DOI:
10.1039/D2TA09613G
(Paper)
J. Mater. Chem. A, 2023,
11, 4170-4182
Ultrathin corrugated nanowire TiO2 as a versatile photoanode platform for boosting photoelectrochemical alcohol and water oxidation†
Received
10th December 2022
, Accepted 26th January 2023
First published on 7th February 2023
Abstract
Ultrathin semiconductor nanowires are promising alternatives to nanoparticle or bulk counterparts for the construction of photoanodes for solar energy conversion, because of their profuse surface reactive sites and excellent charge transport properties. Herein, well-aligned ultrathin corrugated nanowire TiO2 (UCW-TiO2) thin films with a diameter of 10 nm grown on fluorine-doped tin oxide (FTO) substrates by a unique monomicelle-directed assembly method are applied as a versatile photoanode platform. On this platform, photoanodes were used for direct photoelectrochemical (PEC) benzyl alcohol oxidation and can also be dye-sensitized for visible-light driven water oxidation. The conversion of benzyl alcohol (BA) over the pristine UCW-TiO2 photoanode is close to 100%, and the selectivity can reach 90.2% with a faradaic efficiency of 93% after 15 h. With surface anchoring of a suitable chromophore and oxygen evolution catalysts (OECs), the dye-sensitized UCW-TiO2 photoanode achieves 5-fold larger faradaic efficiency for water oxidation than that of a spherically structured TiO2 assembled photoanode. Electrochemical and spectroscopic measurements demonstrate that the ultrathin corrugated nanowire structured TiO2 accelerates electron transport kinetics and suppresses recombination of surface accumulated holes and electrons compared with spherically structured TiO2. Both BA oxidation and water oxidation studies show that the UCW-TiO2 is a versatile photoanode platform for a variety of highly efficient solar energy conversion applications.
Introduction
The technology of converting solar energy into chemical energy is considered to be an environmentally friendly and sustainable way.1,2 Titanium dioxide (TiO2) is considered one of the promising photocatalytic materials, mainly owing to its favorable physical characteristics, including appropriate band-edge positions, high photocorrosion resistance, natural abundance, nontoxicity, and low cost.3–6 TiO2 is often combined with other semiconductors or sensitized with dye molecules to form photoanode assemblies for photoelectrochemical (PEC) organic synthesis and water splitting reactions.7–10 PEC technology for the conversion of biomass alcohols to high-value chemicals, such as corresponding aldehydes is considered one of the most valuable pathways.11,12 The potential of alcohol oxidation reaction is lower than that of water oxidation and the alcohol oxidation reaction necessitates only two holes instead of four compared to water oxidation.13
A dye-sensitized photoelectrosynthesis cell (DSPEC) is particularly useful in that sensitization of wide band gap semiconductors such as TiO2 mitigates the disadvantage of UV-only absorption.14 In DSPEC applications, anatase TiO2 is used as a photoanode support.15 Conventional TiO2 thin films on conductive substrates (FTO) (denoted as NPTiO2 thin films) are prepared using spherical nanocrystalline particulate TiO2 through doctor blading or spin coating.4,16 Although those NPTiO2 thin films have a wide range of applications such as solar cells, organic synthesis and water splitting, those films composed of interconnected spherical TiO2 particles often suffer from charge recombination due to the trap states at grain boundaries. Since electrons randomly walk through the NPTiO2 network, the electron diffusion path becomes longer, which is detrimental to charge transfer and collection, leading to low electron conductivity and PEC performance,17 as shown in Scheme 1A. Molecular dye and oxygen evolution catalysts (OECs) are immobilized on the surface of NPTiO2 thin films to prepare a dye sensitized photoanode for PEC water oxidation. However, according to a previous paper, the dye and OECs co-anchored on the NPTiO2 thin film surface result in fast electron–hole recombination at the molecule/TiO2 interface and grain boundaries, thereby limiting the PEC performance.18–21
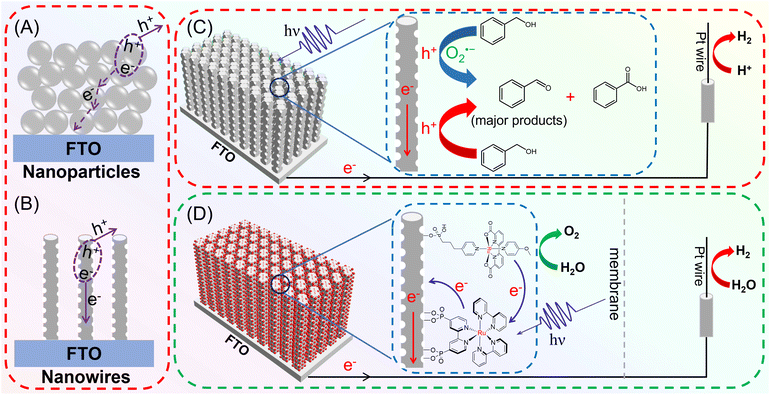 |
| | Scheme 1 Schematic illustrating two typical TiO2-based photoelectrodes and their corresponding charge separation/transport behaviors, (A) nanoparticles and (B) nanowires; UCW-TiO2 as the photoanode platform for efficient charge separation and collection in photoelectrochemical cells: (C) reaction mechanism for the PEC selective aerobic oxidation of benzyl alcohols from the pristine UCW-TiO2 photoanode and simultaneous H2 production; (D) Rubda and RuP sensitized UCW-TiO2 photoanode assembly and the proposed mechanism of electron transfer and charge collection for water oxidation. | |
Considering these drawbacks of NPTiO2 thin films, one-dimensional (1D) nanostructured films possess several advantages such as enlargement of the material surface area, more exposed surface contact between the semiconductor and the electrolyte, etc.22,23 1D nanostructures such as nanowires, nanorods, and nanofibers grown directly on FTO provide preferential electron transport that suppresses charge carrier recombination,24Scheme 1B. However, conventional TiO2 nanowire arrays synthesized by a hydrothermal method usually have a large diameter (∼70–140 nm),25 resulting in a relatively low surface area.
In this work, well-aligned ultrathin corrugated nanowires TiO2 (UCW-TiO2) has been synthesized by a unique monomicelle-directed assembly method. The UCW-TiO2 film (∼800 nm thickness) is composed of vertically aligned corrugated nanowires with an exceptionally small diameter of ∼10 nm and a uniformly adjacent distance of ∼10 nm on the FTO substrate. The highly ordered nanowire structure significantly reduces grain boundaries, which reduces the recombination of photogenerated electrons and holes.26 The UCW-TiO2 was used as a versatile photoanode platform for PEC benzyl alcohol and water oxidation, as shown in Scheme 1C and D. The UCW-TiO2 without any dye sensitization had been employed as a photoanode for aerobic benzyl alcohol (BA) oxidation and it showed excellent performance. BA was efficiently converted into high value-added benzaldehyde with high selectivity, because of the profuse surface reactive sites and excellent charge transport of UCW-TiO2. In order to expand the application of the UCW-TiO2 in PEC cells, with surface anchoring of a suitable chromophore and oxygen evolution catalysts (OEC), the dye-sensitized UCW-TiO2 photoanode was used for visible-light driven water oxidation. The ultrathin corrugated TiO2 nanowires are vertically well-aligned on the FTO substrate and the electrons can be rapidly and directly transferred to the counter electrode at a small electrochemical bias. This means that the ultrathin corrugated TiO2 nanowires simultaneously achieve efficient charge separation, transport, and accumulation of holes in the catalyst. Therefore, the dye-sensitized UCW-TiO2 photoanode achieves 5-fold larger faradaic efficiency for water oxidation than that of a spherically structured TiO2 assembled photoanode. This novel assembled photoanode can be efficiently utilized in a DSPEC for visible light driven water splitting.
Results and discussion
Synthesis and characterization
Fig. 1A shows a schematic illustration of the synthetic process of ultrathin corrugated nanowire TiO2 (UCW-TiO2) on FTO through a unique monomicelle-directed assembly method and calcination treatment (for more details, refer to the Experimental section). In the first step, amphiphilic Pluronic F127/TiO2 spherical composite monomicelles are first formed during the solvent evaporation process at 40 °C, with poly(propylene oxide) (PPO) segments as a core and titania-associated poly(ethylene oxide) (PEO) segments as a shell. In the next array growth process at 150 °C, the spherical monomicelles are linearly arranged vertical to the substrate due to the intermicellar repulsive forces, forming the micelle matrix on the substrate. The micelle matrix confines the growth of the nanowires, leading to the ultrathin diameter and the surface concave structure of the aligned nanowires.27 As shown in Fig. 1B–D, typical scanning electron microscopy (SEM) of the UCW-TiO2 grown on the FTO substrate was carried out. The top-view of SEM images (Fig. 1B) of the as-synthesized UCW-TiO2 film displayed uniform coverage of the FTO substrate by dense and vertically well-aligned TiO2 nanowires. The as-prepared UCW-TiO2 films consisting of vertically well-aligned nanowire TiO2 significantly reduce the reflection of incident light and improve the absorption of photons compared with the tilted orientation of the TiO2 nanorods.28 From the side-view of the SEM images (Fig. 1C and D), the length of the ultrathin corrugated nanowire TiO2 on FTO is ∼800 nm. The uniformly adjacent distance of UCW-TiO2 is ∼10 nm. The nanowire morphology of the TiO2 was further confirmed by transmission electron microscopy (TEM) (Fig. 1E and F). The enlarged TEM image of UCW-TiO2 shows ultrathin nanowires with a diameter of about 10 nm. The corrugated morphology is formed by bowl-like concave structures (∼5 nm diameter) closely arranged along the nanowire axis, Fig. 1F. It is found that the concave structure size can well be tuned by the reaction time. The concave structure size reduced with an increase of the reaction time.27 The TEM (Fig. S1†) images exhibit smooth ultrathin nanowire TiO2 arrays (∼800 nm thickness) without the concave region after the reaction for 48 h (UW-TiO2 arrays). Energy dispersive X-ray spectroscopy (EDS) mapping images (Fig. 1G) demonstrate that Ti and O elements are uniformly distributed within the whole nanowire arrays. The X-ray diffraction (XRD) pattern (Fig. S2†) discloses the rutile phase (JCPDS card no. 03-065-0191) of the UCW-TiO2.
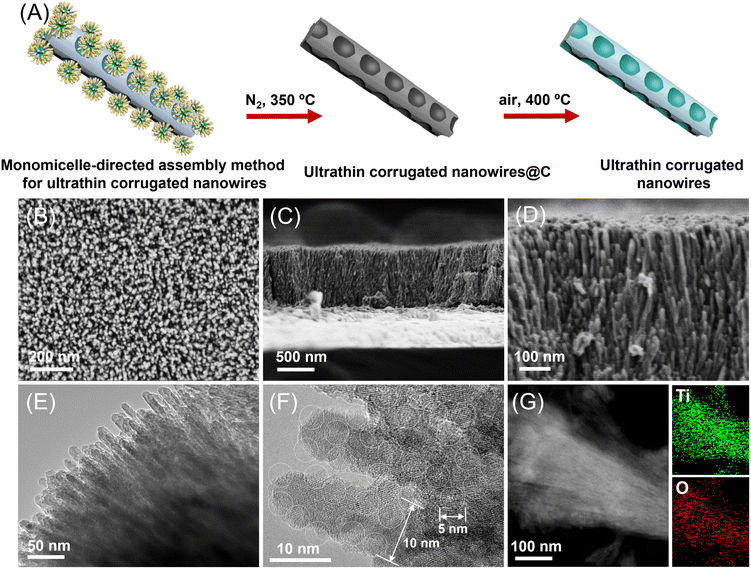 |
| | Fig. 1 Schematic illustration of the synthetic process of UCW-TiO2 (A); morphology characterization of UCW-TiO2, top-view (B) and side-view (C, D) of scanning electron microscopy (SEM) images of UCW-TiO2 on FTO; transmission electron microscopy (TEM) images (E), HRTEM images of UCW-TiO2 (F), and EDS mapping profiles of UCW-TiO2 (G). | |
Benzyl alcohol oxidation
To demonstrate that the synthesized UCW-TiO2 accelerates the electron transfer and improves the charge collection efficiency compared to NPTiO2, different morphologies of TiO2 were used for PEC oxidation of biomass alcohols, such as benzyl alcohol (BA) under mild conditions. The room-temperature PEC catalytic performances of the pristine UCW-TiO2 for selective oxidation of BA to benzaldehyde (BAD) under aerobic conditions were measured at 1.0 V vs. NHE under illumination (100 mW cm−2), as shown in Scheme 1C. The chopped photocurrent density responses of different photoanodes are shown in Fig. 2A. We found that the photocurrent density significantly increased when BA was added to the cell, suggesting that BA was oxidized at the photoanode. Photocurrent for the oxidation of BA based on the UCW-TiO2 photoanode was about 3 times more than that based on the NPTiO2 photoanode (the thickness is about 810 nm, as shown in Fig. S3†) at 1.0 V vs. NHE, under the same conditions, as shown in Fig. 2A. The NPTiO2 photoanode displayed a low photocurrent response throughout the potential window, with a current density of 0.82 mA cm−2 at 1.0 V vs. NHE. In contrast, the photocurrent density of UW-TiO2 and UCW-TiO2 photoanodes increased to 1.9 and 2.2 mA cm−2, respectively. In the process of PEC oxidation of BA, the production of BAD and remaining BA were detected by HPLC. In the first five hours, the UCW-TiO2 photoanode had a high selectivity to BAD (>95%), Fig. 2C. However, the selectivity of BAD over the UW-TiO2 and NPTiO2 photoanodes is ∼91%. As the reaction time progressed, the selectivity of BAD gradually decreased because BAD was further oxidized to benzoic acid by the photogenerated holes. The BA conversion rate of the UCW-TiO2 photoanode was close to 100% after 15 h and the selectivity of BAD was still higher than 90.2%. In the case of the NPTiO2 photoanode, the conversion rate of BA was only 23.6% and the selectivity of BAD was 71.9%. The BAD production of the UCW-TiO2 photoanode was 225.5 μmol after 15 h, and the faradaic efficiency (FE) (calculated by FE (%) = (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 × n(BAD) × 2)/Q) of BAD was close to 90%. However, BAD produced from the NPTiO2 photoanode was only 42.4 μmol with FE = 54.6%, as shown in Fig. 2D. In the PEC experiments for the oxidation of BA, UCW-TiO2 showed better conversion and higher selectivity. Electrochemical impedance spectroscopy (EIS) was used to investigate the interfacial properties of TiO2 based photoanodes under white light illumination (Fig. S4†). According to the Nyquist plots, the semidiameter of the semicircle for UCW-TiO2 was smaller than that of NPTiO2, thereby indicating an improved semiconductor/electrolyte interface charge-transfer rate.
485 × n(BAD) × 2)/Q) of BAD was close to 90%. However, BAD produced from the NPTiO2 photoanode was only 42.4 μmol with FE = 54.6%, as shown in Fig. 2D. In the PEC experiments for the oxidation of BA, UCW-TiO2 showed better conversion and higher selectivity. Electrochemical impedance spectroscopy (EIS) was used to investigate the interfacial properties of TiO2 based photoanodes under white light illumination (Fig. S4†). According to the Nyquist plots, the semidiameter of the semicircle for UCW-TiO2 was smaller than that of NPTiO2, thereby indicating an improved semiconductor/electrolyte interface charge-transfer rate.
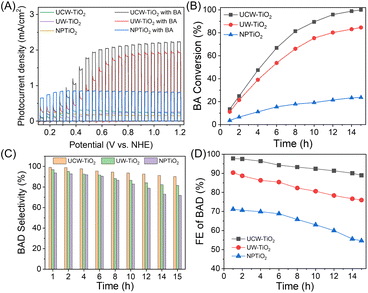 |
| | Fig. 2 PEC oxidation of BA over different photoanodes. (A) Comparison of the chopped photocurrent responses of the UCW-TiO2, UW-TiO2, and NPTiO2 photoanodes in contact with and without BA; (B) BA conversion vs. reaction time at 1.0 V vs. NHE; (C) selectivity of BAD vs. reaction time at 1.0 V vs. NHE. (D) FE of BAD on different photoanodes. | |
Intensity modulated photocurrent spectroscopy (IMPS) measurements were used to further study the interfacial charge transfer kinetics of different morphological TiO2 photoanodes in operational PEC cells for alcohol oxidation. The IMPS spectra were obtained on different photoanodes to compare the relative rates of charge recombination (krec) and transfer (ktrans) occurring at the TiO2/electrolyte interface.29,30Fig. 3A–C show typical IMPS responses of NPTiO2, UW-TiO2 and UCW-TiO2 photoanodes under the applied potential range (0.5–1.0 V vs. NHE). Two semicircles in the lower and upper quadrants of the IMPS spectrum correspond to the resistance-capacitance attenuation and the competition between charge transfer and recombination, respectively.31 The ratio of krec/ktrans is positively proportional to the upper semicircle and a small value could indicate faster charge transfer than charge recombination. As shown in Fig. 3A and B, the radii of upper semicircles of the UW-TiO2 photoanodes were smaller than those of the NPTiO2 photoanodes, indicating that UW-TiO2 as a photoanode retarded charge recombination. At high applied potentials, the upper semicircle of UCW-TiO2 became progressively smaller, Fig. 3C, suggesting that charge recombination was significantly suppressed and charge transfer was significantly accelerated. In contrast, the upper semicircle of the NPTiO2 photoanodes did not change obviously as high applied potentials increased, indicating that rapid charge recombination still existed for the NPTiO2 photoanode under high applied potentials.
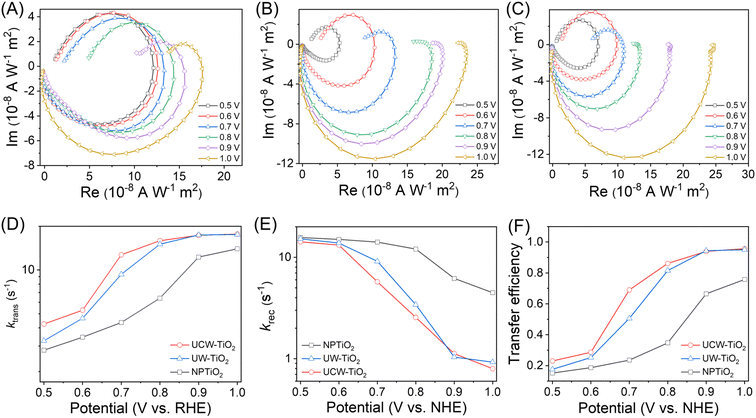 |
| | Fig. 3 IMPS responses of (A) NPTiO2, (B) UW-TiO2 and (C) UCW-TiO2 photoanodes at various applied potentials. (D) The rate constant for charge transfer (ktrans), (E) the rate constant for charge recombination (krec) and (F) the charge transfer efficiency extracted from the IMPS spectra of different photoanodes at various potentials. | |
According to the generalized theory of IMPS, as the frequency increases, the relaxation in the concentration of photogenerated holes at the semiconductor surface is characterized by fmax (at the apex of the upper semicircle), where 2πfmax = ktrans + krec.32,33 The charge transfer efficiency, in terms of ktrans/(ktrans + krec), can be derived from the intersections of the semicircle with the real axis at low and high frequencies (i.e., I1 and I2, respectively), where I1/I2 = ktrans/(ktrans + krec), as shown in Fig. S5.†34 The key parameters krec and ktrans are therefore readily accessible. The values of ktrans and krec for different photoanodes at various potentials are shown in Fig. 3D–E. The ktrans of UCW-TiO2 and UW-TiO2 surpassed that of NPTiO2 at all applied potentials, indicating that the ultrathin nanowire TiO2 greatly promoted the kinetics of charge transfer. At higher potentials, the ktrans of UCW-TiO2 was more than 2.9 times that of NPTiO2 at 0.7 V vs. NHE. The krec reduced slightly with an increase of the potential for NPTiO2, indicating that the NPTiO2 could not effectively suppress charge recombination. However, the krec of UCW-TiO2 and UW-TiO2 reduced obviously, suggesting that the ultrathin nanowire morphology of the TiO2 could effectively suppress charge recombination due to rapid electron transfer. At 1.0 V vs. NHE, the value of krec was 4.5 s−1 for NPTiO2, which is 5.8 times that of UCW-TiO2 (0.78 s−1). As shown in Fig. 3F, it was noteworthy that UCW-TiO2 and UW-TiO2 showed the highest charge transfer efficiency in a wide range of applied potentials, indicating superior charge separation ability. According to the IMPS measurements, we found that the electron transfer in ultrathin nanowire TiO2 was faster than that in NPTiO2, which leads to the improvement of the FE for PEC alcohol oxidation.
A series of control experiments were conducted by adding different scavengers to the system to investigate the reaction mechanism (Fig. S6A†). In the presence of 1,4-benzoquinone (BQ), a superoxide anion radical (O2˙−) scavenger, the conversion of BA decreased significantly, indicating that O2˙− was involved in the PEC BA oxidation. The PEC BA oxidation for different photoanodes under an Ar atmosphere (Fig. S6B–D†) and the chopped photocurrent response (Fig. S7†) also demonstrated that O2 played an important role in accelerating PEC BA oxidation. An obvious decrease in conversion was also observed with the addition of sodium iodate (NaIO3), an e− scavenger, confirming the importance of O2 activation to form O2˙− by photogenerated e− in BA oxidation. Finally, the addition of triethanolamine (TEOA), a h+ scavenger, completely suppressed the BA conversion, indicating that the photogenerated h+ played a key role in the oxidation of BA. According to the above experimental results, h+ and O2˙− are the main active species for the PEC BA oxidation reaction under visible light illumination.
Photoinduced electron transfer dynamics at the interface of UCW-TiO2 with either no BA or 50 mM BA in acetonitrile were investigated by nanosecond transient absorption spectroscopy (TA) measurements. The TA kinetics of the UCW-TiO2 film were measured at 800 nm after pulsed-laser excitation (λex = 355 nm) under an Ar atmosphere. The TA of the UCW-TiO2 film without BA showed a shorter decay half-time (t1/2 = 1.9 × 10−7 s) relative to the UCW-TiO2 film with BA (t1/2 = 2.4 × 10−7 s). Notably, in the presence of BA, the initial TA intensity increased due to a higher yield of e− on the surface of the UCW-TiO2 film. The results indicated that the light-induced h+ in the VB of UCW-TiO2 rapidly transferred to the BA molecule. Electron paramagnetic resonance (EPR) experiments were carried out to confirm that the O2˙− was generated by M-NTiO2 under the illumination, Fig. 4B. Based on these experiments, we propose a mechanism for PEC BA oxidation over the UCW-TiO2 photoanode. The general hypothesis was that the photogenerated holes were transferred to the BA molecules adsorbed on the electrode surface to form the reactive C6H5-CHOH˙* radicals. Subsequently, there are two pathways for the conversion of C6H5-CHOH˙* to BAD in the PEC BA oxidation process. One pathway is that C6H5-CHOH˙* was directly oxidized to BAD through the second photogenerated holes. The other started with dioxygen preferentially being reduced by photogenerated UCW-TiO2 electrons to form O2˙− when the electrolyte was saturated with oxygen gas. The weak oxidant, O2˙−, cleaved the O–H bond in the C6H5-CHOH˙* that was further converted to BAD as the sole oxidized product, resulting in highly selective oxidation of BA to BAD.35–41
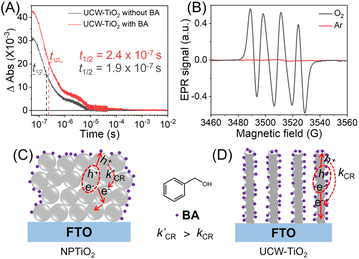 |
| | Fig. 4 (A) Transient absorption changes measured at 800 nm after pulsed 355 nm excitation of UCW-TiO2 with BA or without BA in 0.1 M TBAP acetonitrile solution under an Ar atmosphere. (B) EPR detection of in situ O2˙− formed using DMPO as the spin-trapping agent on UCW-TiO2. Schematic illustration of the effect of different TiO2 for PEC BA oxidation, NPTiO2 (C) and UCW-TiO2 (D). | |
Fig. 4C and D show the reason why the performance of UCW-TiO2 PEC BA oxidation is better than that of NPTiO2. The ultrathin corrugated nanowire structure enhances the refraction of light and improves the utilization rate of light. Meanwhile, it also provides significantly shorter carrier-diffusion paths along the ultrathin wire structure directly on FTO for efficient charge transfer.6,42 The NPTiO2 films composed of interconnected spherical nanocrystalline particulate TiO2 often suffer from charge recombination at grain boundaries and long electron diffusion paths through the TiO2 network with random walk, Fig. 4C. Since the TiO2 nanocrystals are interconnected with each other, the NPTiO2 photoanode gives more limited accessibility of BA molecules at the interface of the NPTiO2. In contrast, for UCW-TiO2 and UW-TiO2, the ultrathin TiO2 nanowire not only significantly enlarges the material surface area extending contact between the semiconductor and electrolyte but also accelerates electron transfer kinetics, further improving charge separation. Then the BA molecules could be uniformly adsorbed on the surface of the UW-TiO2 and UCW-TiO2 photoanodes, Fig. 4D. Because the corrugated structure of UCW-TiO2 can adsorb a large quantity of oxygen molecules, more oxygen molecules were reduced by photogenerated electrons to produce the O2˙−. Such O2˙− accelerates the conversion of BA to BAD. This result brings enhancement in higher photocurrent density leading to better photocatalytic oxidation efficiency for BA oxidation in a PEC cell.
According to the optimal reaction conditions obtained above and understanding the underlying oxidation mechanism, the influence of different para-substituent groups inside BA molecules on the conversion rate and product selectivity of the UCW-TiO2 photoanode were also investigated (Table S1†). The substrate conversion ratio and amount of oxidized products were also determined by HPLC measurements. BA derivatives substituted with electron-donating groups (–CH3, –OCH3) and electron-withdrawing groups (–Br, and –NO2) are efficiently oxidized into the corresponding aldehyde with high selectivity.
Dye-sensitized photoelectrosynthesis cell for water oxidation
Although UCW-TiO2 demonstrates excellent performance in PEC benzyl alcohol oxidation, it is optically transparent in the visible region of the solar spectrum, hindering its further application for solar energy conversion. Water is the electron and proton source of large-scale in nature. Although PEC water oxidation reaction is very promising, it is a challenging transformation since it involves four oxidizing equivalents. Moreover, the potential of water oxidation is 1.23 V vs. NHE at pH = 0 as it is usually catalyzed by oxygen evolution catalysts (OECs). The chromophore and OECs co-anchored on the NPTiO2 thin film surface result in fast electron–hole recombination at the molecule/TiO2 interface and grain boundaries, thereby leading to poor efficiency of PEC water oxidation. Herein, UCW-TiO2 is used as the photoanode support to fabricate a DSPEC for water oxidation. RuP dyes ([Ru(4,4′-(PO3H2)2-2,2′-bipyridine)(2,2′-bipyridine)2]2+) were applied as chromophores for solar energy conversion systems based on their long fluorescence lifetimes, high molar extinction coefficient, and chemical stability. Loading of RuP onto NPTiO2 (the thickness is about 810 nm, as shown in Fig. S3†) and UCW-TiO2 films were accomplished by using the phosphate group.43,44 UV-vis absorption measurements were used to monitor photoelectrode assembly formation. Typical UV-vis data are shown in Fig. S8† for NPTiO2, UCW-TiO2, NPTiO2|RuP, and UCW-TiO2|RuP. The absorption spectral maxima at 460 nm of the NPTiO2|RuP and UCW-TiO2|RuP assemblies are attributed to the metal-to-ligand charge-transfer (MLCT) transition of RuP.
Photocurrent studies
As shown in Fig. 5, the photocurrent density responses to on–off cycles of NPTiO2|RuP, NPTiO2|RuP-Rubda, UCW-TiO2|RuP and UCW-TiO2|RuP-Rubda electrodes were measured using a standard three-electrode photoelectrochemical setup in 0.1 M acetate buffer (pH 4.65) solution, 0.5 M NaClO4 aqueous solution as the supporting electrolyte under visible light illumination (100 mW cm−2, 400 nm cut off filter). NPTiO2|RuP, NPTiO2|RuP-Rubda, UCW-TiO2|RuP and UCW-TiO2|RuP-Rubda photoelectrodes, a platinum wire and an Ag/AgCl were used as working electrodes, the counter electrode and the reference electrode, respectively. A remarkable average photocurrent density response of about 480 μA cm−2 for the UCW-TiO2|RuP-Rubda photoanode was obtained at the end of each cycle. This photocurrent density response was nearly 6 times larger than that measured for an assembly without the catalyst UCW-TiO2|RuP photoelectron under the same conditions, Fig. 5. The chopping photocurrent density response for the UCW-TiO2|RuP-Rubda indicated efficient separation of the electron–hole pairs in the system induced by visible light. However, for NPTiO2|RuP-Rubda, only 200 μA cm−2 photocurrent density was achieved under the same conditions. The results indicated fast electron–hole recombination at the molecule/TiO2 interface and grain boundaries in the NPTiO2|RuP-Rubda photoanode, thereby leading to low PEC water oxidation activity. Fig. 5 shows that, in the absence of Rubda, the transient photocurrent density of NPTiO2|RuP and UCW-TiO2|RuP almost reached ∼500 μA cm−2 at the moment of light-on in the first cycle. From UV-vis measurements (Fig. S8†), we found that the amount of RuP anchored on UCW-TiO2 was lower than that of NPTiO2. However, the transient photocurrent density of UCW-TiO2|RuP is the same as that of UCW-TiO2|RuP, indicating that UCW-TiO2 as the photoanode support has excellent charge transport and separation properties. The electrons from the MLCT excited state of RuP* were injected into the conduction band of TiO2, and the holes remaining on the RuP recombined with electrons leading to the result that the photocurrent density of UCW-TiO2|RuP and NPTiO2|RuP decreased rapidly and stabilized at about 70 μA cm−2. In the presence of Rubda, it was indisputable that the photocurrent density of the NPTiO2|RuP-Rubda photoanode showed a “fast” decreasing trend (48.2%; Fig. 5) at the moment of light-on, thus suggesting that the charge recombination is also a major process on the surface of NPTiO2 co-adsorbed by RuP and Rubda. This suggested that in the NPTiO2 based photoanode, the kinetics of charge recombination was greater than the kinetics of charge transfer due to the presence of a large number of trapping states at the grain boundary as well as long electron diffusion length (random walk) through the NPTiO2 network. More interestingly, the photocurrent density of the UCW-TiO2|RuP-Rubda photoanode showed a “slow” decreasing trend (17.5%; Fig. 5), thus proving that charge recombination was suppressed on the surface of UCW-TiO2. The UCW-TiO2 film has a highly ordered structure that dramatically reduces grain boundaries compared to NPTiO2, which reduces the recombination of photogenerated electrons and holes.
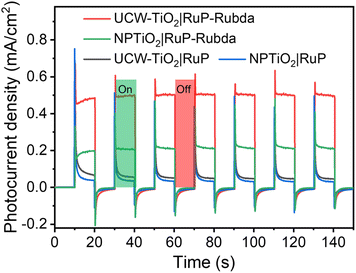 |
| | Fig. 5 Photocurrent density responses to the on–off cycles of illumination by photoanodes, NPTiO2|RuP, NPTiO2|RuP-Rubda, UCW-TiO2|RuP and UCW-TiO2|RuP-Rubda, at 0.4 V vs. NHE in three-electrode PEC cells in 0.1 M acetic acid/acetate buffer (pH 4.65) solutions, 0.5 M NaClO4 aqueous solution as the supporting electrolyte under 1 sun illumination (100 mW cm−2, 400 nm cut off filter). | |
To study the stability and the FE for O2 production, a collector–generator (C–G) setup was used and the results are shown in Fig. 6. In these experiments, O2 generated at the photoanode was monitored at a collector electrode using the FTO separated 1 mm by the generator that has been described earlier.4,45–47 For the UCW-TiO2|RuP-Rubda photoanode, the C-G experiment was performed for 10 min illumination periods with a white light source (100 mW cm−2 and 400 nm cutoff filter). The current responses of the generator electrode (upper) and collector electrode (lower) are shown in Fig. 6A. The current density from the collector electrode slowly rose as O2 generated from the photoanode got detected, when the UCW-TiO2|RuP-Rubda photoanode was under light irradiation. After the light irradiation was turned off, the photoanode immediately dropped to almost zero. At the same time, the collector current gradually decayed as part of the O2 remaining between the two electrodes is gradually reduced by the collector electrode. Fig. 6B shows the charge passed for the UCW-TiO2|RuP-Rubda of generator (black line) and collector (red line) electrodes. The FE for O2 production was calculated from eqn (1), with Qcol and Qgen being the total charge passed at the collector and generator electrodes. The constant 0.7 is the experimentally determined collection efficiency for the cell. For the UCW-TiO2|RuP-Rubda, over 600 s illumination periods, the maximum FE for O2 production reached ∼49.6%.
| | | FE (%) = (Qcol/Qgen)/0.70 × 100% | (1) |
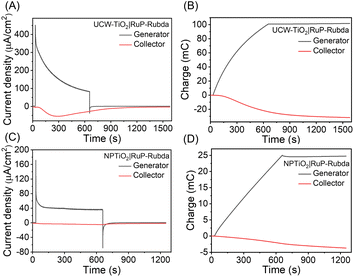 |
| | Fig. 6 O2 measurements for water oxidation detection with the collector–generator setup for UCW-TiO2|RuP-Rubda (A) and NPTiO2|RuP-Rubda (C) photoanodes. The integrated current over time (charge) obtained in the experiment (UCW-TiO2|RuP-Rubda (B); NPTiO2|RuP-Rubda (D)). Eqn (1) was used to calculate faradaic efficiency. All the photoanodes (black) illuminated with 100 mW cm−2 white light with a 400 nm cutoff filter from 30 to 650 s at the potential of 0.4 V vs. NHE. The O2 sensing electrode (red) at a potential of −0.85 V vs. Ag/AgCl. Experiments were performed in 0.1 M acetic acid/acetate buffer at pH 4.65 and 0.5 M NaClO4. | |
The C–G experiments for the NPTiO2|RuP-Rubda photoelectrode in Fig. 6C and D, the photocurrent density showed a rapidly decreasing trend within 50 s, indicating that the charge recombination was also a major process on the surface of NPTiO2 co-adsorbed by RuP and Rubda. A large number of grain boundary defects in NPTiO2 led to the sluggish kinetics of electron transfer and fast charge recombination. A small current was observed from the collector electrode, indicating that little O2 was produced from the photoanode and the FE for O2 production was only 9.7%, which was similar to the results reported in the previous literature.19
Transient absorption
Photoinduced electron transfer dynamics at the interface of UCW-TiO2 (NPTiO2) and RuP were investigated by TA measurements. Transient absorption spectra of different assembled TiO2 based photoanodes at the indicated time delays after pulsed-laser excitation (λex = 532 nm) (5–8 ns fwhm, 1.5 mJ cm−2 per pulse) in argon-saturated 0.1 M acetic acid/acetate buffer at pH 4.65 and 0.5 M NaClO4 solution are shown in Fig. 7A and B. For the photoanode UCW-TiO2|RuP without OEC, excited state RuP* was generated after pulsed-laser excitation (λex = 532 nm), and subsequently an electron was rapidly injected into the conduction band of UCW-TiO2 (kinj > 108 s−1) to form RuIIIP with a bleach at 460 nm in Fig. 7A. The long-lived photoinduced absorption feature over 100 μs indicates the formation of charge separated state, UCW-TiO2(e−)|RuIIIP. The holes on RuP eventually recombine with TiO2(e−)s to return to the ground state, eqn (2).| | 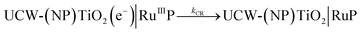 | (2) |
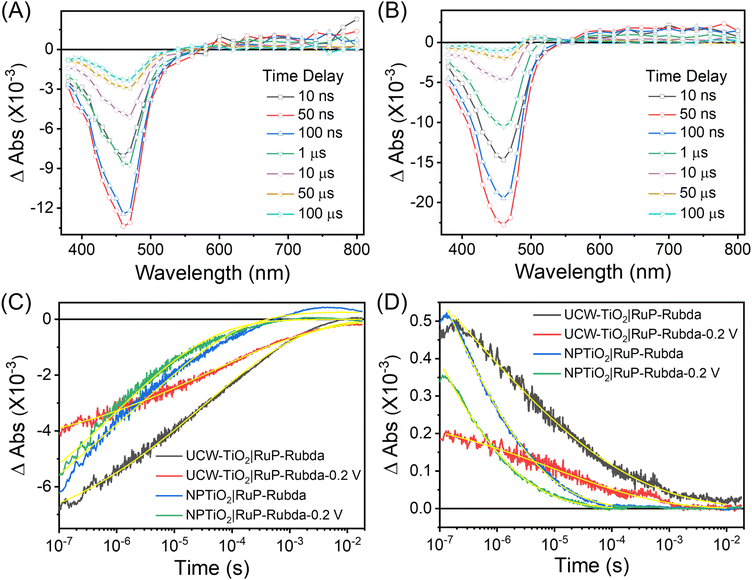 |
| | Fig. 7 Full transient absorption spectra at the indicated time delays after pulsed-laser excitation (λex = 532 nm) for the photoelectrodes: (A) UCW-TiO2|RuP and (B) NPTiO2|RuP. Transient absorption changes measured at 460 nm (C) and 900 nm as a function of delay time, overlaid in yellow are KWW fits. (D) after pulsed-laser excitation (λex = 532 nm) of UCW-TiO2|RuP-Rubda (black), NPTiO2|RuP-Rubda (blue), UW-TiO2|RuP-Rubda (red) and NPTiO2|RuP-Rubda (pink) at 0.2 V vs. Ag/AgCl in 0.1 M acetic acid/acetate buffer at pH 4.65 and 0.5 M NaClO4. | |
In Fig. 7B, for the NPTiO2|RuP photoanode, the bleach of RuP at 460 nm due to the formation of RuIIIP was also observed. In the presence of Rubda, as shown in Fig. 7C, single wavelength decay kinetics for UCW-TiO2|RuP-Rubda and NPTiO2|RuP-Rubda were monitored at 460 nm to investigate the interfacial charge recombination of UCW-TiO2 (or NPTiO2)(e−) with the oxidized sensitizers (RuIIIP). Under the open circuit potential (OCP), the initial TA intensity of UCW-TiO2|RuP-Rubda was almost the same as that of the NPTiO2|RuP-Rubda photoanode. The disappearance of RuIIIP can be ascribed to two electron transfer events that are described in eqn (3) and (4), and the RuIIIP is mainly recombined with TiO2(e−)s under the OCP (eqn (3)). The kinetic decay was non-exponential but could be satisfactorily simulated by a stretched exponential function that was widely used for the charge recombination kinetics at the molecular–semiconductor interface, eqn (5). The beta value was the characteristic stretched exponential factor that was intentionally kept the same for comparative purposes (β = 0.25). By calculating the first moment of the underlying Lévy distribution of the stretched exponential fit, eqn (6),48 the average charge recombination kinetics of NPTiO2(e−) to the RuIIIP was determined to be over ten times 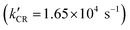 that of UCW-TiO2(e−) to RuIIIP (kCR = 1.24 × 103 s−1). The sluggish interfacial charge recombination could be part of the reasons why the UCW-TiO2 as a photoanode support led to the improved FE of PEC water oxidation.49,50
that of UCW-TiO2(e−) to RuIIIP (kCR = 1.24 × 103 s−1). The sluggish interfacial charge recombination could be part of the reasons why the UCW-TiO2 as a photoanode support led to the improved FE of PEC water oxidation.49,50
| | 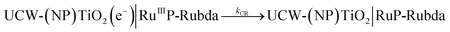 | (3) |
| | 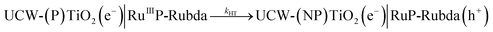 | (4) |
| | | ΔAbs = A0exp[−(kt)β] | (5) |
| | 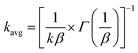 | (6) |
For the PEC water oxidation, interfacial charge recombination is not the only kinetic factor competing with catalytic turnover. How fast can oxidative equivalents accumulate onto the OEC is also crucial. To study the surface hole accumulation during the OEC process, single wavelength decay kinetics for UCW-TiO2|RuP-Rubda and NPTiO2|RuP-Rubda were monitored at 460 nm under a small bias applied potential of 0.2 V vs. Ag/AgCl. As shown in Fig. 7C, the initial TA intensity for UCW-TiO2|RuP-Rubda was obviously decreased, indicating that the UCW-TiO2|RuP-Rubda photoanode had a lower yield of RuIIIP. This suggested that the hole could rapidly transfer from RuIIIP to Rubda, which was beneficial for boosting PEC water oxidation. However, the initial TA intensity for NPTiO2|RuP-Rubda slightly decreased under the same conditions, indicating that the kinetics of hole transfer from RuIIIP to Rubda are sluggish. In order to investigate the kinetics of electron transfer in bulk UCW-TiO2 and NPTiO2, kinetics at 900 nm was chosen as the probe wavelength where TiO2(e−) exclusively absorbs without interference from other species for photoelectrodes, Fig. 7D. The TiO2(e−) signal disappeared from nanoseconds to microseconds and can be described in eqn (3) under OCP. According to eqn (5) and (6), the average charge recombination kinetics of NPTiO2(e−) to the RuIIIP was determined to be over two orders of magnitude 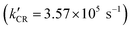 faster than that of UCW-TiO2(e−) to RuIIIP (kCR = 1.35 × 103 s−1). The initial TA amplitude of the UW-TiO2|RuP-Rubda photoanode was decreased by more than half, at the potential of 0.2 V vs. Ag/AgCl, compared to the case of OCP. The results suggested that, after pulsed laser excitation, UCW-TiO2 conduction band electrons were rapidly extracted by a small bias applied potential. However, the initial TA amplitude of the NPTiO2|RuP-Rubda photoanode was decreased slightly under the same conditions, indicating that the density of electrons did not decrease significantly in NPTiO2. In other words, the electrons in NPTiO2 are not quickly extracted to the counter electrode under the applied potential. This result indicates that the kinetics of electron transfer is sluggish in the NPTiO2 film due to the trapping state at grain boundaries. The TA measurements showed that the kinetics of electron transfer in the bulk UCW-TiO2 is faster than that of NPTiO2 and effectively suppresses the recombination of electrons in the conduction band of UCW-TiO2 with RuIIIP under a small bias applied potential. The results are beneficial for UCW-TiO2 as photoanode supports to improve the performance of water oxidation.
faster than that of UCW-TiO2(e−) to RuIIIP (kCR = 1.35 × 103 s−1). The initial TA amplitude of the UW-TiO2|RuP-Rubda photoanode was decreased by more than half, at the potential of 0.2 V vs. Ag/AgCl, compared to the case of OCP. The results suggested that, after pulsed laser excitation, UCW-TiO2 conduction band electrons were rapidly extracted by a small bias applied potential. However, the initial TA amplitude of the NPTiO2|RuP-Rubda photoanode was decreased slightly under the same conditions, indicating that the density of electrons did not decrease significantly in NPTiO2. In other words, the electrons in NPTiO2 are not quickly extracted to the counter electrode under the applied potential. This result indicates that the kinetics of electron transfer is sluggish in the NPTiO2 film due to the trapping state at grain boundaries. The TA measurements showed that the kinetics of electron transfer in the bulk UCW-TiO2 is faster than that of NPTiO2 and effectively suppresses the recombination of electrons in the conduction band of UCW-TiO2 with RuIIIP under a small bias applied potential. The results are beneficial for UCW-TiO2 as photoanode supports to improve the performance of water oxidation.
Our results confirmed that UCW-TiO2 could effectively promote charge separation for water oxidation when it is RuP sensitized and anchored with an OEC. The charge transfer and surface recombination kinetics at the photoanode and electrolyte interface were further quantified by IMPS to study the UCW-TiO2|RuP-Rubda and NPTiO2|RuP-Rubda photoanodes as shown in Fig. 8. The transit time (τd) values of the different samples can be estimated from τd = (2πfmax)−1, in which fmax is the characteristic frequency at the lowest point of the IMPS plot and the τd value of NPTiO2|RuP-Rubda is lower than that of UCW-TiO2|RuP-Rubda.51 Typically, the calculated τd values for NPTiO2|RuP-Rubda and UCW-TiO2|RuP-Rubda are 0.34 ms and 0.62 ms, respectively, which indicates that the UCW-TiO2|RuP-Rubda photoanode shows faster kinetics of charge transfer than that of NPTiO2|RuP-Rubda. The IMPS measurements also prove that the ultrathin nanowire morphology of the TiO2 vertically grown on FTO accelerates electron transfer from the conduction band of UCW-TiO2 to the counter electrode under a small electrochemical bias.
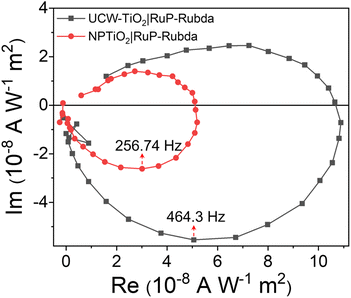 |
| | Fig. 8 IMPS responses of UCW-TiO2|RuP-Rubda and NPTiO2|RuP-Rubda photoanodes. | |
Conclusions
In conclusion, we introduced an ultrathin corrugated nanowire TiO2 (UCW-TiO2) that can be employed as a versatile photoanode platform for photoelectrochemical (PEC) alcohol and water oxidation. Benefiting from vertically grown UCW-TiO2 on the FTO substrate, the photogenerated electrons in the conduction band can rapidly move to the electrode contact, resulting in improved charge separation efficiency. For PEC aerobic alcohol oxidation, the UCW-TiO2 photoanode demonstrates a high conversion rate and high selectivity of corresponding aldehyde. The sensitized photoanode assemblies for PEC water oxidation produced O2 with a faradaic efficiency of 49.6% that is 5-fold larger than that of spherical nanocrystalline particulate TiO2 (NPTiO2). The obtained faradaic efficiency is among the best results of all dye-sensitized TiO2 photoelectrodes reported to date. Transient absorption spectroscopy and intensity-modulated photocurrent spectroscopy demonstrate that either being dye-sensitized for water oxidation or being directly used for alcohol oxidation, the UCW-TiO2 photoanode shows excellent charge transport and separation properties. This work highlights new opportunities to improve the charge separation and charge collection efficiency of PEC photoanodes through ultrathin corrugated TiO2 nanowire semiconductor materials.
Experimental
Materials
All chemicals were purchased from Adamas, Sigma-Aldrich, Beijing Innochem Science & Technology Co., LTD. and used as received unless otherwise noted. Fluorine doped tin oxide (FTO) electrodes (8 Ω sq−1) were purchased from Suzhou Shangyang Solar Technology Co. Ltd. Detailed molecular synthesis is provided in the ESI.†
Instrumentation
Materials characterization.
1H-NMR and 13C-NMR measurements were performed on a Bruker AVANCE III HD spectrometer. Transmission Electron Microscopy (TEM) and Field Emission Transmission Electron Microscopy (HR-TEM) images were taken on a Hitachi HT7700 Exalens and Tecnai G2 F20 S-Twin respectively. Ground state UV-vis absorption spectra were recorded on an Agilent Cary 60 spectrophotometer. Intensity modulated photocurrent spectroscopy (IMPS) and Electrochemical impedance spectroscopy (EIS) measurements were conducted using a potentiostat (IM6ex, Zahner Company) controlled by a Zahner electrochemical workstation.
Nanosecond transient absorption spectroscopy.
Nanosecond transient absorption data in the kinetic mode were obtained on a TSP-2000 (Unisoku) laser flash photolysis system. A Q-switched frequency-tripled pulsed Nd:YAG laser (Quantel Q-Smart 450, 10 Hz) was used to pump an OPO (OPOTek MagicPrism Inline) to output a laser excitation wavelength of 532 nm (5–8 nm full width at half-maximum). A 75 W xenon arc lamp served as the probe beam that was aligned orthogonally to the excitation laser pulse. An R2949 photomultiplier tube (Hamamatsu) coupled to an f/4 monochromator (Acton, Princeton Instrument) was used to achieve signal detection. Transient kinetic data at each wavelength were acquired on a logarithmic time scale on a computer interfaced digital oscilloscope (LeCroy 4024, 12 bit, 200 MHz) with typical 50-100 laser pulse averages. Data were processed in Origin 2021 and fit with least-squares error minimization using the Levenberg–Marquardt iteration method.
Preparation of the photoelectrodes
According to a previous report,52 the synthesis of well-aligned ultrathin corrugated nanowire TiO2 (UCW-TiO2) on a fluorine-doped tin oxide (FTO) substrate was described as follows:
Synthesis of spherical F-127/TiO2 composite monomicelles.
In a typical procedure, 1.6 g of Pluronic F-127 (PEO106PPO70PEO106, Mw = 12600 g mol−1, Sigma-Aldrich Corp.), 2.4 g of acetic acid and 2.4 g of concentrated HCl (36 wt%) were added in 30 mL of tetrahydrofuran (THF). After vigorous stirring for 10 min, 3.4 g of tetrabutyl titanate (TBOT, Sigma-Aldrich Corp.) was added dropwise and 0.20 g of H2O was added subsequently. The formed clear white yellow solution was transferred into two 30 mm × 50 mm volumetric flasks, and left in a drying oven at 45 °C for 10 h.
Synthesis of ultrathin corrugated nanowire TiO2 (UCW-TiO2) on FTO.
In a typical procedure, 2.0 g of the above obtained light yellow gel was transferred into a 20 mL autoclave. Then the pre-cleaned FTO (1 × 3 cm) was placed into the autoclave and leaned on the inner wall of the autoclave. After heating at 150 °C for 20 h in an oven, the autoclave was allowed to cool down to room temperature naturally. The UCW-TiO2 on FTO was washed with water and ethanol, and then dried in an oven. Then, the UCW-TiO2 on FTO was calcined at 350 °C for 3 h in N2 with a heating rate of 1 °C min−1. Finally, in order to remove the excess polymer, the UCW-TiO2 on FTO was obtained by further calcination at 400 °C for 2 h in air at a heating rate of 1 °C min−1. Smooth UW-TiO2 without the concave region was obtained as the same as UCW-TiO2, except for keeping the autoclave at 150 °C for 48 h in an oven.
Loading of RuP and OEC molecular assembly were carried out by immersing the UCW-TiO2 sequentially in 50 mM RuP in MeOH, 5 mM Py-C3-PO(OH)2 in MeOH and 5 mM Ru(2,2′-bipyridine-6,6′-dicarboxylate)(4-methoxypyridine)2 (Ru(bda)(4-MeOPy)2) in MeOH, for 12 h, 12 h, and 10 h, respectively, to form the photoanode UCW-TiO2|RuP-Rubda. The NPTiO2|RuP-Rubda photoanode was prepared as the same as UCW-TiO2|RuP-Rubda.
Photoelectrochemical measurements and O2 detection
Electrochemical and photoelectrochemical experiments were performed using a CH Instruments (CHI) 760E bipotentiostat. White light illumination (400 nm cutoff filter, 100 mW cm−2) was performed using a Thorlabs HPLS 345 light source. Experiments were performed in 0.1 M acetic acid/acetate buffer at pH 4.65 and 0.5 M NaClO4. The collector-generator experiments for O2 detection used a four-electrode setup along with the 760E bipotentiostat.43,44,53 Two working electrodes in conjunction with a Pt counter and Ag/AgCl reference electrode were used. One working (generator) electrode was prepared as described for the photoanodes used in the main text; the other working (collector) electrode was plain FTO. Assembly of the collector-generator setup involved placing the two FTO electrodes with the conductive sides facing with narrow 1 mm thick glass spacers between the lateral edges and sealing the sides with epoxy glue (3M™). Prepared in this way, the space between the two FTO electrodes will fill with electrolyte by capillary action when the cell is placed in solution. To measure the faradaic efficiency for O2 production, the charge passed at the generator electrode under the illumination was compared to the total charge passed at the collector electrode (potential bias at −0.85 V vs. Ag/AgCl) during the entire experiment. The FE was corrected for the collection efficiency of the same collector-generator setup (70%), according to our previous work,16 that was determined with a plain FTO (potential bias at + 1.60 V vs. Ag/AgCl) as the generator electrode and another plain FTO (potential bias at −0.85 V vs. Ag/AgCl) as the collector electrode.54
Photoelectrochemical BA oxidation measurements
All photoelectrochemical tests were conducted on a CHI 760E electrochemical station with a conventional three-electrode system. Ag/Ag+ was used the reference electrode, Pt wire was used as the counter electrode, and UCW-TiO2, UW-TiO2 and NPTiO2 were used as the working electrodes. The target biomass of 50 mM benzyl alcohol (BA) was dissolved in 0.1 M tetrabutylammonium perchlorate (TBAP) acetonitrile solution (5 mL) in a single-chamber photoelectrochemical quartz reactor. The working areas of the electrodes were 1 cm2. Prior to photoelectrochemical measurements, the oxygen gas was bubbled into the anode solution for 15 min until the O2 concentration reached saturation. White light illumination (100 mW cm−2) was performed using a Thorlabs HPLS 345 light source. The conversion ratio of BA, the yield, and the selectivity of benzaldehyde (BAD) from BA were calculated using the following eqn:| | | Conversion = [(C0 − Cr)/C0] × 100% | (7) |
| | | Yield = (Cp/C0) × 100% | (8) |
| | | Selectivity = [Cp/(C0 − Cr)] × 100% | (9) |
where C0 is the primary concentration of BA, and Cr and Cp are the concentrations of left BA and BAD after a certain reaction time, respectively. The faradaic efficiency (FE) was calculated according to the integrated charge (Q) passed during photocatalysis and the amount of aldehyde produced with the eqn:
| FE = 100% × (96485 × n(BAD) × 2)/Q |
The qualitative and quantitative tests of hydrogen produced at the cathode were conducted via headspace gas chromatography (Fuli Instruments Co., GC9790 Plus). The conversion of BA, the yield, and the selectivity of BAD were quantitatively performed using a high-performance liquid chromatograph (HPLC, Agilent 1260, Agilent Co.) equipped with a C18 column (4.6 × 100 nm, 3.5 mm). The test conditions were listed as follows: the wavelength (λ) of the UV detector was 254 nm; the ratio of acetonitrile to water was 70:30; and the flow rate was 0.8 mL min−1.
Detection of O2˙− using EPR
The O2˙− generated by UCW-TiO2 was further detected using the spin-trapping agent, DMPO. 25 μL of DMPO (in DMSO, 0.2 M) was mixed with some UCW-TiO2 powder in 25 μL of acetonitrile under an O2 or Ar atmosphere. While the reaction solution was irradiated with a 500 W Xe lamp (100 mW cm−2), the EPR signal was detected immediately.
Author contributions
FN performed most of the experiments, analyzed the data, and wrote the initial draft of the manuscript. PZ and ZZ performed material characterization. PL measured and analyzed EPR data. KH supervised the study and co-wrote the paper. All the authors discussed the results and reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is sponsored by the National Key R&D Program of China (2018YFA0209401), Natural Science Foundation of Shanghai (21ZR1404400) and National Natural Science Foundation of China (21872037).
References
- J.-B. Pan, S. Shen, L. Chen, C.-T. Au and S.-F. Yin, Adv. Funct. Mater., 2021, 31, 2104269 CrossRef CAS.
- R. Tang, S. Zhou, Z. Zhang, R. Zheng and J. Huang, Adv. Mater., 2021, 33, 2005389 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2010, 46, 755–757 RSC.
- T. Butburee, Y. Bai, H. Wang, H. Chen, Z. Wang, G. Liu, J. Zou, P. Khemthong, G. Q. M. Lu and L. Wang, Adv. Mater., 2018, 30, 1705666–1705674 CrossRef PubMed.
- P. Zhang, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2018, 57, 15076–15080 CrossRef CAS PubMed.
- S. Zhang, Z. Liu, D. Chen, Z. Guo and M. Ruan, Chem. Eng. J., 2020, 395, 125101 CrossRef CAS.
- H. Luo, S. Dimitrov, M. Daboczi, J.-S. Kim, Q. Guo, Y. Fang, M.-A. Stoeckel, P. Samorì, O. Fenwick, A. B. Jorge Sobrido, X. Wang and M.-M. Titirici, ACS Appl. Nano Mater., 2020, 3, 3371–3381 CrossRef CAS.
- Y. Zhu, D. Wang, W. Ni, G. G. Gurzadyan, L. Sun, T. J. Meyer and F. Li, J. Mater. Chem. A, 2022, 10, 9121–9128 RSC.
- L. Luo, Z.-j. Wang, X. Xiang, D. Yan and J. Ye, ACS Catal., 2020, 10, 4906–4913 CrossRef CAS.
- Z. Wu, J. Wang, Z. Zhou and G. Zhao, J. Mater. Chem. A, 2017, 5, 12407–12415 RSC.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- F. Niu, Q. Zhou, R. Liu and K. Hu, ACS Appl. Energy Mater., 2022, 5, 1244–1251 CrossRef CAS.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 RSC.
- B. Shan, A. Nayak, M. K. Brennaman, M. Liu, S. L. Marquard, M. S. Eberhart and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 6493–6500 CrossRef CAS PubMed.
- Y. Zhu, D. Wang, Q. Huang, J. Du, L. Sun, F. Li and T. J. Meyer, Nat. Commun., 2020, 11, 4610 CrossRef CAS PubMed.
- D. Wang, B. D. Sherman, B. H. Farnum, M. V. Sheridan, S. L. Marquard, M. S. Eberhart, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9809–9813 CrossRef CAS PubMed.
- D. Wang, L. Wang, M. D. Brady, C. J. Dares, G. J. Meyer, T. J. Meyer and J. J. Concepcion, J. Phys. Chem. C, 2019, 123, 30039–30045 CrossRef CAS.
- X. Shen, M. Yao, K. Sun, T. Zhao, Y. He, C.-Y. Chi, C. Zhou, P. D. Dapkus, N. S. Lewis and S. Hu, ACS Energy Lett., 2021, 6, 193–200 CrossRef CAS.
- Q. Liu, Z. Li, J. Li, F. Zhan, D. Zhai, Q. Sun, Z. Xiao, H. Luo and D. Zhang, Nano Energy, 2022, 98, 107267 CrossRef CAS.
- R. Zhang, M. Shao, S. Xu, F. Ning, L. Zhou and M. Wei, Nano Energy, 2017, 33, 21–28 CrossRef CAS.
- S. Li, E. W. Shuler, D. Willinger, H. T. Nguyen, S. Kim, H. C. Kang, J.-J. Lee, W. Zheng, C. G. Yoo, B. D. Sherman and G. Leem, ACS Appl. Mater. Interfaces, 2022, 14, 22799–22809 CrossRef CAS PubMed.
- W. Zhang, H. He, H. Li, L. Duan, L. Zu, Y. Zhai, W. Li, L. Wang, H. Fu and D. Zhao, Adv. Energy Mater., 2021, 11, 2003303–2003327 CrossRef CAS.
- P. Zhang, Z. Tian, Y. Kang, B. He, Z. Zhao, C.-T. Hung, L. Duan, W. Chen, Y. Tang, J. Yu, L. Mai, Y.-F. Li, W. Li and D. Zhao, J. Am. Chem. Soc., 2022, 144, 20964–20974 CrossRef CAS PubMed.
- K. Reilly, B. Fang, F. Taghipour and D. P. Wilkinson, ACS Appl. Energy Mater., 2020, 3, 8317–8329 CrossRef CAS.
- D. Zhou, K. Fan, Q. Zhuo, Y. Zhao and L. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 2723–2733 CrossRef CAS PubMed.
- F. Yu, F. Li, T. Yao, J. Du, Y. Liang, Y. Wang, H. Han and L. Sun, ACS Catal., 2017, 7, 1868–1874 CrossRef CAS.
- J. Xie, C. Guo, P. Yang, X. Wang, D. Liu and C. M. Li, Nano Energy, 2017, 31, 28–36 CrossRef CAS.
- R. Peat and L. M. Peter, J. Electroanal. Chem., 1987, 228, 351–364 CrossRef CAS.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- Q. Meng, B. Zhang, H. Yang, C. Liu, Y. Li, A. Kravchenko, X. Sheng, L. Fan, F. Li and L. Sun, Mater. Adv., 2021, 2, 4323–4332 RSC.
- S. Meng, S. Chang and S. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 2531–2538 CrossRef CAS PubMed.
- S. Guo, D. Cao, P. Xiao, G. Zhang, Q. Wang and P. Cui, Inorg. Chem., 2022, 61, 15654–15663 CrossRef CAS PubMed.
- Z. Zhou, Y.-N. Xie, W. Zhu, H. Zhao, N. Yang and G. J. A. C. B. E. Zhao, Appl. Catal., B, 2021, 286, 119868 CrossRef CAS.
- C. Wang, B. Weng, Y. Liao, B. Liu, M. Keshavarz, Y. Ding, H. Huang, D. Verhaeghe, J. A. Steele, W. Feng, B.-L. Su, J. Hofkens and M. B. J. Roeffaers, Chem. Commun., 2022, 58, 10691–10694 RSC.
- M. Hamdy, R. Amrollahi and G. Mul, J. Phys. Chem. C, 2009, 113, 8343 Search PubMed.
- B. Zhang, J. Li, Y. Gao, R. Chong, Z. Wang, L. Guo, X. Zhang and C. Li, J. Catal., 2017, 345, 96–103 CrossRef CAS.
- L. Zhang, D. Jiang, R. M. Irfan, S. Tang, X. Chen and P. Du, J. Energy Chem., 2019, 30, 71–77 CrossRef.
- Y. Dai, G. Xie, X. Jia, B. Guo and J. R. Gong, Appl. Surf. Sci., 2023, 610, 155501 CrossRef CAS.
- D. Wang, Z. Xu, M. V. Sheridan, J. J. Concepcion, F. Li, T. Lian and T. J. Meyer, Chem. Sci., 2021, 12, 14441–14450 RSC.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS PubMed.
- L. Wu, M. Eberhart, A. Nayak, M. K. Brennaman, B. Shan and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 15062–15069 CrossRef CAS PubMed.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- D. Wang, S. L. Marquard, L. Troian-Gautier, M. V. Sheridan, B. D. Sherman, Y. Wang, M. S. Eberhart, B. H. Farnum, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 719–726 CrossRef CAS PubMed.
- C. P. Lindsey and G. D. Patterson, J. Chem. Phys., 1980, 73, 3348–3357 CrossRef CAS.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- P. Xu, C. L. Gray, L. Xiao and T. E. Mallouk, J. Am. Chem. Soc., 2018, 140, 11647–11654 CrossRef CAS PubMed.
- Y. Liu, Y. Jiang, F. Li, F. Yu, W. Jiang and L. J. J. o. M. C. A. Xia, J. Mater. Chem. A, 2018, 6, 10761–10768 RSC.
- K. Lan, Y. Liu, W. Zhang, Y. Liu, A. Elzatahry, R. Wang, Y. Xia, D. Al-Dhayan, N. Zheng and D. Zhao, J. Am. Chem. Soc., 2018, 140, 4135–4143 CrossRef CAS PubMed.
- D. Wang, S. L. Marquard, L. Troian-Gautier, M. V. Sheridan, B. D. Sherman, Y. Wang, M. S. Eberhart, B. H. Farnum, C. J. Dares and T. J. J. J. o. t. A. C. S. Meyer, J. Am. Chem. Soc., 2018, 140, 719–726 CrossRef CAS PubMed.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076–7082 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, additional experimental details, and NMR spectra of relevant molecules. See DOI: https://doi.org/10.1039/d2ta09613g |
| ‡ Fushuang Niu and Pengfei Zhang contributed equally to this paper. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Pengju
Li
a,
Rong
Liu
a,
Wei
Li
b,
Pengju
Li
a,
Rong
Liu
a,
Wei
Li









 that of UCW-TiO2(e−) to RuIIIP (kCR = 1.24 × 103 s−1). The sluggish interfacial charge recombination could be part of the reasons why the UCW-TiO2 as a photoanode support led to the improved FE of PEC water oxidation.49,50
that of UCW-TiO2(e−) to RuIIIP (kCR = 1.24 × 103 s−1). The sluggish interfacial charge recombination could be part of the reasons why the UCW-TiO2 as a photoanode support led to the improved FE of PEC water oxidation.49,50


 faster than that of UCW-TiO2(e−) to RuIIIP (kCR = 1.35 × 103 s−1). The initial TA amplitude of the UW-TiO2|RuP-Rubda photoanode was decreased by more than half, at the potential of 0.2 V vs. Ag/AgCl, compared to the case of OCP. The results suggested that, after pulsed laser excitation, UCW-TiO2 conduction band electrons were rapidly extracted by a small bias applied potential. However, the initial TA amplitude of the NPTiO2|RuP-Rubda photoanode was decreased slightly under the same conditions, indicating that the density of electrons did not decrease significantly in NPTiO2. In other words, the electrons in NPTiO2 are not quickly extracted to the counter electrode under the applied potential. This result indicates that the kinetics of electron transfer is sluggish in the NPTiO2 film due to the trapping state at grain boundaries. The TA measurements showed that the kinetics of electron transfer in the bulk UCW-TiO2 is faster than that of NPTiO2 and effectively suppresses the recombination of electrons in the conduction band of UCW-TiO2 with RuIIIP under a small bias applied potential. The results are beneficial for UCW-TiO2 as photoanode supports to improve the performance of water oxidation.
faster than that of UCW-TiO2(e−) to RuIIIP (kCR = 1.35 × 103 s−1). The initial TA amplitude of the UW-TiO2|RuP-Rubda photoanode was decreased by more than half, at the potential of 0.2 V vs. Ag/AgCl, compared to the case of OCP. The results suggested that, after pulsed laser excitation, UCW-TiO2 conduction band electrons were rapidly extracted by a small bias applied potential. However, the initial TA amplitude of the NPTiO2|RuP-Rubda photoanode was decreased slightly under the same conditions, indicating that the density of electrons did not decrease significantly in NPTiO2. In other words, the electrons in NPTiO2 are not quickly extracted to the counter electrode under the applied potential. This result indicates that the kinetics of electron transfer is sluggish in the NPTiO2 film due to the trapping state at grain boundaries. The TA measurements showed that the kinetics of electron transfer in the bulk UCW-TiO2 is faster than that of NPTiO2 and effectively suppresses the recombination of electrons in the conduction band of UCW-TiO2 with RuIIIP under a small bias applied potential. The results are beneficial for UCW-TiO2 as photoanode supports to improve the performance of water oxidation.