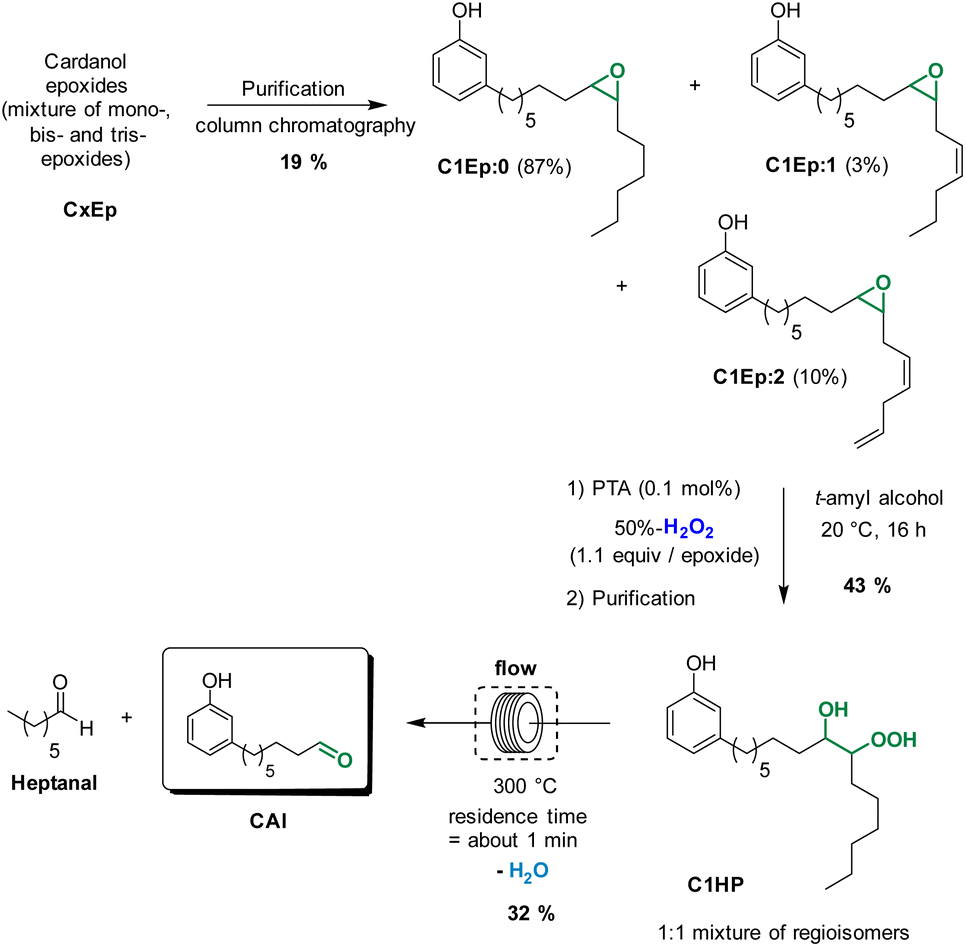 | ||
| Scheme 3 Preparation of 8-(3-hydroxyphenyl)octanal from a purified cardanol mono-epoxide. | ||
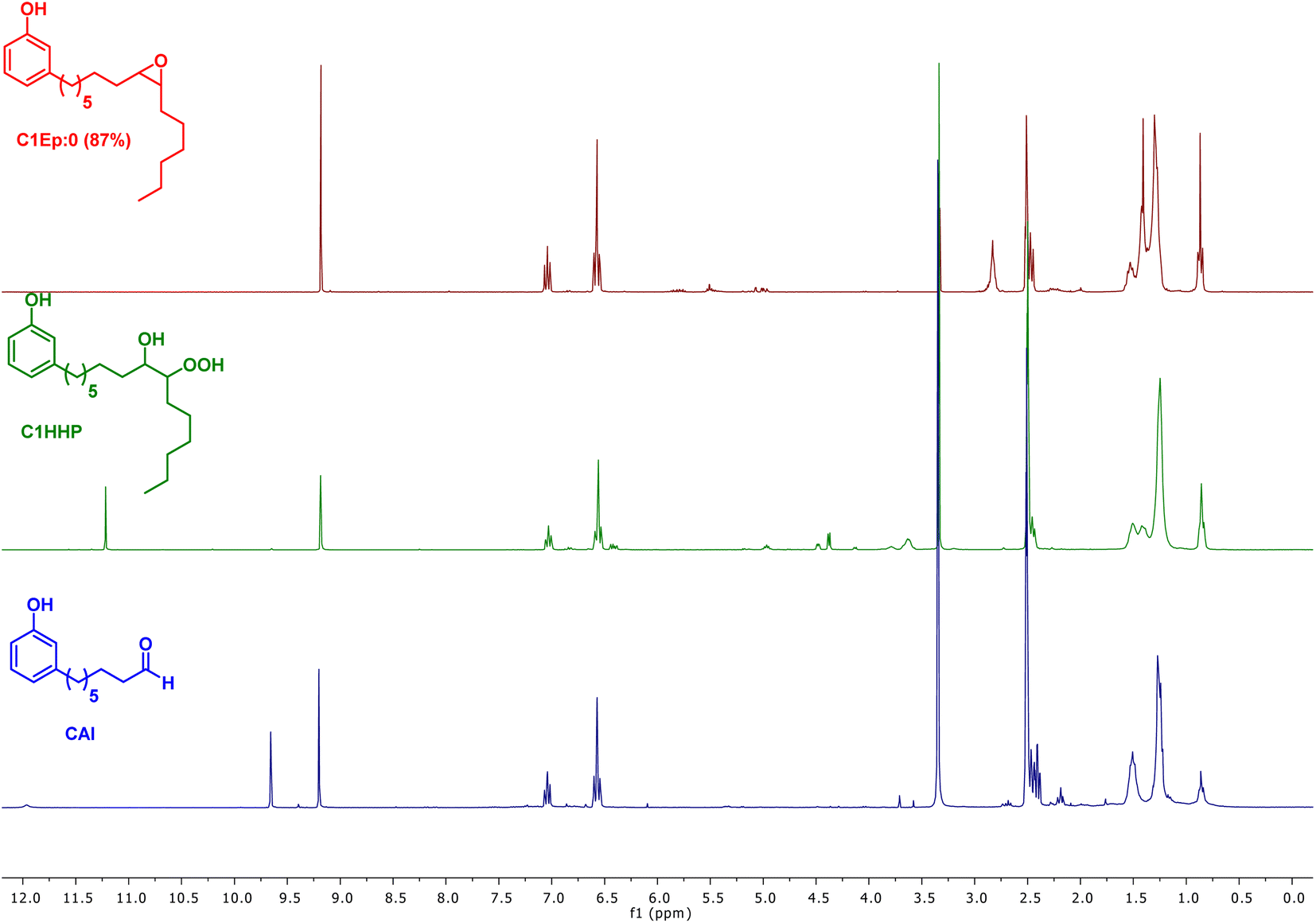 | ||
| Fig. 4 1H NMR (300 MHz, d6-DMSO) of the mixture of cardanol mono-epoxides C1Ep:0 (red, top), cardanol mono-β-hydroxy hydroperoxide C1HP (green, middle) and aldehyde CAl after cleavage and purification (blue, bottom). The DMSO residual peak appears at 2.5 ppm while residual water appears at 3.33 ppm. | ||
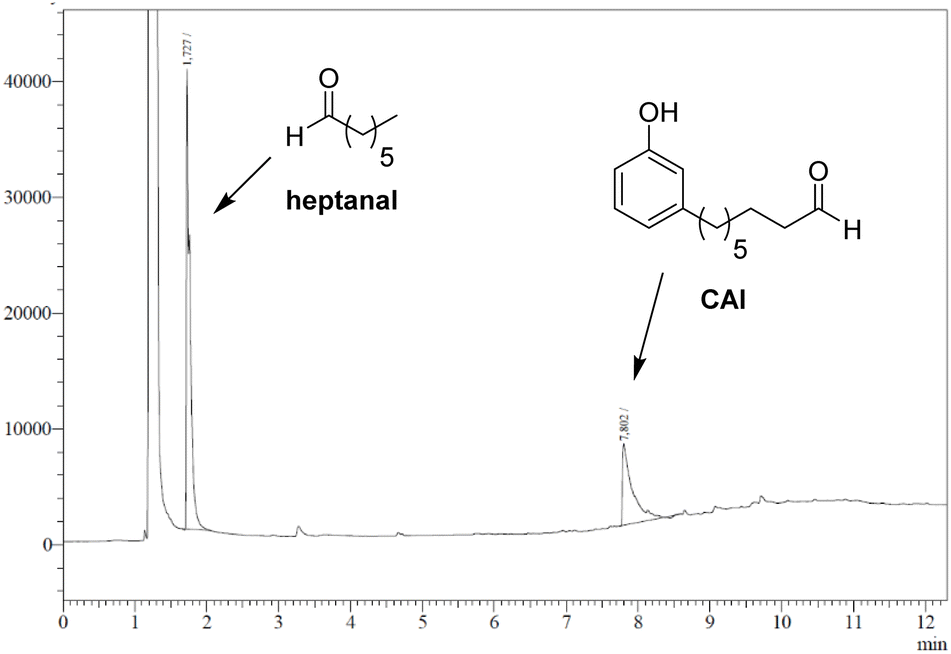 | ||
| Fig. 5 GC chromatogram of cardanol mono-β-hydroxy hydroperoxide (C1HP), showing its thermal cleavage to heptanal and 8-(3-hydroxyphenyl)octanal (CAl) (GC injector temperature = 300 °C). | ||
Finally, β-hydroxy hydroperoxide C1HP was thermally cleaved at 300 °C in a preparative way under flow conditions to give the desired 8-(3-hydroxyphenyl)octanal with 32% yield (Fig. 4, bottom). It should be noted that, in order to simplify the work-up of the reaction, heptanal was not recovered but was removed by distillation (rotary evaporator).
These results show that cardanol β-hydroxy hydroperoxide C1HP can be readily cleaved into 8-(3-hydroxyphenyl)octanal CAl with high selectivity. Therefore, it confirms our hypothesis that the presence of diols prevents the formation of the desired aldehyde. Indeed, the overall selectivity is plummeted by the fact that the ring-opening of the mixture of cardanol epoxides is not selective due to the use of an aqueous solution of H2O2. This problem could be probably solved by using an ethereal solution of H2O2 but this raises safety problems, especially when working on the large scale. The in situ generation of H2O2 would afford – by far – a safer option.
Comparison of methods
To rigorously assess the green potential of the previously presented synthetic routes (Scheme 1), we decided to compare them using classical green metrics, focusing on two important points. The first one insists on the dangerousness and toxicity of the molecules involved in these reactions. The second concerns the atom economy and the reduction of possible waste during the synthesis process. By committing to an eco-responsible approach to replace phenol and formaldehyde in phenolic resins, it is essential for us to evaluate, quantify and choose the most respectful synthesis for people and the environment.In their paper, Graham et al. described ozonization as an effective method for obtaining 8-(3-hydroxyphenyl)octanal by focusing only on the yield of the reaction. They compared another non-optimized oxidative cleavage methods using KIO4. We decided to include these synthetic pathways in our study. We are aware that in order to achieve an oxidative cleavage, strong oxidants should be used, which often leads to some drawbacks such as hazard and toxicity. Therefore, it is important to identify their impact and find promising alternatives or compromises.
Before starting the discussion on the various methods to obtain 8-(3-hydroxyphenyl)octanal, it is worth pointing out that the starting material used in these reactions is cardanol. Cardanol, on its own, is non-toxic, bio-sourced and comes from the recycling of inedible agricultural waste. The use of a cardanol derivative, in addition, as a mono-component, for the synthesis of phenolic resin is already in itself an eco-responsible advance for the substitution of phenol and formaldehyde, both classified as CMR, in the production of these materials.
About the validity of the synthesis pathway, the aim is to find an alternative to ozonization. Although effective, ozone shows a significant potential for danger and needs economical investment to be used without harm. Table 1 compares the routes according to the dangerousness of the molecules used (see ESI† for details). The first thing that can be noticed is that the most dangerous part does not come from reaction solvents (average SC of 12.75) but from reactants (average SC of 30.75). To reduce the SC score of the pathway, the best options are either to use water as a solvent or to use the same solvent for all the steps, if possible. Among reactants, the oxidants are the most dangerous chemicals, the most reactive being ozone (SC 21). It is therefore necessary to find an alternative to the use of ozone in this process of cleavage of cardanol double bonds. However, when comparing each pathway based on their total safety score, the route a exhibits the lowest value (SC of 34) by comparison with other routes that are relatively close (SC of 42–51). In contrast, when comparing the total safety score per number of chemicals, the routes b1, b2 and c (SC/chemicals of 5.3–6.4) appear by far safer than the route a (SC/chemicals of 11.3). This means that the hazard is much more distributed throughout the process for the routes b1, b2 and c than for the route a, where the hazard is concentrated on only one (ozonization) step.
| Entry | Route | Steps | SC of reactants | SC of solvents | Most hazardous molecule (pts) | Number of products | Final SC | Final SC/Nbr of products |
|---|---|---|---|---|---|---|---|---|
| a Synthesis with NaIO4. b Synthesis with KIO4. | ||||||||
| 1 | a | 2 | 28 | 6 | Ozone (21) | 3 | 34 | 11.3 |
| 2 | b1a | 3 | 40 | 11 | Sodium periodate (15) | 8 | 51 | 6.4 |
| 3 | b2b | 3 | 33.5 | 13 | H2O2 (9.5) | 10 | 46.5 | 4.3 |
| 4 | c | 3 | 21.5 | 21 | H2O2 (9.5) | 8 | 42.5 | 5.3 |
In Table 2, each synthesis is compared using different metrics commonly used in green chemistry such as the atom economy (AE), the overall yield, the atom efficacy and the E factor of the synthesis process. Each method is also compared to itself depending on whether or not the aldehyde co-products and the solvents are recycled. Not surprisingly, the metric scores evolve positively when solvents are recycled, which confirms the interest of such action. Despite the concerns of dangerousness of the process, the synthesis method using ozone (route a) is the one that obtains the best scores. Synthesis in a few steps (2), its moderate atom economy (54%) is mainly due to the formation of heptanal and other aldehydes during oxidative cleavage. If recovered at the end of the reaction, considered as co-products and no longer a waste, the AE value of this route would then increase to 89% (Table 2, entry 1).
| Entry | Route | AE (%) | Overall yield (%) | Atom efficacy (%) | E factor | ||||
|---|---|---|---|---|---|---|---|---|---|
| w/o recov. aldehydes | With recov. aldehydes | w/o recov. ald. | With recov. ald. | Standard | With recov. solvents | With recov. solvents & aldehydes | |||
| a Synthesis with NaIO4. b Synthesis with KIO4. | |||||||||
| 1 | a | 54 | 89 | 95 | 51 | 85 | 40 | 6 | 3 |
| 2 | b1a | 26 | 43 | 42 | 11 | 18 | 447 | 13 | 8 |
| 3 | b2b | 17 | 28 | 34 | 6 | 10 | 721 | 28 | 19 |
| 4 | c | 49 | 82 | 3 | 1.5 | 2.5 | 8285 | 70 | 46 |
It should also be noted that all other metrics are also positively impacted by the recovery of aldehyde co-products, mainly heptanal (Table 2, entries 1–4). Indeed, heptanal is a molecule of interest that is used in the perfume industry either as such or as a starting material for the preparation of jasminaldehyde.29–31 Moreover, it can also be used as an alkylating agent for the aldolization32,33 or reductive etherification of polyols34–37 and sugars38–41 to give biobased surfactants. Based on the results presented in Table 2, the method using NaIO4 (route b1) appears to be the best compromise between performances (yield = 42%; AE of 43%; E factor of 8) and safety (SC of NaIO4 = 15), provided the solvents are recycled. The thermal cleavage of β-hydroxy hydroperoxides (route c) is currently the least economically interesting because of its low overall yield (3%). Moreover, the requirement of two purifications by column chromatography on the laboratory scale negatively affects the E factor of the route. However, looking at the atom economy of the route, it is the only one that would be able to compete with reductive ozonolysis (82% vs. 89%). In addition, this route only requires H2O2 as an oxidant, which has be determined as the safer oxidant (SC of 9.5) and the whole process would be amenable to continuous flow synthesis. To conclude, the route c could be envisioned as the most promising solution but the technology is not mature enough. Therefore, future optimization would be necessary.
Synthesis of homo-8-(3-hydroxyphenyl)octanal resin (RCAl)
Contrary to other strategies to prepare cardanol-based phenolic thermosets, our approach relies on the use of a single starting material. The shortening of the C15 chain of cardanol bearing different numbers of unsaturations (i.e., a mixture of 4 compounds) to a single C8 chain considerably simplify the substrate. As a result, the reactivity of the monomer might be more predictable as less side-reactions could be possible. Consequently, the properties of the resulting material might also be more reproducible. In order to observe the possible homopolycondensation of 8-(3-hydroxyphenyl)octanal (CAl), the cardanol-aldehyde single component was heated at 120 °C for 30 min (Scheme 4).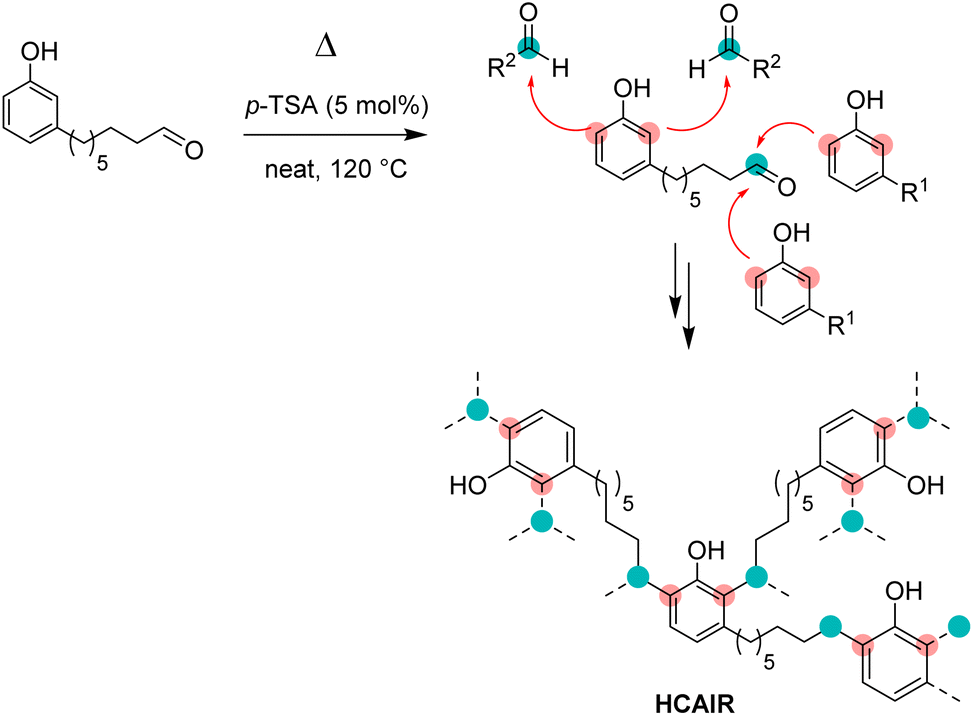 | ||
| Scheme 4 Homopolycondensation of 8-(3-hydroxyphenyl)octonal to obtain RCAl resin. | ||
The change of state (liquid to solid) and of colour (colourless to black) showed that a reaction occurs. The same phenomenon happened, but faster, using CAl mixed with para-toluene sulfonic acid (p-TSA, 0.05 equiv., 5 mol%), a catalyst already used for the synthesis of novolac resins.12 The two reactions were again performed and monitored by differential scanning calorimetry (DSC) from room temperature to 150 °C, at 20 °C min−1. As seen in Fig. 6, for CAl alone, an endothermic peak appears from 100 °C to 140 °C with a peak apex at 129 °C. A similar behavior occurs at lower temperature (from 85 °C to 110 °C, with a peak apex at 110 °C) for CAl mixed with PTSA. This endothermic peak could be explained by the formation and the evaporation of water molecules produced during the polycondensation, confirming that, with or without catalyst, CAl has the ability to homopolycondensate.
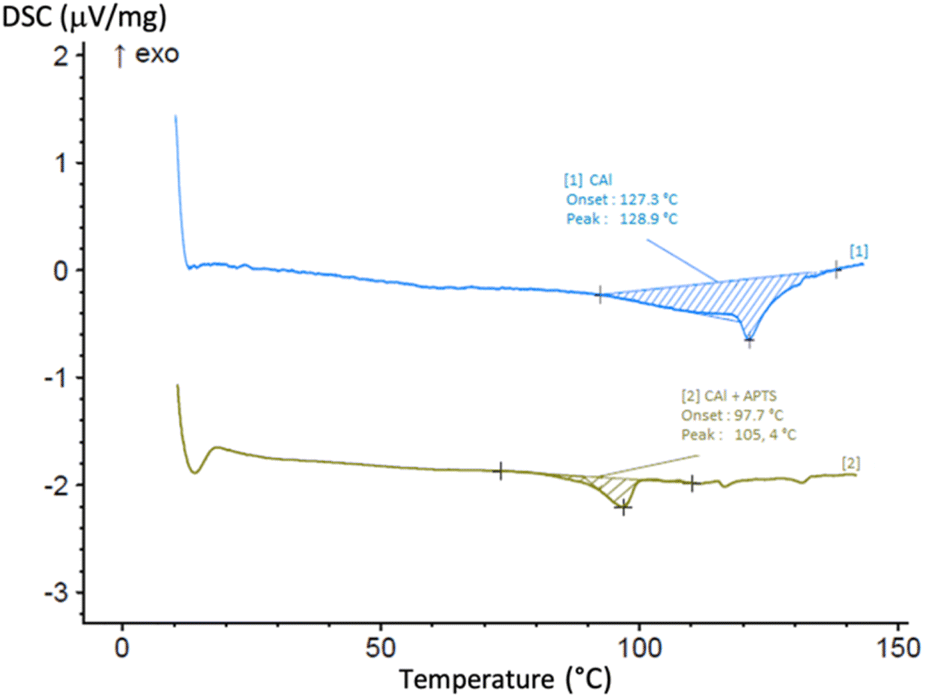 | ||
| Fig. 6 DSC curves from ambient temperature to 150 °C at 20 °C min−1, monitoring the homopolycondensation of [1] CAl alone, [2] CAl mixed with PTSA (5 mol%). | ||
Preliminary characterization of the HCAlR
Glass transition temperature (Tg) of HCAlR was monitored by DSC from −100 °C to 50 °C at 20 °C min−1. The curve (Fig. 7) shows a Tg of −22 °C suggesting a certain flexibility of the resulting material. Indeed, the influence of the dangling chain of cardanol derivatives and their plasticizing effect have already been reported in the synthesis of cardanol-based novolac resins.12,42,43 Even if the length of the cardanol chain have been reduced from 15 to 8 carbons, it seems to still induce a plasticizing effect.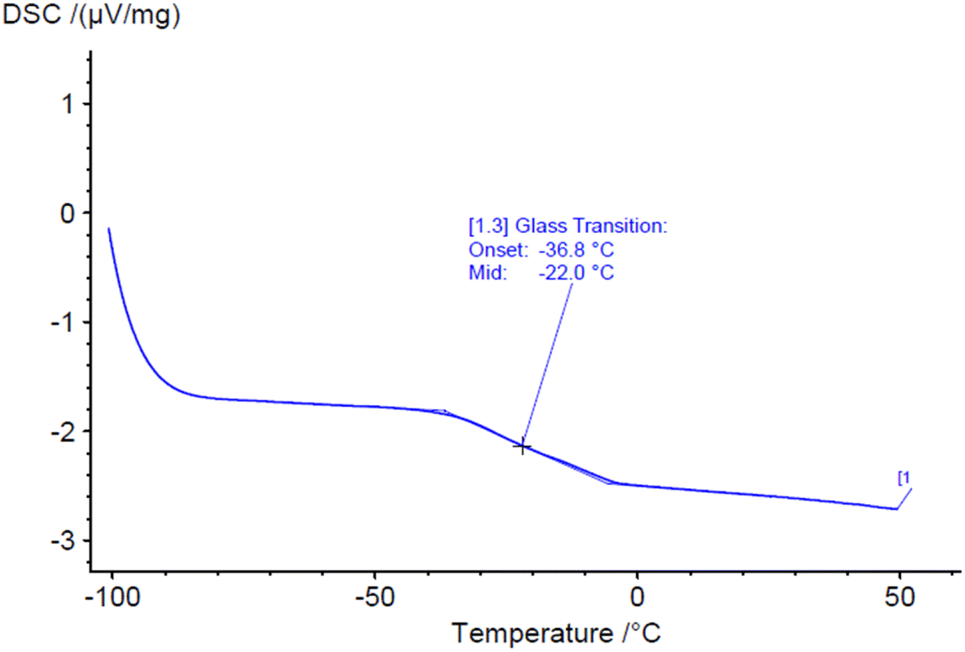 | ||
| Fig. 7 DSC curves of HCAlR for the determination of the glass transition temperature from −100 °C to 50 °C (20 °C min−1). | ||
Phenolic networks are well-known for their good thermal stability often up to 400 °C for 10% weigh loss.6 Even if the dangling chain of CAl are shorter than cardanol, as well as the induced plasticizing effect, it could impact the thermal behaviour of the phenolic material. Thermal properties of HCAlR were determined by thermogravimetric analysis (TGA) under air from 25 °C to 500 °C at 10 °C min−1 as illustrated in Fig. 8.
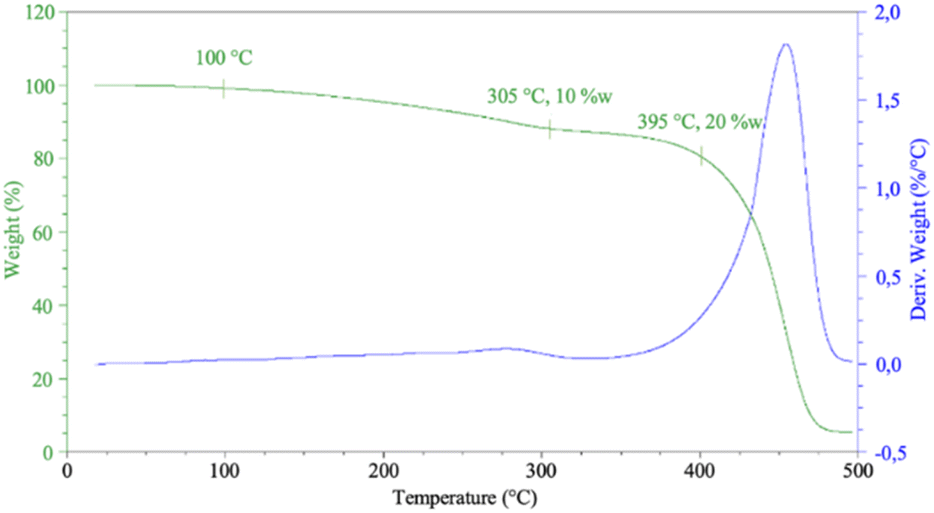 | ||
| Fig. 8 TGA and DTG curve of HCAlR from room temperature to 500 °C, 10 °C min−1. | ||
The TGA and DTG curves show a first low degradation, 10% of weight lost from 100 °C to 305 °C. This degradation could be due to the unfinished polycondensation of CAl and its water evaporation. A second and major degradation step occurs around 400 °C due to the actual degradation of RCAl resin which is a close result to the thermal degradation of cardanol-nonanal-based resin previously reported.12
Conclusions
The synthesis of 8-(3-hydroxyphenyl)octanal (CAl) from cardanol has been studied following two different routes, involving either the cleavage of cardanol diols with NaIO4 (route b1) or the thermal cleavage of cardanol β-hydroxy hydroperoxides (route c). Moreover, their safety score and green metrics were calculated and compared with two other reported routes (ozonization and cleavage of diols with KIO4). Our results show that the method using NaIO4 as an oxidant is today the best compromise between performances and safety impacts. The other synthetic pathway via the thermal cleavage of β-hydroxy hydroperoxides is currently not mature enough but has a great potential to establish itself as the best alternative to reductive ozonolysis, provided further optimization.The use of CAl as a single-component building-block for the synthesis of phenol and formaldehyde-free phenolic thermosets has been demonstrated and followed by differential scanning calorimetry. It led to a promising thermally stable (until 400 °C) and soft material (Tg of −22 °C). A deeper investigation of the formulation of CAl-based novolac resins and their resulting properties is ongoing and will be reported in a near future.
Author contributions
The project was conceptualized by B. Briou and S. Caillol. S. Caillol and N. Duguet acquired the fundings and supervised the project. B. Briou, L. Jégo and T. De Dios Miguel performed the experiments and characterized the materials and products. The manuscript (original draft, review & editing) was handled by B. Briou, S. Caillol and N. Duguet. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The region Auvergne Rhône-Alpes is also acknowledged for a PhD grant to TDDM (Pack Ambition Recherche – project VALCOUPENZ). This project has been labelled by AXELERA, pôle de compétitivité Chimie-environment in Auvergne-Rhône-Alpes. The “Centre Commun de Spectrométrie de Masse” of Lyon (E. Fromentin, A. Vauchez, C. Duchamp) is thanked for HPLC-MS analyses.Notes and references
- K. Hirano and M. Asami, React. Funct. Polym., 2013, 73, 256–269 CrossRef CAS.
- L. Pilato, React. Funct. Polym., 2013, 73, 270–277 CrossRef CAS.
- T. Takeichi, in Polymer Science: A Comprehensive Reference, ed. M. Möller, Elsevier, Amsterdam, 2012, pp. 723–751 Search PubMed.
- G. Astarloa-Aierbe, J. M. Echeverria, J. L. Egiburu, M. Ormaetxea and I. Mondragon, Polymer, 1998, 39, 3147–3153 CrossRef CAS.
- R. A. Pethrick and B. Thomson, Br. Polym. J., 1986, 18, 380–386 CrossRef CAS.
- G. Foyer, B.-H. Chanfi, D. Virieux, G. David and S. Caillol, Eur. Polym. J., 2016, 77, 65–74 CrossRef CAS.
- G. Foyer, B.-H. Chanfi, B. Boutevin, S. Caillol and G. David, Eur. Polym. J., 2016, 74, 296–309 CrossRef CAS.
- E. Erbay, T. Bilgiç, N. Uyanik and Ö. T. Savaşçi, Angew. Makromol. Chem., 1996, 235, 149–160 CrossRef CAS.
- Y. Li, N. Z. Zhi, Y. X. Xiao and X. Long, Advanced Materials Research, Trans Tech Publications, Ltd., 2013, https://doi.org/10.4028/www.scientific.net/amr.634-638.3008 Search PubMed.
- R. Koley, R. Kasilingam, S. Sahoo, S. Chattopadhyay and A. K. Bhowmick, Ind. Eng. Chem. Res., 2019, 58, 18519–18532 CrossRef CAS.
- G. Sui, Y. Cheng, X. Yang, X. Wang and Z. Wang, J. Appl. Polym. Sci., 2019, 136, 47732 CrossRef.
- B. Briou, S. Caillol, J.-J. Robin and V. Lapinte, Eur. J. Lipid Sci. Technol., 2018, 120, 1800175 CrossRef.
- F. Dong, M. Wang and Z. Wang, Int. J. Chem. React. Eng., 2018, 16, 20170107 CrossRef.
- M. Shibata, N. Tetramoto, A. Imada, M. Neda and S. Sugimoto, React. Funct. Polym., 2013, 73, 1086–1095 CrossRef CAS.
- P. Jia, F. Song, Q. Li, H. Xia, M. Li, X. Shu and Y. Zhou, J. Renewable Mater., 2019, 7, 601–619 CAS.
- C. Voirin, S. Caillol, N. V. Sadavarte, B. V. Taxade, B. Boutevin and P. P. Wadgaonkar, Polym. Chem., 2014, 5, 3142–3162 RSC.
- S. Caillol, Curr. Opin. Green Sustainable Chem., 2018, 14, 26–32 CrossRef.
- A. Roy, P. Fajardie, B. Lepoittevin, J. Baudoux, V. Lapinte, S. Caillol and B. Briou, Molecules, 2022, 27, 1443 CrossRef CAS PubMed.
- B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC.
- A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2008, 110, 812–824 CrossRef.
- M. B. Graham and H. P. Tyman, J. Am. Oil Chem. Soc., 2002, 79, 725–732 CrossRef CAS.
- N. D. Vu, S. Bah, E. Deruer, N. Duguet and M. Lemaire, Chem.–Eur. J., 2018, 24, 8141–8150 CrossRef CAS PubMed.
- T. De Dios Miguel, N. D. Vu, M. Lemaire and N. Duguet, ChemSusChem, 2021, 14, 379–386 CrossRef CAS PubMed.
- D. Louvel, T. De Dios Miguel, N. D. Vu and N. Duguet, Eur. J. Org. Chem., 2021, 2990–3014 CrossRef CAS.
- D. B. Menzel, J. Toxicol. Environ. Health, 1984, 13, 183–204 CAS.
- W. A. Pryor, G. L. Squadrito and M. Friedman, Toxicol. Lett., 1995, 82–83, 287–293 CrossRef CAS PubMed.
- K. Koike, M. Nifuku, K. Izumi, S. Nakamura, S. Fujiwara and S. Horiguchi, J. Loss Prev. Process Ind., 2005, 18, 465–468 CrossRef.
- B. Briou, S. Caillol, J.-J. Robin and V. Lapinte, Ind. Crops Prod., 2019, 130, 1–8 CrossRef CAS.
- Z. Tišler, E. Vrbková, J. Kocík, D. Kadlec, E. Vyskočilová and L. Červený, Catal. Lett., 2018, 148, 2042–2057 CrossRef.
- V. S. R. Ganga, S. H. R. Abdi, R. I. Kureshy, N. H. Khan and H. C. Bajaj, J. Mol. Catal. A: Chem., 2016, 420, 264–271 CrossRef CAS.
- M. Pérez-Sánchez and P. D. de María, Catal. Sci. Technol., 2013, 3, 2732–2736 RSC.
- B. Zhu, D. Belmessieri, J. F. Ontiveros, J.-M. Aubry, G.-R. Chen, N. Duguet and M. Lemaire, ACS Sustainable Chem. Eng., 2018, 6, 2630–2640 CrossRef CAS.
- B. Zhu, G.-R. Chen, N. Duguet and M. Lemaire, ACS Sustainable Chem. Eng., 2018, 6, 11695–11703 CrossRef CAS.
- Y. Shi, W. Dayoub, A. Favre-Réguillon, G.-R. Chen and M. Lemaire, Tetrahedron Lett., 2009, 50, 6891–6893 CrossRef CAS.
- Y. Shi, W. Dayoub, G.-R. Chen and M. Lemaire, Green Chem., 2010, 12, 2189–2195 RSC.
- M. Sutter, E. Da Silva, N. Duguet, Y. Raoul, E. Métay and M. Lemaire, Chem. Rev., 2015, 115, 8609–8651 CrossRef CAS PubMed.
- A. Herbinski, E. Metay, E. Da Silva, E. Illous, J.-M. Aubry and M. Lemaire, Green Chem., 2018, 20, 1250–1261 RSC.
- C. Gozlan, R. Lafon, N. Duguet, A. Redl and M. Lemaire, RSC Adv., 2014, 4, 50653–50661 RSC.
- C. Gozlan, E. Deruer, M.-C. Duclos, V. Molinier, J.-M. Aubry, A. Redl, N. Duguet and M. Lemaire, Green Chem., 2016, 18, 1994–2004 RSC.
- D. Belmessieri, C. Gozlan, M.-C. Duclos, V. Molinier, J.-M. Aubry, O. Dumitrescu, G. Lina, A. Redl, N. Duguet and M. Lemaire, Eur. J. Med. Chem., 2017, 128, 98–106 CrossRef CAS PubMed.
- D. Belmessieri, C. Gozlan, M.-C. Duclos, O. Dumitrescu, G. Lina, A. Redl, N. Duguet and M. Lemaire, Bioorg. Med. Chem. Lett., 2017, 27, 4660–4663 CrossRef CAS PubMed.
- Z. Liu, J. Huo and Y. Yu, Mater. Today Commun., 2017, 10, 80–94 CrossRef CAS.
- F. Cardona, A. L. Kin-Tak and J. Fedrigo, J. Appl. Polym. Sci., 2012, 123, 2131–2139 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, detailed calculations of green metrics and safety score, 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d3su00058c |
| This journal is © The Royal Society of Chemistry 2023 |