
Carbon quantum dots aqueous solution as electrolyte for H2O2 production based on photoelectrochemical water splitting†
Na
Wang
,
Yong-Shuai
Zhang
,
Dong-Dong
Wei
,
Hui-Min
Duan
,
Liu-Meng
Mo
and
Hong-Yan
Wang
 *
*
Key Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, 710119, China. E-mail: hongyan-wang@snnu.edu.cn
First published on 19th January 2023
Abstract
Modulation of electrolyte to tailor the activity and selectivity for photoelectrochemical (PEC) water splitting has been long neglected. Herein, we boosted H2O2 accumulation on a BiVO4 photoanode via PEC water oxidation in a series of carbon quantum dots (CQDs) aqueous solutions with aliphatic amino acids as precursors. Mott–Schottky measurements demonstrated CQDs solution can tune catalytic voltage location of the BiVO4 substrate. Open circuit voltage decay (OCVD) demonstrated that the system can achieve efficient charge separation owing to the fabricated dynamic heterojunction between CQDs particles and BiVO4 substrate. Owing to the energy gradient, water oxidation may occur on the more hydrophilic CQDs surface, which regulated H2O2 generation mediated by the formation of hydroxyl radicals (OH˙) from water, as evidenced by electron paramagnetic resonance (EPR). Assisted by a Clark electrode and linear sweep voltammetry, we evidenced that CQDs can efficiently retard H2O2 decomposition. Overall, H2O2 production was performed with a rate averaging 0.33 μmol min−1 cm−2 at 1.23 V with a Faraday efficiency (FE) of 93.5%, in which O2 evolution was nearly completely suppressed. By contrast, the commonly used HCO3− electrolyte only afforded an H2O2 evolution rate of 0.032 μmol min−1 cm−2, corresponding to FE of 18.2%. Here, we executed PEC water oxidative H2O2 accumulation in CQDs solution directly, and the innovation can hopefully draw more attention to catalytic media for improving PEC performance.
1. Introduction
Hydrogen peroxide (H2O2) production on an anode, associated with H2 generation on a cathode, adds much more value for photoelectrochemical (PEC) water splitting.1–4 Until now, many efforts have been devoted to water oxidative H2O2 production,5–16 but practical and effective protocols are still difficult to come by. This dominantly roots from the challenging issue of suppressing the competitive O2 evolution, because the two-electron-mediated H2O2 generation (i.e., H2O2/H2O, E° = 1.76 V) is less thermodynamically favorable than the subsequent four-electron O2 evolution reaction (OER) (i.e., O2/H2O, E° = 1.23 V).3,8,17 Additionally, H2O2 disproportionation into O2 and H2O can be accelerated on the photoanode, which further increases the decomposition of H2O2.2,12,14,18 Therefore, to increase H2O2 accumulation, the desired system should both significantly promote H2O2 generation and effectively prevent it from serious decomposition.7,8 It has been identified that the traditional photoanode fabrication via nanostructuring,13 heteroatom doping,7,10 as well as heterojunction construction13 can furnish efficient H2O2 production from water, but serious degradation of H2O2 on the photoanode still unavoidably occurs. Recently, a SnO2−x overlayer coated BiVO4 photoanode achieved great success in water oxidative H2O2 generation, accompanied by the efficient inhibition of H2O2 decomposition, which endorses such a strategy.8As compared with photoanode fabrication, selection of an appropriate electrolyte is a much more convenient and effective way to try to improve PEC water splitting.3,4,19 This is because the electrolyte used in PEC and other electrochemistry (EC) systems plays a vital role in charge migration and mass diffusion, and thus has a remarkable influence on catalytic behavior.20–22 However, until now, little attention has been paid to tailoring PEC performance through the modulation of electrolyte medium. Recently, we demonstrated that a carbon quantum dots (CQDs) aqueous solution generated from EDTA·2Na can act as an electrolyte for PEC water oxidative O2 production on α-Fe2O3 photoanodes with robust efficiency and durability.23 It was found that the negatively charged CQDs particles and α-Fe2O3 substrate can form a dynamic heterojunction to trigger efficient charge separation during the PEC process. Moreover, the CQDs solution can regulate and stabilize the water splitting product by tuning the catalytic voltage location via the interaction between the interface of the reaction medium and catalytic material. Therefore, we proposed that operating PEC water splitting in an appropriate CQDs solution in combination with an appropriate photoanode can direct water oxidation into H2O2 production rather than the competitive 4-electron transfer for O2 evolution. More importantly, the degradation of H2O2 on the photoanode can hopefully also be prevented. These features are considered to regulate the PEC water oxidation selectivity and improve H2O2 accumulation by guiding the pronounced synergy of electrolyte and photocatalyst. Here, we investigated a series of new CQDs aqueous solutions with linear aliphatic amino acids as precursors, in which PEC water oxidation on a BiVO4 photoanode exhibited far superior ability for H2O2 accumulation compared with the commonly used HCO3− electrolyte. The related mechanism was also studied in detail.
2. Results and discussion
2.1 Fabrication and characterization of CQDs
The preparation of CQDs was carried out via hydrothermal treatment, in which 3-aminopropanoic acid, 4-aminobutanoic acid and 6-aminocaproic acid were selected as precursors. After purification and freeze-drying, three CQDs samples were isolated, designated 3-CQDs, 4-CQDs and 6-CQDs, respectively. High-resolution transmission electron microscopy (HR-TEM) indicates the fabricated CQDs consisted of spherical nanoparticles with uniform diameters of 3–6 nm. Both graphitic and amorphous forms of carbon were detected, typically with lattice fringes in the graphitic regions corresponding to (100) interlayer spacing of 2.1 Å in CQDs (Fig. 1a).24 In powder X-ray diffraction (XRD) spectra, each sample showed a broad peak centered around 24.9°, 23.7° and 23.4°, respectively, which was related to the respective lattice spacing of 3.5 Å in 3-CQDs, 3.7 Å in 4-CQDs and 3.8 Å in 6-CQDs, consistent with the (200) reflection of disordered graphitic-like material (Fig. 1b).25–27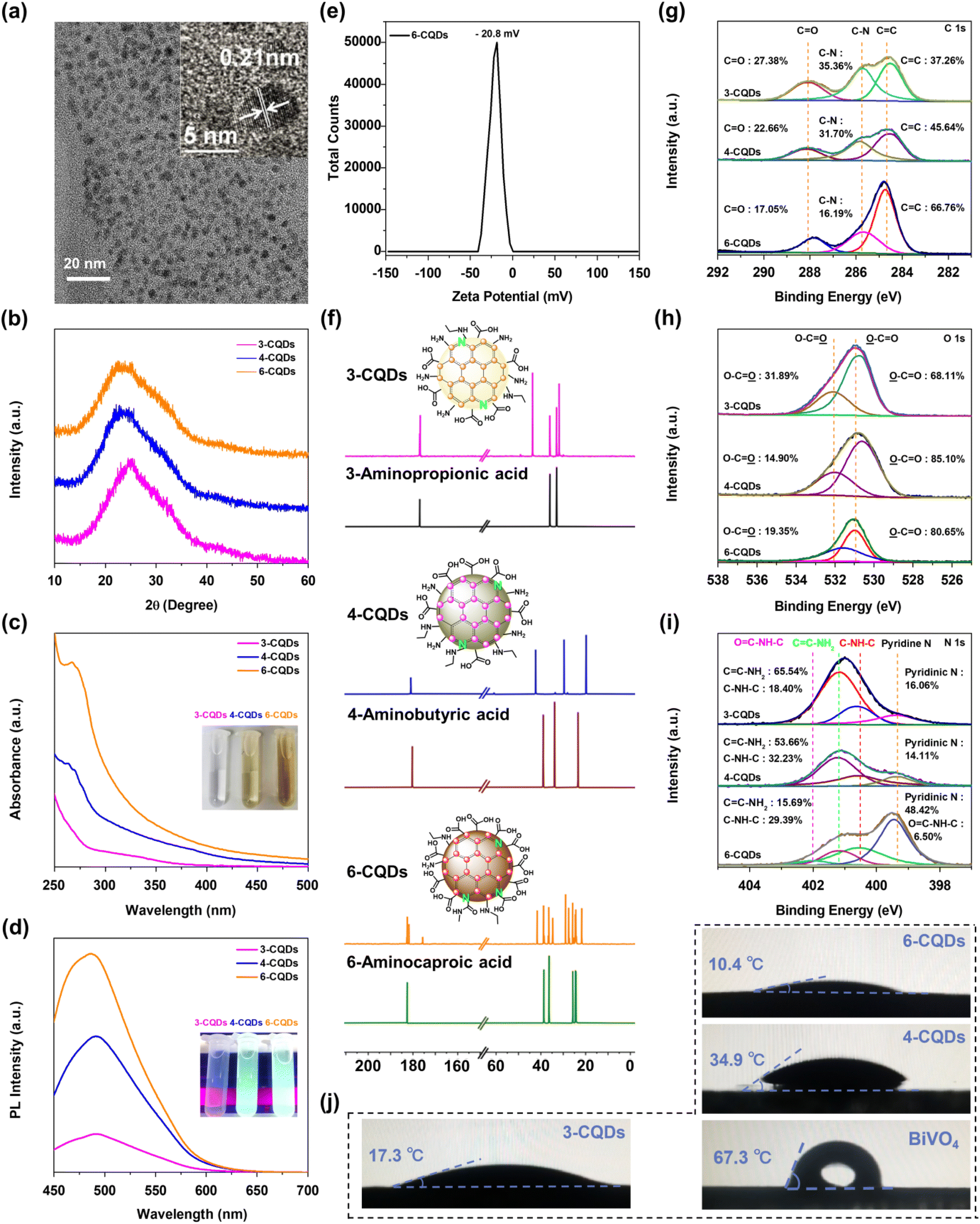 | ||
| Fig. 1 (a) HR-TEM image of 6-CQDs, and inset showing lattice spacing of 0.21 nm, which corresponds to the (100) plane of graphitic structure; (b) the XRD spectra of CQDs samples; (c) UV-vis spectra and (d) photoluminescence spectra of CQDs samples in water; (e) the zeta potential of 6-CQDs; (f) the 13C NMR spectra of CQDs and their corresponding parent precursors in D2O, with insets showing the proposed surface components of each CQDs compound; high-resolution C 1s (g), O 1s (h) and N 1s (i) XPS spectra of CQDs samples; (j) contact angles of CQDs samples and BiVO4 substrate in water droplets experiments. | ||
When the CQDs samples were dispersed in water, transparent solutions were obtained, indicating the great hydrophilic nature of CQDs. Typically, the aqueous solution of 6-CQDs displayed a characteristic absorption band ranging from 300 to 400 nm ascribed to π→π* (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) conjugated structure transition, followed by an absorption tail extending to the visible light region (Fig. 1c).27,28 Impressively, 6-CQDs exhibited the widest absorption band in the visible light region as compared with 3-CQDs and 4-CQDs. The fluorescence emission profile is further displayed in Fig. S1,† which exhibits typical excitation-wavelength-dependent behavior for CQDs.26,29 Along with the excitation wavelength moving from 350 to 410 nm, the maximum of 3-CQDs shifted from λ = 402 to 478 nm. With the extension of the carbon chain in the corresponding parent precursor, CQDs displayed gradually stronger luminescence intensity, accompanied by a red-shift of the emission spectra. Typically, under identical excitation conditions, 6-CQDs exhibited the strongest luminescence (Fig. 1d). In combination with UV-vis absorption, this implied that 6-CQDs can hold the greatest charge carrier concentration, which may indicate great influence on visible light-driven reaction activity.30–32 Additionally, the zeta potential of the samples was examined, showing negative values (Fig. 1e and S2†), which implied the negatively-charged CQDs particles can move directionally to the hole-accumulated photoanode plate under electrostatic force during the PEC process.23,33
C) conjugated structure transition, followed by an absorption tail extending to the visible light region (Fig. 1c).27,28 Impressively, 6-CQDs exhibited the widest absorption band in the visible light region as compared with 3-CQDs and 4-CQDs. The fluorescence emission profile is further displayed in Fig. S1,† which exhibits typical excitation-wavelength-dependent behavior for CQDs.26,29 Along with the excitation wavelength moving from 350 to 410 nm, the maximum of 3-CQDs shifted from λ = 402 to 478 nm. With the extension of the carbon chain in the corresponding parent precursor, CQDs displayed gradually stronger luminescence intensity, accompanied by a red-shift of the emission spectra. Typically, under identical excitation conditions, 6-CQDs exhibited the strongest luminescence (Fig. 1d). In combination with UV-vis absorption, this implied that 6-CQDs can hold the greatest charge carrier concentration, which may indicate great influence on visible light-driven reaction activity.30–32 Additionally, the zeta potential of the samples was examined, showing negative values (Fig. 1e and S2†), which implied the negatively-charged CQDs particles can move directionally to the hole-accumulated photoanode plate under electrostatic force during the PEC process.23,33
It was noted that CQDs samples were difficult to keep totally dry, which implied their surfaces were likely capped with abundant hydrophilic groups.34 In their infrared (IR) spectra, 6-CQDs exhibited two strong features at ∼1550 and ∼1411 cm−1, corresponding to anti-symmetric and symmetric stretches of CO groups, with stretching vibrations of O–H at ∼3128 and ∼2938 cm−1 (Fig. S3a†).26,27 Another clear signal at ∼3689 cm−1 belonged to the stretching vibrations for the N–H bond in –NH–C or –CONH–.35 Similar signals were observed in 4-CQDs (Fig. S3b†), although with slight shift. The IR spectrum of 3-CQDs displayed a stretching vibration band for O–H at ∼3028 cm−1, accompanied by anti-symmetric and symmetric stretches of CO centered at ∼1590 cm−1 (Fig. S3c†).26 Significantly, the N–H signal in 3-CQDs was much broader than that in 4-CQDs and 6-CQDs, implying NH2 groups were incorporated. The 13C NMR spectra of CQDs and their corresponding precursors are shown in Fig. 1f. Clearly, the spectra profiles are different in each group, suggesting the successful generation of CQDs. In 13C NMR, 6-CQDs exhibited a variety of carbon environments, including –COOH and –CONHC groups, and 3-CQDs displayed two signals close together in the –COOH range. In the same range, 4-CQDs showed a single signal ascribed to –COOH groups. X-ray photoelectron spectroscopy (XPS) in Fig. 1g depicts the high-resolution C 1s spectra, which are deconvoluted into three signals. The lowest energy contribution peaks centered at 285.8 eV and 288.1 eV represented the presence of C–N and CO groups, respectively. The signals at 284.7 eV belonged to CC environment, corresponding to sp2-hybridized graphitic carbon atoms.36–38 Clearly, 6-CQDs can incorporate more conjugated CC groups than 3-CQDs and 4-CQDs. It is accepted that π-conjugated structures can show excellent hole-transporting properties, which implies that 6-CQDs enables the best hole migration to be achieved in the system.39 In the O 1s spectra, the peaks at 531.0 eV and 531.7 eV belonged to OC and O–C species, respectively (Fig. 1h).31,40,41 In the high-resolution N 1s spectra, the peak at 399.3 eV corresponded to pyridine-like nitrogen, indicating the doped N was exposed on the surface of CQDs.42 The signal situated at binding energy 400.5 eV corresponded to C–NH–C, and that at 401.2 eV demonstrated the presence of CC–NH2 (Fig. 1i).29,40 Typically, a band centered at 402 eV was found in the spectrum of 6-CQDs, which was correlated with the –CONHC group.43 As a comparison, the proportion of CC–NH2 bonding was decreased in the order from 3-CQDs, 4-CQDs to 6-CQDs, and 4-CQDs incorporated a slightly higher proportion of the hydrophobic C–NH–C component. It should be noted that 3-CQDs and 6-CQDs presented a smaller contact angle than 4-CQDs in the water droplets experiments exhibited in Fig. 1j, featuring a better hydrophilic character of the surfaces, which implied that 6-CQDs and 3-CQDs expose more abundant –COOH or –NH2 groups than 4-CQDs. In line with our combined characterization results, the surface components of each material are proposed in the inset of Fig. 1i. As exhibited in Fig. 1j, all three CQDs samples indicated much better hydrophilic properties than the BiVO4 substrate. It has been perceived that a more hydrophilic surface facilitates confinement of water molecules, and further exerts a serious influence on the generation and conversion of the related intermediates, which thus can tune the PEC activity and even selectivity.44,45
2.2 PEC water oxidative activity and selectivity for H2O2 production on BiVO4 photoanode in CQDs solution
The performance of PEC H2O2 generation on BiVO4 photoanode in CQDs solution was examined with linear sweep voltammetry (LSV) under the irradiation of commercial LEDs (λ = 420 nm). As expected, the photo-electronic response in each CQDs solution greatly surpassed that in the aqueous solution containing the corresponding parent compounds, and showed a distinct cathodic shift of onset potential (Fig. 2a and S4). Furthermore, KHCO3 aqueous solution with identical electro-conductibility to CQDs was employed for reference. It is generally recognized that concentrated HCO3− electrolyte is so far the most efficient catalytic medium for the direct production of H2O2 from water oxidation,8,11–14,16,19 although recent studies have pointed out that the HCO3− anion can react with H2O2 to prompt its consumption.2,46,47Fig. 2a depicts the LSV curves of the BiVO4 photoanode in three CQDs solutions and in KHCO3 aqueous solution. In the darkness, the current ascribed to the EC process for water oxidation was negligible. Under irradiation, 3-CQDs presented 0.91 mA cm−2 photocurrent density at 1.23 V (vs. reversible hydrogen electrode, RHE), which was slightly decreased to 0.64 mA cm−2 when using 4-CQDs instead. 6-CQDs can achieve a more robust activity, with 1.14 mA cm−2 at 1.23 V, and the photocurrent displayed a sustained increase over the entire potential range. HCO3− electrolyte initiated a photocurrent density of 0.49 mA cm−2 at 1.23 V, which was far less than the CQDs solution. We noted that CQDs can sustain a stable photocurrent over long periods of time, suggesting robust stability and durability in the PEC system (Fig. S5†).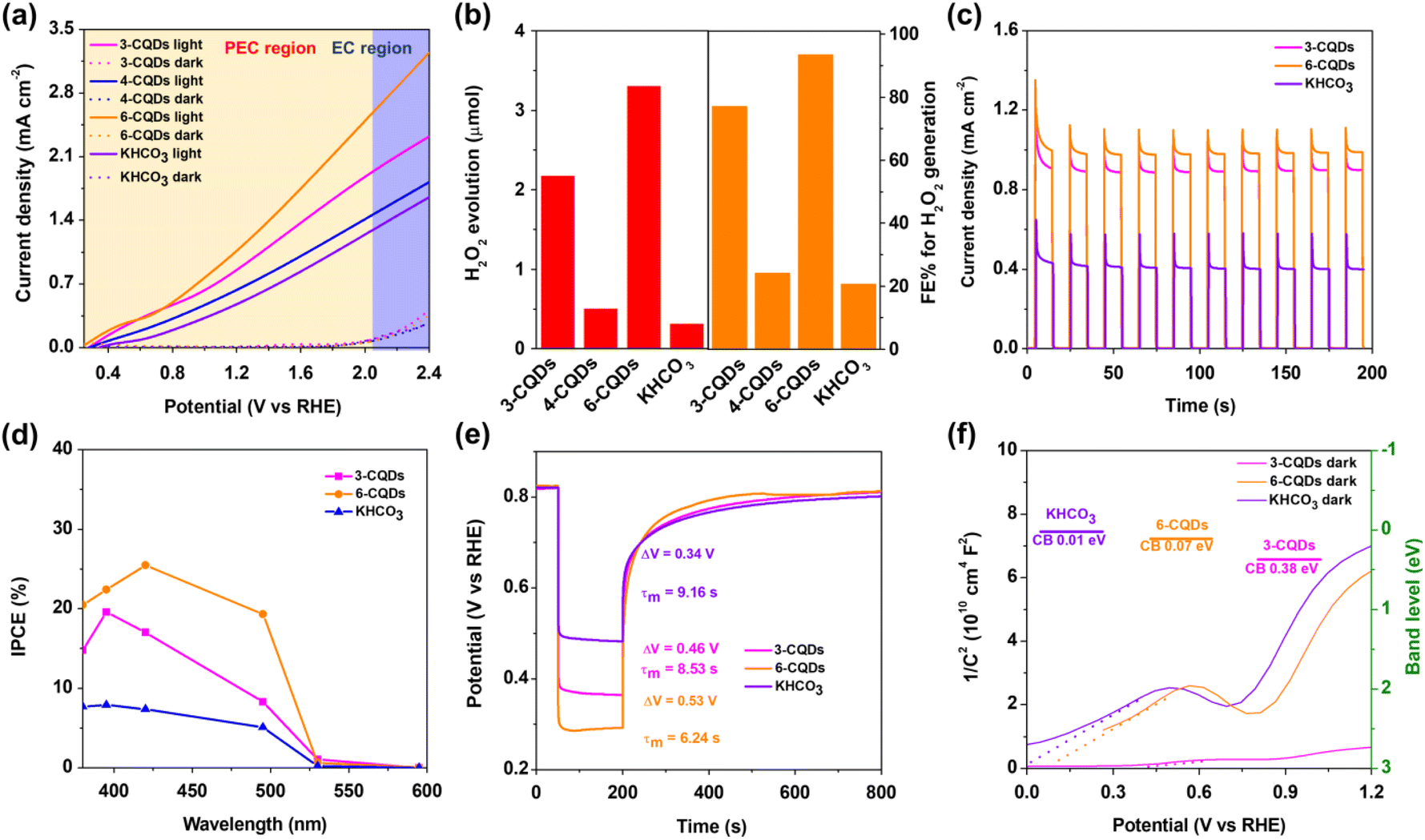 | ||
| Fig. 2 (a) The LSV of BiVO4 photoanode in CQDs aqueous solution and KHCO3 electrolyte in the dark or under irradiation of 420 nm LED lamp (the input optical power is 8.37 mW cm−2); (b) the yield of H2O2 and FE% for H2O2 generation; (c) chopped light irradiation curves with 420 nm LED of BiVO4 photoanode in CQDs aqueous solution and KHCO3 electrolyte; (d) IPCE of BiVO4 photoanode in 3-CQDs, 6-CQDs and KHCO3 aqueous solution under different excitation wavelengths; (e) OCVD plots of BiVO4 photoanode in 3-CQDs, 6-CQDs and KHCO3 aqueous solution in the dark or under irradiation of 420 nm LED lamp from the electrolyte–electrode side (ΔV is the difference in voltage between dark and illumination conditions); (f) Mott–Schottky plots of BiVO4 photoanode in 3-CQDs, 6-CQDs and KHCO3 aqueous solution under irradiation. | ||
Since the competitive 4-electron transfer water oxidative O2 evolution always occurs, the ability of CQDs solution for H2O2 generation on BiVO4 photoanode was examined. As prolonged photo-electrolysis could inevitably lead to the decomposition of H2O2, the PEC system was placed in baths of ice (below 5 °C) with continuous illumination for 600 s at 1.23 V. The identification and quantification of H2O2 were based on the N,N-diethyl-1,4-phenylene-diamine (DPD) method (Fig. S6 and S7†).8 The amounts of O2 were detected and evaluated by gas chromatography (GC). The results indicated that H2O2 generation in 3-CQDs solution was dominant, showing a total amount of 2.18 μmol. 4-CQDs solution was active for both O2 and H2O2 generation, in which the evolution of H2O2 was evaluated as 0.51 μmol, accompanied by O2 production of 0.06 μmol (Fig. 2b). However, since 4-CQDs can react with DPD indicator directly for an unclear reason (Fig. S7b†), the following research was concentrated on 3-CQDs and 6-CQDs. Importantly, 6-CQDs performed better for H2O2 production, and the O2 evolution was nearly completely suppressed. This system produced 3.31 μmol H2O2, at a rate of up to 0.33 μmol min−1 cm−2. Increasing the bias to 1.5 V, the rate could be improved to 0.42 μmol min−1 cm−2 (Fig. S8†). Typically, 6-CQDs afforded a Faraday efficiency (FE) of 93.5% for H2O2 production at 1.23 V. In sharp contrast, the H2O2 evolution rate in HCO3− electrolyte was only 0.032 μmol min−1 cm−2, corresponding to an FE of 18.2% (Fig. 2b).
2.3 Mechanistic study for PEC water oxidative H2O2 production in CQDs solution
Fig. 2c illustrates the transient photocurrent responses versus time with chopped light irradiation, which brings out the reproducible photocurrent with almost identical value for each on/off cycles, highlighting the satisfied stability for PEC performance. Moreover, the photo-response was very sensitive to the light, regularly increasing or falling off to the baseline as the light was switched on or off, which suggests the facile charge transport.29,37Incident photon-to-current conversion efficiency (IPCE) profiles were recorded with the bias at 1.23 V under continuous illumination. In both CQDs and KHCO3 solutions, the photo-response extended to 530 nm, corresponding to a band gap for BiVO4 of 2.35 eV.48 Significantly, BiVO4 in 6-CQDs solution yielded a much higher value at each wavelength (Fig. 2d). Based on a broad arsenal of related reports, the improvement in IPCE originates from the efficient separation of photo-generated electron–hole pairs.48,49 Therefore, the photovoltage–time (V–t) spectrum was recorded based on open circuit voltage decay (OCVD),50 from which photovoltage (ΔV) was extracted.51 As shown in Fig. 2e, BiVO4 presented ΔV of 0.34 V in KHCO3 solution, which was increased to 0.46 V in 3-CQDs and 0.53 V in 6-CQDs solutions, respectively. This means that CQDs solutions enable better charge separation.51 The average charge decay lifetime of the V–t profile provides the carrier lifetime in the structures of semiconductor devices, here demonstrating that the harmonic mean of lifetime τm for BiVO4 was 9.16 s in KHCO3, 8.53 s in 3-CQDs and 6.24 s in 6-CQDs, respectively. Clearly, CQDs solution has the better ability to deactivate the surface trap state,52 suggesting that the photo-generated charge carriers can survive to participate in the water splitting reaction instead being deactivated by surface traps, and thus PEC performance was dramatically enhanced.51
Furthermore, the band location of the photoanode in CQDs and KHCO3 was determined based on Mott–Schottky plots (Fig. 2f). Notably, the flat band potential (Vfb) of BiVO4 was pinned under dark and illumination conditions.8 Under irradiation, BiVO4 exhibited positive slopes in Mott–Schottky plots, as for n-type semiconductor.53,54 The plots indicated that Vfb of BiVO4 in HCO3− electrolyte was 0.01 V, which was positively shifted to 0.38 V and 0.07 V in 3-CQDs and 6-CQDs solution, respectively. Since an n-type semiconductor has negligible gap between Vfb and the bottom edge of the conduction band (CB),55 it is evident that CQDs solution can cause the CB level of BiVO4 to shift downward.56–58 As characterized in IPCE, the BiVO4 photoanode showed an energy gap (Eg) of 2.35 eV. Accordingly, the valence band (VB) of BiVO4 in 3-CQDs and 6-CQDs solution was located at 2.73 and 2.42 eV, respectively. Also from CV measurement, the HOMO and LUMO levels for 3-CQDs were evaluated as 2.15 eV and 0.23 eV, respectively (Fig. S9a†).59 Switching into 6-CQDs, the levels were 1.97 eV and 0.04 eV, respectively (Fig. S9b†). In this sense, the fabricated band edge alignment between BiVO4 and CQDs allows establishment of a type II heterojunction configuration, in which the electrons can migrate from the LUMO band of CQDs to the CB of BiVO4, and holes can move from the VB of BiVO4 to the HOMO of CQDs (Fig. 3a).59–61 As a result, the charge separation can be efficiently improved.
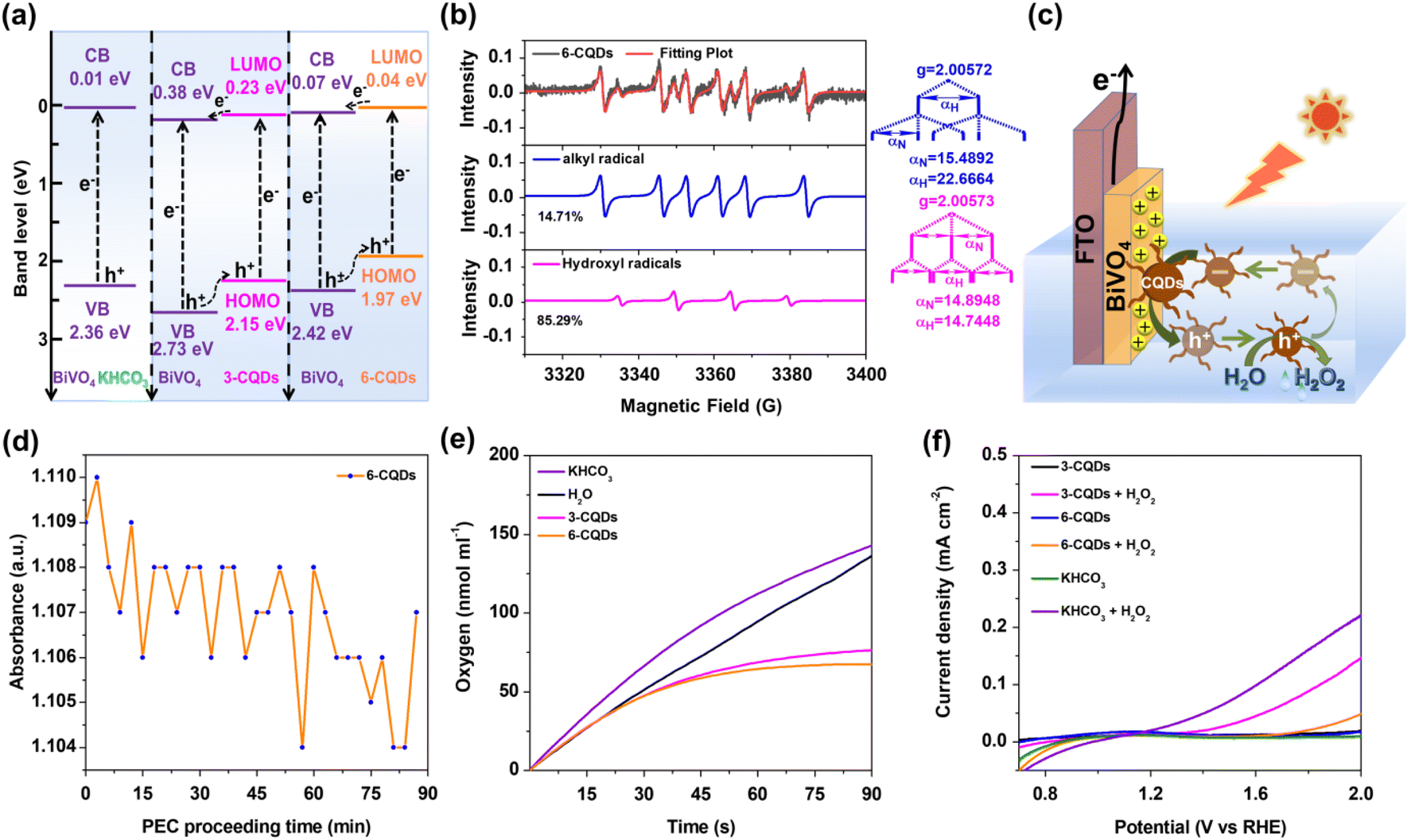 | ||
Fig. 3 (a) The determined energy levels of BiVO4 photoanode in KHCO3 electrolyte and CQDs aqueous solutions; (b) experimental EPR spectrum (black) and simulated spectra (blue and pink) of the system after PEC process on BiVO4 photoanode in 6-CQDs solution at 1.23 V for 10 min; (c) the proposed process for water oxidative H2O2 production on BiVO4 photoanode in CQDs solution; (d) in situ UV-vis absorption at 266 nm for the system with PEC water oxidation on BiVO4 photoanode in 6-CQDs solution; (e) O2 evolution in a mixture of H2O2 and CQDs or KHCO3 aqueous solution (V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 1:1) determined by Clark electrode under 420 nm irradiation; (f) LSV of BiVO4 photoanode in a mixture of H2O2 and CQDs solution (V:V = 7:1). :V = 1:1) determined by Clark electrode under 420 nm irradiation; (f) LSV of BiVO4 photoanode in a mixture of H2O2 and CQDs solution (V:V = 7:1). | ||
Further, electron paramagnetic resonance (EPR) measurements were used to monitor the potential radicals for H2O2 generation. With the help of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), EPR signals were captured from 6-CQDs solution under identical PEC conditions (Fig. 3b). Simulated by Simfonia software, a sextet featuring an approximate intensity ratio of 1:1:1:1:1:1 (αN = 15.4 G, αH = 22.7 G) was identified, suggesting the formation of DMPO-C adduct.62 This agrees with the above proposal that photo-generated holes can migrate to CQDs and then be exhausted, yielding carbon radicals. The proportion of DMPO-C species is 85.3%, and the other 14.7% response comprises a quartet of peaks with an approximate intensity ratio of 1:2:2:1 (αN = 14.9 G, αH = 14.7 G), which corresponds to a DMPO-OH adduct.8,63 That CQDs can keep integrity after a long period of photoelectrolysis demonstrated the high stability and durability of CQDs during the PEC process (Fig. S5 and S10†). More importantly, combined with the fact that the ˙OH signal was correlated with the formation of H2O2,15,17,64 this implied that in the CQDs system, water oxidation may occur on the CQDs surface and the formed carbon radical can be recovered to CQDs after oxidizing water. A similar EPR response was measured when 3-CQDs were used (Fig. S11†), but the intensity of ˙OH was weaker, with the proportion decreased to 10.4%. For the KHCO3 system, no clear peak was assigned to ˙OH. This trend is in line with the activity for H2O2 production, with the order 6-CQDs > 3-CQDs > KHCO3. Since water oxidation occurred on the BiVO4 photoanode in KHCO3 electrolyte, we proposed that, besides much better charge separation, the higher activity for CQDs is due to the much more hydrophilic surface compared with the BiVO4 substrate. This is because more H2O molecules can be confined, thus water oxidation was facilitated, leading to an improved performance for H2O2 generation.44,45
Based on these results, we propose a mechanism for PEC water oxidative H2O2 production in CQDs solution (Fig. 3c). As a typical n-type semiconductor, photo-generated electrons on BiVO4 are repelled from the surface into the bulk solid and then diffuse to the counter-electrode through the external circuit for hydrogen production. Holes are left and accumulated on the BiVO4 photoanode, leading to a positively-charged surface,65 which can further attract negatively-charged CQDs particles. The holes migrate along the energy gradient to the CQDs, which are oxidized by the holes and repulsed from the BiVO4 substrate into solution for water oxidation, and finally recover to their original states and stay in the solution. Owing to such a cycle going back and forth, a dynamic balance of CQDs adsorbing onto or desorbing off the BiVO4 surface can be established. In order to confirm the proposal, an in situ UV-vis spectroelectrochemical investigation was conducted, as shown in Fig. 3d and S12,† in which the absorption of CQDs solution varied periodically, manifesting the CQDs can dynamically move out of and into the solution.
Moreover, the decomposition of H2O2 in the presence of CQDs or KHCO3 was probed with a Clark electrode, which is a generally used method to quantify O2 evolution of a system in real time.66,67 This indicated that HCO3− unavoidably accelerated degradation of H2O2 into O2 either in the dark or under irradiation (Fig. 3e and S13†). This is consistent with previous observations in the literature that one of the difficulties in H2O2 accumulation results from the consumption of H2O2 by reacting with HCO3− anions.2,46,47 In stark contrast, O2 evolution in 3-CQDs or 6-CQDs solution was remarkably suppressed under the same conditions, suggesting CQDs can retard the H2O2 decomposition. Additionally, Fig. 3f records the LSV curves of BiVO4 with addition of H2O2 in different solutions. Compared with HCO3− electrolyte, in CQDs solutions the currents that arose from H2O2 decomposition were noticeably decreased, further evidencing the sluggish degradation of H2O2. Clearly, CQDs can set up a key counterbalance between boosting water oxidation into H2O2 generation and retarding H2O2 decomposition, which benefits the H2O2 accumulation.
3. Conclusions
In summary, we achieved H2O2 accumulation on a BiVO4 photoanode via PEC water oxidation in a series of CQDs aqueous solutions. Our observations demonstrated that CQDs can feature as both electrolyte and semiconductor, with the following advantages: (1) band edge configuration between CQDs and BiVO4 photoanode allows fabrication of a heterojunction, which enables efficient charge separation; (2) an improved hydrophilic surface provided by CQDs favors confinement of H2O molecules, which improves PEC water oxidative selectivity to produce H2O2; (3) as compared with HCO3−, CQDs can prevent H2O2 from serious decomposition. As a result, H2O2 production in CQDs solution was performed with an average rate of 0.33 μmol min−1 cm−2 at 1.23 V with a Faraday efficiency of 93.5%, and the competitive O2 evolution was nearly completely suppressed. Such performance greatly surpassed that which occurred in the commonly used HCO3− electrolyte. As a non-parallel example to execute PEC water oxidative H2O2 generation in CQDs aqueous solution directly without any other traditional supporting electrolyte, our present work highlights a more striking and facile strategy to tailor the selectivity and activity for PEC water splitting. Subsequently, we have expanded our research to other CQDs systems, aiming for efficient hydrogen production or oxygen reduction based on PEC operation, which work is on-going.Author contributions
Na Wang: data curation, validation, visualization, formal analysis, writing; Yong-Shuai Zhang: visualization, data curation, formal analysis; Dong-Dong Wei: validation, writing and correcting; Hui-Min Duan: validation, writing and correcting; Liu-Meng Mo: validation, writing and correcting; Hong-Yan Wang: supervision, visualization, formal analysis, conceptualization, writing, editing, reviewing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support from National Natural Science Foundation of China (22072082, 21402113), National Natural Science Foundation of Shaanxi Province (2019JQ-175), the Fundamental Research Funds for the Central Universities (GK202103026).References
- S. C. Perry, D. Pangotra, L. Vieira, L. L. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- C. R. Dong, Y. L. Yang, X. M. Hu, Y. J. Cho, G. Y. Jang, Y. H. Ao, L. Y. Wang, J. Y. Shen, J. H. Park and K. Zhang, Nat. Commun., 2022, 13, 4982 CrossRef CAS PubMed.
- Y. D. Xue, Y. T. Wang, Z. H. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS PubMed.
- J. L. Liu, Y. S. Zou, B. J. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- Y. C. Miao and M. F. Shao, Chin. J. Catal., 2022, 43, 595–610 CrossRef CAS.
- T. H. Jeon, B. Kim, C. Kim, C. Xia, H. T. Wang, P. J. J. Alvarez and W. Y. Choi, Energy Environ. Sci., 2021, 14, 3110–3119 RSC.
- T. H. Jeon, H. Kim, H. I. Kim and W. Y. Choi, Energy Environ. Sci., 2020, 13, 1730–1742 RSC.
- K. Zhang, J. L. Liu, L. Y. Wang, B. J. Jin, X. F. Yang, S. L. Zhang and J. H. Park, J. Am. Chem. Soc., 2020, 142, 8641–8648 CrossRef CAS PubMed.
- Y. J. Liu, Y. Z. Han, Z. Y. Zhang, W. Zhang, W. Z. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- J. H. Baek, T. M. Gill, H. Abroshan, S. Park, X. J. Shi, J. Nørskov, H. S. Jung, S. Siahrostami and X. L. Zheng, ACS Energy Lett., 2019, 4, 720–728 CrossRef CAS.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2019, 9, 1801294 CrossRef.
- Y. Miyase, S. Takasugi, S. Iguchi, Y. Miseki, T. Gunji, K. Sasaki, E. Fujita and K. Sayama, Sustainable Energy Fuels, 2018, 2, 1621–1629 RSC.
- X. J. Shi, L. L. Cai, I. Y. Choi, M. Ma, K. Zhang, J. H. Zhao, J. K. Kim, J. K. Kim, X. L. Zheng and J. H. Park, J. Mater. Chem. A, 2018, 6, 19542–19546 RSC.
- K. Fuku, Y. Miyase, Y. Miseki, T. Funaki, T. Gunji and K. Sayama, Chem.–Asian J., 2017, 12, 1111–1119 CrossRef CAS PubMed.
- X. J. Shi, S. Siahrostami, G. L. Li, Y. R. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. L. Zheng and J. K. Norskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- K. Fuku and K. Sayama, Chem. Commun., 2016, 52, 5406–5409 RSC.
- S. Siahrostami, G. L. Li, V. Viswanathan and J. K. Norskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Energy Environ. Sci., 2012, 5, 9496 RSC.
- D. Pangotra, L. I. Csepei, A. Roth, C. Ponce de León, V. Sieber and L. Vieira, Appl. Catal., B, 2022, 303, 120848 CrossRef CAS.
- P. T. Xu, T. J. Milstein and T. E. Mallouk, ACS Appl. Mater. Interfaces, 2016, 8, 11539–11547 CrossRef CAS PubMed.
- S. E. Koops, B. C. O'Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- H. Kusama, H. Orita and H. Sugihara, Langmuir, 2008, 24, 4411–4419 CrossRef CAS PubMed.
- H. Y. Wang, R. Hu, N. Wang, G. L. Hu, K. Wang, W. H. Xie and R. Cao, Appl. Catal., B, 2021, 296, 120378 CrossRef CAS.
- J. Y. Li, X. R. Yun, Z. L. Hu, L. J. Xi, N. Li, H. Tang, P. C. Lu and Y. R. Zhu, J. Mater. Chem. A, 2019, 7, 26311–26325 RSC.
- Y. M. Hu, Z. L. Zhao, R. Ahmad, M. Harb, L. Cavallo, L. M. Azofra, S. P. Jiang and X. Y. Zhang, J. Mater. Chem. A, 2022, 10, 12713 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- X. M. Li, S. L. Zhang, S. A. Kulinich, Y. L. Liu and H. B. Zeng, Sci. Rep., 2014, 4, 4976 CrossRef CAS.
- S. J. Zhu, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- X. Chen, W. W. Zhang, L. X. Zhang, L. P. Feng, C. X. Zhang, J. Jiang, T. J. Yan and H. Wang, J. Mater. Chem. A, 2020, 8, 18816–18825 RSC.
- J. C. Wang, T. S. Zhou, Y. Zhang, L. Li, C. H. Zhou, J. Bai, J. H. Li, H. Zhu and B. X. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 45392–45402 CrossRef CAS PubMed.
- B. Q. Wang, Z. R. Deng, X. Z. Fu and Z. H. Li, J. Mater. Chem. A, 2018, 6, 19735–19742 RSC.
- J. Di, J. X. Xia, M. X. Ji, B. Wang, S. Yin, Q. Zhang, Z. G. Chen and H. M. Li, ACS Appl. Mater. Interfaces, 2015, 7, 20111–20123 CrossRef CAS PubMed.
- S. Wang, L. P. Li, Z. H. Zhu, M. L. Zhao, L. M. Zhang, N. N. Zhang, Q. N. Wu, X. Y. Wang and G. S. Li, Small, 2019, 15, 1804515 CrossRef PubMed.
- R. Miao, S. F. Zhang, J. F. Liu and Y. Fang, Chem. Mater., 2017, 29, 5957–5964 CrossRef CAS.
- Y. X. Li, Y. Yuan, X. Liang and L. S. Zhao, ACS Appl. Nano Mater., 2022, 5, 14507–14519 CrossRef CAS.
- H. J. Yu, Y. F. Zhao, C. Zhou, L. Shang, Y. Peng, Y. H. Cao, L. Z. Wu, C. H. Tung and T. R. Zhang, J. Mater. Chem. A, 2014, 2, 3344 RSC.
- Y. Q. Dong, H. C. Pang, H. B. Yang, C. X. Guo, J. W. Shao, Y. W. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- Z. Iwan, U. M. Javier, G. Paul, A. Juan, G. Giulia, M. O. Agustín, O. Enrique, M. Nazario and K. N. Mohammad, Adv. Energy Mater., 2017, 7, 1601674 CrossRef.
- H. J. Jian, R. S. Wu, T. Y. Lin, Y. J. Li, H. J. Lin, S. G. Harroun, J. Y. Lai and C. C. Huang, ACS Nano, 2017, 11, 6703–6716 CrossRef CAS PubMed.
- J. Q. Pan, Y. Z. Sheng, J. X. Zhang, J. M. Wei, P. Huang, X. Zhang and B. X. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 RSC.
- R. X. Hu, L. L. Li and W. J. Jin, Carbon, 2016, 111, 133–141 CrossRef.
- R. M. S. Sendão, D. M. A. Crista, A. C. P. Afonso, M. D. V. Martínez de Yuso, M. Algarra, J. C. G. Esteves da Silva and L. P. D. Silva, Phys. Chem. Chem. Phys., 2019, 21, 20919–20926 RSC.
- Y. Ding, S. Maitra, C. H. Wang, R. T. Zheng, M. Y. Zhang, T. Barakat, S. Roy, J. Liu, Y. Li, T. Hasan and B. L. Su, J. Energy Chem., 2022, 70, 236–247 CrossRef CAS.
- Y. L. Yang, S. C. Wang, Y. L. Jiao, Z. L. Wang, M. Xiao, A. J. Du, Y. L. Li, J. S. Wang and L. Z. Wang, Adv. Funct. Mater., 2018, 28, 1805698 CrossRef.
- X. J. Yang, Y. H. Duan, J. L. Wang, H. L. Wang, H. L. Liu and D. L. Sedlak, Environ. Sci. Technol. Lett., 2019, 6, 781–786 CrossRef CAS PubMed.
- D. E. Richardson, H. R. Yao, K. M. Frank and D. A. Bennett, J. Am. Chem. Soc., 2000, 122, 1729–1739 CrossRef CAS.
- Y. Pihosh, I. Turkevych, K. Mawatari, T. Asai, T. Hisatomi, J. Uemura, M. Tosa, K. Shimamura, J. Kubota, K. Domen and T. Kitamori, Small, 2014, 10, 3692–3699 CrossRef CAS PubMed.
- S. L. Xie, H. Su, W. J. Wei, M. Y. Li, Y. X. Tong and Z. W. Mao, J. Mater. Chem. A, 2014, 2, 16365–16368 RSC.
- B. Mukherjee, W. Wilson and V. R. Subramanian, Nanoscale, 2013, 5, 269–274 RSC.
- Q. S. Ruan, M. K. Bayazit, V. Kiran, J. J. Xie, Y. Wang and J. W. Tang, Chem. Commun., 2019, 55, 7191–7194 RSC.
- Y. C. Pu, G. M. Wang, K. D. Chang, Y. H. Ling, Y. K. Lin, B. C. Fitzmorris, C. M. Liu, X. H. Lu, Y. X. Tong, J. Z. Zhang, Y. J. Hsu and Y. Li, Nano Lett., 2013, 13, 3817–3823 CrossRef CAS PubMed.
- W. Q. Fang, Y. M. Lin, R. Z. Xv and L. Fu, ACS Appl. Energy Mater., 2022, 5, 11402–11412 CrossRef CAS.
- M. Y. Huang, J. C. Bian, W. Xiong, C. Huang and R. Q. Zhang, J. Mater. Chem. A, 2018, 6, 3602–3609 RSC.
- J. Premkumar, Chem. Mater., 2004, 16, 3980–3981 CrossRef CAS.
- R. Katoh, M. Kasuya, S. Kodate, A. Furube, N. Fuke and N. Koide, J. Phys. Chem. C, 2009, 113, 20738–20744 CrossRef CAS.
- D. F. Watson and G. J. Meyer, Coord. Chem. Rev., 2004, 248, 1391–1406 CrossRef CAS.
- D. Kuciauskas, J. E. Monat, R. Villahermosa, H. B. Gray, N. S. Lewis and J. K. McCusker, J. Phys. Chem. B, 2002, 106, 9347–9358 CrossRef CAS.
- C. Mahala, M. D. Sharma and M. Basu, Inorg. Chem., 2020, 59, 6988–6999 CrossRef CAS PubMed.
- H. Ma, Z. J. Wang, W. Zhao, H. Ren, H. Y. Zhu, Y. H. Chi and W. Y. Guo, J. Phys. Chem. Lett., 2022, 13, 8484–8494 CrossRef CAS PubMed.
- J. W. Xia, N. Karjule, L. Abisdris, M. Volokh and M. Shalom, Chem. Mater., 2020, 32, 5845–5853 CrossRef CAS.
- L. Chen, J. Duan, P. H. Du, W. L. Sun, B. Lai and W. Liu, Water Res., 2022, 221, 118747 CrossRef CAS PubMed.
- X. W. Wang, Z. Han, L. H. Yu, C. T. Liu, Y. F. Liu and G. Wu, ACS Sustainable Chem. Eng., 2018, 6, 14542–14553 CrossRef CAS.
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. - Eur. J., 2018, 24, 5016–5031 CrossRef CAS PubMed.
- C. A. Bignozzi, S. Caramori, V. Cristino, R. Argazzi, L. Meda and A. Tacca, Chem. Soc. Rev., 2012, 42, 2228–2246 RSC.
- J. W. Huang, Y. Zhang and Y. Ding, ACS Catal., 2017, 7, 1841–1845 CrossRef CAS.
- H. Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499–14502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09453c |
| This journal is © The Royal Society of Chemistry 2023 |