
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting the interaction between halloysite and charged PNAs for their controlled release†
Serena
Riela
 a,
Ana
Borrego-Sánchez
b,
Silvia
Cauteruccio
*c,
Raquel
de Melo Barbosa
d,
Marina
Massaro
*a,
C. Ignacio
Sainz-Díaz
e,
Rita
Sánchez-Espejo
d,
César
Viseras-Iborra
de and
Emanuela
Licandro
c
a,
Ana
Borrego-Sánchez
b,
Silvia
Cauteruccio
*c,
Raquel
de Melo Barbosa
d,
Marina
Massaro
*a,
C. Ignacio
Sainz-Díaz
e,
Rita
Sánchez-Espejo
d,
César
Viseras-Iborra
de and
Emanuela
Licandro
c
aDepartment of Biological, Chemical and Pharmaceutical Sciences and Technologies, University of Palermo Viale delle Scienze, Ed. 17 90128 Palermo, Italy. E-mail: marina.massaro@unipa.it
bInstituto de Ciencia Molecular, Universitat de València, Carrer del Catedrátic José Beltrán Martinez 2, 46980 Paterna, Spain
cDepartment of Chemistry, University of Milan, via Golgi 19 20133 Milan, Italy. E-mail: silvia.cauteruccio@unimi.it
dDepartment of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, University of Granada, Campus of Cartuja, 18071 s/n, Granada, Spain
eAndalusian Institute of Earth Sciences, CSIC-UGR, Avenida de las Palmeras 4, 18100, Armilla, Granada, Spain
First published on 8th June 2023
Abstract
The design and development of nanomaterials that could be used in nanomedicine are of fundamental importance to obtain smart nanosystems for the treatment of several diseases. Halloysite, because of its interesting features, represents a suitable nanomaterial for the delivery of different biologically active species. Among them, peptide nucleic acids (PNAs) have attracted considerable attention in recent decades for their potential applications in both molecular antisense diagnosis and as therapeutic agents, although up to now, the actual clinical applications have been very limited. Herein we report a systematic study on the supramolecular interaction of three differently charged PNAs with halloysite. Understanding the interaction mode of charged molecules with the clay surfaces represents a key factor for the future design and development of halloysite based materials which could be used for the delivery and subsequent intracellular release of PNA molecules. Thus, three different PNA tetramers, chosen as models, were synthesized and loaded onto the clay. The obtained nanomaterials were characterized using spectroscopic studies and thermogravimetric analysis, and their morphologies were studied using high angle annular dark field transmission electron microscopy (HAADF/STEM) coupled with Energy Dispersive X-ray spectroscopy (EDX). The aqueous mobility of the three different nanomaterials was investigated by dynamic light scattering (DLS) and ζ-potential measurements. The release of PNA tetramers from the nanomaterials was investigated at two different pH values, mimicking physiological conditions. Finally, to better understand the stability of the synthesized PNAs and their interactions with HNTs, molecular modelling calculations were also performed. The obtained results showed that PNA tetramers interact in different ways with HNT surfaces according to their charge which influences their kinetic release in media mimicking physiological conditions.
Introduction
Nanotechnology is developing rapidly in various fields, seeking natural or synthetic materials that can be easily adhered to on a large scale and at a low cost. The morphology of such nanomaterials appears as fibrous1 or consisting of sheets with tubular structures that are based on different compositions, such as carbon, or phyllosilicate-based materials. The particular structure of the nanomaterials, indeed, has provided outstanding advantages because of their unique properties such as the nanoscale size, high surface area, and tunable surface chemistry. In this context, clay minerals, phyllosilicates having nanometric dimensions used in healthcare since ancient times due to their intrinsic properties, are one of the most promising materials.2,3Halloysite is an aluminosilicate clay, belonging to the kaolin group, and has attracted considerable attention for application in biology because of its interesting features.4 Halloysite, with a chemical formula of Al2Si2O5(OH)4 × nH2O is a natural and low-cost material that possesses a predominantly hollow tubular structure. Due to this feature, it is commonly known as halloysite nanotubes (HNTs). Structurally, HNTs are constituted by an external surface composed of siloxane (Si–O–Si) groups, an internal surface, a lumen, that consists of a gibbsite-like array of aluminol (Al–OH) groups, and the presence of some Al–OH and Si–OH groups at the edges of the material or as structural defects at the external surface. Furthermore, ζ-potential measurements have shown that HNTs are ionized in a wide pH range, presenting a negative charge at the external surface and a positively charged lumen.
The different chemical composition at the inner and outer surfaces allows the chemical modification of the nanotubes by grafting suitable functional groups which have allowed the synthesis of different nanomaterials with hierarchical structures.5 In addition, the opposite charges at surfaces were exploited for the immobilization, by electrostatic attraction interactions, of different molecules to obtain innovative nanomaterials for different purposes.6,7 In this context, proteins or enzymes were selectively immobilized at the external surface or loaded into the lumen by working in pH ranges above or below their isoelectric points.8 In this way, efficient nanocontainers for drug delivery or biomimetic nanoreactors were successfully synthesized.
HNTs, in contrast to other tubular nanomaterials, present suitable dimensions for macrophage removal. Different studies assessed the biocompatibility of HNTs9,10 and established that they exert relevant toxicity in vitro at a concentration of 1000 μg mL−1;11 whereas in vivo experiments highlighted that the oral administration limit is ca. 20 mg kg−1 BW.11 They also possess the intrinsic capacity to cross cellular membranes localizing in the perinuclear region.12,13
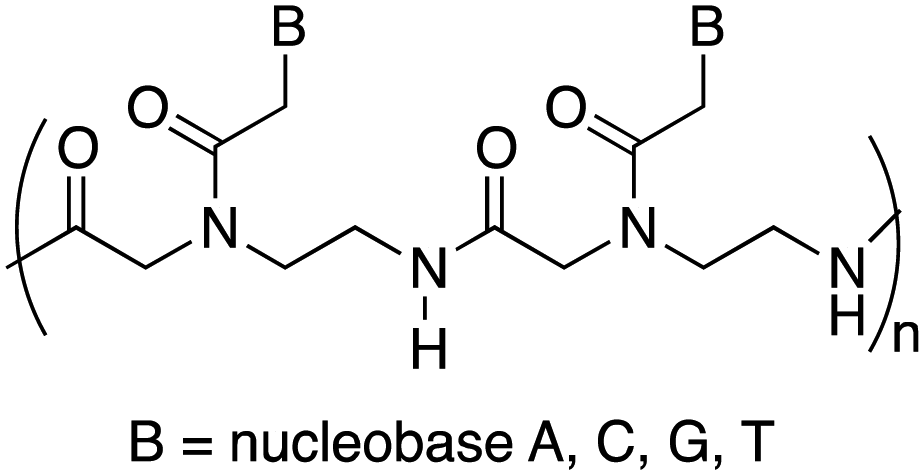
Peptide nucleic acids (PNAs) can be considered one of the most powerful analogues of natural oligonucleotides and have attracted considerable attention in recent decades for their potential applications in both molecular antisense diagnosis and as therapeutic agents. Compared with natural oligonucleotides, they offer some advantages such as greater biological stability against nuclease and protease enzymes, higher specificity, and stability in interactions with complementary RNA or DNA. This is due to their chemical structure, completely different compared to that of natural nucleic acids, made of a backbone of electrostatically neutral N-(2-aminoethyl)glycine on which nucleobases are appended through amide bonds (Fig. 1).14 However, despite the remarkable potentialities possessed by PNAs, their poor water solubility, low uptake by eukaryotic cells, and rapid renal clearance have so far prevented the application of PNAs in the biomedical field. To overcome these limitations several strategies have been considered15 even though the cellular uptake remains an unsolved issue and there are currently no drugs available based on PNA, even though a recent publication mentions one PNA drug in clinical trials, and others in development.16
 | ||
| Fig. 1 General structure of a portion of the polyamide backbone of the peptide nucleic acid: a dimer. | ||
Recently, some of us reported the covalent linkage of PNA tetramers, chosen as a model, to HNTs to develop nanomaterials for intracellular PNA delivery.17 The characterization of the obtained nanomaterial highlighted that the PNA tetramer retains its capability to bind complementary single-stranded DNA when immobilized onto HNTs.
To the best of our knowledge, to date, there have been no reports in the literature dealing with the supramolecular loading of PNA molecules onto halloysite. This approach, being simpler than the covalent one, could represent a promising strategy for the delivery of PNA based drugs.
Herein we report a systematic study on the supramolecular interaction of three different PNAs with HNTs. Understanding the interaction mode of charged molecules with HNT surfaces represents a key factor for the future design and scale up of the synthesis of HNT based materials which can be used for the delivery and subsequent intracellular release of PNA molecules.
Therefore, a model PNA tetramer, containing the four nucleobases A, C, G, and T, was selected for this study, and three differently charged derivatives of this PNA tetramer were prepared: the positively charged PNA 1, the negatively charged PNA 2 and the neutral PNA 3 (Fig. 2).
 | ||
| Fig. 2 Structure of PNA tetramers 1–3. | ||
After the PNA tetramer synthesis, they were loaded onto HNTs and the obtained nanomaterials were investigated using spectroscopic characterization and thermogravimetric analysis, and their morphologies were studied using high angle annular dark field transmission electron microscopy (HAADF/STEM) coupled with Energy Dispersive X-ray (EDX) spectroscopy. The aqueous mobility of the three different nanomaterials was investigated using dynamic light scattering (DLS) and ζ-potential measurements. The release of PNA tetramers from the nanomaterials was investigated at two different pH values, mimicking physiological conditions.
Finally, computational calculations were also carried out in order to better understand and analyze the stability of the PNAs synthesized and their interactions with HNTs.
Results and discussion
The preparation of the different HNTs–PNA nanomaterials was accomplished in a two-step procedure: (i) synthesis of three PNA tetramers 1–3 with appropriate terminal groups, (ii) conjugation of PNAs 1–3 on HNTs by exploiting the electrostatic interactions between PNAs and HNTs.Synthesis of PNA tetramers 1–3
PNA tetramers 1–3 were prepared manually according to standard protocols for solid-phase synthesis of PNAs.18 More in detail, the positively charged PNA 1 was synthesized through two different procedures, namely the Boc/Z and Fmoc/Bhoc strategies, involving the use of the commercially available MBHA and the ChemMatrix® resins, respectively, and the appropriately protected PNA monomers. The MBHA resin supported PNA tetramer 4 was subjected to cleavage by stirring it in the presence of a mixture of TFA/TFMSA/m-cresol/thioanisole for 1.5 h.Otherwise, the ChemMatrix® resin supported PNA tetramer 5 was treated with a solution of piperidine in NMP to remove the Fmoc protecting group, and then with a mixture of TFA/m-cresol for 1.5 h (Scheme 1).
 | ||
Scheme 1 Cleavage of the positively charged PNA 1: (i) TFA/TFMSA/m-cresol/thioanisole (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1, v/v/v/v), 1.5 h; (ii) piperidine in NMP (1:5 v/v); (iii) TFA/m-cresol (9:1, v/v), 1.5 h. :2:1:1, v/v/v/v), 1.5 h; (ii) piperidine in NMP (1:5 v/v); (iii) TFA/m-cresol (9:1, v/v), 1.5 h. | ||
Both procedures provided the crude tetramer 1 that was purified by means of the RP-HPLC and analyzed by UPLC-HR-ESI+ mass analysis (see the ESI†). The resin supported PNA 4 was used as a starting building block to obtain the negatively charged tetramer 2 and the neutral tetramer 3, which were synthesized through a three-step protocol, that involved the removal of the Boc group from 4 under acidic conditions followed by neutralization with DIPEA and coupling with a large molar excess of succinic anhydride and acetic anhydride, to give 2 and 3, respectively (Scheme 2). After the cleavage, crude PNA tetramers 2 and 3 were purified by RP-HPLC and analyzed by UPLC-HR-MS analysis (see the ESI†). Both tetramers 2 and 3 were also prepared starting from the resin supported PNA tetramer 5 according to the Fmoc/Bhoc strategy, achieving similar results.
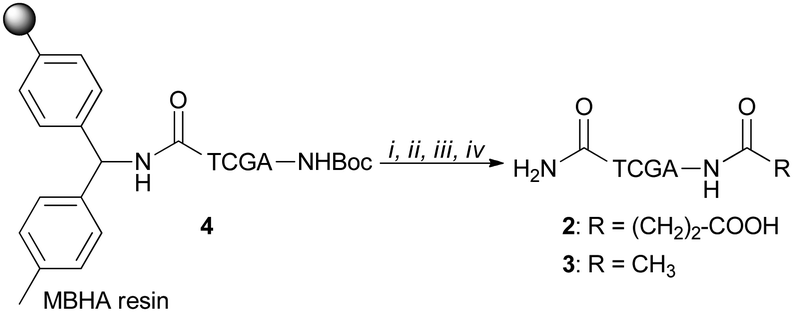 | ||
| Scheme 2 Synthesis of PNAs 2 and 3: (i) TFA/m-cresol (95:5); (ii) DIPEA (5% in DCM); (iii) succinic anhydride (20 equiv. for 2) or acetic anhydride (20 equiv. for 3), NMP, 4 h; (iv) TFA/TFMSA/m-cresol/thioanisole (6:2:1:1), 1 h. | ||
Synthesis of HNTs/PNA nanomaterials
The loading of the three different PNA tetramers 1–3 onto halloysite was carried out by following a standard procedure adopted for the supramolecular loading of organic molecules onto HNTs.19 In detail, a dispersion of halloysite (50 mg) in water (2.5 mL) was mixed with a solution of PNA tetramers in water (10 mM, 0.5 mL). Afterward, the dispersions were evacuated for 30 min (200 torr) and left to stir at room pressure for 15 min, and this cycle was repeated twice. Finally, the dispersions were stirred at room temperature for ca. 16 h. After work-up, the amount of PNA tetramers loaded on HNTs was ca. 5 wt%, 6.3 wt% and 5.8 wt% for HNTs-1, HNTs-2 and HNTs-3, respectively, as estimated by TGA. Based on the electrostatic interactions which can occur between HNTs and the three PNA tetramers, three different nanomaterials can be obtained where the PNA molecules are loaded inside the HNT lumen (HNTs-2), on the external surface (HNTs-1) and on both surfaces (HNTs-3), respectively (Scheme 3).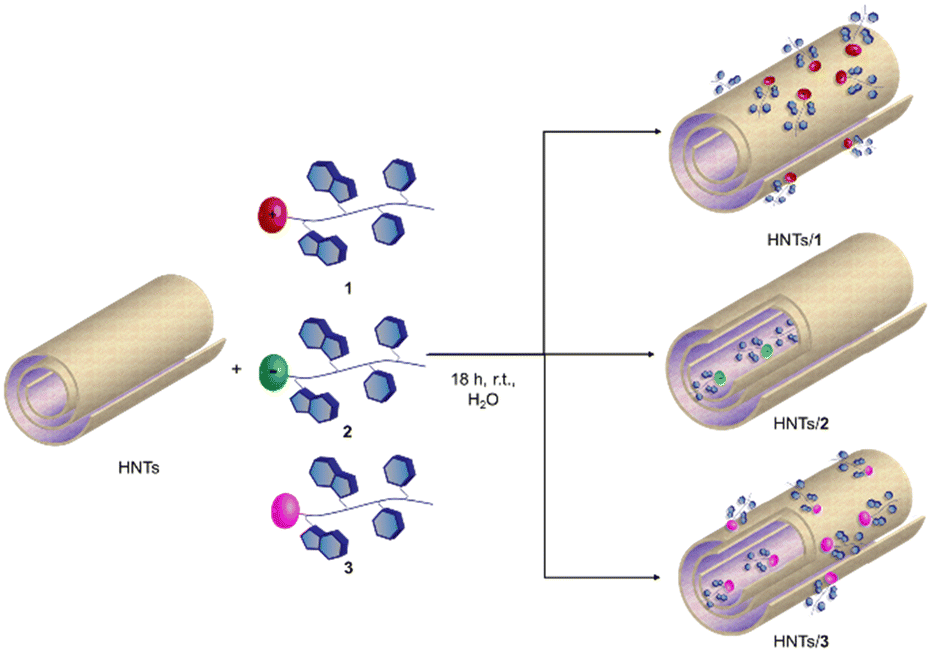 | ||
| Scheme 3 Schematic representation of PNA tetramers 1, 2 and 3 onto HNTs. | ||
The HNTs/PNA nanomaterials were characterized by FT-IR spectroscopy and TGA, and the colloidal properties were estimated by DLS and ζ-potential measurements. Furthermore, the morphology of the nanomaterials was imaged by TEM and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) coupled with an EDX probe.
Firstly, the nanomaterials were characterized by thermogravimetric analysis/derivative thermogravimetry (TGA/DTG) under an air atmosphere.
PNA molecules 1–3 totally decompose in a multi-step process occurring in a temperature range between 200 and 600 °C (Fig. 3a).
 | ||
| Fig. 3 Thermoanalytical curves and their derivatives of (a) PNA tetramers 1, 2 and 3; (b) HNTs/1, HNTs/2 and HNTs/3 nanomaterials. | ||
In contrast, HNTs exhibit relatively good stability up to ca. 450 °C, and then between 450 and 600 °C a mass loss occurs, due to the expulsion of the interlayer water molecules of HNTs (black line in Fig. 3b). By the comparison of the TGA curves (Fig. 3b) of pristine HNTs with those of HNTs/1, HNTs/2 and HNTs/3 nanomaterials, it is possible to observe that in all cases investigated, the nanomaterials, according to the presence of organic molecules, start to be degraded earlier than the pristine HNTs. Analysis of the DTG curves showed, indeed, that the HNTs/PNA nanomaterial degradation starts at ca. 240 °C, reaching stability up to 800 °C. The amount of PNA tetramers loaded into HNTs was calculated using the rule of a mixture by considering the mass losses at 25–150 °C (ML150) and the residual masses at 800 °C (MR800) for the pristine components (HNTs and PNA tetramers) and the HNTs/PNA nanomaterials.20,21 From these data it was possible to calculate the loading efficiencies reported above.
The FT-IR spectra of pristine HNTs, HNTs/1, HNTs/2 and HNTs/3 nanomaterials are reported in Fig. 4. As it is possible to observe, besides the typical vibration bands of halloysite, all nanomaterials present some additional vibration features attributable to the supramolecularly interacting PNA tetramers. In detail, it is possible to observe the stretching vibration of the amide bond at ca. 1650 cm−1 and the peaks in the range 1600–1350 cm−1 related to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N groups. In addition, the bands at ca. 1583, 1508, 1440, 1414 and 1290 cm−1 related to the stretching vibrations of the carbonyl and aromatic groups of the tetramers are also observable.
C and C–N groups. In addition, the bands at ca. 1583, 1508, 1440, 1414 and 1290 cm−1 related to the stretching vibrations of the carbonyl and aromatic groups of the tetramers are also observable.
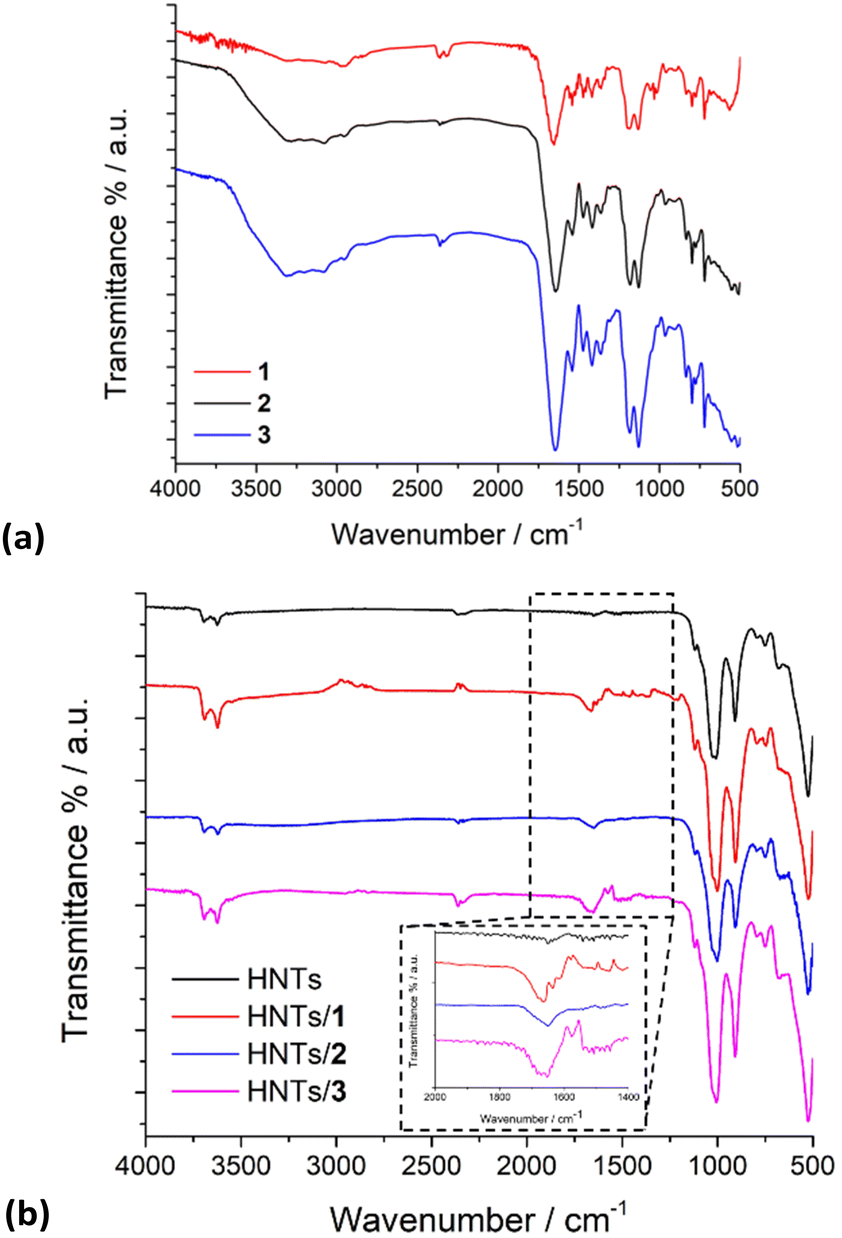 | ||
| Fig. 4 FT-IR spectra of (a) PNA tetramers 1, 2 and 3; (b) HNTs, HNTs/1, HNTs/2 and HNTs/3 nanomaterials. | ||
The colloidal properties of the obtained HNTs nanomaterials were estimated by DLS and ζ-potential measurements. DLS measurements allow the determination of the structural characteristics of the nanomaterials by monitoring their mobility in water and by measuring the average translational diffusion coefficient. This coefficient considers the dimension, shape and hydration of the diffusing particles and also the existence of aggregation phenomena. By applying the Stokes–Einstein equation it is possible to calculate the average diameter of the equivalent sphere, which can be considered as an index to follow the changes in particle dimensions and interparticle aggregation. The results obtained are reported in Table 1 and Fig. 5. As it is possible to note pristine HNTs showed an average size of ca. 422 nm with a polydispersity index (PDI) of ca. 0.31. Since the PDI is a measure of the heterogeneity of a certain sample based on its size, it is possible to conclude that in this case the value obtained indicates a broad size distribution of diffusing objects. After loading of positively charged PNA onto HNTs the Z-average size of the nanomaterial HNTs/1 decreases according to the introduction of a hydrophilic moiety onto HNTs, which allows better diffusion in aqueous medium.
| Nanomaterial | Z-Average size (nm) | PDI | ζ-Potential value (mV) |
|---|---|---|---|
| HNTs | 422 ± 30 | 0.31 ± 0.04 | −34 |
| HNTs/1 | 329 ± 10 | 0.23 ± 0.01 | −31.9 |
| HNTs/2 | 484 ± 16 | 0.43 ± 0.04 | −39 |
| HNTs/3 | 320 ± 10 | 0.25 ± 0.03 | −30.9 |
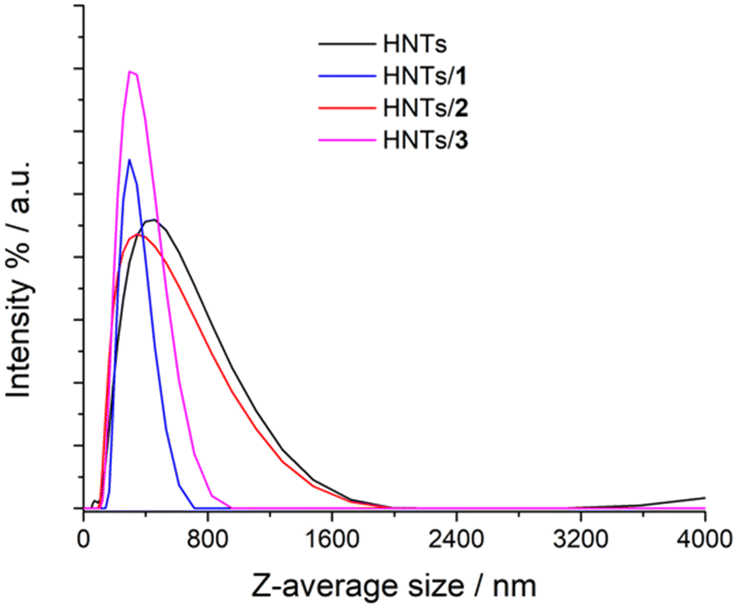 | ||
| Fig. 5 Distribution functions of the Z-average size values of HNTs, HNTs/1, HNTs/2 and HNTs/3 nanomaterials. | ||
The HNTs/2 nanomaterial showed a Z-average size value very similar to that of pristine HNTs, further confirming the loading in the lumen. The interaction of the clay with the uncharged PNA tetramer 3 led to the formation of nanoparticles in the dispersion which possess a Z-average size smaller than that of HNTs. This finding led us to hypothesize that the loading of 3 could occur both in the lumen and on the HNT external surface (see infra). Therefore, based on these findings, it is possible to conclude that the introduction of PNA tetramers onto HNT surfaces did not induce relevant variation in the diffusion mode of pristine HNTs so, it could be expected, for future biological applications, that the HNTs/PNA nanomaterials show the same cellular uptake of halloysite.7
The effect of the different PNA loading onto HNTs was investigated by ζ-potential measurements in water and the obtained results are reported in Table 1. As it is possible to observe, all nanomaterials present a negative ζ-potential value. The nanomaterials HNTs/1 and HNTs/3 showed ζ-potential values close to the one of pristine HNTs. The slight increase in their values in comparison to HNTs further confirms the presence of PNA tetramers on the external surface. The HNTs/2 nanomaterial presents a ζ-potential value of ca. −39 mV, more negative of HNTs. In the nanomaterial HNTs/2 indeed, PNA molecules interact with the HNT lumen leading to a partial neutralization of the internal positive charge as demonstrated by the lower ζ-potential value in comparison to pristine HNTs.
The morphology of the different nanomaterials was imaged by TEM and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM). The TEM images of all HNTs/PNA nanomaterials (Fig. 6a–c and Fig. S1, ESI†) showed the characteristic tubular structure of halloysite. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping showed that the PNA molecules were present on the overall surface of the tubes, as highlighted by the distribution of N atoms (highlighted in red in all samples). Moreover, close observation of the tubes showed the presence of N atoms localized in different portions of the tubes depending on the PNA tetramer loaded into HNTs.
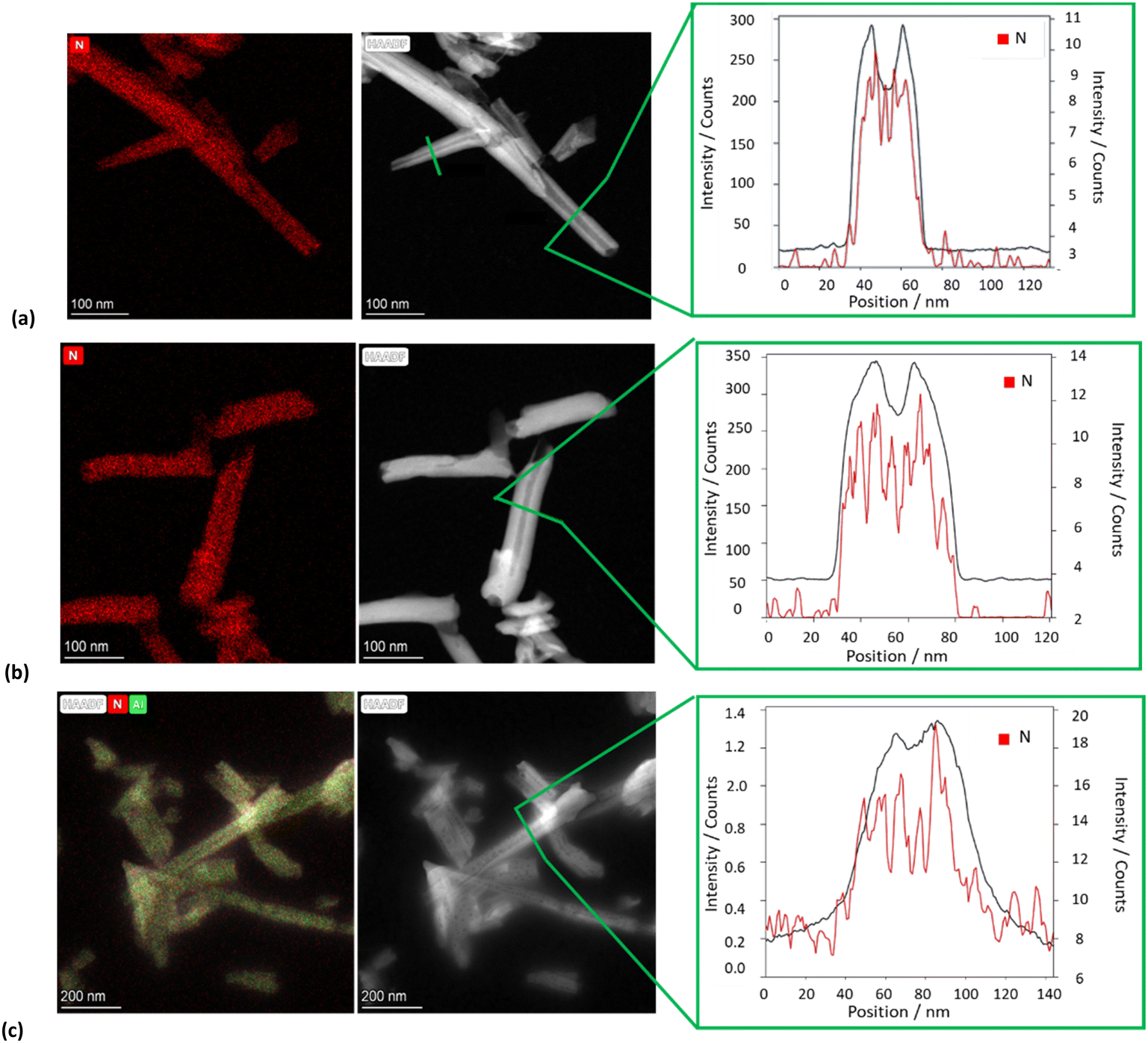 | ||
| Fig. 6 TEM, HAADF-STEM images and EDS elemental mapping of N atoms along the selected area of the HAADF-STEM image of (a) HNTs/1, (b) HNTs/2 and (c) HNTs/3 nanomaterials. | ||
In particular, EDS elemental mapping performed along the HNT section highlighted the presence of N atoms at the external surface in the case of nanomaterial HNTs/1 (Fig. 6a), in the lumen in the case of HNTs/2 (Fig. 6b) and colocalized at both surfaces in HNTs/3 (Fig. 6c) confirming the aforementioned hypotheses. It should also be noted that in the case of the nanomaterial HNTs/3 (Fig. 6c) the external surface of HNTs appears to be less defined probably due to the low contrast of C atoms due to their low atomic number Z, further confirming the presence of organic matter interacting with it.
Kinetic release
To evaluate the performances of the obtained nanomaterials for application in the biomedical field, the kinetic release of the different PNA tetramers from HNTs/1, HNTs/2 and HNTs/3 nanomaterials was evaluated by the dialysis bag method at pH 7.0 and pH 3.0 to mimic physiological conditions. In particular pH 3.0 was chosen to simulate gastric pH for potential oral administration of the obtained nanomaterials. The obtained kinetic data are reported in Fig. 7. As it is possible to observe, the release of PNA tetramer 1 from the HNTs/1 nanomaterial was very fast reaching the 100 wt% of molecules released after ca. 300 min at both pH values investigated (Fig. 7a). This behavior agrees with the desorption of molecules from the HNT external surfaces.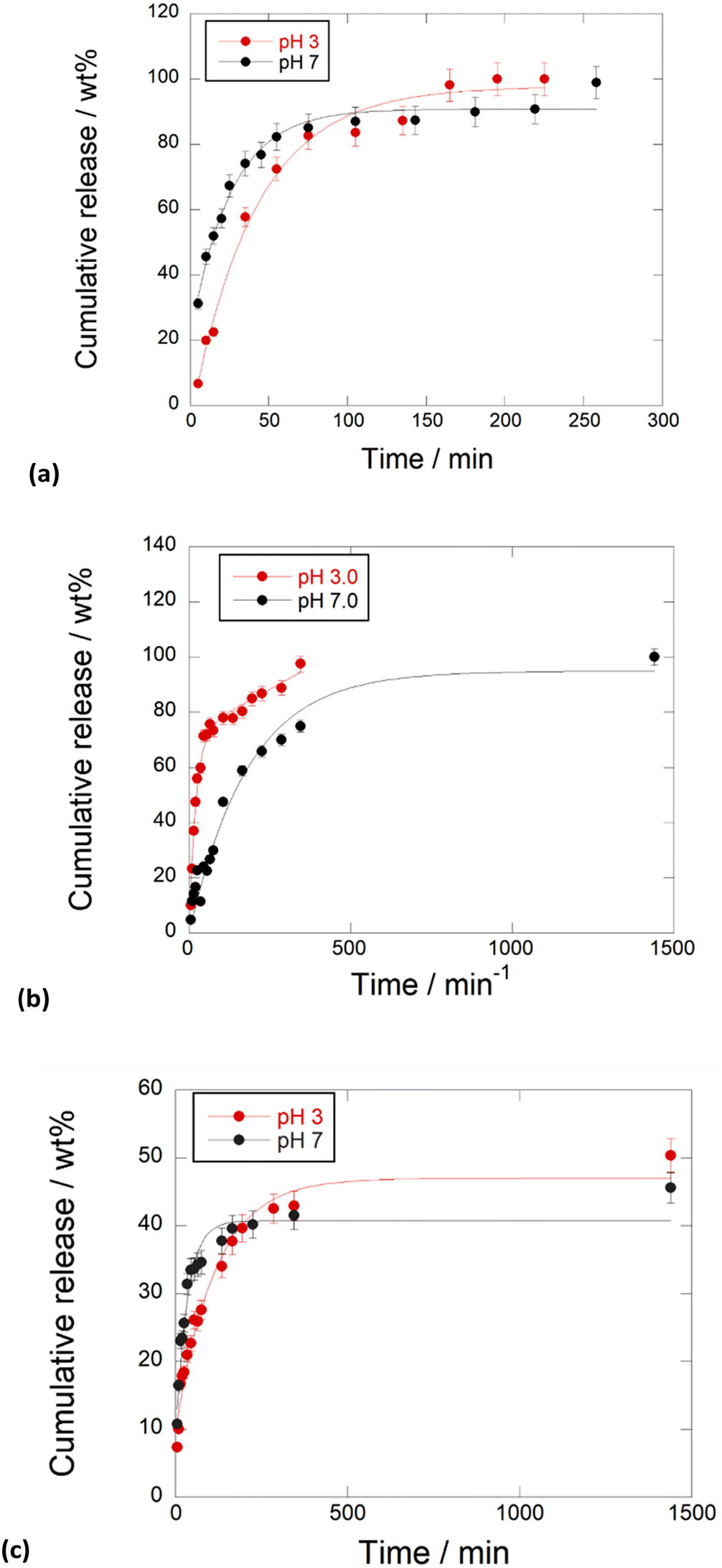 | ||
| Fig. 7 Kinetic release of PNA tetramers from (a) HNTs/1, (b) HNTs/2 and (c) HNTs/3 nanomaterials at pH 3.0 and pH 7.0, 37 °C. Reported are the mean ± standard deviation values. | ||
Regarding the release of 2 from the HNTs/2 nanomaterial, it showed different behavior depending on the release medium. In particular, it was observed that, in acidic medium, the total amount of 2 loaded into HNTs was released from the carrier in ca. 350 min, while a slow release at pH 7.0 was observed, and in this case the total amount of PNAs loaded was released after 24 h. These findings could be explained by the existence of electrostatic attraction interactions existing between the negatively charged tetramer 2 and the positively charged HNT lumen, that are missing at pH 3.0. The HNTs/3 nanomaterial showed different behavior. In this case both at pH 3.0 and pH 7.0 a slow release of tetramer 3 was observed, where ca. 40 wt% of the total tetramer loaded is released after 500 min, reaching a plateau after 24 h (Fig. 7b).
This is in agreement with the presence of 3 at both HNT surfaces, indeed, it was possible to hypothesize that some attraction interactions could exist between the PNA molecules which slow down the overall release as observed for other kinds of molecules released by the tubes.22
Similar results were found by Bertucci et al. who reported that by tuning the net charge of specific PNA loaded into porous silicon nanoparticles it is possible to modulate the temporal profile of the delivery process.23
The kinetic data were analyzed by different mathematical models to obtain information about the release mode. In particular different models, namely zero-order, first-order, Power Fit and the combination of two different models, namely zero and first order models, were applied. The obtained fits revealed that the release of 1 from HNTs/1 follows the first order at both pH models (k = 0.021 ± 0.002 min−1, R2 = 0.9899 and k = 0.043 ± 0.003 min−1, R2 = 0.9967 at pH 3.0 and 7.0, respectively) indicating fast desorption from the HNT external surface. In contrast, the release of 2 is better explained by the sum of an exponential and a linear term (k1 = 0.05 ± 0.01 min−1, k2 = 0.048 ± 0.004 min−1, R2 = 0.9852) at pH 3.0, due to the diffusion of 2 from the lumen which could probably interact with the HNT external surface by hydrogen bonding interactions and thus fast released in the medium. Conversely, under neutral conditions, the experimental data are well fitted by a first order model (k = 0.017 ± 0.003 min−1, R2 = 0.9846) indicating slow diffusion from the HNT lumen.
Regarding PNA 3, its release from the HNTs/3 nanomaterial at pH 3.0 is better described by a first order model (k = 0.008 ± 0.001 min−1, R2 = 0.9748) in agreement with diffusion/desorption from HNT surfaces.
Conversely, at pH 7.0, the kinetic data are better fitted by the sum of an exponential and a linear term (k1 = 0.051 ± 0.004 min−1, k2 = 0.005 ± 0.001 min−1, R2 = 0.9748). The latter results could agree with the interaction of 3 with both HNT surfaces.
Molecular modelling
On the other hand, the tetramer structures optimized with DMol3 in an aqueous environment have all heterocyclic rings in a parallel orientation to each other, except the adenine ring in the chloride salt of PNA 2 (Fig. 8).
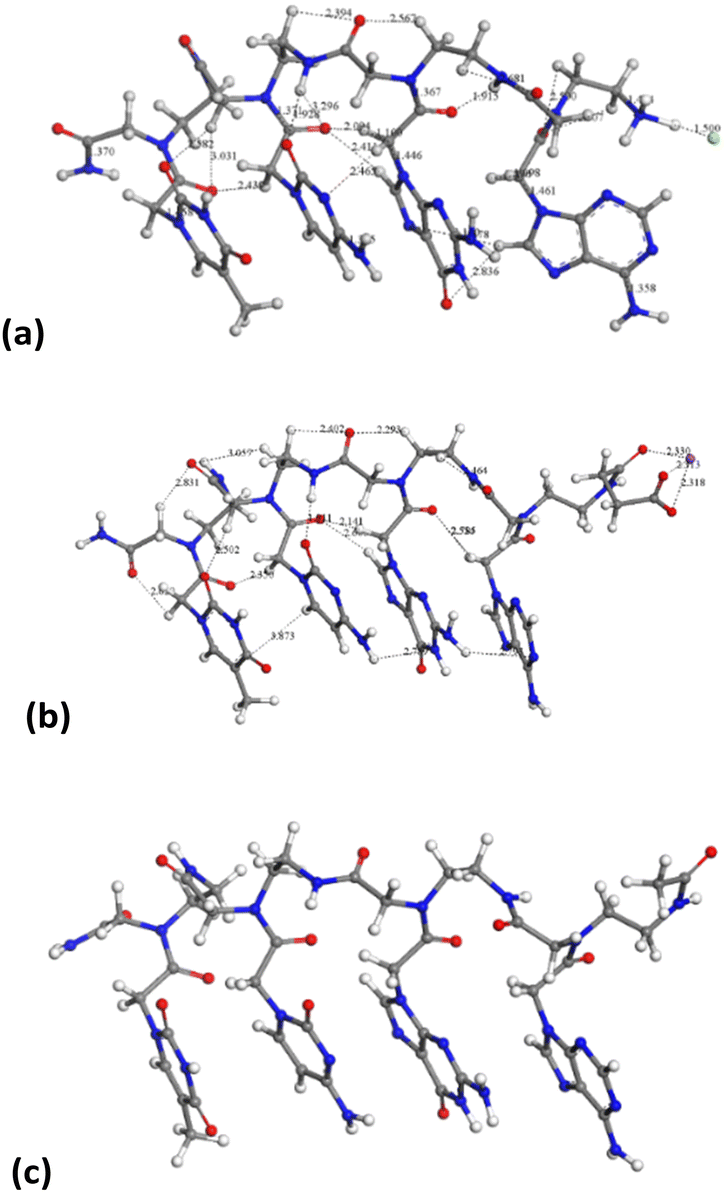 | ||
| Fig. 8 Molecular structures optimized by DFT in aqueous media PNA tetramer 1 (chloride salt) (a), PNA tetramer 2 (Na salt) (b), and PNA tetramer 3 (c). The main intramolecular interactions are described by dashed lines. The H, C, N, and O atoms are in white, gray, blue and red colors, respectively. This criterium is applied to the rest of the atomic models in this work. | ||
The average distance between heterocyclic rings is 2.4–3.0 Å owing to π–π interactions and some hydrogen bonds CO⋯HC. The peptidic chain is maintained by hydrogen bonds between carbonyl groups and NH and CH groups, d(CO⋯HN) = 1.915–2.600 Å, d(CO⋯HC) = 2.39–3.03 Å. This orientation is similar to that observed experimentally in the crystal structure of a peptide nucleic acid duplex from X-ray diffraction,24 where there is an aqueous environment with the crystallization of water molecules. Hence, the presence of water molecules facilitates the co-planarity of all nucleotide rings in these tetramers.
For confirming the validation of these calculations, the IR frequencies of the main vibrational modes were calculated and compared with the values of the experimental spectra (Table 2).
| Mode | PNA tetramer 1 | PNA tetramer 2 | PNA tetramer 3 |
|---|---|---|---|
| a Cytosine. b Chain terminal amide group. c Adenine. d Guanine. e Peptidic chain. f Thymine. g Ammonium cation. h Carboxylate. | |||
| ν(NH2)as | 3650a, 3644b, 3639c, 3598d | 364cg, 3641a, 3639b, 3585d, 3529e | 3661c, 3648b, 3644a, 3585d |
| ν(NH2)s | 3518a, 3506b, 3504c, 3494d | 3511ac, 3499b, 3480d | 3527c, 3513a, 3505b |
| ν(NH) | 3524e, 3507f, 3498a, 3468g, 3348–3344e, 3173g | 3517f, 3509d, 3496–3462e, 3330e | 3518b, 3516f, 3508d, 3482–3320e |
| ν(CH) | 3227c, 3221c, 3211–3194a, 3188f, 3153b, 3117c | 3268c, 3218d, 3215a,3206f, 3181a, 3150c | 3222c, 3220f, 3219ad, 3157a, 3127c |
| ν(CH2) | 3144–3024 | 3149–3071, 3054–2993s | 3165–3158, 3148–3138, 3126–2989 |
| ν(CH3) | 3079, 3018–3005sf | 3126, 3012s | 3107, 3000sf |
|
ν(CO) |
1764d, 1749b, 1738e, 1735f, 1727e, 1725f, 1708–1678e | 1746d, 1742b, 1718f, 1711b, 1706f, 1698–1655b, 1646b, 1551h | 1757d, 1735b, 1709e, 1707b, 1706f, 1705e, 1697f, 1673–1646e |
| Ring | 1573c, 1566–1518d, 1509–1480a | 1651af, 1580c, 1576–1552d, 1513a, 1480c | 1667f, 1650a, 1580c |
| δ(NH2)s | 1633dg, 1629e, 1623c, 1584a, 1553b | 1639–1589, 1565–1654 | 1630–1560 |
| δ(NH) | 1542–1533b, 1451, 1385–1310 | 1525–1520, 1495, 1451, 1380–1376 | 1526–1521 |
| δ(CH) | 1676f, 1649a, 1489–1483c, 1486–1454e, 1446–1403se | 1475–1452, 1449–1399, 1381 | 1475–1405 |
| ν(C–NH2) | 1488 | ||
| δ(CH3) | 1457f | 1436, 1394s | |
| γ(CH) | 1385–1310, 880–758 | 1374–1316, 1312–1180, 1165–774, 697–640 | 1235–1209e, 775d |
| γ(NH) | 961–926g, 720–667e | 760f, 744d, 705f | 739–684s |
The calculated frequencies are consistent with the experimental spectra in all cases. The broad multiple bands that appear at 3650–2900 cm−1 in the experimental spectra correspond to the overlapping result of the stretching modes of NH and CH bonds. The bands higher than 3300 cm−1 are assigned to the stretching vibration modes of N–H bonds. The shoulder observed at frequencies higher than 3520 cm−1 in the experimental spectra can be assigned to the asymmetric stretching mode of the NH2 group, ν(NH2)as. Our calculations allow a distinction between the vibration modes of different amino groups, from adenine, cytosine, guanine, the terminal amide, and those from the peptidic chain. The ν(NH2)as mode of adenine shows higher frequency in 2 and 3, except in 1 due to the different torsional position of the heterocyclic ring. In contrast, guanine shows the lowest frequencies probably due to the interactions with vicinal heterocycles. A similar effect is observed in the symmetric mode of ν(NH2)s whose bands appear at slightly lower frequencies and they can be within the high-frequency shoulder of the experimental band. The ν(NH) bands of the N–H groups appear at slightly lower frequencies than the above ones. The NH groups of the peptide chain show lower frequencies due to intramolecular interactions of vicinal CO groups. In 1 the ν(NH) mode of the ammonium group appears at a lower frequency than other NH groups. The bands that appear at the frequency range 3268–2989 cm−1 are assigned to the stretching mode of the C–H bond of the CH, CH2, and CH3 groups.
The intense band observed experimentally at 1760–1550 cm−1 is the overlapping of bands assigned to the stretching ν(CO), bending δ(NH2)s and ring deformation modes. The shoulder at a higher frequency corresponds to ν(CO) where small frequency differences between CO groups can be observed in our calculations, owing to different electronic and intramolecular interactions. In 2 the ν(CO) of carboxylate appears at a lower frequency of 1551 cm−1. The small band observed experimentally at 1525–1500 cm−1 can be assigned to the δ(NH) mode confirmed by our calculations. The bands appearing at the 1480–1350 cm−1 range of the experimental spectra are assigned by our calculations to several δ(NH) and δ(CH) modes. The bands at 1100–1230 cm−1 of the experimental spectra are assigned to the out-of-plane deformation mode, γ(CH). In 1, the experimental spectrum shows a band at 930–980 cm−1 assigned to γ(NH) of the ammonium group by our calculations, whereas the γ(NH) mode of the rest of the groups appears at 850–640 cm−1 along with some bands of γ(CH).
The IR spectra of these molecules were simulated (Fig. 9), being consistent with the experimental ones (Fig. 4a).
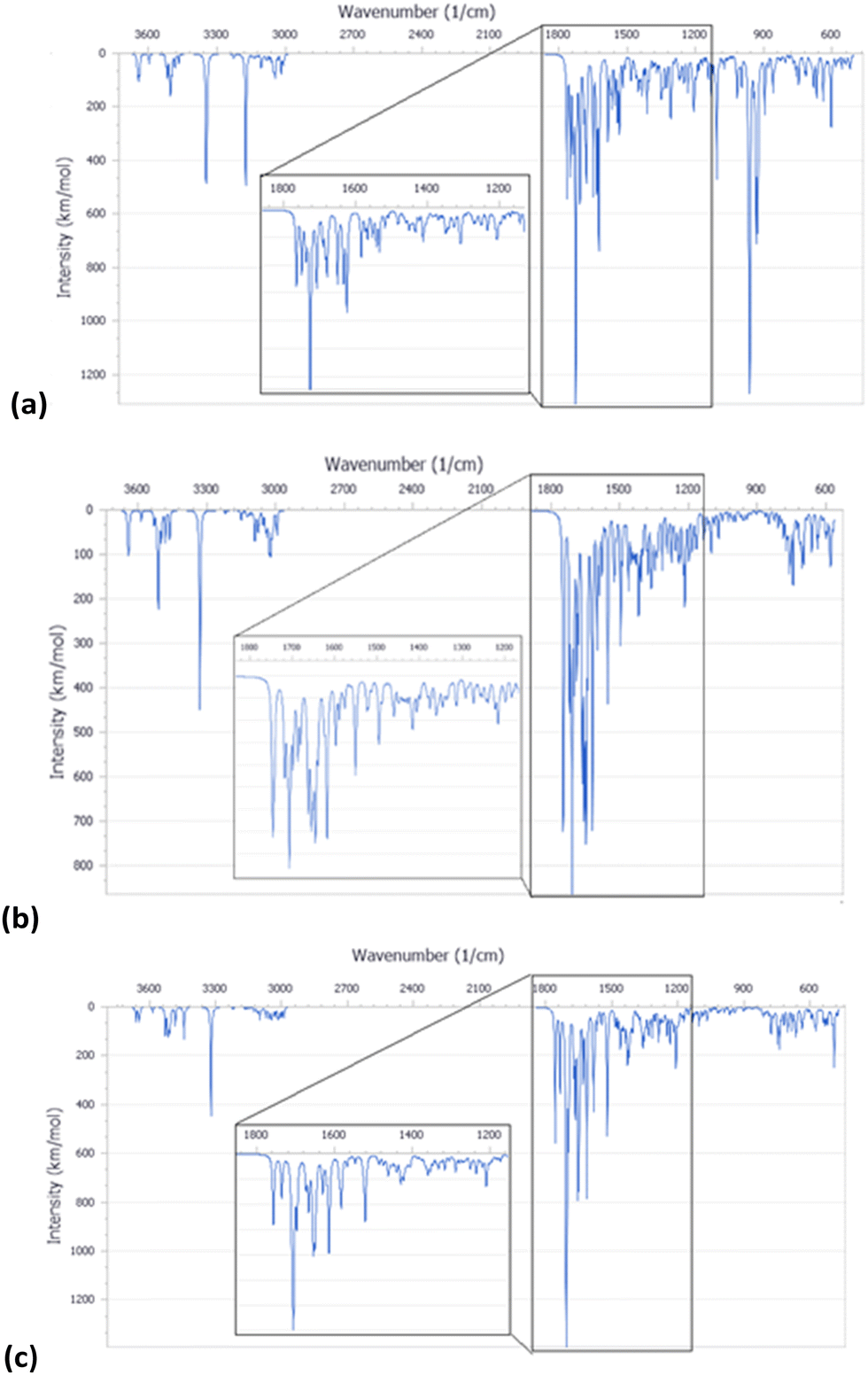 | ||
| Fig. 9 IR spectra simulated from the calculated molecular structure with DFT for PNA tetramer 1 (chloride salt) (a), PNA tetramer 2 (Na salt) (b), and PNA tetramer 3 (c). | ||
PNA tetramers’ adsorption on the internal or/and external surfaces of halloysite
Following the experimental data, we studied the adsorption of three tetramers of PNA on halloysite. Specifically, we generated four models: halloysite with tetramer 1 adsorbed on the external surface; halloysite with tetramer 2 adsorbed on the internal surface; halloysite with tetramer 3 adsorbed on the external surface; and halloysite with tetramer 3 inside the nanotube. After the equilibration and optimization of these complexes with the COMPASS force field, the resultant structures were analyzed (Fig. 10).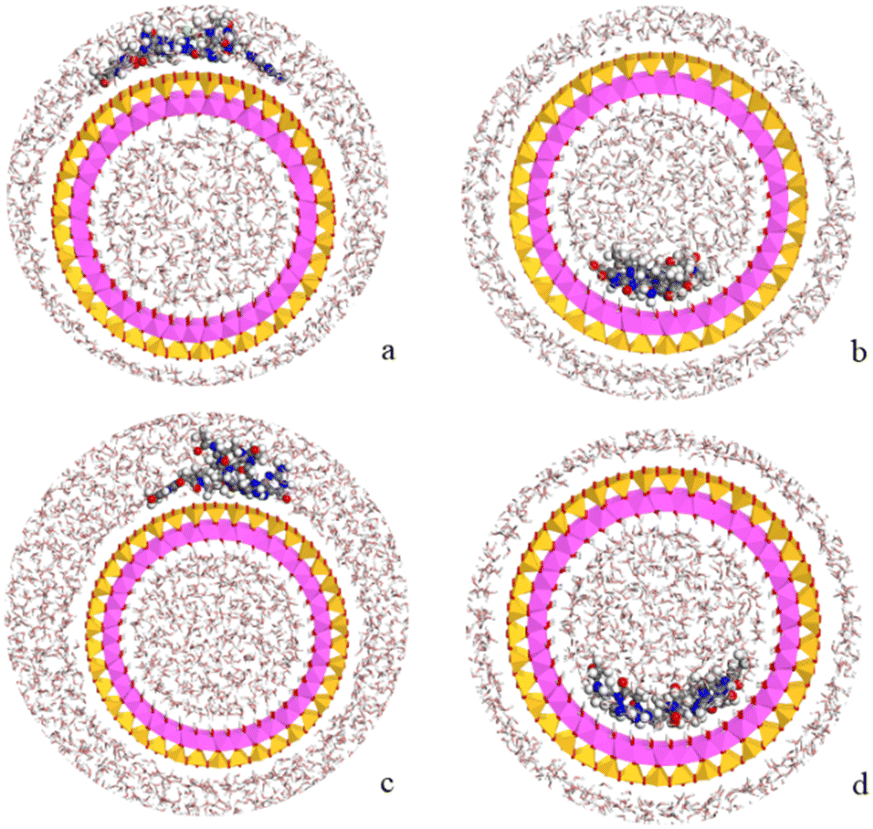 | ||
| Fig. 10 Complexes of PNA tetramer 1 adsorbed on the external surface of halloysite (a), of PNA tetramer 2 adsorbed on the internal surface (b) and of PNA tetramer 3 adsorbed on the external (c) and internal (d) surfaces of the nanotube in water solution. The atoms of silicon, aluminum, oxygen, hydrogen, carbon, nitrogen, chlorine and sodium are presented in yellow, pink, red, white, grey, blue and light green and purple, respectively. | ||
In all the adsorption complexes (Fig. 10), the PNA tetramers are positioned with an orientation parallel to the surface of the nanotube with a mainly flat conformation. Moreover, when the PNA tetramers are adsorbed on the external surface of the nanotube, hydrogen bond interactions between the hydrogen atoms of the tetramers and the oxygen atoms of the surface around 2.5 Å were found. When the PNA tetramers are adsorbed on the internal surface of halloysite, the main interactions were hydrogen bonds between the oxygen of the carbonyl groups and the hydrogens of the internal surface around 2.0 Å.
Lastly, the adsorption energies of the models were compared according to the equation Eads = (ECOMPLEX) – (EPNA + EHNTs). The adsorption energy of the complex with the PNA tetramer 1 (Fig. 10a) was −157.7 kcal mol−1, of the complex with PNA tetramer 2 (Fig. 10b) was −222.4 kcal mol−1, of the complex with PNA tetramer 3 adsorbed on the external surface (Fig. 10c) was −127.3 kcal mol−1 and inside the nanotube (Fig. 10d) was −51.3 kcal mol−1. The results showed that all the complexes presented negative adsorption energy. Therefore, the loading of the PNA tetramers is favorable on the halloysite nanotube.
Experimental
Materials and methods
All the reagents and solvents were purchased from commercial sources and used without further purification.Halloysite nanotubes used in this study were obtained from Merck and used as received.
The monomers Boc–PNA–T–OH (>95%), Boc–PNA–C(Z)–OH (>95%), Boc–PNA–G(Z)–OH (>95%), Boc–PNA–A(Z)–OH (>95%), Fmoc–PNA–T–OH (>95%), Fmoc–PNA–C(Bhoc)-OH (>95%), Fmoc–PNA–G(Bhoc)–OH (>95%), Fmoc–PNA–A(Bhoc)-OH (>95%) were purchased from ASM Research Chemicals GmbH (Hannover, Germany). HNTs (lyophilized powder, MQ200) were purchased from Merck (Darmstadt, Germany). Polystyrene MBHA resin (0.88 mmol g−1) was purchased from Novabiochem, and the H-rink amide ChemMatrix® (0.60 mmol g−1) was purchased from Merck (Darmstadt, Germany). Polypropylene one-way syringes and the corresponding polytetrafluoroethylene (PTFE) frits, used as reactors for the manual solid phase synthesis, were purchased from Alltech Associates Inc. (Lokeren, Belgium). The reverse-phase RP-HPLC purification of PNA tetramers 1–3 was performed on an Agilent 1200 Series system, using the semi-preparative Luna C18 column (25 cm × 10 nm, 5 μm). A binary mixture of solvents A (water + 0.1% TFA) and B (acetonitrile + 0.1% TFA) was used as the mobile phase. Chromatographic purifications were performed at room temperature, with a constant flow rate of 3 mL min−1 and linear gradients. The absorbance was measured at 260 and 280 nm. UPLC-MS analyses were recorded on an Acquity UPLC I-class instrument (Waters) equipped with an Acquity PDA detector (Waters). Chromatographic separation was performed using an ACQUITY UPLC BEH C18 column (100 × 2.1 mm, 1.7 μm) (Waters) fitted with a VanGuard cartridge (Waters) for tetramer 3, and an ACQUITY UPLC HSS T3 C18 column (100 × 2.1 mm, 1.8 μm) (Waters) fitted with a VanGuard cartridge (Waters) for tetramers 1 and 2 at a constant flow rate of 0.4 mL min−1. A binary mixture of solvents A (water + 0.1% formic acid) and B (acetonitrile + 0.1% formic acid) was used as the mobile phase. The absorbance was measured at 260 nm. HR-ESI+ MS(/MS) analyses were recorded with a Synapt G2-Si QToF instrument (Waters) interfaced through a Zspray™ ESI-probe for electrospray ionization (Waters).
The concentration of PNA solutions in aqueous solution was determined by measuring the absorbance at 260 nm with an Agilent 8453 UV/Vis spectrophotometer. The molar extinction coefficient of PNA tetramers was calculated according to the literature.25 Water HPLC Plus grade (Merck) was used to prepare PNA solutions and for the loading experiments of PNAs on HNTs.
A suspension of HNTs in water was sonicated with an Elmasonic S30H sonicated at T = 30 °C. The vacuum/air cycles were performed using a vacuum chamber connected to a diaphragm vacuum pump and a VC-900 vacuum regulator. The FT-IR spectra were registered using a Jasco Mod FT-IR 4600 Infrared spectrometer in transmittance mode. The DLS and Zeta Potential analyses were registered using a Malvern Zetasizer Nano instrument at 25 °C, equipped with a 633 nm solid state He–Ne laser at a scattering angle of 173°. TGA analyses were performed by means of a PerkinElmer TGA thermogravimetric analyser, under air in the temperature range of 30 °C to 800 °C at a heating rate of 10 °C min−1.
Transmission Electron Microscopy (TEM) was performed by means of an FEI Titan G2 60–300 ultra-high resolution transmission electron microscope coupled with Analytical Electron Microscopy (AEM) performed with a SUPER-X silicon-drift windowless X-ray Energy-Dispersive Spectroscopy (XEDS) detector. AEM spectra were saved in mode STEM (Scanning Transmission Electron Microscopy) with a HAADF (High Angle Annular Dark Field) detector. X-ray chemical elemental maps were also collected.
Synthetic procedures
:5, v/v, 2 × 8 min). The resin was then washed with CH2Cl2 and NMP. In a vial, a solution of DIPEA (10 equiv., 0.45 mmol, 78 μL) and the Fmoc/Bhoc protected monomer (5.0 equiv., 0.239 mmol) in NMP was added to a solution of HATU (5.0 equiv., 0.216 mmol, 82 mg) in NMP, and the resulting mixture was shaken for 2 min. The activated acid was then transferred to the resin and shaken for 2 h at room temperature. The resin was then washed with NMP and CH2Cl2. The two-step cycle, Boc-deprotection and coupling, was repeated for each PNA monomer to obtain the resin-supported PNA tetramer 5. The resin was treated with a solution of piperidine in NMP (1:5, v/v, 2 × 8 min), and then washed with NMP and CH2Cl2. Subsequently, the resin was dried under vacuum and treated with a cleavage mixture of TFA/m-cresol (9/1, v/v) for 1.5 h at room temperature. After filtration and partial evaporation of TFA, PNA 1 was precipitated in ice-cold diethyl ether, and the precipitate was dried, dissolved in water, and purified by RP-HPLC. PNA sample 1 was isolated in a similar amount and purity to those obtained following the Boc/Z strategy.
Kinetic release
The release of the three PNA tetramers from HNTs/PNA nanomaterials was done as follows: 15 mg of the sample were dispersed in 1 mL of dissolution medium (HCl 1 × 10−3 N and phosphate buffers at pH 3.0 and 7.4, respectively) and transferred into a sealed dialysis membrane (Medicell International Ltd MWCO 12–14000 with a diameter of 21.5 mm). Subsequently the membrane was put in a round bottom flask containing 9 mL of the release medium at 37 °C and stirred. At fixed time, 1 mL of the release medium was withdrawn and analyzed by UV-vis measurements at a λ value of 260 nm. To ensure sink conditions, 1 mL of fresh solution was used to replace the collected one. Total amounts of drug released (Ft) were calculated as follows: | (1) |
Models
PNA tetramers 1–3 models were generated sketching by hand using the Materials Studio package26 taking into account the experimental information. The terminal amino group of PNA tetramer 1 was protonated and neutralized with a chloride anion according to experimental conditions. Analogously, the carboxylate terminal group of PNA tetramer 2 was neutralized with a Na+ cation following the experimental conditions. A zwitterionic form of a neutral PNA tetramer 3 model was also generated and inserted in a periodical box of 15 × 15 × 35 Å along with 226 water molecules filled by the Monte Carlo method reaching a final density of 1 g cm−3. In the same way, the water molecule was previously generated by hand.On the other hand, a slide of halloysite nanotube, with the stoichiometry Al2Si2O5(OH)4, was obtained from previous work.27 Subsequently, a periodical crystal structure of this nanotube was generated using periodic boundary conditions22,28,29 The unit cell of halloysite has the formula Al76Si76O190(OH)152 with 646 atoms, and an internal diameter of 27 Å. This structure is a suitable model to study the drug-clay adsorption process. To carry out the adsorption of a PNA tetramer, a 1 × 1 × 4 supercell of halloysite was generated with the formula Al304Si304O760(OH)608 and with 2584 atoms.
Molecular modeling methodology
The geometry optimization of PNA tetramers 1–3 was performed with the COMPASS force field30 based on empirical interatomic potentials and using the Forcite code.26 The water molecule was also optimized with the COMPASS force field. Additionally, the solvation free energy of PNA tetramer 3 was calculated by means of molecular dynamics simulations of the periodical box with the zwitterionic model of the neutral PNA tetramer 3 and water molecules. After a simulation with the NVE ensemble with 1 fs steps for 5 ps, an additional simulation was performed at the NVT ensemble for 50 ps. Moreover, quantum mechanical calculations based on Density Functional Theory (DFT) were performed using the DMol3 code31 with GGA and PBE26 functionals. Double numerical plus polarization functions were used for the atomic orbital basis sets with a cut-off of 3.5 Å. The Grimme semiempirical dispersion correction was used32 and DFT semi-core pseudopotentials (DSPPs)33 were applied. The COSMO (COnductor like Screening MOdel) approach34 was used for simulating a solvent environment of an aqueous medium (dielectric constant = 78.54). The IR frequencies of the main normal vibration modes were calculated by analyzing the Hessian matrix produced by finite atomic displacements.On the other hand, the optimization of the unit cell of the halloysite nanotube structure was performed with quantum mechanical calculations using DFT with the CASTEP code.26 The functionals used were GGA and PBE. On-the-fly generated (OTFG) ultrasoft pseudopotentials were used with Koelling–Harmon relativistic treatment,35 and the cut off energy of the calculation was 300 eV.26 After the optimization of the halloysite unit cell, a 1 × 1 × 4 supercell was created to study the adsorption of the PNA tetramers and this halloysite structure was fixed, except for the hydrogen atoms of the internal surface.
PNA tetramers 1–3 were placed on the internal and/or external surface of halloysite, following the experimental data. Four complexes were prepared: halloysite with tetramer 1 adsorbed on the external surface; halloysite with tetramer 2 adsorbed on the internal surface; halloysite with tetramer 3 adsorbed on the external surface; and halloysite with tetramer 3 inside the nanotube. These drug-halloysite complexes were optimized and, subsequently, 100 ps of NVT molecular dynamic simulation was performed to equilibrate the systems using Forcite with COMPASS.26 Later, the last structure of the dynamics was again optimized and filled with the previously optimized water molecules using the Monte Carlo method with COMPASS.26 In this way, water with a density of 1 g cm−3 filled the adsorption complexes (∼3444 water molecules). After that, the resultant adsorption complexes were optimized with COMPASS to obtain the most stable structure and calculate its energy.
The adsorption energy of the complexes was estimated using the equation Eads = (ECOMPLEX) – (EPNA + EHNTs). For this, the energy of the optimized adsorption complexes was calculated (ECOMPLEX), as well as the tetramers and halloysite with the water of the optimized complexes were isolated and the optimized energy was calculated (EPNA and EHNTs), with the COMPASS force field.26
Conclusions
Halloysite is a versatile nanomaterial, and has shown promising features for application in nanomedicine. Because of its peculiar structure, different surface charges and the presence of an empty lumen, it is efficiently used as a nanocontainer for the delivery of active species.In the present work, the supramolecular interaction of the clay with three differently charged PNA tetramers, chosen as models, was carried out.
Firstly, PNA tetramers 1–3 were prepared manually according to standard protocols for the solid-phase synthesis of PNAs by adopting two different procedures, namely Boc/Z and Fmoc/Bhoc strategies, depending on the charge that would be introduced onto them.
Afterwards, they were loaded onto halloysite by following a standard procedure adopted for the supramolecular loading of organic molecules onto HNTs.
The obtained nanomaterials were characterized using FT-IR spectroscopy and thermogravimetric analyses that allowed calculating the loading percent of each PNA into HNTs. DLS and z-potential measurements highlighted that the three different nanomaterials possess different aqueous diffusion features and different surface charges in agreement with the different HNT surfaces interacting with the PNA tetramers. Indeed, the interaction of negatively charged PNA 2 with HNTs led to the synthesis of a nanomaterial with a similar Z-average size of HNTs and a more negative surface charge in agreement with its lumen confinement. In contrast, the interaction of halloysite with the positively charged PNA and the neutral one did not lead to any variation in the charge of the nanomaterials in comparison to HNTs, but they showed a slight increase in the average size of the nanomaterials. These findings were explained by considering the interaction of the positively charged PNA 1 with the HNT external surface and that of neutral 3 with both surfaces.
Morphological characterization using HAADF/STEM investigations highlighted the presence of the PNA tetramers at the external or inner surface or on both depending on their charge, as proved by the EDS elemental mapping along the tube section. Kinetic release experiments under different pH mimicking physiological conditions showed that PNA 3 is slowly released over time, whereas the release of PNA 1 and 2 is ruled by electrostatic forces.
Finally, theoretical calculations were performed to study the structures of the tetramers and to obtain the IR spectra of these molecules, being consistent with the experimental ones. Furthermore, the simulations confirmed the adsorption of the tetramers on halloysite, showing hydrogen bonds and electrostatic interactions taking place between the tetramers and the internal and/or external surfaces of the HNTs. The favorable adsorption processes observed in the experimental results were also confirmed.
In summary, this study reports the possibility to combine the PNA features with a low cost, naturally available in large amounts and biocompatible carrier such as halloysite. Furthermore, using the supramolecular approach it is possible to scale up the synthesis of these nanomaterials for future potential clinical applications of PNA based systems. The present study opens the doorway for the design of diversified PNA molecules with specific sequences able to interact with halloysite surfaces for the treatment of different pathologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mass spectrometry analyses of PNA tetramers were performed at the Mass Spectrometry facility of Unitech COSPECT at the University of Milan (Italy). The authors are thankful to Dr Emma Lambert.Notes and references
- S. Pawłowska, T. A. Kowalewski and F. Pierini, Soft Matter, 2018, 14, 8421–8444 RSC.
- M. I. Carretero, Appl. Clay Sci., 2002, 21, 155–163 CrossRef CAS.
- D. Peixoto, I. Pereira, M. Pereira-Silva, F. Veiga, M. R. Hamblin, Y. Lvov, M. Liu and A. C. Paiva-Santos, Coord. Chem. Rev., 2021, 440, 213956 CrossRef CAS.
- A. Stavitskaya, M. Rubtsova, A. Glotov, V. Vinokurov, A. Vutolkina, R. Fakhrullin and Y. Lvov, Nanoscale Adv., 2022, 4, 2823–2835 RSC.
- M. Massaro, R. Noto and S. Riela, Molecules, 2020, 25, 4863 CrossRef CAS PubMed.
- M. Massaro, S. Pieraccini, S. Guernelli, M. L. Dindo, S. Francati, L. F. Liotta, G. C. Colletti, S. Masiero and S. Riela, Appl. Clay Sci., 2022, 230, 106719 CrossRef CAS.
- M. Massaro, M. Notarbartolo, F. M. Raymo, G. Cavallaro, G. Lazzara, M. M. A. Mazza, C. Viseras-Iborra and S. Riela, ACS Appl. Nano Mater., 2022, 5, 13729–13736 CrossRef CAS.
- J. Tully, R. Yendluri and Y. Lvov, Biomacromolecules, 2016, 17, 615–621 CrossRef CAS PubMed.
- A. C. Santos, C. Ferreira, F. Veiga, A. J. Ribeiro, A. Panchal, Y. Lvov and A. Agarwal, Adv. Colloid Interface Sci., 2018, 257, 58–70 CrossRef CAS PubMed.
- M. V. Gorbachevskii, A. V. Stavitskaya, A. A. Novikov, R. F. Fakhrullin, E. V. Rozhina, E. A. Naumenko and V. A. Vinokurov, Appl. Clay Sci., 2021, 207, 106106 CrossRef CAS.
- F. R. Ahmed, M. H. Shoaib, M. Azhar, S. H. Um, R. I. Yousuf, S. Hashmi and A. Dar, Colloids Surf., B, 2015, 135, 50–55 CrossRef CAS PubMed.
- E. Rozhina, A. Panchal, F. Akhatova, Y. Lvov and R. Fakhrullin, Appl. Clay Sci., 2020, 185, 105371 CrossRef CAS.
- A. Stavitskaya, G. Fakhrullina, L. Nigamatzyanova, E. Sitmukhanova, E. Khusnetdenova, R. Fakhrullin and V. Vinokurov, Materials, 2021, 14, 5469 CrossRef CAS PubMed.
- S. Volpi, U. Cancelli, M. Neri and R. Corradini, Pharmaceuticals, 2021, 14, 14 CrossRef CAS PubMed.
- M. Terracciano, F. Fontana, A. P. Falanga, S. D'Errico, G. Torrieri, F. Greco, C. Tramontano, I. Rea, G. Piccialli, L. De Stefano, G. Oliviero, H. A. Santos and N. Borbone, Small, 2022, 18, 2204732 CrossRef CAS PubMed.
- R. Brazil, ACS Centr. Sci., 2023, 9, 3–6 CrossRef CAS PubMed.
- M. Massaro, E. Licandro, S. Cauteruccio, G. Lazzara, L. F. Liotta, M. Notarbartolo, F. M. Raymo, R. Sánchez-Espejo, C. Viseras-Iborra and S. Riela, J. Colloid Interface Sci., 2022, 620, 221–233 CrossRef CAS PubMed.
- P. E. Nielsen, Peptide Nucleic Acids: Methods and Protocols, Springer Science + Business Media, LLC, part of Springer Nature, Humana New York, NY, 3rd edn, 2020 Search PubMed.
- M. Massaro, G. Barone, V. Barra, P. Cancemi, A. Di Leonardo, G. Grossi, F. Lo Celso, S. Schenone, C. Viseras Iborra and S. Riela, Int. J. Pharm., 2021, 599, 120281 CrossRef CAS PubMed.
- L. Lisuzzo, G. Cavallaro, P. Pasbakhsh, S. Milioto and G. Lazzara, J. Colloid Interface Sci., 2019, 547, 361–369 CrossRef CAS PubMed.
- L. Lisuzzo, G. Cavallaro, S. Milioto and G. Lazzara, Polymers, 2020, 12, 1766 CrossRef CAS PubMed.
- M. Massaro, A. Borrego-Sánchez, R. Sánchez-Espejo, C. Viseras Iborra, G. Cavallaro, F. García-Villén, S. Guernelli, G. Lazzara, D. Miele, C. I. Sainz-Díaz, G. Sandri and S. Riela, Appl. Clay Sci., 2021, 215, 106310 CrossRef CAS.
- M. Neri, J. Kang, J. M. Zuidema, J. Gasparello, A. Finotti, R. Gambari, M. J. Sailor, A. Bertucci and R. Corradini, ACS Biomater. Sci. Eng., 2022, 8, 4123–4131 CrossRef CAS PubMed.
- H. Rasmussen, J. S. Kastrup, J. N. Nielsen, J. M. Nielsen and P. E. Nielsen, Nat. Struct. Biol., 1997, 4, 98–101 CrossRef CAS PubMed.
- https://www.pnabio.com/pdf/PNA_oligomer_handling_PNABio.pdf .
- M. S. Biovia, Discovery Studio Modeling Environment, Version 2018, Biovia, San Diego, CA, USA, 2018 Search PubMed.
- A. Borrego-Sánchez, C. I. Sainz-Díaz, L. Perioli and C. Viseras, Molecules, 2021, 26, 4392 CrossRef PubMed.
- A. Borrego-Sánchez, A. Hernández-Laguna and C. I. Sainz-Díaz, J. Mol. Model., 2017, 23, 106 CrossRef PubMed.
- C. Valentino, T. Martínez Rodríguez, A. Borrego-Sánchez, P. Hernández Benavides, F. Arrebola Vargas, J. M. Paredes, S. Rossi, C. I. Sainz Díaz, G. Sandri, P. Grisoli, M. D. M. Medina Pérez and C. Aguzzi, Pharmaceutics, 2023, 15, 1140 CrossRef CAS PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- B. Delley, Mol. Simul., 2006, 32, 117–123 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00637a |
| This journal is © The Royal Society of Chemistry 2023 |