
Sodium trithiocarbonate cathode for high-performance sodium–sulfur batteries†
Hyunki
Sul
 ,
Amruth
Bhargav
and
Arumugam
Manthiram
*
,
Amruth
Bhargav
and
Arumugam
Manthiram
*
Walker Department of Mechanical Engineering, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: rmanth@mail.utexas.edu
First published on 25th November 2022
Abstract
The high abundance and low cost of sodium and sulfur make room-temperature sodium–sulfur (RT Na–S) batteries an attractive technology compared to the current lithium-ion batteries for large-scale grid-storage applications. However, the commercialization of RT Na–S batteries is impeded by the slow kinetics of Na–S chemistry, severe sodium polysulfide shuttling, and uncontrollable growth of dendritic Na. Herein, sodium trithiocarbonate (Na2CS3) is applied as a cathode material to facilitate concurrent improvement in both electrodes, leading to a high-rate performance with an extended cycle life. The conductive characteristic of the carbon–sulfur resonance bond enables fast ion and electron transfer throughout the cathode, resulting in superior electrochemical reactivity. At the cathode, the presence of Na2CS3 forms an oligomer-structured layer to suppress the dissolution and shuttling of active materials. Meanwhile, when small portions of Na2CS3 intermediates migrate to the anode, a stable solid electrolyte interphase (SEI) layer with uniform Na-ion flux is formed, enabling improved Na stripping and plating performance. A series of electrochemical and material characterization techniques, accompanied by density functional theory calculations, demonstrate that Na2CS3 is a promising candidate to realize high-rate performance long cycle life RT Na–S batteries.
Introduction
The rapid growth in the global energy demand drives the development of clean energy technologies with sustainable sources, such as wind and solar energies. To resolve the challenges coming from the intermittence of these renewable energy sources, secondary Li-ion battery technologies have become one of the most attractive strategies.1–4 However, the intrinsic barriers of traditional Li-ion batteries, including limited energy density of <250 W h kg−1 and less abundant resources of some elements5 hinder their use in large-scale applications. Among various alternative energy storage systems, metal–sulfur batteries have become a promising strategy due to their high energy density, low cost, and high natural abundance.6 Especially, sodium–sulfur (Na–S) electrochemistry is gaining attention since it delivers a high theoretical energy density of 1274 W h kg−1, while the price of sodium is only 1/25 of that of lithium (Li: 20 mg kg−1vs. Na: 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 mg kg−1 in the Earth's crust).7
600 mg kg−1 in the Earth's crust).7
The history of Na–S batteries dates back to the 1960s, when the molten states of sodium and sulfur (operating in a temperature range of 300–350 °C) were proposed and developed for stationary energy-storage applications.8–10 However, the high operating temperatures and the reactive nature of Na and S in their molten state cause severe durability, safety, and maintenance issues.11 Hence, room-temperature Na–S (RT Na–S) battery systems have been pursued over the past decade as a promising alternative for both stationary grid storage and transportation applications.
RT Na–S batteries were introduced in 2006 with high capacity and well-defined charge/discharge curves,12 but they suffer from poor cycling stability. Similar to lithium–sulfur (Li–S) batteries, the critical challenge is the intrinsic soluble behavior of the intermediate charge/discharge species, the so-called sodium polysulfides, in liquid electrolytes.13,14 Although the chemistries of Na–S and Li–S are analogous, the more reactive characteristic of Na metal and the extreme solubility of sodium polysulfides in liquid electrolytes aggravate capacity fade. Long-chain glyme-based ethers, such as tetraethylene glycol dimethyl ether (TEGDME), have been considered as promising electrolytes because of their high boiling point and good electrochemical stability towards polysulfide anions.15–17 Nevertheless, fast capacity decay is still encountered as the dissolved sodium polysulfides freely shuttle between the cathode and the anode. The migrated sodium polysulfides spontaneously react with Na metal and get reduced to insulating Na2S, leading to a loss of active lithium and sulfur species.
Furthermore, Na metal with a highly reducing nature can undergo an irreversible reaction with solvent molecules or salts, which leads to a loss of Na inventory and depletion of the electrolyte. Above all, conventional RT Na–S batteries could not maintain a stable solid-electrolyte interface (SEI) that would prevent the degradation of the Na-metal anode.18 Accordingly, preventing sodium polysulfide shuttling and protecting the Na-metal anode have been the critical strategies for extending the cycle life and improving the performance of RT Na–S batteries.
One of the most promising solutions to mitigate the long-chain sodium polysulfide shuttling is the adsorption and immobilization of the polysulfide species. Strong polysulfide adsorption sites could effectively confine the sodium polysulfides on the cathode side and reduce the shuttle effect. Metal-based sulfiphilic sites are known to effectively immobilize the polysulfides.19–21 Metals, such as Ti, Mn, and Cu, polarize the cathode surface and provide strong polysulfide adsorption ability through polar–polar interactions. Other inorganic compounds, such as MXenes, have also been reported as promising materials owing to their outstanding polysulfide immobilization ability and high areal sulfur loading.22 Another promising strategy is to utilize electrocatalysts to promote a facile conversion of long-chain polysulfides into insoluble short-chain polysulfides, which precipitate out and remain on the cathode side. Transition metal-based electrocatalysts (Co, Fe, Cu, and Ni) combined with carbon hosts have been reported to be effective in facilitating fast electrochemical kinetics for the sodium polysulfide redox reaction.23–26 Thus, the aforementioned strategies, including the adsorption and conversion of long-chain polysulfides, are well-known strategies to prevent the dissolution and migration of polysulfides.
To overcome the challenges of reactive Na-metal anode degradation, the importance of homogeneous Na surface morphology control has been emphasized. The formation of a smooth and dense Na surface minimizes the reaction sites that can undergo parasitic reactions with the electrolyte and polysulfides. It is well known that the chemical composition of the SEI layer critically impacts the Na morphology since Na ions are plated onto and stripped from the underlying Na metal during cycling. The stripping and plating of Na metal take place through the following steps: (1) electron transfer at the bulk Na metal/SEI interface; (2) diffusion of Na ions through the SEI layer; (3) Na-ion transfer at the SEI/electrolyte interface.27,28 Based on various experimental results, step 2 is recognized as the rate-determining step that controls the nucleation and morphology of the Na-metal anode. Therefore, the formation of a kinetically favorable SEI layer with uniform Na-ion flux is crucial in facilitating a homogeneous Na deposition. Artificial SEI layers, including NaBr, NaxSny, and ionic polymers, have been introduced to reduce the Na-ion diffusion barrier.29–31 Electrolyte additives to modify the physical and chemical properties of SEI layers have also been reported.32–34
Recently, some reports have shown that trithiocarbonates enhance the cycling performance of Li–S batteries by simultaneously stabilizing both the cathode and the anode sides.35–37 In this work, inspired by the outstanding performance of trithiocarbonates in Li–S systems, sodium trithiocarbonate (Na2CS3) was synthesized and applied as a cathode material to improve the electrochemical performance of RT Na–S cells. Evidence from experimental results and theoretical calculations indicates several intriguing characteristics: (i) the highly covalent character of Na2CS3 molecules provides an efficient electron and ion conductive network, which improves the utilization of active materials and rate performance. (ii) Oligomer-structured intermediates formed upon cell charge have less tendency to dissolve into the electrolyte, thus reducing the polysulfide shuttle effect. (iii) A unique composition of the SEI is formed on the Na-metal surface, facilitating a uniform Na-ion flux and homogeneous Na morphology. The half cells with the Na2CS3 cathode show remarkable improvements in their initial discharge capacity and cycling performance (55% retention after 100 cycles) compared to the Na2S cathode (14% retention after 100 cycles). A series of material characterization methodologies support the notion that trithiocarbonates are applicable in the Na–S system, and they have enormous potential for high-energy-density, long cycle life RT Na–S batteries.
Experimental
Synthesis and preparation of Na2CS3 and Na2S cathodes
Na2CS3 was synthesized by a simple one-step wet ball-milling method. Commercial sodium sulfide powder (Na2S, Sigma Aldrich) was initially refined with zirconia (YSZ) grinding media in a zirconia milling container at 400 rpm. The total milling duration was 24 h with intervals of 30 min milling followed by 30 min resting. The refined Na2S powder and carbon disulfide (CS2, Alfa Aesar) were transferred into a PTFE bottle in a 1:3 mole ratio. 15 mL of 1,2-dimethoxyethane (DME) was added as a slurry medium and zirconia ceramic balls were used as grinding media. The slurry was wet ball milled for 24 h with a long roll jar milling apparatus (US Stoneware 802 CVM). The resulting slurry was dried at 50 °C for 2 h to remove the solvents and obtain Na2CS3 powder.
Na2CS3 cathodes were prepared by wet ball-milling the synthesized Na2CS3 powder and commercial multi-walled carbon nanotubes (MWCNTs, Nanostructured & Amorphous Materials Inc.) in a 7:3 weight ratio. The homogeneously mixed slurry was drop cast between two pieces of carbon nanotube papers (AvCarb P50, 7/16-inch diameter) and fully dried to make a binder-free Na2CS3 cathode. The loading of Na2CS3 in the cathode is 1.5 mg cm−2 or 3.0 mg cm−2.
Binder-free Na2S cathodes were obtained with an analogous procedure. The refined Na2S powder was wet ball milled with MWCNTs in a 7:3 weight ratio for 24 h. The slurry was then drop cast into carbon nanotube papers at a Na2S loading of 1.5 mg cm−2 or 3.0 mg cm−2.
Materials characterization
X-ray diffraction (XRD) was performed with a Rigaku Miniflex 600 diffractometer. XRD patterns were collected at a scan rate of 0.5° min−1 with a step size of 0.02°. Fourier transform infrared spectra (FTIR) were collected with a Thermo Scientific Nicolet iS5 FTIR spectrometer. Nuclear magnetic resonance (NMR) spectroscopy was performed with a Bruker Avance III 500 MHz NMR spectrometer. D6-Acetone was used as a solvent medium. X-ray photoelectron spectroscopy (XPS) measurements were carried out with a Kratos Analytical spectrometer with monochromatic Al Kα as a radiation source. The data processing and peak fitting were conducted with CasaXPS software. The surface morphologies of the synthesized materials and electrodes were examined with a FEI Quanta 650 field emission scanning electron microscope (SEM). Thermogravimetric analysis (TGA) was conducted with a Mettler TGA/DSC 1 in an argon atmosphere. The temperature ramp rate was 10 °C min−1. Ultraviolet-visible (UV-vis) spectroscopy data were collected with a Cary 5000 spectrophotometer.Electrochemical cell assembly
Na metal half cells were assembled in CR-2032-type coin cells inside an argon-filled glovebox. The cells consist of a Na-metal foil anode, 1.5 M sodium perchlorate (NaClO4) and 0.2 M sodium nitrate (NaNO3) in tetraethylene glycol dimethyl ether (TEGDME) electrolyte, ceramic coated Celgard separator, and binder-free cathodes. The cells contained active material loadings of 1.5 mg cm−2 or 3.0 mg cm−2 with an electrolyte to an active material ratio of 25 μL mg−1. The Na‖Na symmetric cells were assembled with an identical Na metal foil as the working and counter electrodes and the abovementioned electrolyte.Electrochemical performance measurements
The electrochemical cell cycling was conducted with an Arbin battery cycler at room temperature. A voltage window of 1.2 to 2.6 V was employed at a C/10 rate. The stability of the symmetric cells was evaluated at a current density of 1 mA cm−2 and an areal capacity of 1 mA h cm−2. Cyclic voltammetry (CV) data were collected with a Biologic VMP potentiostat at a rate of 0.05 mV s−1. The electrochemical impedance spectroscopy (EIS) measurements were performed with a Biologic VMP potentiostat at a frequency of 1 MHz to 0.1 kHz.Optical H-type cell assembly
In situ visible electrochemical tests were conducted in an H-type glass cell. The H-type cell was tested inside a glovebox within a voltage range of 1.2 to 2.6 V with a VoltaLab potentiostat. 7.7 mg of active material was uniformly drop-cast onto a CNT paper and dried overnight to obtain binder-free cathodes. The cathodes were combined with Na metal foil and cycled at a low current of 40 μA.Density functional theory-based calculations
To evaluate the electron density distribution and electronic conductivity of the molecules, first principles-based calculations were conducted with the Vienna ab initio simulation package (VASP). The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) correlation functional was used as a baseline theory.38,39 An energy cutoff of 600 eV was applied with a Monkhorst–Pack reciprocal space grid of 2 × 2 × 2. The molecular structures were prepared with Avogadro 1.2.0 software and relaxed until the residual force on the entire system is below 0.02 eV. The density of states (DOS) was calculated with the Heyd–Scuseria–Ernzerhof functional. The average net charge was calculated with the Bader charge analysis F90 open-source code.Results and discussion
Synthesis and material characterization
Na2CS3 has been reported as an effective precipitating agent to remove heavy metals from galvanic wastewater for decades.40,41 Trithiocarbonates can be easily formed from a reaction between carbon disulfide (CS2) and other sulfide sources.42–44 Here, the synthesis of Na2CS3 was carried out by a one-step wet ball milling of sodium sulfide (Na2S), and an excess amount of CS2 in a mole ratio of 1:3. 1,2-Dimethoxyethane (DME) was used as the slurry medium to facilitate a homogeneous reaction between solid Na2S and liquid CS2 (Fig. 1a). After drying out the solvent and surplus CS2, the orange-red colored powder was collected. The scanning electron microscopy (SEM) image of the synthesized product reveals the surface morphology change from granular micron-sized (2–8 μm) Na2S to a largely clustered (20 μm) compound, which indicates the chemical transformation taking place at the surface (Fig. S1†). The bulk crystalline structure of the obtained compound was examined with X-ray diffraction (XRD), as shown in Fig. 1b. Kapton tape was used to prevent the sample from air exposure. Regardless of the interference of the Kapton tape peak below 30° (Fig. S2†), the rest of the peaks correlate well with the reference peaks of Na2CS3. The broadened peaks indicate that even though the secondary particles are micron-sized, primary crystallites seem to be nano-sized Na2CS3. The disappearance of the Na2S peaks reveals the complete conversion of the sulfides into trithiocarbonates. The thermal stability of the synthesized Na2CS3 is shown in Fig. S3.†
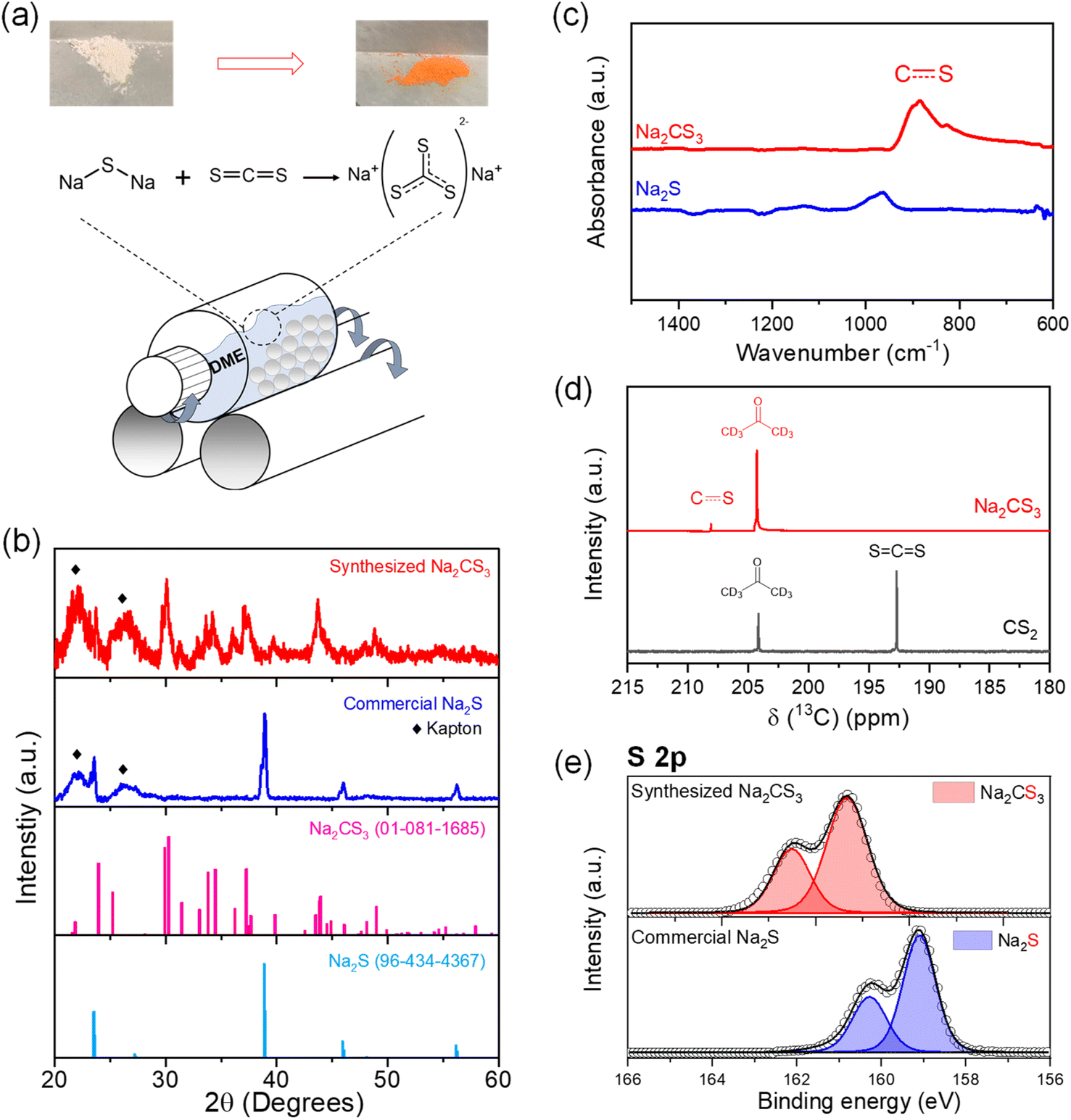 | ||
| Fig. 1 (a) Synthesis schematic of Na2CS3. (b) XRD patterns of the synthesized Na2CS3 and commercial Na2S, reference peaks of Na2CS3, and reference peaks of Na2S. (c) FTIR spectra of Na2CS3 and Na2S. (d) 13C NMR spectra of Na2CS3 and CS2. (e) S 2p XPS data of the synthesized Na2CS3 and commercial Na2S. | ||
To investigate the chemistry and molecular structure of Na2CS3, Fourier transform infrared (FTIR) spectroscopy was conducted (Fig. 1c). Distinctive to Na2S, the carbon–sulfur resonance bond stretching is reflected as a broad adsorption band at 820–930 cm−1. The peak position correlates with the previously reported carbon–sulfur resonance bond of trithiocarbonates.37,45 To further confirm the bonding environment surrounding the carbon nuclei, 13C nuclear magnetic resonance (NMR) spectroscopy was employed. As shown in Fig. 1d, a peak affiliated with the carbon–sulfur resonance bond appears at 207 ppm. A clear downfield shift is detected compared to the peak of CS2 at 192 ppm, due to the decreased diamagnetic shielding effect expected from the heightened number of surrounding sulfur atoms. The chemical state of sulfur is revealed by X-ray photoelectron spectroscopy (XPS) in Fig. 1e. Compared to the sulfide peak (S2−) appearing at 159 eV, the carbon–sulfur resonance peak appears at 161.3 eV. The peak shift towards higher binding energy confirms the oxidation of sulfur, while forming Na2CS3. Hence, the series of material characterization techniques confirm the complete reaction of Na2S and CS2 to form Na2CS3.
Electrochemical cell performances
With a thorough understanding of the formation of Na2CS3 and its molecular structure, Na metal half cells were assembled and tested at a C/10 rate. An ether-based electrolyte, tetraethylene glycol dimethylether (TEGDME), was chosen since it is known to suffer from high solubility of long-chain polysulfides, severe shuttling effect, and failure to form a stable SEI.46–48 The active materials were dissolved in a DME solvent along with multi-walled carbon nanotubes (MWCNTs) in a weight ratio of 7:3. This solution was drop-cast between two carbon papers and fully dried to be used as a binder-free cathode. Ceramic-coated separators were utilized to ensure sufficient wetting, and the cells were constructed in the form of Na‖Na2CS3 and Na‖Na2S. An excess electrolyte amount (25 μL mg−1 with respect to the active material) was injected to avoid additional variables of lean-electrolyte conditions. The specific capacities were calculated based on the active material weight for the ease of comparison. The cells were charged up to 2.6 V and discharged down to 1.2 V, which will be discussed in detail in the later section.
Fig. 2a shows the third cycle galvanostatic charge–discharge curves of Na‖Na2CS3 and Na‖Na2S cells with an active material loading of 1.5 mg cm−2. The discharge mechanism of Na–S is similar to that of Li–S but not identical, due to the intrinsic property differences between sodium and lithium, such as standard reduction potential (−2.71 V for Na and −3.04 V for Li vs. SHE), ionic radius, and reactivity. Similar to previous reports in the literature,49 the Na‖Na2S cells show a conventional multi-step reduction reaction with an average discharge voltage of 1.79 V. In the high-discharge plateau region (2.31–2.20 V), highly soluble Na2S8 is produced. In the subsequent sloping region (2.20–1.68 V), it is converted into less soluble Na2S4. In the low-discharge plateau region (1.68–1.62 V), transitional products of insoluble Na2S3 and Na2S2 are formed, and the final sloping region (1.62–1.20 V) corresponds to the solid–solid conversion into Na2S.
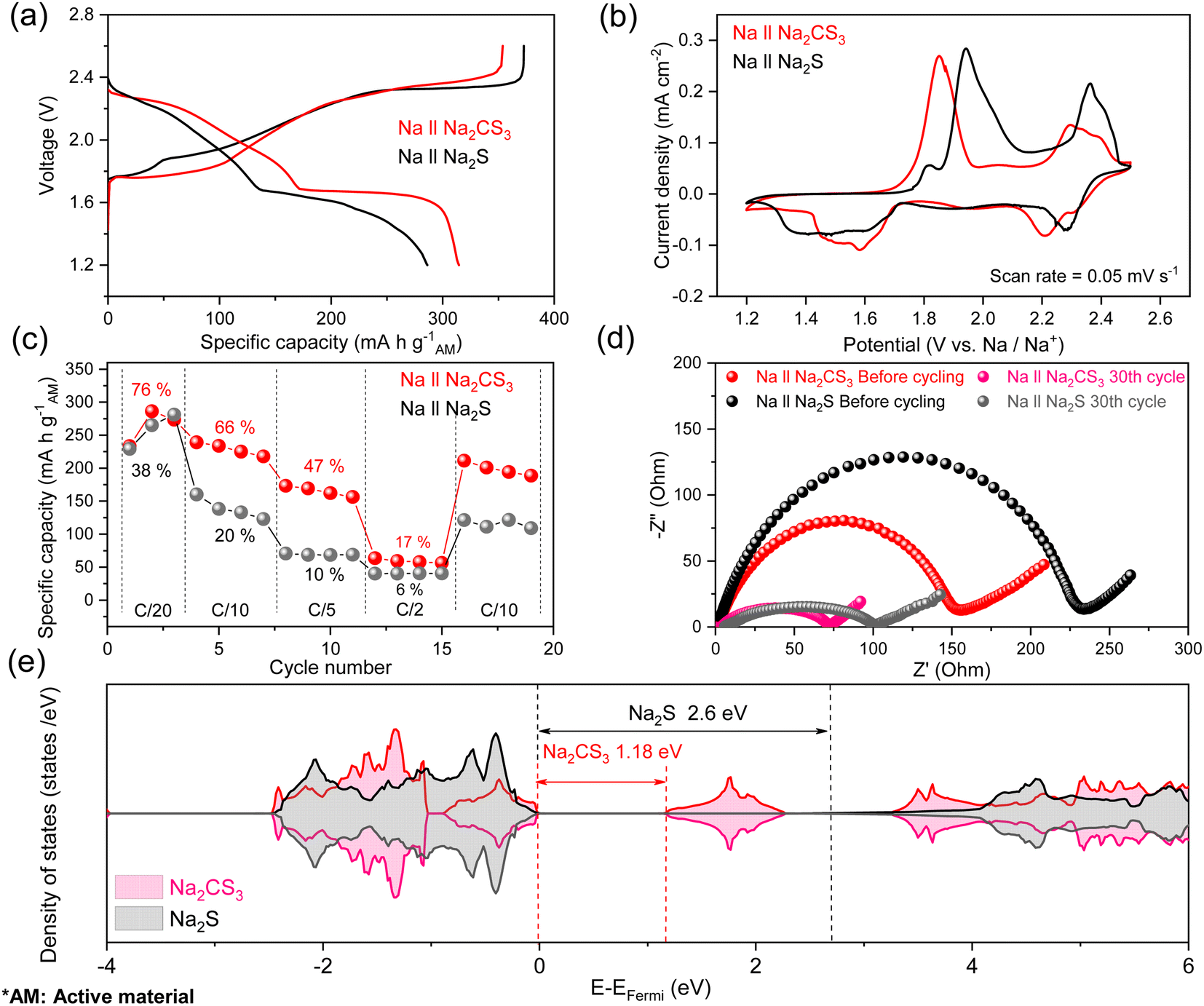 | ||
| Fig. 2 (a) Voltage curves of the Na‖Na2CS3 and Na‖Na2S cells in the 3rd cycle with an active material loading of 1.5 mg cm−2. (b) CV curves of Na‖Na2CS3 and Na‖Na2S cells. (c) Rate performance of Na2CS3 and Na2S cathodes at various C rates. The percentage number corresponds to the active material utilization based on the theoretical capacity. All specific capacities are based on the active material loading. (d) Nyquist plots before and after 30 cycles. (e) Total electronic density of states of Na2CS3 and Na2S. | ||
While the discharge curve of the Na‖Na2CS3 cell shows an analogous two-plateaued trend, it exhibits a much higher average discharge voltage of 1.88 V. The high-discharge plateau becomes prominent at a voltage of ∼2.28 V, and the sloping region (2.28–1.68 V) is shifted towards a higher potential compared to the Na2S cathode. The low-discharge plateau at 1.68 V also becomes more noticeable with an enhanced total discharge capacity of 314 mA h g−1 compared to the control cell (285 mA h g−1). It is expected that the different molecular coordination of Na2CS3 alters the conventional sulfur reaction pathway and shifts the discharge voltage curve. Furthermore, there are significant differences in the charge profiles. Specifically, much less overpotential is detected on the lower-charge plateau (1.88 V for Na2S and 1.75 V for Na2CS3). The reduced polarization between charge and discharge curves signifies that the reaction of Na2CS3 might be kinetically more favorable compared to that of Na2S.
The cyclic voltammetry (CV) curves of the Na‖Na2CS3 and Na‖Na2S cells (Fig. 2b) show consistent results with the voltage curves. Na‖Na2S cells show an upper cathodic peak at 2.29 V and a broad lower cathodic peak centered at 1.47 V, whereas Na‖Na2CS3 cells show two sharp cathodic peaks at 2.21 and 1.56 V. Furthermore, it is clearly shown that the anodic peaks of Na‖Na2CS3 cells get shifted towards lower potentials compared to Na‖Na2S cells. From the voltage profiles and CV curves, it is observed that the altered reaction pathway of Na2CS3 leads to both a reduced polarization and higher total capacity.
The reduced polarization at the early stages of the cell cycling can be attributed to the enhanced reaction kinetics of the cathode. To verify this hypothesis, galvanostatic charge/discharge at various current rates was evaluated as shown in Fig. 2c. The Na‖Na2CS3 cells achieved an average capacity of 264, 229, 165, and 59 mA h g−1 at, respectively, C/20, C/10, C/5, and C/2 rates, which are much higher than those of the Na‖Na2S cells (258, 139, 69, and 40 mA h g−1). When the rate is recovered to C/10, the average capacity recovers back to 198 mA h g−1, which indicates the superior rate performance and stability of the Na2CS3 cathode. It is important to note that the Na2CS3 cathode displays an extremely high material utilization of 76% (the theoretical specific capacity of Na2CS3 is 348 mA h g−1) at a C/20 rate, whereas the Na2S cathode exhibits only 38% utilization (the theoretical specific capacity of Na2S is 687 mA h g−1). The remarkable material utilization and rate performance support the facile reaction kinetics of the Na2CS3 cathode.
It is well accepted that conjugated structures facilitate the electron transport process during the electrochemical reaction. Since the CS32− anion contains a carbon–sulfur resonance bond, which is highly conductive, delocalized electrons provide sufficient ionic and electronic pathways to maintain facile reaction kinetics, higher material utilization, and low resistance of the cell. Furthermore, the similar electronegativity between carbon (2.55) and sulfur (2.58) forms a covalent bond, which acts as a good electron conductor compared to the highly ionic Na2S cathode. The density of states calculation was conducted to compare the band gap of single molecule Na2CS3 and Na2S. The narrower band gap of Na2CS3 (1.18 eV) rather than Na2S (2.6 eV), as shown in Fig. 2e, well supports the highly conductive nature of Na2CS3.
With a firm understanding of the facile electrochemical process taking place in Na2CS3, the cycling stability of the cells at an active material loading of 1.5 mg cm−2 is compared. Surprisingly, as shown in Fig. 3a, the Na‖Na2CS3 cells show a remarkable cycling performance improvement compared to the conventional Na‖Na2S cells. The peak discharge capacity is 306 mA h g−1 on the active material basis, and a high capacity of 178 mA h g−1 is maintained (58% retention) after 50 cycles. In addition, throughout the entire cycling process, the Na2CS3 cathode shows a higher coulombic efficiency than the Na2S cathode, indicating that the trithiocarbonate-based cathode material effectively mitigates the polysulfide shuttle effect. In contrast, the conventional Na2S cathode delivers a peak discharge capacity of 300 mA h g−1 and experiences a sharp capacity decay to 82 mA h g−1 (27% retention). The rapid capacity drop is mainly caused by the severe shuttling effect of polysulfides in glyme-based electrolytes.50
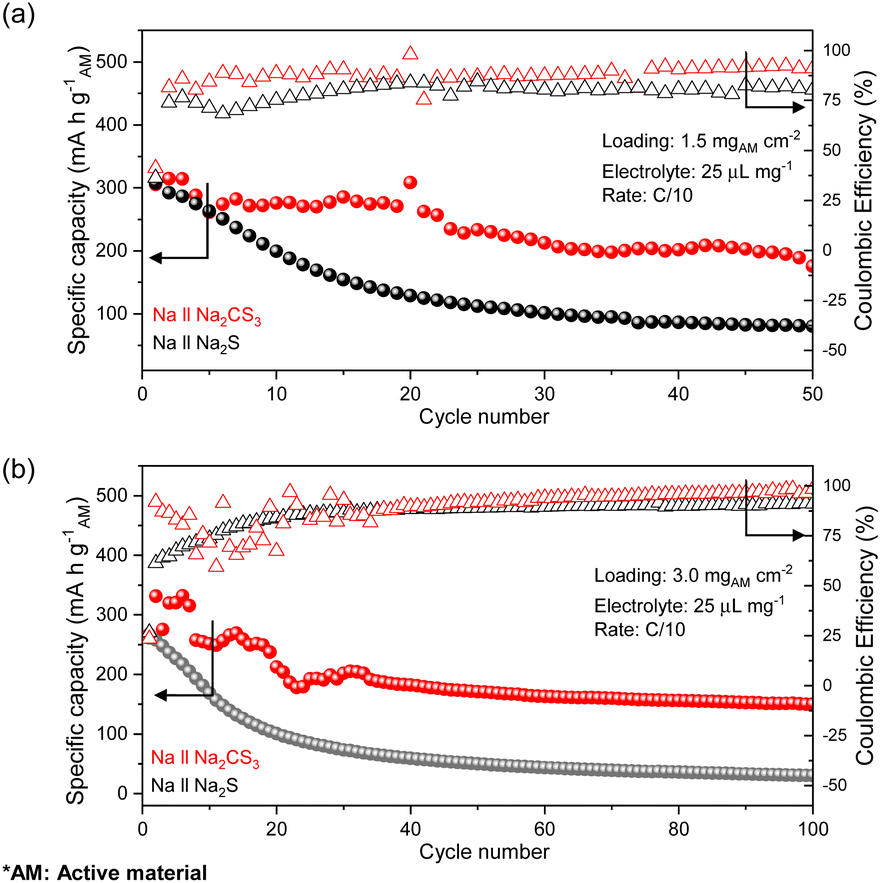 | ||
| Fig. 3 Cycling performances of half cells with the Na2CS3 cathode (red) and Na2S cathode (black) at an active material loading of (a) 1.5 mg cm−2 and (b) 3.0 mg cm−2. | ||
To further improve the energy density, cells with a higher active material loading of 3.0 mg cm−2 were also tested. As shown in Fig. 3b, the Na2CS3 cathode still outperforms (151 mA h g−1 and 55% retention) the conventional Na2S cathode (34 mA h g−1 and 14% retention) at the end of 100 cycles. At a high loading, the discharge capacity and coulombic efficiency of Na‖Na2CS3 cells become unstable at the early stages of cycling, which might be mainly due to the formation of an interphase layer between the electrodes and the electrolyte. The prolonged cycling stability and enhanced coulombic efficiency of the Na2CS3 cathode signify that trithiocarbonates are an attractive cathode material for RT Na–S batteries. Now, how Na2CS3 contributes to the improvement in the cell performance needs to be investigated in detail and understood.
Reaction mechanisms and suppressed active material dissolution of Na2CS3
To unveil the reaction mechanisms of Na2CS3 and its contribution to the cell performance improvement, XPS analysis was conducted on the cathode at different stages of charge (Fig. 4a). When the pristine Na‖Na2CS3 cells are charged up to 2.2 V, the S 2p3/2 peak at 162.9 eV emerges, which represents the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond.51 Small peaks detected beyond 166 eV come from the thiosulfate and sulfate complexes originating from the oxidized sulfide species.52,53 On the cell charged up to 2.4 and 2.6 V, the constant attenuation of the carbon–sulfur resonance bond (161.3 eV) and increment of the CS bond are observed. During the de-sodiation process, it is explored that the intermediate products start to form [–CS3–CS3–] oligomer-like compounds, as shown in Scheme 1. Interestingly, when the cell is charged up to 2.8 V, a sudden increase in the peak of sulfate complexes is detected. It is assumed that at this potential, severe electrolyte and salt decomposition is triggered with the presence of an intermediate charged product of Na2CS3.
S bond.51 Small peaks detected beyond 166 eV come from the thiosulfate and sulfate complexes originating from the oxidized sulfide species.52,53 On the cell charged up to 2.4 and 2.6 V, the constant attenuation of the carbon–sulfur resonance bond (161.3 eV) and increment of the CS bond are observed. During the de-sodiation process, it is explored that the intermediate products start to form [–CS3–CS3–] oligomer-like compounds, as shown in Scheme 1. Interestingly, when the cell is charged up to 2.8 V, a sudden increase in the peak of sulfate complexes is detected. It is assumed that at this potential, severe electrolyte and salt decomposition is triggered with the presence of an intermediate charged product of Na2CS3.
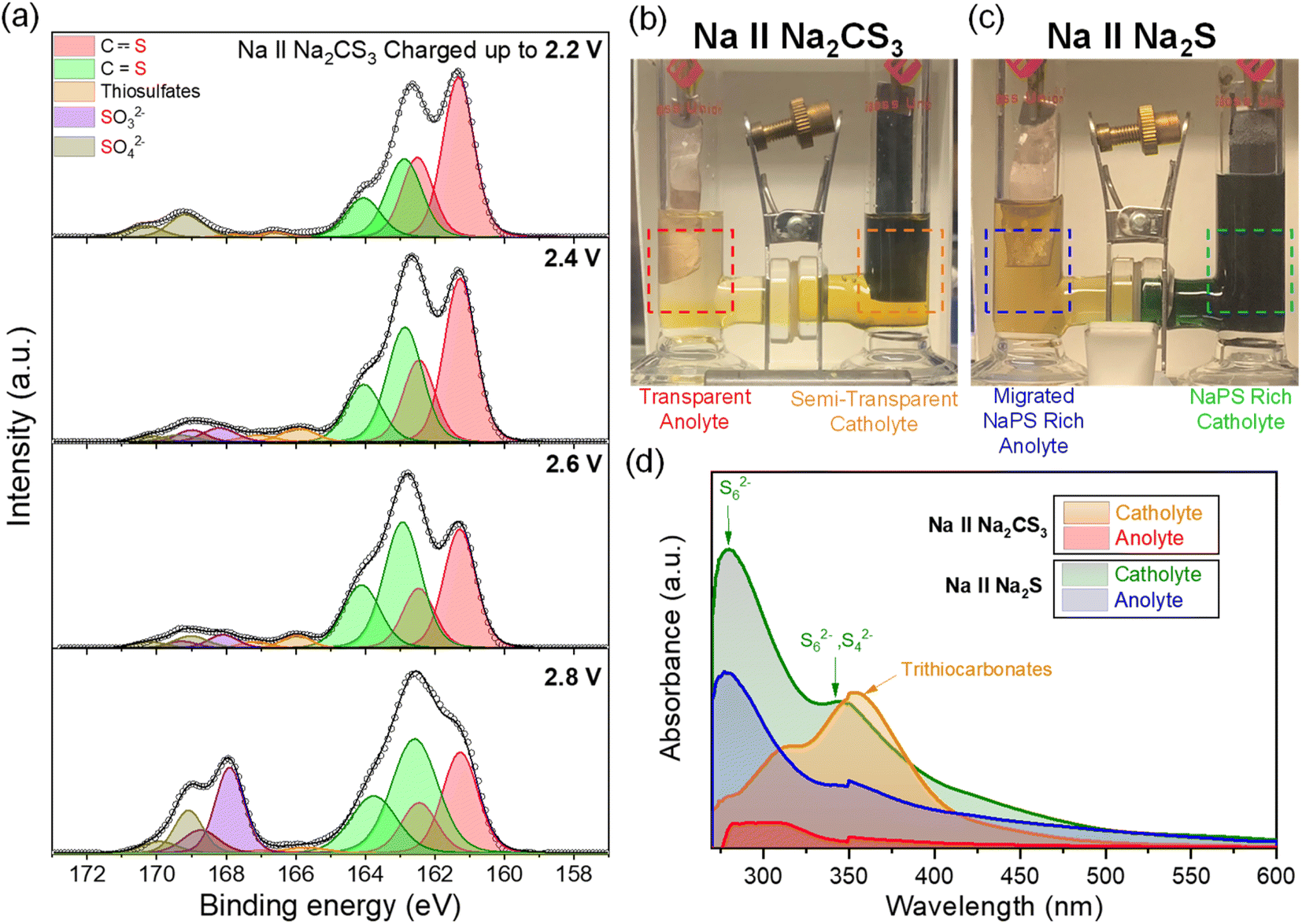 | ||
| Fig. 4 (a) S 2p XPS data of the Na2CS3 cathode charged up to different states. Image of optical H-cells after a single charge and discharge: (b) Na‖Na2CS3 and (c) Na‖Na2S cells. (d) UV-visible spectra of the catholytes and anolytes of Na‖Na2CS3 and Na‖Na2S H-cells. Electrolytes were collected at the discharged state. | ||
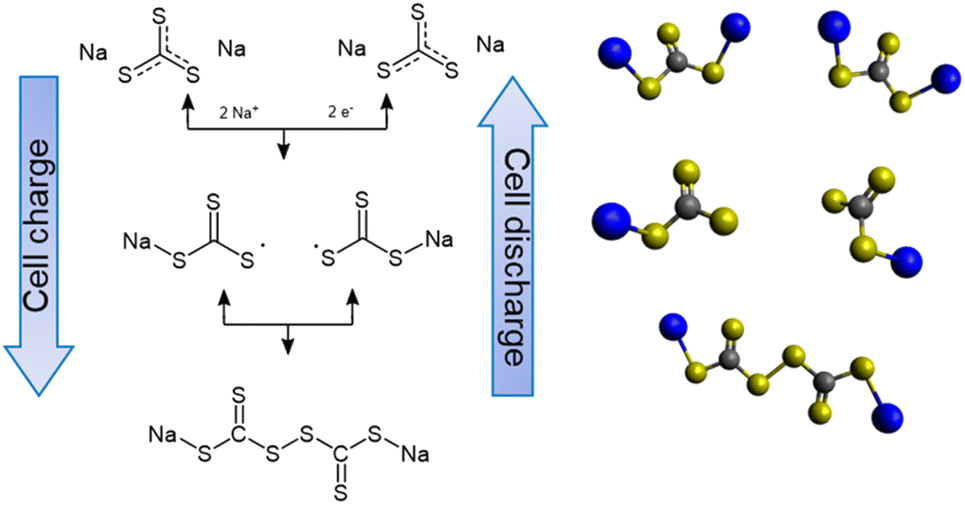 | ||
| Scheme 1 Reaction mechanism of Na2CS3 during charge and discharge. | ||
The SEM image of the electrode reveals well the charging process of Na2CS3, as shown in Fig. S4.† At the early stages of charge (2.2 V), the electrode is mainly covered with sharp-edged Na2CS3. Upon charging (2.4 V), the transformation from the carbon–sulfur resonance bond to CS takes place at the boundaries of Na2CS3, and the sharp edges start to become blunt. At 2.6 V, most of the surface material got transformed into CS containing species, whereas at 2.8 V the electrode is suddenly covered with the electrolyte and salt decomposed products. The SEM image provides well the visual image of active material transformation and electrolyte/salt decomposition upon cell charge, which correlates with the XPS data. The effect of the electrolyte and salt decomposition when charging the cells up to 2.8 V is examined in Fig. S5.† A sharp capacity decay within 3 cycles is observed when the Na‖Na2CS3 cells are charged up to 2.8 V, whereas a stable cycling performance is shown when the cells are charged to 2.6 V. It is expected that the irreversible reactions between Na2CS3 species and the electrolyte take place above 2.6 V, resulting in a fast capacity decay. To avoid this type of reaction, all the electrochemical cells were charged only up to 2.6 V.
It is also worthwhile to explore how the [–CS3–CS3–] oligomer layers formed upon charge can improve the cell cycling performance. Compared to sodium polysulfides, oligomer structured Na2CS3 compounds might have a heavier molar mass and bulky structure. This could prevent the dissolution and migration of the active materials, thus effectively inhibiting the shuttling of active materials. The effect of Na2CS3 preventing the active material dissolution is evidenced by the optical H-type electrochemical cell. The Na-metal anode was combined with Na2CS3 and Na2S cathodes and galvanostatically cycled inside an argon-filled glovebox. As shown in Fig. 4b, the Na2CS3 cathode exhibits a semi-transparent yellow catholyte after a single charge and discharge step due to the suppressed active material dissolution. The transparent anolyte and fresh sodium-metal surface correspond to the mitigated shuttle effect. In contrast, the Na2S cathode in Fig. 4c shows a severe dissolution and diffusion of sodium polysulfides with an obvious catholyte color change into opaque green. The color of dissolved sodium polysulfides in TEGDME is known to be green, as previously reported.54 The anolyte tuned semi-transparent orange and the discoloration of the sodium metal surface was detected, which clearly reveals the parasitic side reaction of Na metal and migrated sodium polysulfides taking place at the surface. The optical H-cell qualitatively confirms the prominent suppression of active-material dissolution and the shuttle effect of the Na2CS3 cathode.
To quantitatively measure the dissolved active materials in the electrolyte from the optical H-cell, ultraviolet-visible (UV-vis) spectroscopy was conducted. Fig. S6a† shows the UV-vis absorption spectra of electrolytes at the charged and discharged states of the H-cell, when using Na2CS3 as a cathode. During the initial charge of the cell, a negligible amount of active material gets dissolved into both the catholyte and the anolyte. The subsequent discharging step reveals some amount of active material dissolution into the catholyte. The broad peak in the 300–400 nm region corresponds to the intermediate oligomer structured trithiocarbonate species.55 It is important to note that the anolyte still shows no peaks, which indicates that the intermediate Na2CS3 species barely diffuse into the anode side.
In contrast, Fig. S6b† demonstrates the high absorbance of polysulfide peaks in both the catholyte and the anolyte when using Na2S as a cathode. The peak at 280 nm represents the long chain polysulfides (S62−), which aligns with the synthesized Na2S6 peak shown in Fig. S7.† The broad band at 350–400 nm corresponds to S42− and S62−, which exhibits the co-existence of multiple forms of polysulfides in equilibrium.48,56 The high intensity of polysulfide signals at both the catholyte and the anolyte exhibits severe sodium polysulfide dissolution and migration throughout the cell cycling. Fig. 4d compares the absorption spectra of Na2CS3 and Na2S cathodes at their discharged state in a single plot. It is clearly observed that the oligomer-structured Na2CS3 intermediates prominently hamper active material dissolution and the shuttle effect compared to the conventional Na2S cells. These H-cell and UV-vis data are consistent with the cycling data, showing that the Na2CS3 cathode performs with superior coulombic efficiency.
Sodium-metal anode morphology and SEI chemistry
It is necessary to further investigate the effect of Na2CS3 on the Na-metal anode side. The Na‖Na2CS3 and Na‖Na2S cells after 20 cycles were disassembled, and the Na metal was rinsed with TEGDME solvent. The macroscopic morphology of the Na metal was compared based on its optical photographs. Surprisingly, as shown in Fig. S8,† the Na-metal anode paired with the Na2CS3 cathode still maintains the shiny surface with a partial presence of discolored spots. In contrast, Na metal assembled with the Na2S cathode shows a thick discolored layer covering the entire surface. The detailed microscopic morphology of the Na-metal anode was investigated with SEM. As shown in Fig. 5a, smooth and non-dendritic growth behavior is detected in the Na metal combined with the Na2CS3 cathode. It might be the consequence of stable SEI layer formation in the presence of Na2CS3, which protects well the Na metal from side reactions and helps improve the sodium stripping and plating performance. In contrast, the Na metal assembled with the Na2S cathode exhibits a bumpy and porous surface morphology (Fig. 5b). Such irregular growth of Na metal led to severe side reactions with electrolytes and polysulfides, which causes a depletion of the electrolyte and consumption of the active materials.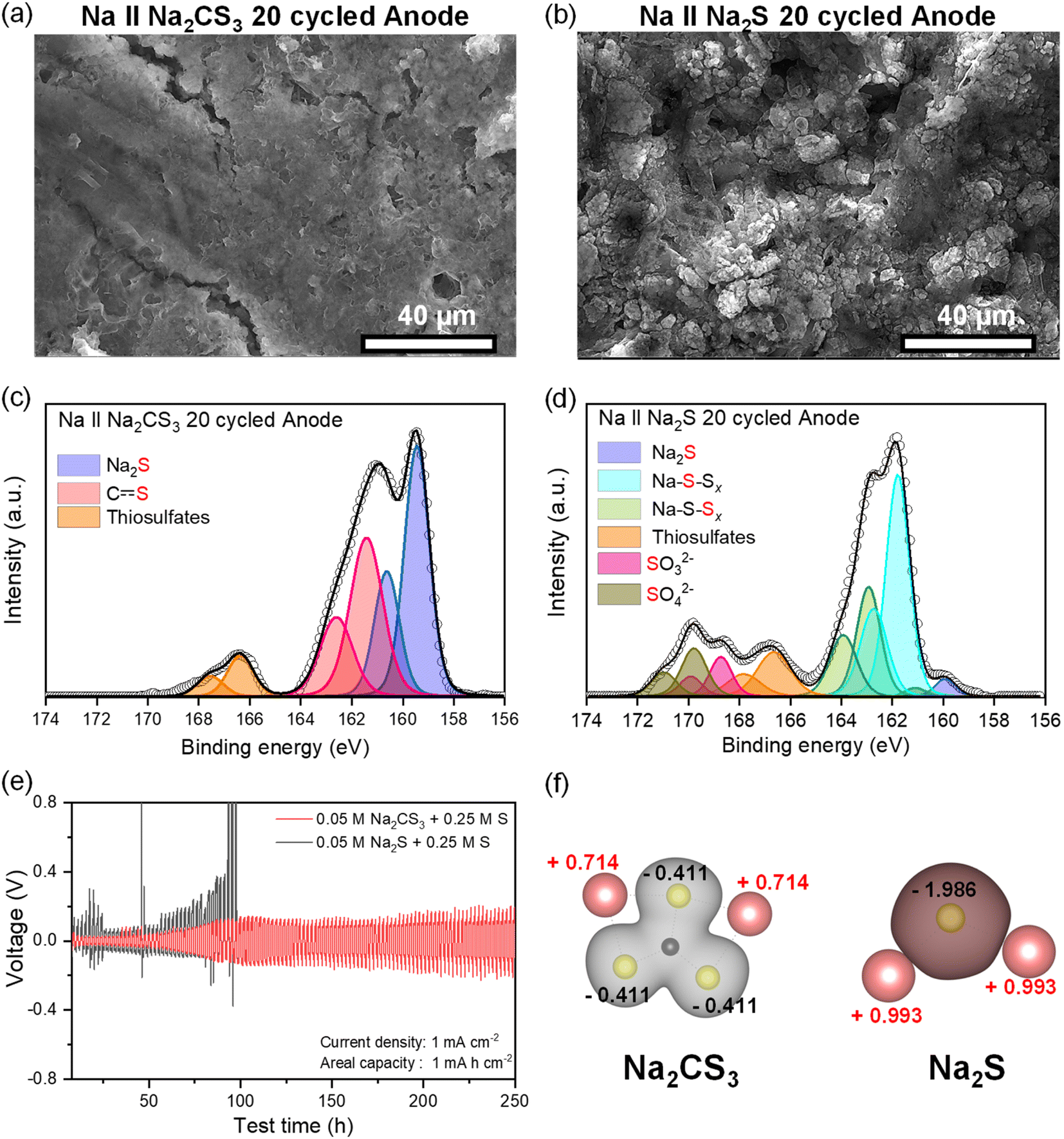 | ||
| Fig. 5 SEM image of the Na-metal anode after 20 cycles: (a) Na‖Na2CS3 and (b) Na‖Na2S cells. S 2p XPS data of the Na-metal anode after 20 cycles: (c) Na‖Na2CS3 and (d) Na‖Na2S cells. (e) Cycling stability of Na‖Na symmetric cells with polysulfide forms of Na2CS3 and Na2S6. (f) Bader charge analysis of Na2CS3 and Na2S. | ||
To gain insights into the homogeneous Na-metal anode morphology in the presence of Na2CS3, XPS analysis was employed to understand the chemical composition of the surface of the SEI layer. Interestingly, the major component of the SEI layer in the Na‖Na2CS3 cell turns out to be a reduced sulfur species of S2− from Na2S (159.5 eV) and the carbon–sulfur resonance bond from Na2CS3 (161.3 eV) (Fig. 5c). A small intensity of thiosulfates (166.3 eV) signifies the minimized side reactions of Na metal with the electrolyte, owing to the uniform morphology of sodium with a low surface area. To further identify the species in the bulk of the deposited sodium metal, Ar+ sputtering was conducted. In general, the surface of the SEI layer may not have a complete reduction of the material due to the limited access to metallic Na and electron conduction pathways. However, such a kinetic hindrance will be attenuated on the bulk side of sodium metal, so more severe reducing conditions could be fostered on the bulk side of sodium metal. Fig. S9† shows the S 2p XPS data after sputtering the surface for 10 min (∼250 nm of material removal), and the carbon–sulfur resonance bond is still detected at 161.3 eV. This implies that trithiocarbonates are stable toward reduction, which makes them an ideal SEI component. In sharp contrast, the SEI layer of the Na‖Na2S cell is dominated by the sodium polysulfide signals of bridging sulfur (163 eV) and terminal sulfur species (161.9 eV). The high intensity of the oxidized sulfur species, such as thiosulfates, SO32− (168.6 eV), and SO42− (169.8 eV), indicates the reactive Na metal undergoing severe side reactions with the electrolyte, which implies that it does not form a stable SEI layer.
From the cycled Na-metal anode analysis, it is confirmed that the inclusion of Na2CS3 in the system clearly induces a homogeneous Na metal morphology and forms a unique carbon–sulfur resonance bond containing a SEI layer. Also, from the previous section, the effective inhibition of polysulfide shuttling from the Na2CS3 cathode has been verified. It is widely known that the parasitic reactions between corrosive Na metal and migrated polysulfides lead to a complex, unstable, and fragile SEI layer that causes roughened Na deposition. Now it is important to clarify if the improved Na metal surface morphology is solely the consequence of the suppressed polysulfide shuttling, or the formation of a unique carbon–sulfur resonance bond containing SEI layer also contributes to the enhanced stripping and plating performance.
To answer this question, the long-term cycling stability of Na‖Na symmetric cells was tested at a current density of 1 mA cm−2 and an areal capacity of 1 mA h cm−2. The 0.05 M polysulfide forms of Na2CS3 and Na2S were dissolved into the electrolyte. As shown in Fig. 5e, the cell containing the polysulfide form of Na2CS3 shows a much lower initial overpotential (27 mV) compared to the conventional sodium polysulfides (86 mV) at the early stages of cycling. The voltage hysteresis gradually increases to 110 mV until 80 h, and the cell maintains its overpotential values for more than 250 h. However, the cell including conventional sodium polysulfides shows a sudden increase in overpotential after 60 h. It showed cell failure at around 100 h, indicating unstable SEI formation and dendritic growth of sodium. The Na‖Na symmetric cell experiment demonstrates that the cell polarization can be greatly reduced by introducing Na2CS3 compounds into the system. Generally, the reduced voltage hysteresis and stable cycle life of Na‖Na symmetric cells are due to the formation of a favorable Na-ion conducting SEI layer, suppressing the dendritic growth of Na. Thus, it has been proven that the homogeneous morphology of the Na-metal anode and prolonged cycle life are not only the consequence of the suppressed shuttle effect, but also due to the formation of a unique, stable SEI layer. The result is in good agreement with a previous report that trithiocarbonates can act as a SEI modifying agent to enhance the Li morphology in Li-ion batteries.35
To understand the underlying mechanism of Na2CS3, including the SEI layer showing the enhanced stripping and plating performance, first-principles density functional theory (DFT) calculation was conducted. Bader charge analysis shown in Fig. 5f reveals an electron density spread on Na2CS3 and Na2S molecules. For ternary sulfides (Na2CS3), which contain carbon–sulfur resonance bonds, the covalent characteristics between carbon and sulfur bonds effectively spread the electron density. Consequently, the sulfur atom, which is the constituent with the highest electron density in the molecule, only contains a net electron charge of −0.411. The positively charged Na ions might experience a low diffusion barrier when traveling through the Na2CS3-rich SEI layer. This can facilitate a homogeneous and facile Na-ion flux without local charge accumulation. In contrast, the binary Na2S and other sodium polysulfides (Fig. S10†), which are the main SEI components of the conventional Na‖Na2S cells, have a highly ionic bond characteristic due to the large electronegativity difference between Na (0.93) and S (2.58). The sulfur atom in Na2S possesses a high electron density, resulting in a net electron charge of −1.986. The terminal sulfur of Na2S2 and Na2S4 sodium polysulfides also showed a high electron charge of −0.706 and −0.612. The locally concentrated charge causes an uneven electric field and Na-ion flux, leading to poor inhomogeneous Na deposition. Bader charge analysis provides a good explanation of a Na2CS3-containing SEI layer promoting a uniform and dense morphology of the Na metal surface, minimizing the reaction sites of corrosive sodium with electrolytes and polysulfides.
Conclusions
In summary, we have shown that trithiocarbonates applied as a cathode material in RT Na–S batteries enable a multi-functional improvement in the cell performance, both from a viewpoint of reaction kinetics and from a cycling stability perspective. Owing to its conjugate structure and highly covalent characteristics, Na2CS3 enables efficient active material utilization and superior rate performance compared to the Na2S cathode. From a viewpoint of cell cyclability improvement, Na2CS3 is used to modulate both the cathode and the anode dynamics. It is recognized that the [–CS3–CS3–] oligomer structures are formed at the cathode during cell cycling, which suppresses active material dissolution and shuttling. Furthermore, a unique carbon–sulfur resonance bond containing a SEI layer is formed on the Na-metal anode surface, which enables a uniform Na-ion flux. Consequently, the Na2CS3 cathode displays a higher average discharge voltage (1.88 V) compared to the Na2S cathode (1.79 V) with lower charge-transfer resistance and cell polarization. An outstanding active material utilization of 76% is realized, while Na2S shows only 38% utilization at the C/20 rate. In the well-designed Na‖Na2CS3 cells, a superior cycling performance is achieved with an active material loading of 1.5 mg cm−2 (58% retention over 50 cycles) and 3.0 mg cm−2 (55% retention over 100 cycles). This work provides a rational cathode design strategy to realize high-energy density long cycle life RT Na–S batteries.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under award number DE-SC0005397.References
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- A. Manthiram, A. Vadivel Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621–638 RSC.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, Adv. Funct. Mater., 2020, 30, 2004084 CrossRef CAS.
- D. Lide, CRC Handbook of Chemistry and Physics, 85th edn, 2004 Search PubMed.
- A. Manthiram and X. Yu, Small, 2015, 11, 2108–2114 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- N. Tanibata, M. Deguchi, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 5232–5238 CrossRef CAS.
- C. W. Park, J. H. Ahn, H. S. Ryu, K. W. Kim and H. J. Ahn, Electrochem. Solid-State Lett., 2006, 9, A123–A125 CrossRef CAS.
- Y. Wang, Y. Zhang, H. Cheng, Z. Ni, Y. Wang, G. Xia, X. Li and X. Zeng, Mol, 2021, 26, 1535 CrossRef CAS PubMed.
- H. Liu, W.-H. Lai, Y. Lei, H. Yang, N. Wang, S. Chou, K. Liu, S. X. Dou, Y.-X. Wang, H. Liu, W.-H. Lai, Y. Lei, H. Yang, N. Wang, S. Chou, H. K. Liu, S. X. Dou and Y.-X. Wang, Adv. Energy Mater., 2022, 12, 2103304 CrossRef CAS.
- W. Du, Y. Wu, T. Yang, B. Guo, D. Liu, S. J. Bao and M. Xu, Chem. Eng. J., 2020, 379, 122359 CrossRef CAS.
- X. Yu and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1943–1947 CrossRef CAS PubMed.
- Y. M. Chen, W. Liang, S. Li, F. Zou, S. M. Bhaway, Z. Qiang, M. Gao, B. D. Vogt and Y. Zhu, J. Mater. Chem. A, 2016, 4, 12471–12478 RSC.
- N. Chawla and M. Safa, Electron, 2019, 8, 1201 CrossRef CAS.
- B. W. Zhang, Y. D. Liu, Y. X. Wang, L. Zhang, M. Z. Chen, W. H. Lai, S. L. Chou, H. K. Liu and S. X. Dou, ACS Appl. Mater. Interfaces, 2017, 9, 24446–24450 CrossRef CAS PubMed.
- N. Wang, Y. Wang, Z. Bai, Z. Fang, X. Zhang, Z. Xu, Y. Ding, X. Xu, Y. Du, S. Dou and G. Yu, Energy Environ. Sci., 2020, 562, 562–570 RSC.
- D. Ma, Y. Li, J. Yang, H. Mi, S. Luo, L. Deng, C. Yan, M. Rauf, P. Zhang, X. Sun, X. Ren, J. Li and H. Zhang, Adv. Funct. Mater., 2018, 28, 1705537 CrossRef.
- W. Bao, C. E. Shuck, W. Zhang, X. Guo, Y. Gogotsi and G. Wang, ACS Nano, 2019, 13, 11500–11509 CrossRef CAS.
- H. Liu, W. Pei, W. H. Lai, Z. Yan, H. Yang, Y. Lei, Y. X. Wang, Q. Gu, S. Zhou, S. Chou, H. K. Liu and S. X. Dou, ACS Nano, 2020, 14, 7259–7268 CrossRef CAS.
- Z. Yan, J. Xiao, W. Lai, L. Wang, F. Gebert, Y. Wang, Q. Gu, H. Liu, S. L. Chou, H. Liu and S. X. Dou, Nat. Commun., 2019, 10, 4793 CrossRef PubMed.
- B. W. Zhang, T. Sheng, Y. D. Liu, Y. X. Wang, L. Zhang, W. H. Lai, L. Wang, J. Yang, Q. F. Gu, S. L. Chou, H. K. Liu and S. X. Dou, Nat. Commun., 2018, 9, 4082 CrossRef PubMed.
- B. W. Zhang, S. Li, H. L. Yang, X. Liang, W. H. Lai, S. Zhao, J. Dong, S. Q. Chu, Q. F. Gu, J. Liang, Y. Du, X. Xu, L. Cao, Y. X. Wang, F. Pan, S. L. Chou, H. K. Liu and S. X. Dou, Cell Rep. Phys. Sci., 2021, 2, 100531 CrossRef CAS.
- X. Zhang, A. Wang, X. Liu and J. Luo, Acc. Chem. Res., 2019, 52, 3223–3232 CrossRef CAS PubMed.
- Y. He, X. Ren, Y. Xu, M. H. Engelhard, X. Li, J. Xiao, J. Liu, J. G. Zhang, W. Xu and C. Wang, Nat. Nanotechnol., 2019, 14, 1042–1047 CrossRef CAS PubMed.
- S. Wei, S. Choudhury, J. Xu, P. Nath, Z. Tu and L. A. Archer, Adv. Mater., 2017, 29, 1605512 CrossRef PubMed.
- V. Kumar, A. Y. S. Eng, Y. Wang, D. T. Nguyen, M. F. Ng and Z. W. Seh, Energy Storage Mater., 2020, 29, 1–8 CrossRef.
- S. Choudhury, S. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef.
- X. Zheng, H. Fu, C. Hu, H. Xu, Y. Huang, J. Wen, H. Sun, W. Luo and Y. Huang, J. Phys. Chem. Lett., 2019, 10, 707–714 CrossRef CAS PubMed.
- Y. Lee, J. Lee, J. Lee, K. Kim, A. Cha, S. Kang, T. Wi, S. J. Kang, H. W. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2018, 10, 15270–15280 CrossRef CAS.
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 9069–9072 CrossRef CAS PubMed.
- Y. Ein-Eli, J. Electroanal. Chem., 2002, 531, 95–99 CrossRef CAS.
- Y. Chu, X. Cui and Q. Pan, ACS Appl. Energy Mater., 2018, 1, 6919–6926 CrossRef CAS.
- H. Sul, A. Bhargav and A. Manthiram, Adv. Energy Mater., 2022, 2200680 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035109 CrossRef.
- M. Thomas, D. Zdebik and B. Białecka, Pol. J. Environ. Stud., 2018, 27, 1753–1763 CrossRef CAS PubMed.
- M. Thomas, V. Kozik, A. Bąk, K. Barbusiński, J. Jazowiecka-rakus and J. Jampilek, Materials, 2021, 14, 1 Search PubMed.
- B. D. Stone and M. L. Nielsen, US Pat., 2893835A, 1959 Search PubMed.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- C. M. James, PhD thesis, University of Cologne, 2021.
- H. Seidel and R. Meyn, Z. Naturforsch. B, 1971, 26, 1192 CrossRef CAS.
- L. Wang, T. Wang, L. Peng, Y. Wang, M. Zhang, J. Zhou, M. Chen, J. Cao, H. Fei, X. Duan, J. Zhu and X. Duan, Natl. Sci. Rev., 2022, 9, 3 Search PubMed.
- X. Yu and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1943–1947 CrossRef CAS PubMed.
- J. Pampel, S. Dörfler, H. Althues and S. Kaskel, Energy Storage Mater., 2019, 21, 41–49 CrossRef.
- X. Yu and A. Manthiram, Chem.–Eur. J., 2015, 21, 4233–4237 CrossRef CAS PubMed.
- D. Liu, Z. Li, X. Li, X. Chen, Z. Li, L. Yuan and Y. Huang, ACS Appl. Mater. Interfaces, 2022, 14, 6658–6666 CrossRef CAS PubMed.
- A. Bhargav and A. Manthiram, Adv. Energy Mater., 2020, 10, 2001658 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- I. Kim, J.-Y. Park, C. Kim, J.-W. Park, J.-P. Ahn, J.-H. Ahn, K.-W. Kim and H.-J. Ahn, J. Electrochem. Soc., 2016, 163, A611–A616 CrossRef CAS.
- J. A. Venter, PhD thesis, University of Pretoria, 2007.
- X. Yu and A. Manthiram, Adv. Energy Mater., 2015, 5, 1500350 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07918f |
| This journal is © The Royal Society of Chemistry 2023 |