
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly crosslinked polyesters prepared by ring-opening copolymerization of epoxidized baru nut and macaw palm oils with cyclic anhydrides†
Aaron L.
Vermiglio
a,
Rafael T.
Alarcon
 b,
Éder T. G.
Cavalheiro
b,
Gilbert
Bannach
c,
Thomas J.
Farmer
*a and
Michael
North
*a
b,
Éder T. G.
Cavalheiro
b,
Gilbert
Bannach
c,
Thomas J.
Farmer
*a and
Michael
North
*a
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK. E-mail: michael.north@york.ac.uk; thomas.farmer@york.ac.uk
bSão Carlos Institute of Chemistry, USP – University of São Paulo, 13566-590, São Carlos, SP, Brazil
cSchool of Sciences, Department of Chemistry, UNESP – São Paulo State University, 17033-260, Bauru, SP, Brazil
First published on 15th May 2023
Abstract
Epoxidised Brazilian vegetable oils obtained from the baru nut (Dipteryx alata Vogel) and the macaw palm (Acrocomia aculeata) react with bioderivable anhydrides (succinic, glutaric, itaconic or citraconic) to give highly crosslinked polyesters. The polymerisation is initiated by a metal-free catalyst system comprising dicyclohexyl urea and bis(triphenylphosphine)iminium chloride. The resulting polymers can be obtained as powders or rubbery disks and are chemically and physically characterised. On treatment with aqueous sodium hydroxide, the polymers degrade to water soluble glycerol and sodium carboxylates.
Sustainability spotlightThe work in this paper aligns primarily with UNSDG 12: sustainable consumption and production as it is concerned with the synthesis of highly crosslinked polyesters from epoxides obtained by epoxidation of triglycerides obtained from Brazilian biomass and bioderivable anhydrides. The resulting polymers are shown to be degradable at end of life. Hence, the polymers address sustainability issues at both start and end of life. The overview of UNSDG 12 notes that unsustainable patterns of consumption and production are root causes of climate change, biodiversity loss and pollution. Our approach to a major class of chemicals (polymers) will help to address this. |
Introduction
Polymers are ubiquitous in modern life and have been produced commercially for over 100 years. Their global production currently exceeds 360 million metric tonnes per annum.1 However, both the production and end of life disposal of polymers give rise to serious sustainability issues. Commercial polymers are almost entirely derived from petrochemicals by processes which consume 8% of global oil and gas production, making them second only to transport fuel in the use of oil and gas. Currently, less than 1% of commercial polymers are sustainably sourced.1 The end of life treatment of polymers is equally problematic with just 9% being recycled. The remainder is either incinerated (12%) resulting in carbon dioxide emissions or discarded on land or in the oceans2 (79%) causing major pollution problems.3 Thus a major challenge in green chemistry is the development of new polymers which are sustainably sourced and degradable at end of life. This will allow the development of a sustainable and potentially circular economy for polymers,4 but requires the integrated consideration of feedstocks, production methodology and design for degradation.Polyesters have great potential to address these sustainability issues. They are potentially chemically degradable at end of life by hydrolysis of their backbone ester bonds, making them more recyclable than many other classes of widely used polymers.5 In addition, the oxygenated functional groups needed to produce monomers for polyester synthesis are commonly found in biomass, giving polyesters the potential to be biosourced. There are three main routes for the production of polyesters. Commercially, the most developed synthesis is the thermally induced condensation polymerization between a diol and a diacid or diester (Scheme 1a)6 which, for example, is used in the production of poly(ethylene terephthalate) (PET).7 Since many diacids and diols are readily available from biomass, it should be possible to prepare sustainably sourced polymers in this way. However, the harsh thermal conditions8 needed to drive this process to high molecular weights (220–260 °C for PET9) limit the functional groups that can be present in the monomers. An alternative approach to polyester synthesis involves the ring-opening polymerisation of lactones or lactides (Scheme 1b)10 which is used in the commercial production of poly(caprolactone) and poly(lactic acid).11 This is a lower temperature, catalysed process which is already being used with biomass derived monomers, but the range of potential monomers is rather limited. The third approach to polyester synthesis is the ring-opening, alternating copolymerization (ROCOP) of epoxides and cyclic anhydrides (Scheme 1c).12 ROCOP is a lower temperature catalysed process, so it is also compatible with functionalized monomers. Cyclic anhydrides are readily prepared from diacids and epoxides can be formed by the epoxidation of alkenes; so the ROCOP route to polyesters is also compatible with sustainably sourced monomers.
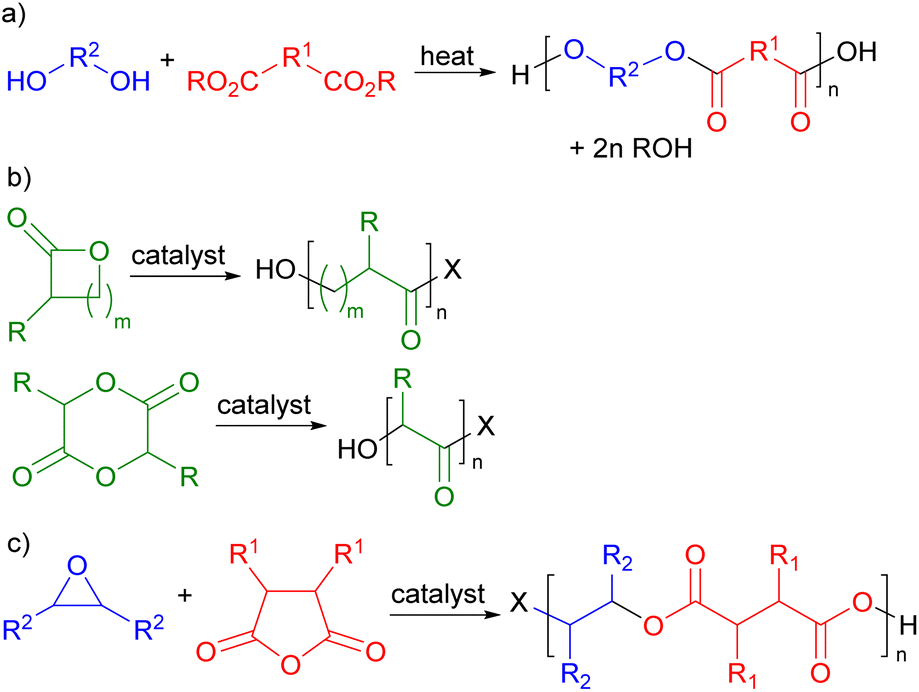 | ||
| Scheme 1 Routes for the synthesis of polyesters. (a) Condensation polymerisation. (b) Ring-opening polymerization. (c) Ring-opening copolymerization. | ||
ROCOP is a living polymerization process that is capable of producing polymers with narrow molecular weight distributions. However, it is also very susceptible to chain-transfer by adventitious moisture which reduces the molecular weight of the polymer. As a result, ROCOP is typically carried out under rigorously inert and anhydrous conditions involving the use of a glove box. Recently, we reported that ROCOP of functionalised biomass derivable anhydrides and epoxides could be carried out with both metal-based and metal-free initiators without the need for a glove box or other specialised equipment.13 Although the linear polyesters formed had rather low molecular weights due to moisture induced chain-transfer, the functionality they possessed (alkenes and alkyl halides) allowed them to be crosslinked to produce insoluble, polyester derived resins. This approach worked well, but did require two steps and a non-bioderived crosslinking agent. We realised that both these factors could be avoided if instead of using simple mono-epoxides as monomers, a biomass derived monomer containing multiple epoxides was used. This would allow the one-step synthesis of completely sustainably sourced crosslinked polyester resins. In addition, the detrimental effect of chain-transfer would be avoided as each crosslinking polyester chain can be short without compromising the formation of the crosslinked resin.
In previous work we have also reported the synthesis of epoxidised triglycerides obtained from baru nut (Dipteryx alata Vogel) and macaw palm (Acrocomia aculeata) oils (Fig. 1).14 Baru nuts and macaw palms are grown commercially in the Cerrado region of Brazil15 and produce vegetable oils which are high in polyunsaturated fatty acids. Hence, epoxidation of baru nut oil gave epoxidised baru nut oil 1 containing on average 3.5 epoxides per triglyceride and epoxidation of macaw palm oil gave epoxidised macaw palm oil 2 containing on average 4 epoxides per triglyceride. Therefore, in this paper, we show that ROCOP applied to epoxidised vegetable oils 1 and 2 and biomass derivable anhydrides leads directly to highly crosslinked, insoluble polyester resins. Both the chemical and physical properties of the resins are determined and it is shown that they can be degraded to water soluble sodium carboxylates on treatment with aqueous sodium hydroxide.
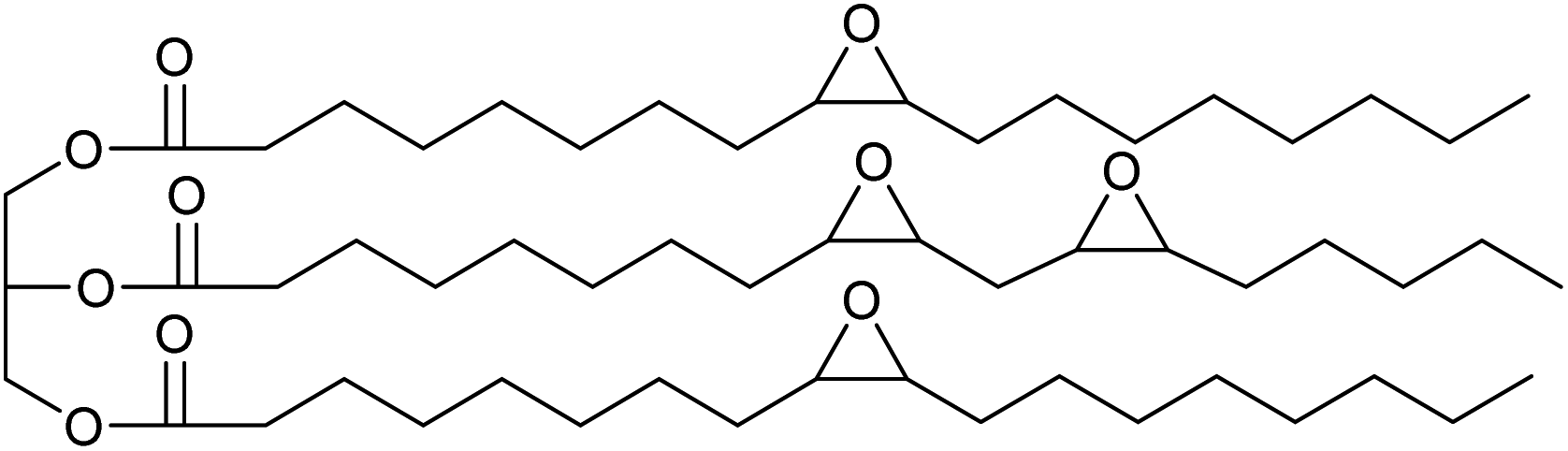 | ||
| Fig. 1 Simplified structural representations of epoxidised oils 1 and 2. In reality each fatty acid chain may contain 0–2 epoxides and fewer or more than the 18 carbon atoms shown. On average, 1 contains 3.5 epoxides per triglyceride and 2 contains 4 epoxides per triglyceride. | ||
Results and discussion
In previous work,11 we showed that both metal-based catalysts (aluminium and chromium salophen complexes) and the organocatalyst dicyclohexyl urea16 (DCHU, 3, Fig. 2) were capable of inducing the ROCOP of mono-epoxides and anhydrides in the presence of bis(triphenylphosphine)iminium chloride (PPNCl, 4) at 100 °C for 15–60 minutes under solvent-free, standard laboratory conditions. Of these catalysts, DCHU is particularly attractive as it is an industrial by-product17 and will not introduce coloured and/or toxic metal residues into the polymer. Therefore, in this work polymerisations were carried out using the DCHU/PPNCl initiator system. DSC analysis (ESI, Fig. S1†) of a mixture of macaw oil 2, succinic anhydride 5, DCHU 3 and PPNCl 4 (250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000:1:1 ratio) showed that 100 °C was the lowest temperature at which exothermic polymerisation occurred, so this was used as the polymerisation temperature throughout the study (Scheme 2). Reactions were left for 18 hours as initial studies showed that the reaction mixture became highly viscous and then solidified as the crosslinking polymerisation progressed.
:1000:1:1 ratio) showed that 100 °C was the lowest temperature at which exothermic polymerisation occurred, so this was used as the polymerisation temperature throughout the study (Scheme 2). Reactions were left for 18 hours as initial studies showed that the reaction mixture became highly viscous and then solidified as the crosslinking polymerisation progressed.
 | ||
| Fig. 2 Structural formulas of initiators 3 and 4 and anhydrides 5–8. | ||
 | ||
| Scheme 2 Synthesis of cross-linked polyesters 9a–h. | ||
Epoxidised macaw palm oil 2 is a liquid at room temperature whilst epoxidised baru nut oil 1 is a low melting point solid. Each of these epoxidised triglycerides were polymerised with four potentially bioderivable anhydrides 5–8. Succinic anhydride 5 can be obtained from succinic acid which is produced commercially by fermentation of sugars.18 Itaconic anhydride 6 can be produced either from itaconic acid (manufactured by fungal fermentation of sugars19) or by thermolysis of citric acid.20 Citraconic anhydride 7 is a regioisomer of itaconic anhydride 6. It is a natural product found in coffee and can be manufactured by thermolysis of itaconic or citric acids.17 Glutaric anhydride can be obtained from the natural product glutaric acid.21 Thus a set of eight highly crosslinked polyesters 9a–h was produced as shown in Scheme 2. After polymerisation, the solid polymer mass produce was ground to a powder and subjected to Soxhlet extraction (using dichloromethane, ethyl acetate or ethanol) to constant weight to remove initiators 3 and 4 along with any unreacted monomers, giving polymers 9a–h as white–yellow powders with the percentage yields shown in Table 1. These percentage yields are based on the theoretical incorporation of all of the epoxidised oil (1, 2) and all of anhydrides 5–8. In reality, this cannot be achieved for two reasons. As the polymerisation progresses, the reaction mixture becomes more viscous, so the ROCOP process becomes slower, resulting in unreacted monomers and low molecular weight oligomers being formed which are subsequently removed during the Soxhlet extraction. In addition, ether formation (involving only epoxidised oils 1 or 2) can occur and will result in unreacted anhydride 5–8 being present and subsequently removed during the Soxhlet extraction. Whilst intermolecular reaction of epoxides to form polyethers is only a minor side-reaction in ROCOP using the 3/4 initiator system,13 in the case of epoxidised oils 1 and 2, ether formation can occur intramolecularly and this is likely to occur at a rate that is more competitive with ROCOP, especially as the rate of ROCOP is reduced in the latter stages of the polymerisation.
| Polymer | Yielda (%) | T s (°C) | T D10 (°C) | T g (°C) |
|---|---|---|---|---|
| a Percentage yield after Soxhlet extraction and based on the theoretical incorporation of all epoxidised oil and anhydride. b Limit of thermal stability temperature (Ts) and temperature at which the polymer has lost 10% of its weight (TD10) determined by thermogravimetric analysis under argon with a heating rate of 10 °C min−1. | ||||
| 9a | 67 | 273 | 340 | −14.9 |
| 9b | 61 | 264 | 325 | −5.5 |
| 9c | 59 | 253 | 304 | |
| 9d | 46 | 271 | 341 | −14.9 |
| 9e | 35 | 295 | 352 | |
| 9f | 54 | 274 | 339 | |
| 9g | 59 | 261 | 334 | |
| 9h | 46 | 290 | 356 | −13.6 |
Thermogravimetric analysis (ESI, Fig. S2–S10†) showed that all of the powdered polymers 9a–h were thermally stable up to ca. 250 (9c); 260 (9b, g); 270 (9a, d, f) or 290 °C (9e, h). Polymers derived from baru nut oil (9e–h) all had higher thermal stability and decomposition temperatures (by 12–30 °C) than the corresponding polymer made from the same anhydride and macaw palm oil (9a–d). The polymers derived from unfunctionalised anhydrides 5 and 8 had similar decomposition temperatures (TD10); 340 ± 1 °C for 9a, d and 354 ± 2 °C for 9e, h and these were higher than the corresponding decomposition temperatures of polymers derived from unsaturated anhydrides 6 and 7 (304–325 °C for 9b, c; 334–339 °C for 9f, g). The polymers obtained from anhydrides 5 and 8 also presented a higher thermal stability (271 ± 2 °C for 9a, d; 290 ± 5 °C for 9e, h).
Polymers 9a–h were also analysed by differential scanning calorimetry (DSC) between −60 and 200 °C (ESI, Fig. S11–S18†). Polymers 9a, b, d, h had glass transition temperatures between −5 and −15 °C (0.3–0.4 J g−1), no glass transition temperature was detected for the other four polymers and no other phase changes were detected, suggesting that all of polymers 9a–h were amorphous.
IR analysis of polymers 9a–h (ESI, Fig. S21–S28†) revealed that in the functional group region (4000–1600 cm−1), the spectra were very similar to those of the epoxidised vegetable oil from which they were prepared (ESI, Fig. S19 and S20†), but there were clear differences in the fingerprint region (1600–600 cm−1). However, detailed analysis of the 2000–1500 cm−1 region did show the presence of a weak alkene stretching band at 1653 cm−1 in polymers derived from macaw oil and unsaturated anhydrides 6 and 7 as shown in Fig. 3. In the corresponding baru nut oil derived polymers (9e–h), this alkene stretching band was weaker and only clearly visible in polymer 9g derived from anhydride 7 (ESI, Fig. S27†).
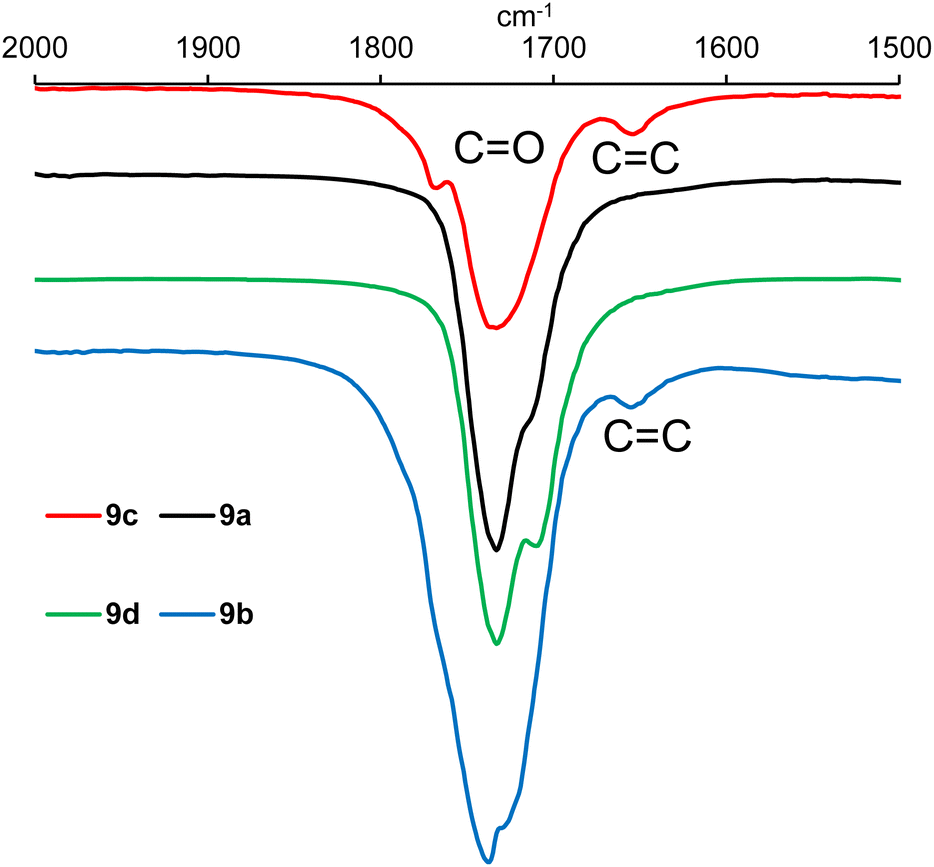 | ||
| Fig. 3 Expansion of the IR spectra of polymers 9a–d between 1500 and 2000 cm−1. | ||
Polymers 9a–h were also analysed by solid-state 13C NMR spectroscopy (ESI, Fig. S29–S36†). Fig. 4 shows a comparison of the solution state 13C NMR spectrum of epoxidised macaw palm oil 2 and the solid-state 13C NMR spectrum of polymer 9a derived from 2 and succinic anhydride 5 (the corresponding spectral comparison of epoxidised baru nut oil 1 and polymer 9e is given in the ESI, Fig. S37†). The peaks between 10 and 35 ppm in both spectra correspond to the aliphatic chains of the macaw palm oil and show that the oil has remained intact during the thermal polymerisation. Ester carbonyl peaks are also clearly visible at 170–180 ppm in both spectra. The most significant difference between the two spectra is in the 50–90 ppm range. In the spectrum of epoxidised oil 2, a series of signals corresponding to C–O groups are present. Those in the range 50–60 ppm can be assigned to carbons within epoxide rings, whilst the two signals between 60 and 70 ppm correspond to the esterified glycerol carbons. In the spectrum of polymer 9a, the epoxide carbons are clearly absent, showing that the polymerisation has gone to completion and a broadened ester C–O signal is present at 70–90 ppm corresponding to both the glycerol esters and the newly formed esters arising from reaction of the epoxides with anhydride 5.
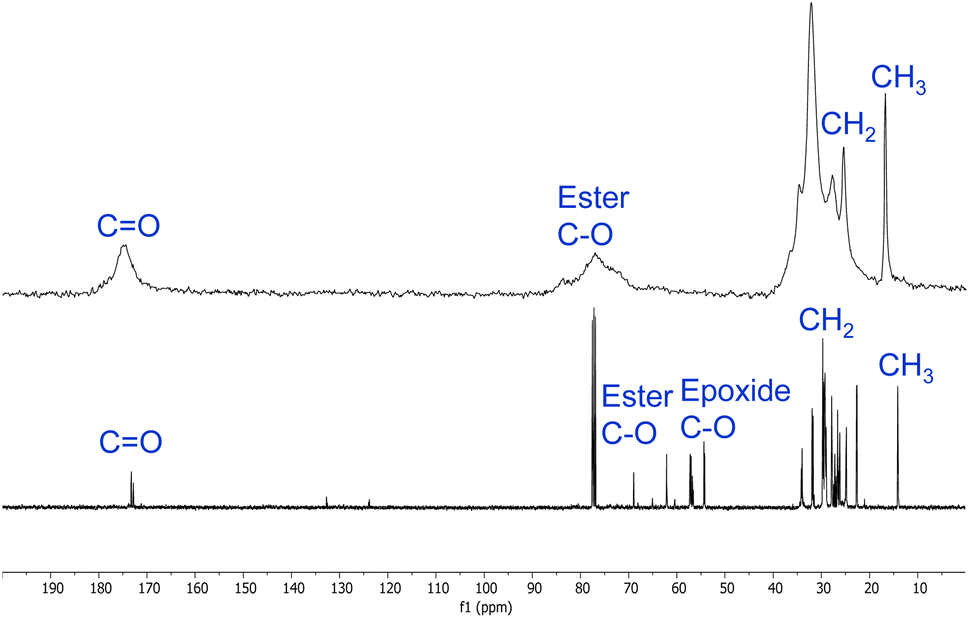 | ||
| Fig. 4 Comparison of the 13C NMR spectra of epoxidised macaw palm oil 2 (bottom) and polyester 9a (top). | ||
Whilst all eight polymers 9a–h displayed very similar solid state 13C NMR spectra (ESI, Fig. S29–S36†), there were subtle differences between the spectra as illustrated in the 60–200 ppm expansions of polymers 9a–d shown in Fig. 5. Thus, polymers 9a and 9d derived from unfunctionalised anhydrides 5 and 8 displayed almost identical spectra, though the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak of polymer 9d was broader than that of polymer 9a. The spectra of polymers 9b and 9c derived from α,β-unsaturated anhydrides 6 and 7 both showed an additional CO peak at 165–170 ppm corresponding to a conjugated carbonyl system. This was further supported by the presence of conjugated alkene signals between 120 and 155 ppm. It is known22 that itaconic and citraconic groups can equilibrate under thermal conditions which accounts for both the complexity of the alkene region of the spectra of polymers 9b and 9c and their similarity.
O peak of polymer 9d was broader than that of polymer 9a. The spectra of polymers 9b and 9c derived from α,β-unsaturated anhydrides 6 and 7 both showed an additional CO peak at 165–170 ppm corresponding to a conjugated carbonyl system. This was further supported by the presence of conjugated alkene signals between 120 and 155 ppm. It is known22 that itaconic and citraconic groups can equilibrate under thermal conditions which accounts for both the complexity of the alkene region of the spectra of polymers 9b and 9c and their similarity.
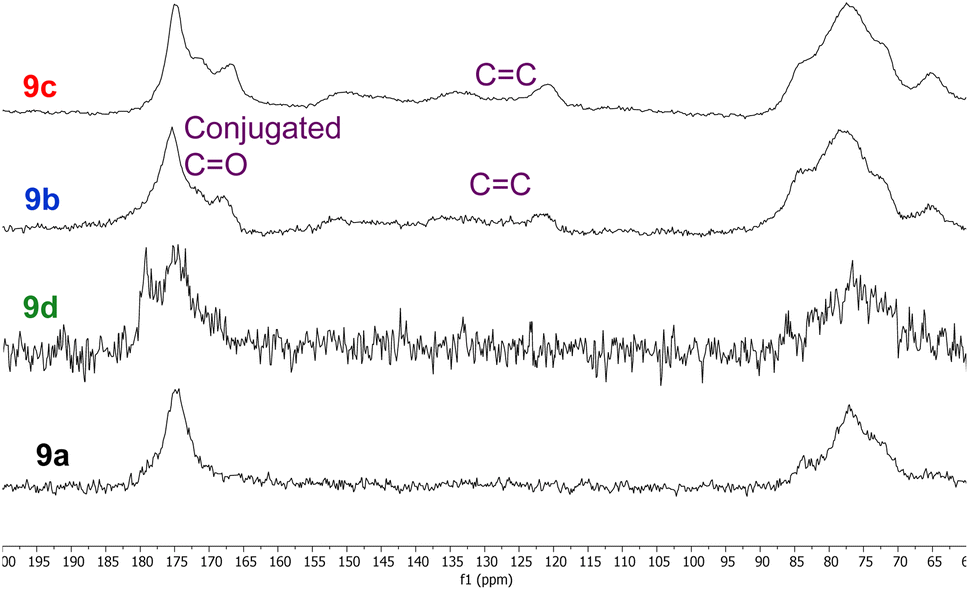 | ||
| Fig. 5 Comparison of the 60–200 ppm region of the solid-state 13C NMR spectra of polymers 9a–d. | ||
The synthesis of polymers 9a–d could be modified to produce polymeric disks (ca. 3 cm diameter and 3 mm depth) rather than powders. Epoxidised macaw palm oil 2 was chosen for this study as it is a liquid at room temperature. Addition of polymerisation initiators 3 and 4 and an anhydride (5–8) followed by warming under nitrogen to 50 °C gave a free flowing homogeneous solution. At this temperature, no polymerisation occurs as shown by DSC analysis of the polymerisation mixture (ESI, Fig. S1†). The solution of monomers and initiators could then be poured into an aluminium mould placed within a flat-bottomed flange flask and heated to 100 °C under nitrogen for 18 hours to induce the polymerisation. After cooling to room temperature and removal of the aluminium mould, transparent, yellow–orange disks were obtained as shown in Fig. 6. The Shore A hardness,23 of the disks were determined using a durometer, giving the results shown in Table 2. All the disks had Shore A hardness values of 60–80 which are comparable to those of car tyre treads (70) and shoe heels (80).20
 | ||
| Fig. 6 Polymeric disk obtained from triglyceride 2, anhydride 5 and initiators 3 and 4 (250:1000:1:1 ratio). | ||
DSC analysis (−80 to 200 °C) of samples of the polymeric disks identified a glass transition temperature at between −18 and 1 °C for all four polymers as detailed in Table 2 (ESI, Fig. S38–S41†). These values are comparable with those obtained for powdered samples of polymers 9a, b and d. Polymer 9a showed a second glass transition at 90 °C. Tanδ values obtained by differential mechanical analysis (DMA) are equivalent to Tg temperatures obtained by DSC. All the disks exhibited tanδ temperatures below room temperature indicating that they are in a rubbery state at 25 °C (ESI, Fig. S42†). These results are consistent with the Shore A hardness values, indicating that the disks are soft and have low rigidity. Below the tanδ temperature, the polymers are in the glassy region, turning into a brittle and rigid material. Polymer 9c has the highest temperature of tanδ, which can be related to a higher cross-linking density (ν, 1738.1 mol m−3).
Tensile strength analysis (ESI, Fig. S43†) indicates that all the polymer disks support stresses below 1 MPa, which can be related to their rubbery state. Furthermore, the strain for each sample did not surpass 20%, as expected for non-linear and cross-linked polymers.24,25
Finally, the ability to chemically degrade the polymers at the end of their life to prevent them accumulating in the environment was investigated. Samples of polymers 9a–d were exposed to 1 M aqueous sodium hydroxide. Polymers 9a, b, d completely dissolved in less than 8 hours whilst polymer 9c required 24 hours to give a homogeneous solution (ESI, Fig. S44†).
Experimental
Materials and methods
Epoxidized baru nut and macaw palm oils were obtained as previously reported.12 Other chemicals were obtained from Merck, Sigma-Aldrich or Alfa Aesar and used as received.Infrared spectra were recorded on a PerkinElmer Spectrum 400 FTIR spectrometer. Solution-state 13C NMR spectra were recorded at 100 MHz on a JEOL ECS400 spectrometer and solid-state 13C NMR spectra were recorded at 100 MHz on a Bruker AVIII HD 400 WB spectrometer.
DSC traces on powdered samples were acquired using a TA Instruments Q2000 modulated Differential Scanning Calorimeter. DSC curves for disk samples were obtained using a DSC-Q10 modulus controlled by a Thermal Advantage® for Q-Series software v.5.5.24 (both from TA Instruments) using 11.0 ± 0.2 mg of sample, analysed in a 40 μL closed aluminium crucible with a 0.7 mm pin hole in the centre of the lid in cooling/heating stages (−80 to 200 °C) with a dynamic N2 atmosphere (50.0 mL min−1) and setting a heating rate of 10.0 °C min−1. Measurements of thermal transitions were taken from the final heat-cool cycle (4th or 5th) in all instances. TGA analyses were acquired using a Stanton Redcroft STA 625 instrument.
DMA analyses were carried out using a Q800 apparatus controlled by a Thermal Advantage® for Q-Series software v.5.5.24 (both from TA instruments). The polymer samples were cut into strips with ca. 20 mm length, 6 mm width and 2 mm thickness (±0.02 mm) prior to analysis. Samples were analysed from −80 °C to 80 °C under a N2 atmosphere, using a frequency of 1 Hz and a heating rate of 3 °C min−1. The DMA provided curves which were used to obtain storage modulus (E′) and tanδ values. The cross-link density (ν) was determined from the equation (ν = E′/3RT) where E′ is the rubbery region storage modulus, R is the gas constant (8.314 J K−1 mol−1), and T (K) is the rubbery temperature (Tg +40 °C). The tensile strength test (stress–strain curves) for each polymer was determined in the same DMA equipment with similar specimens at 25 °C.
Shore A hardness measurements were made using a Sauter 100-0 HBA Shore A Durometer. The needle of the durometer was pushed into polymer disks (9a–d) until the hardness value had peaked. In all cases, the peak Shore A is reported and measurements were taken under ambient conditions.
General procedure for the synthesis of powdered polymers 9a–h
To an oven-dried 15 mL sample vial were added DCHU 3 (2.24 mg, 0.01 mmol, 1 eq.), PPNCl 4 (5.7 mg, 0.01 mmol, 1 eq.), triglyceride 1 or 2 (2.5 mmol, 250 eq.) and anhydride 5–8 (10 mmol, 1000 eq.). The vial was sealed, purged with N2, then heated to 100 °C for 18 hours. After cooling to room temperature, the polymerisation mixture was ground to a powder and placed in a cellulose Soxhlet extraction thimble. Extraction of initiators and unreacted monomers using CH2Cl2 until the polymer reached constant weight left polymers 9a–h as white–yellow powders. EtOAc or EtOH could also be used as greener extraction solvents and were only marginally less effective than CH2Cl2 (4–5%, by weight, less material extracted after 18 hours).General procedure for the synthesis of polymer disks 9a–d
To an oven-dried 15 mL sample vial were added DCHU 3 (2.2 mg, 0.01 mmol, 1 eq.), PPNCl 4 (5.7 mg, 0.01 mmol, 1 eq.) and epoxidised macaw palm oil 2 (2.4 g, 2.5 mmol, 250 eq.). The vial was sealed, purged with N2, then warmed to 50 °C for 30 minutes. After this time, anhydride 5–8 (10.0 mmol, 1000 eq.) was added. The vial was resealed, purged with N2, then warmed to 50 °C until a homogeneous solution formed (ca. 5 minutes). The pre-polymerisation solution was poured into a circular aluminium mould (3 cm diameter) which was placed into a flat-bottomed flange flask. The flask was sealed and flushed with nitrogen, then heated to 100 °C for 18 hours. After cooling to room temperature, the flange flask was opened and the polymer removed from the aluminium mould to give polymers 9a–d as yellow–orange disks.δ 0.4 °C, stress 0.4 MPa, strain 11.6%, cross-linking density 794.8 mol m−3.
δ 0.5 °C, stress 0.4 MPa, strain 18.4%, cross-linking density 408.7 mol m−3.
δ 17.5 °C, stress 0.8 MPa, strain 11.3%, cross-linking density 1738.1 mol m−3.
δ −9.4 °C, stress 0.0005 MPa, strain 7.6%, cross-linking density 699.0 mol m−3.
Conclusions
Ring-opening copolymerisation of epoxidised Brazilian triglycerides and biomass derivable anhydrides can be used to produce bio-derivable, highly crosslinked polyesters. The polymerisation is induced by non-metallic initiators and requires no special precautions (glove box etc.) to avoid chain-transfer due to adventitious water. The polymers can be produced as either powders or disks and their spectral and physical properties are dominated by those of the triglyceride from which they were prepared. The polymers were totally degraded to soluble components in 8–24 hours on treatment with 1 M aqueous sodium hydroxide. Thus, the polymers are both sustainably sourced and at end of life can be treated to prevent their accumulation in the environment.Author contributions
Aaron L. Vermiglio: investigation. Rafael T. Alarcon: investigation, resources, writing – original draft. Éder T. G. Cavalheiro: data curation, funding acquisition, writing – review and editing. Gilbert Bannach: supervision, funding acquisition, project administration, writing – review and editing. Thomas J. Farmer: supervision, writing – original draft. Michael North: conceptualisation, data curation, supervision, project administration, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank São Paulo Research Foundation – FAPESP (grant 2019/22217-8; 2021/14879-0, and 2021/02152-9) and National Council for Scientific and Technological Development-CNPq (grant 303247/2021-5).Notes and references
- https://www.european-bioplastics.org/market/, accessed 1 November, 2022.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. Eriksen, L. C. M. Lebreton, H. S. Carson, M. Thiel, C. J. Moore, J. C. Borerro, F. Galgani, P. G. Ryan and J. Reisser, PLoS One, 2014, 9, e111913 CrossRef CAS PubMed; E. van Sebille, C. Wilcox, L. Lebreton, N. Maximenko, B. D. Hardesty, J. A. van Franeker, M. Eriksen, D. Siegel, F. Galgani and K. L. Law, Environ. Res. Lett., 2015, 10, 124006 CrossRef; J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC; J.-P. Lange, Energy Environ. Sci., 2021, 14, 4358–4376 RSC; S. A. Backer and L. Leal, Acc. Chem. Res., 2022, 55, 2011–2018 CrossRef CAS PubMed.
- M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed; G. X. De Hoe, T. Şucu and M. P. Shaver, Acc. Chem. Res., 2022, 55, 1514–1523 CrossRef PubMed; R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860–9871 CrossRef; Y. Liu, Z. Yu, B. Wang, P. Li, J. Zhu and S. Ma, Green Chem., 2022, 24, 5691–5708 RSC.
- J. H. Park, J. Y. Jeon, J. J. Lee, Y. Jang, J. K. Varghese and B. Y. Lee, Macromolecules, 2013, 46, 3301–3308 CrossRef CAS.
- W. A. MacDonald, Polym. Int., 2002, 51, 923–930 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, vol. A21, pp. 233–238 Search PubMed.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- A. Stopper, T. Rosen, V. Venditto, I. Goldberg and M. Kol, Chem.–Eur. J., 2017, 23, 11540–11548 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC; J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed; D. R. G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC; X. Liang, F. Tan and Y. Zhu, Front. Chem., 2021, 9, 647245 CrossRef PubMed; A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef PubMed; W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef PubMed.
- M. N. D. Haslewood, T. J. Farmer and M. North, J. Polym. Sci., 2023, 61, 311–322 CrossRef CAS.
- R. T. Alarcon, C. Gaglieri, K. J. Lamb, M. North and G. Bannach, Ind. Crops Prod., 2020, 154, 112585 CrossRef CAS.
- R. T. Alarcon, K. J. Lamb, G. Bannach and M. North, ChemSusChem, 2021, 14, 169–188 CrossRef CAS PubMed.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef; N. L. Benoiton, Chemistry of Peptide Synthesis, CRC Press, Boca Raton, FL, 2016 Search PubMed.
- M. Jiang, J. Ma, M. Wu, R. Liu, L. Liang, F. Xin, W. Zhang, H. Jia and W. Dong, Bioresour. Technol., 2017, 245, 1710–1717 CrossRef CAS PubMed.
- M. Zhao, X. Lu, H. Zong, J. Li and B. Zhuge, Biotechnol. Lett., 2018, 40, 455–464 CrossRef CAS PubMed.
- J. H. Crowell, Production of itaconic and citraconic anhydrides, US Pat. 2258947, 1941.
- A. Besrat, C. E. Polan and L. M. Henderson, J. Biol. Chem., 1969, 244, 1461–1467 CrossRef CAS PubMed.
- A. Takasu, M. Ito, Y. Inai, T. Hirabayashi and Y. Nishimura, Polym. J., 1999, 31, 961–969 CrossRef CAS.
- https://www.smooth-on.com/page/durometer-shore-hardness-scale/, accessed on 21 November, 2022.
- C. Gaglieri, R. T. Alarcon, R. Magri, M. North and G. Bannach, J. Appl. Polym. Sci., 2021, 139, e52990 Search PubMed.
- C. Di Mauro, S. Malburet, A. Genua, A. Graillot and A. Mija, Biomacromolecules, 2020, 21, 3923–3935 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00088e |
| This journal is © The Royal Society of Chemistry 2023 |