
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Marine-origin polysaccharides-based free-standing multilayered membranes as sustainable nanoreservoirs for controlled drug delivery
Cristiana F. V.
Sousa
 ,
Luís P. G.
Monteiro
,
João M. M.
Rodrigues
,
João
Borges
* and
João F.
Mano
*
,
Luís P. G.
Monteiro
,
João M. M.
Rodrigues
,
João
Borges
* and
João F.
Mano
*
CICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: joaoborges@ua.pt; jmano@ua.pt
First published on 21st June 2023
Abstract
The layer-by-layer (LbL) assembly technology has been widely used to functionalise surfaces and precisely engineer robust multilayered bioarchitectures with tunable structures, compositions, properties, and functions at the nanoscale by resorting to a myriad of building blocks exhibiting complementary interactions. Among them, marine-origin polysaccharides are a sustainable renewable resource for the fabrication of nanostructured biomaterials for biomedical applications owing to their wide bioavailability, biocompatibility, biodegradability, non-cytotoxicity, and non-immunogenic properties. Chitosan (CHT) and alginate (ALG) have been widely employed as LbL ingredients to shape a wide repertoire of size- and shape-tunable electrostatic-driven multilayered assemblies by exploring their opposite charge nature. However, the insolubility of CHT in physiological conditions intrinsically limits the range of bioapplications of the as-developed CHT-based LbL structures. Herein, we report the preparation of free-standing (FS) multilayered membranes made of water-soluble quaternised CHT and ALG biopolymers for controlled release of model drug molecules. The influence of the film structure in the drug release rate is studied by assembling two distinct set-ups of FS membranes, having the model hydrophilic drug fluorescein isothiocyanate-labelled bovine serum albumin (FITC-BSA) either as an intrinsic building block or added as an outer layer after the LbL assembly process. Both FS membranes are characterised for their thickness, morphology, in vitro cytocompatibility, and release profile, with those having FITC-BSA as an intrinsic LbL ingredient denoting a more sustained release rate. This work opens up new avenues for the design and development of a wide array of CHT-based devices for biomedical applications, overcoming the limitations associated with the insolubility of native CHT under physiological conditions.
 João Borges | Dr João Borges is a Senior Researcher at CICECO – Aveiro Institute of Materials in the Department of Chemistry at the University of Aveiro, Portugal. He graduated and received his PhD in Chemistry from the University of Porto in 2008 and 2013, respectively. From 2013 to 2018, he was a postdoctoral fellow at the University of Minho and University of Aveiro. His research focuses on the molecular design, synthesis and development of supramolecular multicomponent biomaterials, by combining polysaccharides, self-assembling peptides and nucleic acids, to be used as bioinstructive matrices to control cell functions and as platforms for controlled drug/therapeutics delivery. |
1. Introduction
Over the last decades, there have been unprecedented advances on the design and synthesis of new drug molecules and polymeric drug carriers aiming to enhance the loading, protection, transport, controlled release, and targeting efficacy of bioactive agents to improve human health.1–5 Among the drug vehicles, nanosized polymeric micelles,6–8 polymer particles,9–11 polymersomes,12–17 or liposomes7,8 have been receiving considerable attention mainly due to their high drug loading capacity. However, such nanoreservoirs entail major hurdles mainly in which concern the encapsulation of hydrophobic cargoes and their on-demand site specific controlled release within a desired therapeutic window.11,18Having this in mind, the functionalisation of virtually any surface with multicomponent multilayered thin films by resorting to the Layer-by-Layer (LbL) assembly technology has been widely regarded as a promising methodology to engineer size- and shape-tunable multifunctional biomaterial nanocoatings to be used as nanoreservoirs for the efficient loading and sustained release of either hydrophilic or hydrophobic proteins/drugs/therapeutics.11,19–25 In fact, the simplicity, cost-effectiveness, reproducibility, and high versatility imparted by the LbL assembly technology in terms of the vast array of template surfaces that can be functionalised irrespectively on the size, shape and surface chemistry, materials to be assembled, and mild processing conditions turn it into a very appealing bottom-up approach to produce tailored (bio)functional materials with applicability in biology, biotechnology and biomedical field.26–32 Those include the assembly of template-free robust multilayered devices with size, geometry and topography reminiscent of the template surface, including free-standing (FS) membranes,33–40 tube-like structures,41–44 multilayered particles/capsules,45–52 hierarchical (multi)compartmentalized systems,53–55 and constructs56–58 by assembling a multitude of building blocks exhibiting complementary interactions,59 thus being very appealing to be used as implantable or injectable biomaterials. To date, the LbL assembly technology has been mostly employed to produce electrostatic-driven supramolecular multilayered assemblies by exploiting the attractive electrostatic interactions between oppositely charged polymeric materials.21,26,30,59–61 Among them, polysaccharides are very appealing building blocks for assembling multilayered devices for biomedical applications owing to their biocompatibility, biodegradability, non-cytotoxicity, and non-immunogenic properties, as well as high chemical diversity and versatility, and structural similarity to the native extracellular matrices of living tissues.62 Polysaccharides extracted from marine-origin organisms, widely discarded as waste by the food industry, are particularly attractive renewable natural biopolymers for enabling high added-value sustainable multifunctional biomaterials owing to their wide and ready bioavailability, biocompatibility and physicochemical properties. Chitosan (CHT), obtained through the deacetylation of chitin extracted mainly from the crustaceans’ shells,63 and alginate (ALG), extracted from brown algae,64 have been commonly assembled into a wide repertoire of electrostatic-driven 2D and 3D multilayered devices for bioapplications making use of their opposite charge nature.36–40,44,50–52,54–58,65–68 However, the insolubility of CHT in physiological conditions (pH < pKa ∼6–6.5)68,69 hinders the incorporation of therapeutic molecules in the CHT-derived LbL nanostructures, thus limiting their bioapplications.
Recently, considerable attention has been devoted to the modification of CHT by quaternary amine groups aiming to increase its solubility in neutral pH (i.e., physiological media) and enable the incorporation of bioactive agents under non-denaturing conditions, thus preserving their biological activity.70 Although N-(2-hydroxypropyl)-3-trimethylammonium chitosan chloride (HTCC) has been proposed as a promising alternative to the native CHT biopolymer with proven biocompatibility and safety,71–74 there are only a few studies in the literature reporting the LbL build-up of HTCC-derived multilayered thin films for applications in the biomedical field, namely in protein/drug delivery. In this regard, Kumorek et al. showed that [HTCC/heparin (Hep)]3 multilayered thin films could be loaded with the fibroblast growth factor 2 (FGF-2) and model proteins, such as albumin and lysozyme driven by the diffusion of the proteins into the film.75 Likewise, Urbaniak et al. adsorbed (HTCC/Hep)n multilayered thin films onto an anchoring HTCC/tannic acid bilayer functionalised glass substrates and studied the influence of the film structure on the protein release profile. It was shown that the release rate of Hep-binding proteins was higher when the proteins were incorporated into the LbL thin films as an integral part of the Hep layers (i.e., Hep-protein complexes) than as individual protein layers.76 Moreover, multilayered thin coatings encompassing HTCC/ALG bilayers have demonstrated to support wound healing.77,78
However, these works report the assembly of non-template free multilayered thin nanocoatings, which cannot be easily detached from the underlying substrates, thus hindering their capacity to be used as implantable biomaterials to fulfil biomedical and healthcare needs. As such, the development of fully biocompatible, biodegradable, and easily detachable self-standing multilayered films, which can retain the size, geometry and topographical features of the underlying substrate without requiring a physical support when implanted, is highly desired.
Herein, we report the preparation of thicker and robust electrostatic-driven FS multilayered membranes made of water-soluble HTCC and ALG biopolymers, at physiological pH, for the controlled release of model drug molecules. Two different set-ups of FS membranes having the model drug fluorescein isothiocyanate-labelled bovine serum albumin (FITC-BSA) incorporated either as an intrinsic building block [i.e., (HTCC/ALG/HTCC/FITC-BSA)100] or added as an outer layer into the FS membranes [i.e., (HTCC/ALG)200/HTCC/FITC-BSA] assembled onto hydrophobic low surface energy poly(vinyl carbonate) substrates were prepared aiming to assess the influence of the film structure in the drug release profile (Fig. 1). The growth of the HTCC/ALG multilayered thin nanofilms was monitored in situ by quartz crystal microbalance with dissipation monitoring (QCM-D), and the production of the FS membranes, encompassing virtually any number of layers, attempted on low surface energy substrates using an automatic dipping robot. The morphology, wettability, and mechanical properties of the as-produced self-standing membranes were evaluated by scanning electron microscopy (SEM), water contact angle (WCA), and universal mechanical testing machine, respectively. Moreover, the release rate and the in vitro cytocompatibility of the HTCC/ALG membranes towards human umbilical cord-mesenchymal stem cells (hUC-MSCs) were also studied. These biocompatible and biodegradable FS membranes represent advanced nanocarrier vehicles for the encapsulation and sustained release of therapeutics in in vitro and in vivo scenarios, holding great promise to be used as implantable biomaterials for modular tissue engineering and regenerative medicine strategies.
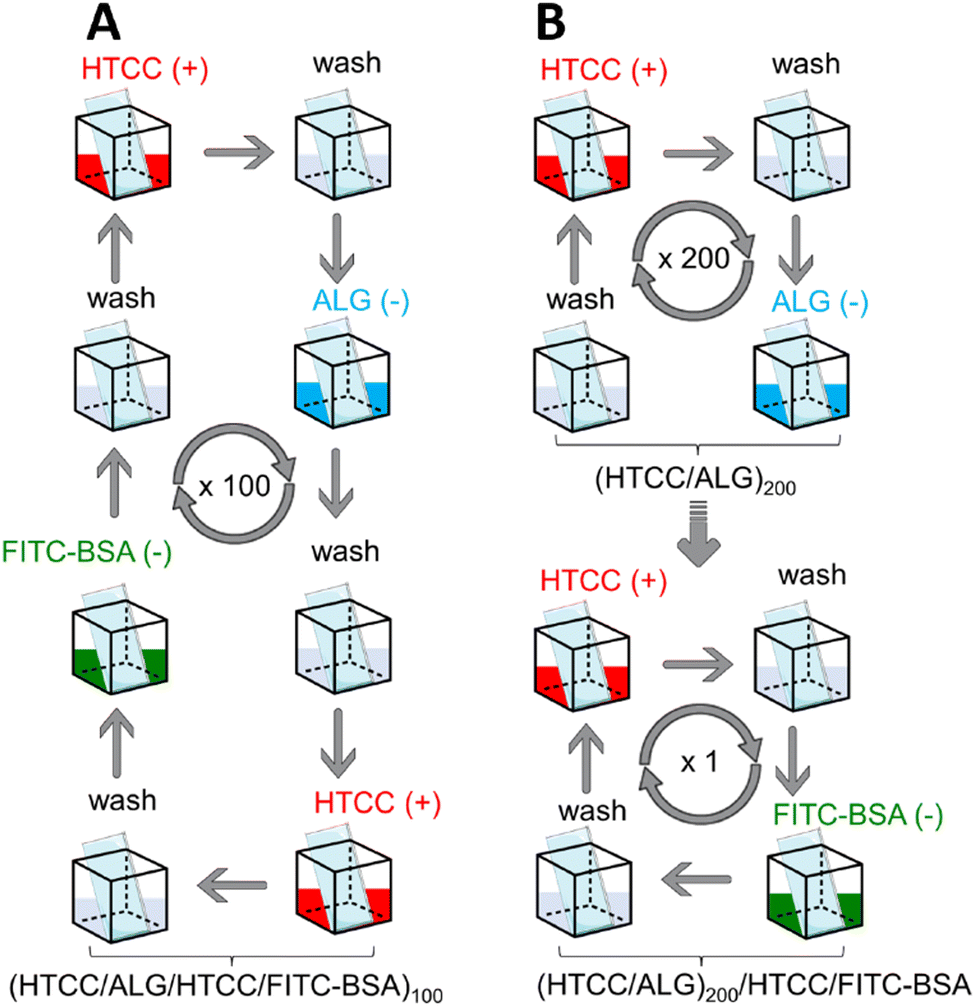 | ||
| Fig. 1 Schematic illustration of the preparation of the two set-ups of FS multilayered membranes: (A) (HTCC/ALG/HTCC/FITC-BSA)100 and (B) (HTCC/ALG)200/HTCC/FITC-BSA assembled onto poly(vinyl carbonate) substrates by resorting to attractive electrostatic interactions between HTCC, ALG and FITC-BSA. | ||
2. Materials and methods
2.1 Materials
CHT with a molecular weight (Mw) of 184.84 kDa, viscosity ∼122 cP, and a degree of deacetylation (DA) of 94% was kindly provided by Primex ehf® (Siglufjordur, Iceland) and used without further purification. Low viscosity sodium ALG derived from brown algae (Mw = 538 kDa, viscosity ∼250 cP), deuterium chloride [20 wt% solution in deuterium oxide (D2O, 100.0 atom%)], glycidyltrimethylammonium chloride (GTMAC), phosphate buffered saline (PBS), hydrochloric acid (HCl), FITC-BSA, alginate lyase (powder, ≥10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per g solid), Triton X-100 and paraformaldehyde were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Deuterium oxide (99.8%) was acquired from TCI Chemicals (Montgomeryville, PA, USA) and sodium hydroxide (NaOH) was purchased from LabChem (Zelienople, PA, USA). Glacial acetic acid (CH3COOH) and hydrogen peroxide (H2O2) were purchased from JMGS (Odivelas, Lisboa, Portugal) and Carlo Erba (Sabadell, Barcelona, Spain), respectively. All other reagents, namely ammonium hydroxide (NH4OH), MEM alpha medium, fetal bovine serum (FBS), penicillin, streptomycin, live/dead kit, phalloidin tetramethylrhodamine B isothiocyanate and 4,6-diaminidino-2-phenylindole-dilactate (DAPI) were purchased from Thermo Fischer Scientific (Fair Lawn, NJ, USA). All the aqueous solutions were prepared using ultrapure water from a Milli-Q Plus water purification system (resistivity >18.2 MΩ cm) from Merck Millipore (Burlington, MA, USA).
000 units per g solid), Triton X-100 and paraformaldehyde were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received. Deuterium oxide (99.8%) was acquired from TCI Chemicals (Montgomeryville, PA, USA) and sodium hydroxide (NaOH) was purchased from LabChem (Zelienople, PA, USA). Glacial acetic acid (CH3COOH) and hydrogen peroxide (H2O2) were purchased from JMGS (Odivelas, Lisboa, Portugal) and Carlo Erba (Sabadell, Barcelona, Spain), respectively. All other reagents, namely ammonium hydroxide (NH4OH), MEM alpha medium, fetal bovine serum (FBS), penicillin, streptomycin, live/dead kit, phalloidin tetramethylrhodamine B isothiocyanate and 4,6-diaminidino-2-phenylindole-dilactate (DAPI) were purchased from Thermo Fischer Scientific (Fair Lawn, NJ, USA). All the aqueous solutions were prepared using ultrapure water from a Milli-Q Plus water purification system (resistivity >18.2 MΩ cm) from Merck Millipore (Burlington, MA, USA).
2.2 Synthesis of quaternised chitosan
Quaternised chitosan (HTCC) was prepared by reacting CHT with GTMAC, as previously described.79 Briefly, 600 mg of CHT (3.7 mmol) was suspended in 40 mL of distilled water and the suspension was stirred for 1 h at room temperature (RT). Then, GTMAC (44.7 mmol, 12 equiv.) was added in three equal portions (3 × 2 mL) with 1 h intervals. The reaction temperature was raised to 80 °C for 48 h after the last addition of GTMAC. Once the reaction was completed, the reaction mixture was filtered and transferred into a dialysis bag with a molecular weight cut-off of 3.5 kDa and dialyzed against distilled water for 5 days under stirring, with the dialysis solvent being exchanged twice a day. The obtained product was frozen at −80 °C and freeze-dried to obtain the purified CHT derivative (i.e., HTCC) as a white foam, which was characterised by liquid proton nuclear magnetic resonance (1H NMR), solid-state carbon nuclear magnetic resonance (13C NMR) and attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy. The degree of quaternisation (DQ) was determined according to the 1H NMR spectra, following eqn (1), as described elsewhere.80CHT: 1H NMR (300.13 MHz, D2O) – δ (ppm): 2.04 (s, 3H, N-Acetyl), 2.98–3.15 (m, 35H, H2), 3.58–4.02 (m, 120H, H3-H6), 4.72 (s, 18H, H1).
HTCC: 1H NMR (300.13 MHz, D2O) – δ (ppm): 2.04 (s, 3H, N-Acetyl), 2.46–2.82 (m, 36H, Ha), 2.88–2.97 (m, 20H, H2), 3.21 [s, 162H, C(CH3)3], 3.36–3.79 (m, 104H, H3-H6), 4.23–4.35 (m, 36H, Hc), 4.61 (s, 18H, Hb), 4.69 (s, 20H, H1).
CHT: 13C NMR (400 MHz) – δ (ppm): 23.3 (CH3N-Acetyl), 56.9 (C2), 60.7 (C6), 75.0 (C5 + C3), 81.1–85.7 (C4), 104.7 (C1), 173.9 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O N-Acetyl).
O N-Acetyl).
HTCC: 13C NMR (400 MHz) – δ (ppm): 23.4 (CH3N-Acetyl), 54.9 [N(CH3)3], 61.8–64.8 (C2 + C6), 69.5 (Ca + Cc), 75.2 (C5 + C3), 82.4 (C4), 104.1 (C1), 173.5 (CO N-Acetyl).
2.3 Liquid and solid-state NMR spectroscopy
1H NMR spectra of the native CHT and HTCC were recorded on a Bruker Avance II 300 spectrometer (300.13 MHz; Bruker, Germany) using D2O as the internal reference.13C NMR spectra of the native CHT and HTCC were recorded on a Bruker Avance III 400 wide-bore spectrometer (400 MHz; Bruker, Germany) operating at a B0 field of 9.4 T with a Larmor frequency of 100.6 MHz. All experiments were performed on a triple-resonance 4 mm Bruker magic-angle spinning probe. The samples were packed into ZrO2 rotors with Kel-F caps (4 mm).
The deconvolution and simulation of all NMR spectra were carried out using the MestreNova® 9.0.1 software and the chemical shifts (δ) are quoted in ppm.
2.4 ATR-FTIR spectroscopy
The ATR-FTIR spectra of the native CHT and HTCC were acquired in the absorbance mode using a Bruker TENSOR 27 FTIR spectrometer (Thermo Scientific, USA) fitted with a “Golden Gate” ATR module equipped with a diamond crystal. The ATR-FTIR spectra were measured in the spectral range of 4000–400 cm−1 by averaging 256 individual scans per sample at a resolution of 4 cm−1. All data were linear baseline corrected and normalised using the OPUS software supplied with the instrument.2.5 Zeta (ζ)-potential measurements
Prior to the build-up of the electrostatic-driven multilayered assemblies, the net electrical charge of freshly prepared 0.1 mg mL−1 HTCC, ALG and FITC-BSA aqueous solutions in 10 mM PBS at pH 7.4 was assessed by measuring their ζ-potentials. The ζ-potentials of 0.1 mg mL−1 HTCC, CHT and ALG aqueous solutions in 0.1 M acetate buffer at pH 5.5 were also measured to compare the build-up of HTCC/ALG multilayered assemblies with their CHT/ALG counterparts. Moreover, the ζ-potentials of 0.1 mg mL−1 HTCC and CHT aqueous solutions were measured in a broad pH range (pH 1 to 13 for HTCC and pH 1 to 6 for CHT), by adjusting the solution pH with either 0.1 M HCl or 0.1 M NaOH, to demonstrate the permanent positive charge nature of the HTCC biopolymer regardless of the solution pH. The ζ-potentials of the individual solutions were determined at 25 °C using a Zetasizer Nano-ZS (Malvern Instruments Ltd., Royston, Hertfordshire, UK). The electrophoretic mobility (u) was converted into a ζ-potential value following the Smoluchowski equation (ζ = uη/ε; η and ε refer to the viscosity and permittivity of the solution, respectively).81 The measurements were performed in triplicates and averaged for each sample.2.6 Build-up of the multilayered thin films by QCM-D
The build-up of the HTCC/ALG multilayered thin films onto the gold (Au)-coated 5 MHz AT-cut quartz crystal sensors (QSX301 Gold, QSense, Sweden) was monitored in situ by QCM-D (QSense Pro, Biolin Scientific, Gothenburg, Sweden). Prior to the build-up of the films, the Au-coated quartz crystal sensors were submitted to UV/ozone treatment (UV/Ozone ProCleaner 220, BioForce Nanosciences, Inc.) for 10 min, followed by immersion in a 5:1:1 ultrapure water: ammonia (NH4OH, 25%): hydrogen peroxide (H2O2, 30%) (v/v) cleaning solution in an ultrasound bath at 70 °C for 10 min. Then, the quartz crystal sensors were rinsed with ultrapure water, dried under a soft stream of N2, and resubmitted to UV/ozone treatment for another 10 min. The freshly cleaned quartz crystal sensors were inserted in the QCM-D apparatus and equilibrated in a 10 mM PBS aqueous solution at pH 7.4 until a stable baseline was achieved. Then, the Au-coated quartz crystals substrates were alternately exposed to a 0.1 mg mL−1 HTCC (6 min adsorption time) and 0.1 mg mL−1 ALG (6 min adsorption time) aqueous solutions in 10 mM PBS at pH 7.4. In-between the adsorption of the polycationic and polyanionic aqueous solutions, the substrate was rinsed with 10 mM PBS aqueous solution at pH 7.4 for 4 min to remove weakly adsorbed molecules. The assembly process was repeated five times until reaching (HTCC/ALG)5 multilayered thin films. Afterwards, the (HTCC/ALG)5 nanofilms were dried under a soft stream of N2. The (HTCC/ALG/HTCC/FITC-BSA)5 multilayered nanofilms were prepared in a manner reminiscent of the (HTCC/ALG)5 multilayered thin films, i.e., by the alternate exposure of the substrate to the HTCC, ALG, HTCC and FITC-BSA aqueous solutions for 6 min each, with 4 min of rinsing time in-between the adsorption of each layered material. The (HTCC/ALG)5/HTCC/FITC-BSA multilayered thin films followed the same procedure used for the build-up of the (HTCC/ALG)5 multilayered thin films with the deposition of two additional layers encompassing HTCC (6 min adsorption time) and FITC-BSA (2 h adsorption time). Besides, the build-up of (HTCC/ALG)5 and (CHT/ALG)5 multilayered thin films (6 min adsorption time for each polymer and 4 min for washing) in 0.1 M acetate buffer at pH 5.5 was attempted to compare the film growth. All experiments were performed at a constant flow rate of 50 μL min−1 and at 25 °C. The Au-coated quartz sensors were excited at multiple overtones (1, 3, 5, 7, 9 and 11 corresponding to 5, 15, 25, 35, 45 and 55 MHz, respectively) and the changes in the frequency (Δf) and dissipation (ΔD) were monitored in real time. The frequency of each overtone was normalised to the fundamental resonant frequency (5 MHz) of the quartz crystal substrate (Δfn/n, n denotes the overtone number). The results presented herein correspond to the frequency and energy dissipation shifts associated to the 7th overtone (n = 7; 35 MHz) owing to their lowest level of noise. However, the results are representative of the other overtones. The hydrodynamic thickness of the multilayered thin films at each deposition cycle was estimated using the Voigt-based viscoelastic model,82 implemented in the Dfind software (Broadfit function) from QSense, assuming a fluid density of 1000 kg m−3, a layer density of 1000 kg m−3, and a fluid viscosity of 1 mPa s.
2.7 Production and characterisation of the FS membranes
The build-up of the (HTCC/ALG)200, (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes in 10 mM PBS aqueous solution at pH 7.4, as well as (HTCC/ALG)200 and (CHT/ALG)200 in 0.1 M acetate buffer at pH 5.5 was performed by repeating the alternate immersion of a hydrophobic low surface energy poly(vinyl carbonate) substrate into the oppositely charged polymeric material solutions using a home-made automatic dipping robot (CORPUS®, Guimarães, Portugal). Briefly, the poly(vinyl carbonate) substrate was alternatively and repetitively immersed in aqueous solutions of HTCC (+) and ALG (−) for 6 min each to build-up (HTCC/ALG)200 FS membranes. In-between the adsorption of each oppositely charged polymer, the substrate was rinsed in a 10 mM PBS aqueous solution at pH 7.4 for 4 min to remove the weakly adsorbed layers and avoid the cross-contamination of the biopolymeric solutions. The deposition cycles were repeated two hundred times until reaching a (HTCC/ALG)200 FS membrane. The (HTCC/ALG/HTCC/FITC-BSA)100 FS membranes were prepared in a reminiscent manner by the alternate immersion of the substrate onto HTCC (+), ALG (−), HTCC (+) and FITC-BSA (−) aqueous solutions for 6 min each with intermediate rinsing steps after the adsorption of each layered material (4 min), and the assembly process was repeated one hundred times. The preparation of the (HTCC/ALG)200/HTCC/FITC-BSA FS membranes was performed in a reminiscent manner of the one described for the preparation of the (HTCC/ALG)200 membranes with the additional immersion of the functionalised substrate in HTCC (6 min) and FITC-BSA (2 h) aqueous solutions (10 mM PBS at pH 7.4), with rinsing steps of 4 min after the adsorption of each material. The concentration of all the biopolymers used was 2 mg mL−1, while the one for FITC-BSA was 0.5 mg mL−1.The (HTCC/ALG)200 and (CHT/ALG)200 FS membranes prepared in 0.1 M acetate buffer at pH 5.5 were produced in a reminiscent manner of the (HTCC/ALG)200 counterparts prepared in 10 mM PBS at pH 7.4 to compare their morphological, wettable and mechanical properties.
After the easy detachment of the membranes from the underlying poly(vinyl carbonate) substrate, their dry thickness was measured using a digital micrometer (Acha, Eibar, Spain). The thickness values represent the average of five measurements for each sample.
2.8 WCA measurements
The wettability of the FS membranes was measured in air through the static sessile drop method using a home-made experimental apparatus equipped with a wide-angle webcam. The WCA measurements were carried out at RT by creating ultrapure water drops of 15 μL at the tip of the syringe, followed by placing them over the FS membranes. The ImageJ software (Fiji 1.52n) was used for the analysis of the contact angles. The measurements were repeated three times per sample and averaged for each FS membrane formulation.2.9 SEM
The surface morphology of the different FS membranes was analysed in a field emission gun scanning electron microscope (FEG-SEM Hitachi SU-3800, Tokyo, Japan) operated in high vacuum in the secondary electrons mode at an acceleration voltage of 10 kV and working distances between 4.7 and 5.4 mm. Prior to the SEM analysis, all samples were fixed to aluminum stubs by double-sided carbon conductive adhesive tape and then sputtered coated with a conductive Au/palladium layer using a Polaron E5000 sputter coater (Quorum Technologies, Laughton, UK).2.10 Mechanical properties of the FS membranes
The mechanical properties of the (HTCC/ALG)200 and (CHT/ALG)200 FS membranes prepared in 0.1 M acetate buffer at pH 5.5, and (HTCC/ALG)200 membranes produced in 10 mM PBS at pH 7.4 were assessed by performing uniaxial tensile measurements on an universal mechanical testing machine (Instron, Bluehill® Universal, Illinois Tool Works Inc.) equipped with a 50 N load cell, a gauge length established to 20 mm and a tensile speed of 1 mm min−1. Before the assay, the membranes were sectioned into 30 mm length and 5 mm width. The studies were performed in the dry state.The elastic modulus was determined by the tangent method using the initial linear region of the stress–strain curves (tensile-strain less than 1.25%). The ultimate tensile strength and elongation-at-break were obtained at the maximum peak determined for each individual stress–strain curves.
2.11 FITC-BSA loading capacity and in vitro release profile
The FITC-BSA loading capacity was assessed by the incubation of the (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes, in triplicates, into a 10 mg mL−1 alginate lyase aqueous solution in PBS (pH 7.4), at 37 °C, for 7 days to promote the complete degradation of the FS membranes and release the total amount of encapsulated FITC-BSA. The in vitro release profile of FITC-BSA from (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes was determined by the incubation of the membranes, in triplicates, into 1 mL of 10 mM PBS aqueous solution at pH 7.4 at 50 rpm and 37 °C for 4 days. At each predetermined time-point (0.1, 0.5, 1, 2, 4, 7, 24, 48, 72 and 96 h), 0.5 mL of the release medium was collected and freeze-dried, and 0.5 mL of fresh PBS was added to the sample. The fluorescence of the sample solutions (loading studies) and lyophilised samples (release assays) was measured at excitation (λexc) and emission (λem) wavelengths of 495 and 520 nm, respectively, using a microplate reader (Synergy HTX Biotek, Izasa Scientific, Carnaxide, Portugal). A standard calibration curve of FITC-BSA (0–80 μg mL−1) in PBS (pH 7.4) was used to determine the amount of the cumulative released mass over time.2.12 Metabolic activity and viability assays
The cytotoxicity of the as-produced FS membranes was evaluated by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4- sulfophenyl)-2H-tetrazolium (MTS) cell proliferation assay and live/dead fluorescence study, according to the manufacturer's instructions, using hUC-MSCs as a model of adherent cells. Briefly, the (HTCC/ALG)200, (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes were incubated, in triplicates, in 1 mL of MEM alpha medium with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin in 24-well plates at 37 °C, 5% CO2 and fully humified for 3 days, as previously described.83 The resulting mediums with the compounds released from the FS membranes were used for the metabolic and viability assays.For the metabolic assays, upon reaching 70–80% of confluence, hUC-MSCs were seeded in 96-well plates at a density of 5 × 103 cells per well and allowed to adhere for 24 h at 37 °C in a humified atmosphere containing 5% CO2. Afterwards, the medium was replaced by the resulting medium released from the FS membranes. After 1- and 3-days of post-transfection, the medium was replaced by dPBS containing MTS (6:1) (100 μL per well) and incubated for 4 h to determine the metabolic activity of hUC-MSCs. The absorbance was measured at 490 nm, using a microplate reader. hUC-MSCs cultured with normal culture medium were used as positive control. The metabolic activity assays were performed in three independent experiments with triplicates.
The cell viability assays were performed in a manner reminiscent of the MTS cell proliferation assays, i.e., by incubating 50 × 103 cells per well in 24-well plates for 24 h at 37 °C in a humified atmosphere containing 5% CO2. The cells were then exposed to the culture medium containing the extracts released from the FS membranes and, after 1 and 3 days of culture, the hUC-MSCs were stained with calcein-AM (1:500 in dPBS) and propidium iodide (1:1000 in dPBS) for 30 min at 37 °C, protected from the light. Afterwards, the cells were washed twice with dPBS and immediately visualized by fluorescence microscopy.
2.13 Cell morphology
For the assessment of the cell morphology, 50 × 103 cells per well were seeded in 24-well plates for 24 h at 37 °C in a humified atmosphere containing 5% CO2. After 24 h, the culture medium was replaced by the culture medium containing the extracts released from the FS membranes. After 1- and 3-days of post-transfection, the culture medium was removed, and the cells were washed with dPBS and fixed with 4% (v/v) paraformaldehyde solution in PBS for 1 h at RT. The fixed samples were then rinsed with PBS and incubated with 0.5% Triton X-100 for 5 min at RT to permeabilize cells. Afterwards, the samples were washed with dPBS and blocked with 5% FBS for 1 h at RT. The cells were washed again with dPBS and solutions of phalloidin tetramethylrhodamine B isothiocyanate (25:1000 in dPBS) and DAPI (1:1000 in dPBS) were added to the samples for 30 and 5 min, respectively, to stain the F-actin cytoskeleton and nuclei of the hUC-MSCs, respectively. Then, the cells were washed with dPBS and immediately visualized by fluorescence microscopy.
2.14 Fluorescence microscopy
Fluorescence microscopy micrographs of the cells cultured with the medium containing the extracts released from the (HTCC/ALG)200, (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes were acquired in an upright motorized widefield fluorescence microscope (Axio Imager M2, Carl Zeiss, Jena, Germany) equipped with a 200 W HXP lamp, a 3.0 Mpix monochromatic camera (Axiocam 105 mono; Carl Zeiss, Jena, Germany), and a 5x objective (Carl Zeiss, Jena, Germany). Acquired data were processed in the Zeiss ZEN v2.3 blue edition software.2.15 Statistical analysis
Unless otherwise noted, all experiments were performed in triplicates (n = 3) and the results presented as mean ± standard deviation (SD). The statistical analysis was performed by one-way ANOVA followed by Tukey's post-hoc multiple comparison test using the GraphPad Prism 9.4.0 (GraphPad Inc.) software. Statistically significant differences were considered for ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05.3. Results and discussion
3.1 Synthesis and characterisation of the chitosan derivative
The insolubility of CHT under physiological conditions is a major challenge when aiming for bioapplications. In this regard, quaternary chitosan derivatives have been proposed due to their solubility in water at neutral pH.In this work, water-soluble HTCC was synthesised by reacting native CHT with the quaternary agent, GTMAC, in aqueous media (Fig. 2).
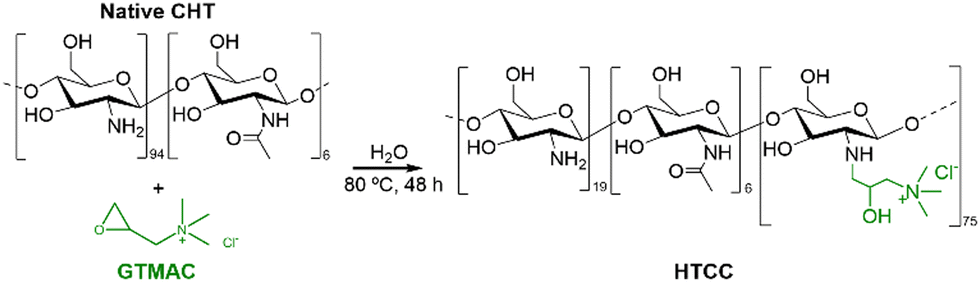 | ||
| Fig. 2 Schematic representation of the reaction between the native CHT biopolymer and GTMAC to obtain the HTCC biopolymer. | ||
The chemical modification of the native CHT was confirmed by 1H NMR. Fig. 3A reveals the presence of an intense peak at 3.21 ppm, assigned to the methyl protons of the quaternary ammonium groups [−N+(CH3)3], and three new peaks assigned to Ha, Hc and Hb, from the CH2, CH, and CH2 protons at 2.64, 4.30 and 4.69 ppm, respectively. The integration of the protons of the newly identified peaks confirmed the successful modification of the native CHT at a ratio of 2:9:2:1 [Ha:N+(CH3)3:Hc:Hb]. Moreover, the successful chemical modification of the native CHT biopolymer was also assessed by 13C NMR (Fig. 3B), revealing a characteristic intense peak at 54.9 ppm, assigned to the methyl carbons of the quaternary ammonium groups. Furthermore, two new peaks were identified at 63.3 and 69.5 ppm, corresponding to the carbons Cb and Ca + Cc, respectively, corroborating the 1H NMR data. The DQ was found to be 75%, calculated following the eqn (1):80
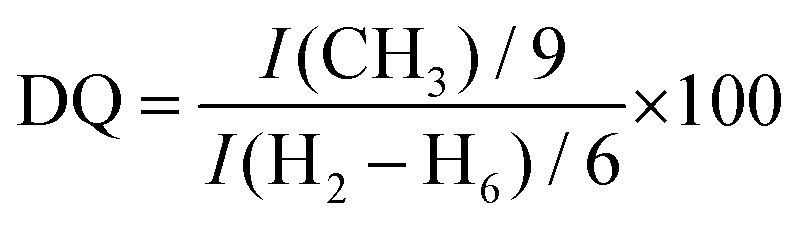 | (1) |
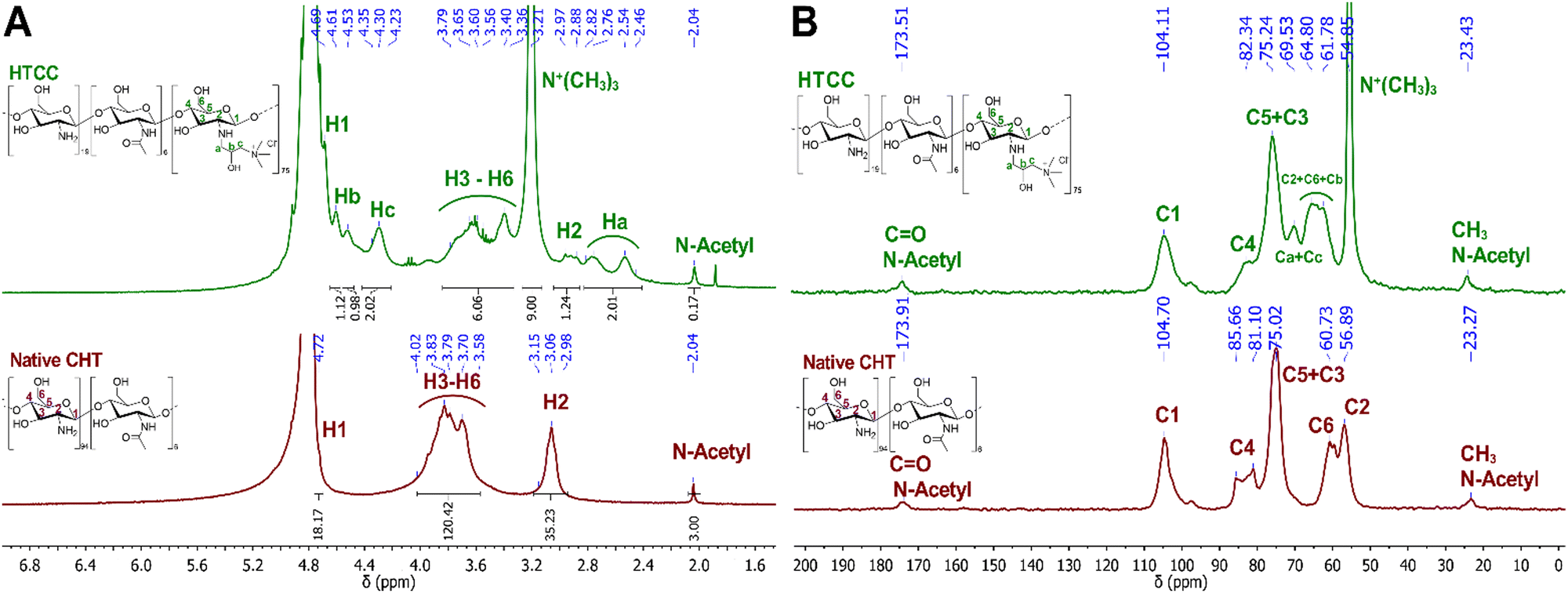 | ||
| Fig. 3 (A) 1H NMR and (B) 13C NMR spectra of HTCC and native CHT biopolymers. | ||
The DQ is influenced by key reaction parameters, including reaction time, temperature, and the molar ratio of GTMAC to CHT-free amino groups, and increases while increasing the temperature and reaction time, as previously described.80 Moreover, it has been reported that the water solubility of HTCC is improved while increasing the DQ.74 As such, based on the conditions described in the previous studies and aiming for a high DQ, the synthesis of the HTCC biopolymer was performed at 80 °C using a reaction time of 48 h.
Complementary to the NMR analysis, ATR-FTIR measurements were attempted on the native CHT and HTCC powders to assess the successful modification of the native CHT biopolymer. The ATR-FTIR spectrum of the HTCC powder revealed a well-defined absorption band at 1477 cm−1, attributed to the methyl groups in the quaternary ammonium side chains. Furthermore, it is also evident the presence of a peak at 1647 cm−1, which is characteristic of the GTMAC molecule.84,85 In addition, the absence of the peaks exhibited by the native CHT biopolymer at 1645 and 1587 cm−1 confirms that the –NH2 groups of the CHT biopolymer were replaced by N-(2-hydroxypropyl)-3-trimethylammonium chloride groups, suggesting the successful modification of the native CHT biopolymer (Fig. 4A).
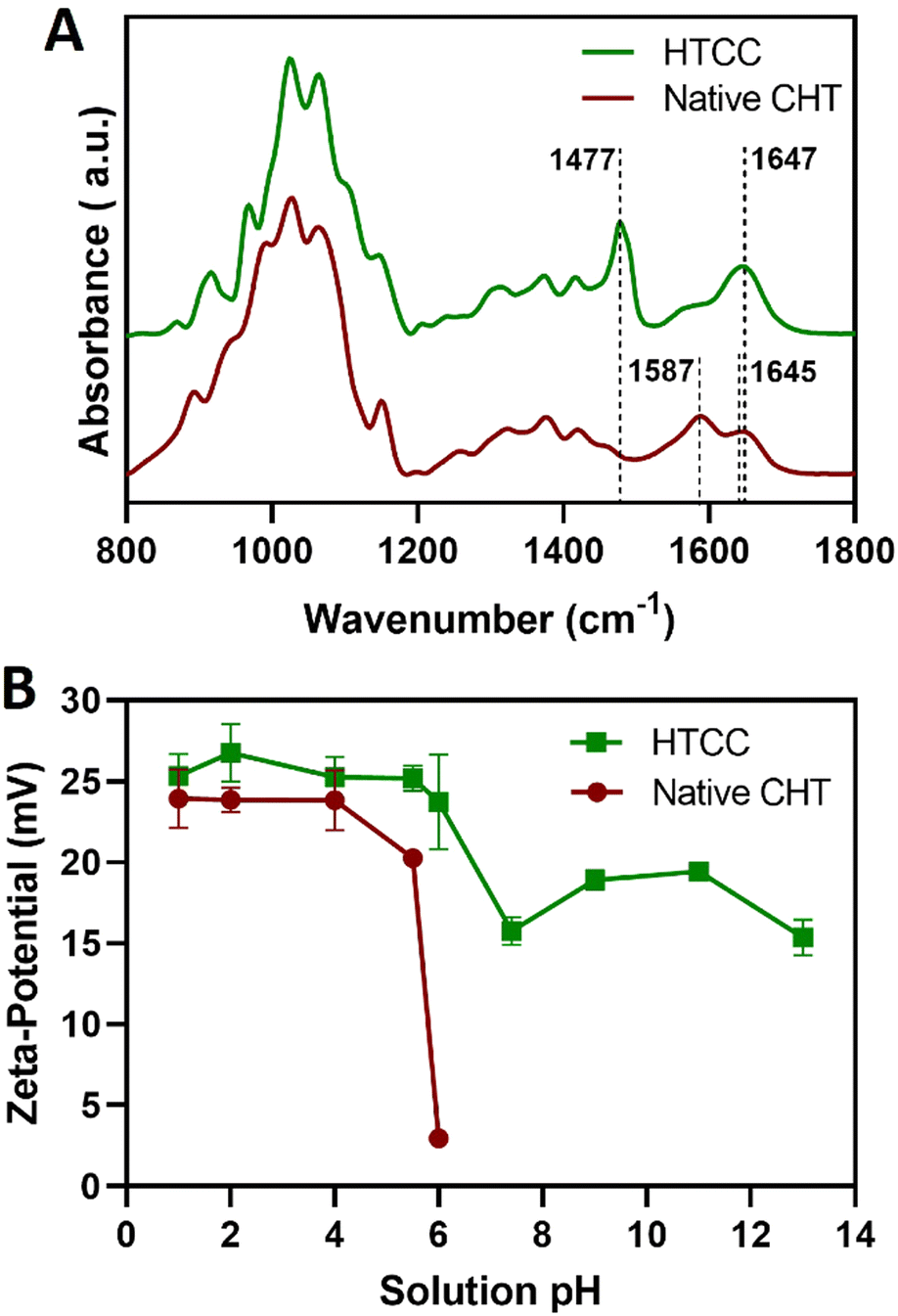 | ||
| Fig. 4 (A) ATR-FTIR spectra of HTCC and native CHT powders. (B) Evolution of the ζ-potential for 0.1 mg mL−1 HTCC and CHT aqueous solutions as a function of the solution pH presented as mean ± SD of three independent experiments (n = 3, per condition) performed in triplicates. | ||
3.2 Build-up of the multilayered thin films
Prior to the build-up of the multilayered thin films and robust FS membranes, the net electrical charges of the freshly prepared 0.1 mg mL−1 HTCC, ALG and FITC-BSA aqueous solutions in 10 mM PBS at pH 7.4, and 0.1 mg mL−1 HTCC, CHT and ALG aqueous solutions in 0.1 M acetate buffer at pH 5.5 were assessed by measuring their ζ-potentials. Moreover, to confirm the permanent cationic nature of HTCC over a wide pH range, we have measured the ζ-potentials of 0.1 mg mL−1 HTCC aqueous solutions ranging from pH 1 to 13 and compared with those obtained for CHT aqueous solutions ranging from pH 1 to 6 (Fig. 4B). While the HTCC biopolymer revealed to be positively charged in all solution pH and, thus, soluble in water, with no significant differences in-between the pH range tested, as expected the ζ-potential of the CHT solution dropped significantly at pH 6 owing to the vicinity of its pKa.68,69 As such, the HTCC biopolymer holds great promise in the biomedical field. The ζ-potentials of 0.1 mg mL−1 HTCC, ALG and FITC-BSA aqueous solutions in 10 mM PBS at pH 7.4 were found to be +15.7 ± 1.81 mV, −17.4 ± 2.05 mV and −2.54 ± 0.374 mV, respectively, thus revealing the cationic and anionic nature of HTCC and ALG biopolymers, respectively, and the slight anionic nature of FITC-BSA. The ζ-potentials of 0.1 mg mL−1 HTCC, CHT and ALG aqueous solutions in 0.1 M acetate buffer at pH 5.5 were found to be +25.2 ± 1.07 mV, +20.3 ± 1.97 mV, and −24.1 ± 1.53 mV, respectively. In this regard, we hypothesised that electrostatic-driven multilayered thin films and FS multilayered membranes could be developed by the LbL alternate deposition of oppositely charged materials. Thus, the LbL growth of (HTCC/ALG)5 (Fig. 5A), (HTCC/ALG/HTCC/FITC-BSA)5 (Fig. 5B) and (HTCC/ALG)5/HTCC/FITC-BSA (Fig. 5C) multilayered thin films in PBS at pH 7.4, and (HTCC/ALG)5 (Fig. 5D) and (CHT/ALG)5 (Fig. 5E) in acetate buffer at pH 5.5 was monitored in situ by the QCM-D technique by applying an alternating electric field across the Au-coated quartz crystal sensor. The sequential decrease in the Δf7/7 over time after the deposition of each building block onto the Au-plated quartz crystal surface reveals the increase in the hydrodynamic mass per layer and the effective interaction between the adsorbed materials, thus confirming the successful and stable LbL growth of the multilayered thin films. While the multilayered thin films encompassing either HTCC/ALG or CHT/ALG bilayers are built mainly due to attractive electrostatic interactions between the oppositely charged biopolymers, the adsorption of the slightly negatively charged FITC-BSA layers onto the previously adsorbed positively charged HTCC is expected to be predominantly due to hydrophobic and hydrogen bonding intermolecular interactions. | ||
| Fig. 5 QCM-D monitoring of the normalized frequency (Δf7/7) and dissipation (ΔD7) shifts at the 7th overtone as a function of time for the build-up of (A) (HTCC/ALG)5, (B) (HTCC/ALG/HTCC/FITC-BSA)5 and (C) (HTCC/ALG)5/HTCC/FITC-BSA multilayered thin films in 10 mM PBS at pH 7.4, and (D) (HTCC/ALG)5 and (E) (CHT/ALG)5 in 0.1 M acetate buffer at pH 5.5, onto Au-coated quartz crystal sensors. The inset numbers in: (A) and (D) refer to the adsorption of HTCC (1), ALG (3), and rising steps (2 and 4); (B) and (C) refer to the adsorption of HTCC (1 and 5), ALG (3), FITC-BSA (7) and rising steps (2, 4, 6 and 8); (E) refer to the adsorption of CHT (1), ALG (3), and rinsing steps (2 and 4). (F) Cumulative hydrodynamic thickness evolution for the (HTCC/ALG)5 and (CHT/ALG)5 films as a function of the number of deposited layers, estimated using the Voigt-based viscoelastic model. The red and green lines represent the linear and exponential trend lines, respectively. | ||
On the other hand, the increase in the ΔD7 values reveals the soft character and damping properties, i.e., the viscoelastic nature of the multilayered films, which is a common feature of soft polymeric films.30,86,87 Moreover, the Δf7/7 and ΔD7 values remain unchanged after the rinsing steps, thus revealing the strong interaction between the biopolymers and the irreversible nature of the adsorption process. However, one could denote differences in the extend of the adsorption and viscoelastic properties, i.e., Δf7/7 and ΔD7 changes, respectively, of both the chitosan and the ALG layers while resorting to either HTCC or native CHT biopolymers for the build-up of the distinct multilayered thin film formulations. A direct comparison of the (HTCC/ALG)5 (Fig. 5D) and (CHT/ALG)5 (Fig. 5E) multilayered films built at pH 5.5 revealed higher Δf7/7 and ΔD7 shifts for the adsorption of both the chitosan and ALG layers when resorting to the HTCC biopolymer. After the assembly of the five bilayered films, the Δf7/7 and ΔD7 shifts increased by ∼4- and 2.7-fold on the (HTCC/ALG)5 multilayered film when compared to the (CHT/ALG)5 film counterpart, respectively, thus indicating the higher softness, viscoelastic, and water-rich, i.e., hydrated nature of the HTCC-based film. Interestingly, a close look into the ΔD7 shifts obtained for the (HTCC/ALG)5 multilayered film (Fig. 5D) enabled us to observe differences in the viscoelastic properties of the HTCC and ALG layers at each deposition step. While the ALG layers denoted a similar increment in the ΔD7 values irrespectively on the ALG deposition cycle, the HTCC ones exhibited an exponential-like growth with the increase in the number of adsorbed HTCC layers. Such results were corroborated by the cumulative hydrodynamic thickness evolution of both multilayered thin nanofilms at each adsorption step, estimated using the QCM-D data via the Voigt-based viscoelastic model (Fig. 5F).82 While the (CHT/ALG)5 multilayered film revealed a linear growth trend reaching a maximum thickness of 40 nm by the end of the assembly process, the (HTCC/ALG)5 film exhibited an exponential growth regime with a maximum thickness of 125 nm, similar to what has been reported for the growth of soft biopolymeric multilayered films encompassing poly(L-lysine) (PLL)/hyaluronic acid (HA), PLL/poly(L-glutamic acid) or CHT/HA bilayers.28,86,88–90 Such behaviour suggests the diffusion of the HTCC chains “in” and “out” of the whole film during the LbL film growth, as previously reported for the aforementioned multilayered films encompassing PLL layers.
3.3 Production and characterisation of the FS multilayered membranes
The multilayered thin films were translated into thicker, robust, and easy to handle FS membranes by repeating the alternate immersion of hydrophobic, low surface energy poly(vinyl carbonate) substrates into oppositely charged polymeric aqueous solutions, followed by their easy detachment, without the need for any chemical or physical stimulus, due to the weak interaction between the first assembled layer and the substrate. The FS membranes were characterised for their thickness using a micrometer, for their morphological properties by SEM, as well as for their wettability by WCA measurements.The membranes having FITC-BSA as an intrinsic protein layer of the multilayered film, i.e., (HTCC/ALG/HTCC/FITC-BSA)100 revealed the highest dry thickness (27.4 ± 3.2 μm), whereas the (HTCC/ALG)200/HTCC/FITC-BSA counterparts exhibited a thickness of 17.6 ± 2.3 μm. The (HTCC/ALG)200 FS membranes revealed a thickness of 17.3 ± 1.5 μm. Thus, the incorporation of FITC-BSA into the multilayered film provide us with the possibility to control the thickness of the LbL film. The (HTCC/ALG)200 and (CHT/ALG)200 membranes prepared in acetate buffer at pH 5.5 exhibited a dry thickness of 27.5 ± 1.3 μm and 32.4 ± 2.0 μm, respectively. The dry thickness obtained for the (CHT/ALG)200 FS membranes is consistent with the one reported previously for similar CHT/ALG membranes encompassing 200 bilayers and produced following the same experimental assembly conditions.36 Moreover, the morphology of the upper part of the detached FS membranes was analysed by SEM. While all FS membranes containing HTCC revealed a rather smooth and homogenous surface, the ones containing native CHT exhibited a rougher surface, corroborating the previous findings (Fig. 6).37 However, it is interesting to note differences in the FS membranes assembled with the same building blocks but resorting to different solvents and solution pH. For instance, the (HTCC/ALG)200 membranes assembled in PBS at pH 7.4 (Fig. 6C and D) are non-porous and smoother than the ones assembled in acetate buffer at pH 5.5 (Fig. 6I and J), which revealed some degree of porosity. The FS membrane formulations were further inspected for their wettability, revealing wettable properties (Fig. 7). Although the WCA of all membranes was in the range from 43.6 ± 1.3° to 56.3 ± 1.4°, the introduction of the BSA as an intrinsic building block of the LbL membrane denoted a slightly more wettable state than the ones having BSA as an individual outer layer. Moreover, the as-prepared CHT/ALG membranes assembled at pH 5.5 denoted the highest WCA, thus revealing their slightly more hydrophobic nature. It has been claimed that an increase in the surface roughness has been typically associated with an increase in the surface hydrophobicity.30,90 Such behaviour corroborates the higher roughness observed by SEM for the native CHT/ALG FS membranes.
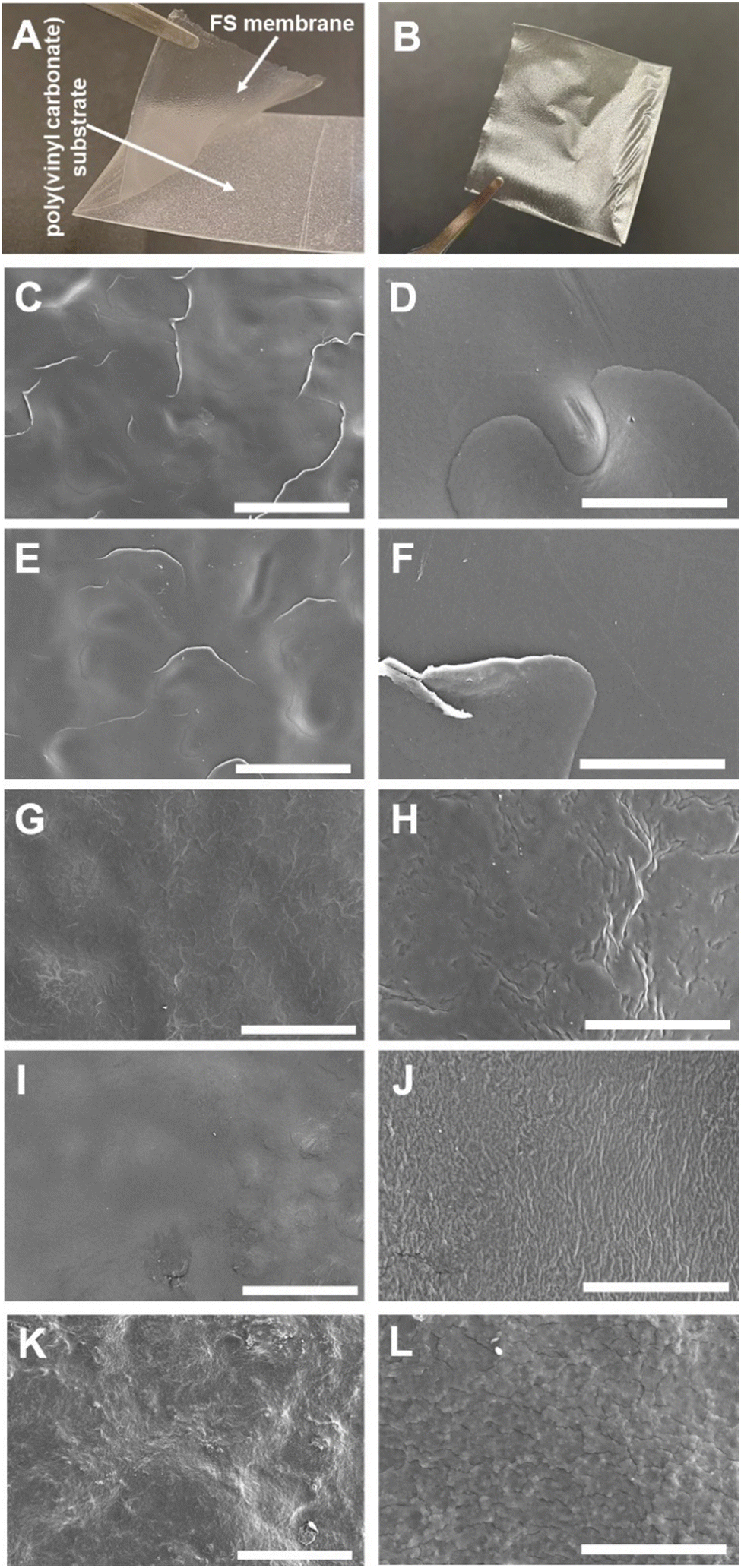 | ||
| Fig. 6 Representative optical images of HTCC/ALG FS membranes (A) produced on poly(vinyl carbonate) substrate and (B) after detachment. Representative SEM micrographs of the (C and D) (HTCC/ALG)200, (E and F) (HTCC/ALG/HTCC/FITC-BSA)100 and (G and H) (HTCC/ALG)200/HTCC/FITC-BSA FS membranes prepared in PBS at pH 7.4, and (I and J) (HTCC/ALG)200 and (K and L) (CHT/ALG)200 FS membranes produced in acetate buffer at pH 5.5. Scale bars represent 200 μm in the panels C, E, G, I and K, and 30 μm in the panels D, F, H, J and L, respectively. | ||
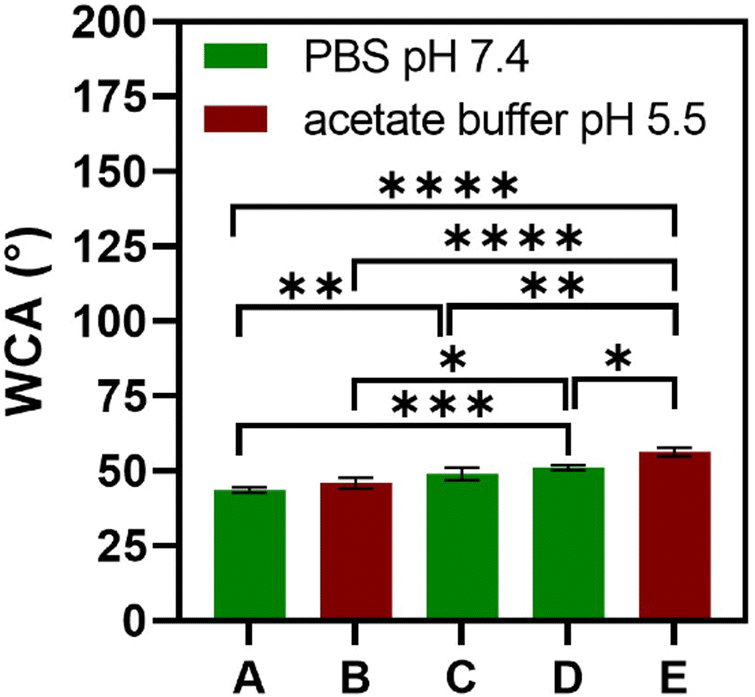 | ||
| Fig. 7 WCA of all FS membrane formulations in PBS (pH 7.4) and acetate buffer (pH 5.5): (A) (HTCC/ALG/HTCC/FITC-BSA)100, (B) (HTCC/ALG)200, (C) (HTCC/ALG)200/HTCC/FITC-BSA, (D) (HTCC/ALG)200, and (E) (CHT/ALG)200. Data are presented as mean ± SD of three independent experiments (n = 3, per condition) performed in triplicates. Significant differences were found for **** p ≤ 0.0001, *** p ≤ 0.001, ** p ≤ 0.01, and * p ≤ 0.05. | ||
3.4 Mechanical properties of the FS multilayered membranes
The mechanical properties of the BSA-free FS multilayered membranes were evaluated in the dry state and in the tensile mode using a universal mechanical testing machine. For comparison with the native CHT-based FS membranes, the mechanical properties of the HTCC-derived membranes were assessed at pH 7.4 and pH 5.5.Fig. 8A shows the representative stress–strain curves for the HTCC and native CHT-based FS membranes. The results showed that the buffer pH in which the HTCC membranes are constructed did not significantly change their Young modulus (Fig. 8B) with values ranging from 1251 ± 199 MPa to 982 ± 200 MPa for the membranes constructed at pH 7.4 and 5.5, respectively. Additionally, there were no significant differences in the Young modulus between the membranes constructed with native CHT (1006 ± 213 MPa) or HTCC under acidic environments. A similar trend was observed for the elongation-at-break (Fig. 8C), in which no significant differences were found for all studied membrane formulations. Nevertheless, regarding the ultimate tensile strength (Fig. 8D), the membranes comprising HTCC showcased a significantly higher tensile strength (24.1 ± 0.7 and 28.3 ± 4.5 MPa for FS membranes constructed at pH 7.4 and 5.5, respectively), irrespectively on the buffer pH, when compared with their counterparts containing native CHT constructed at pH 5.5 (16.8 ± 2.3 MPa). Furthermore, there were no significant differences in the ultimate tensile strength between the HTCC FS membranes assembled at pH 5.5 and 7.4. These findings reveal that the quaternisation of the native CHT enables the assembly of FS multilayered membranes under physiological conditions with enhanced mechanical properties when compared to the membranes constructed with native CHT under acidic environments. This represents a significant advancement in overcoming a major challenge associated to the insolubility of CHT under physiological pH when aiming for biomedical applications.
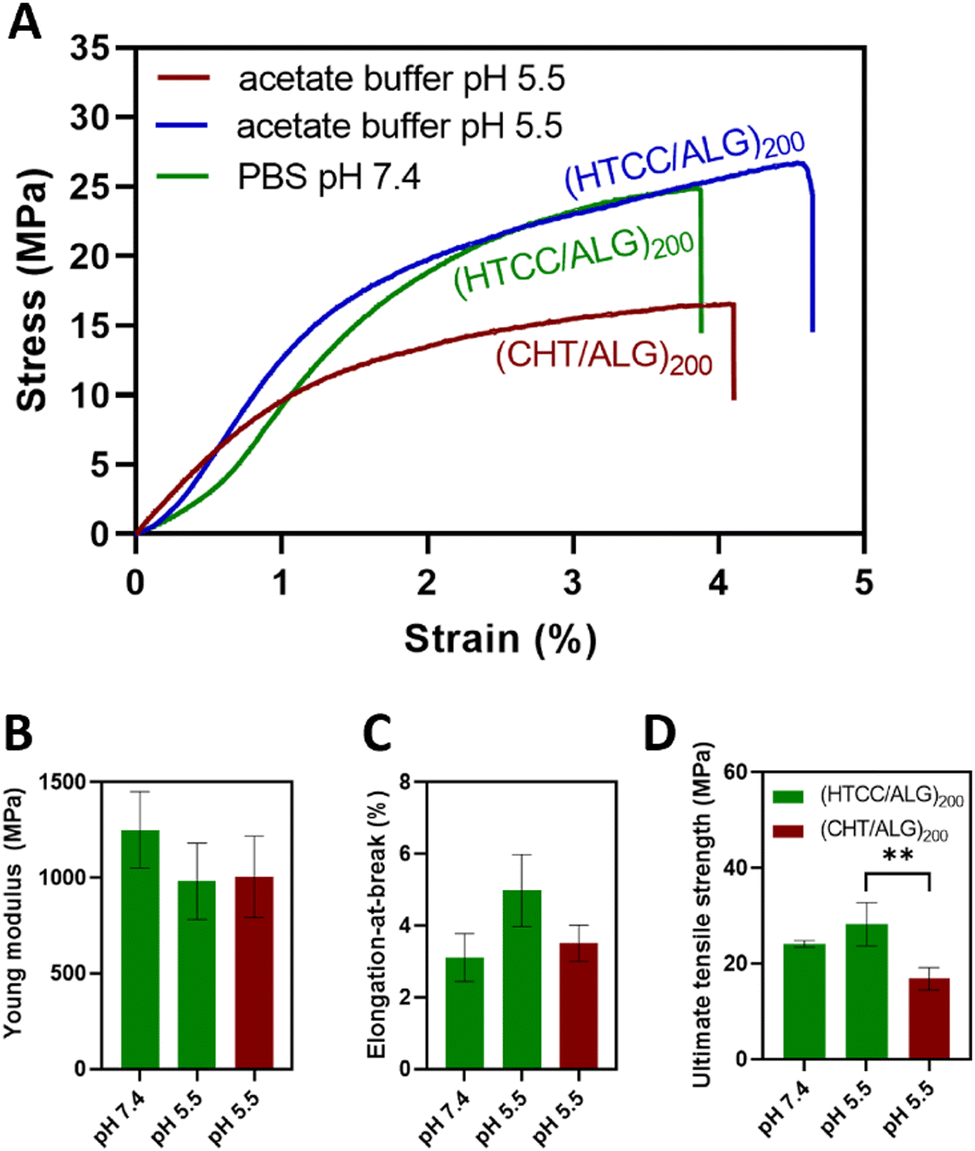 | ||
| Fig. 8 Mechanical properties of the FS membranes in PBS (pH 7.4) and acetate buffer (pH 5.5). (A) Representative stress-strain curves, (B) Young modulus, (C) elongation-at-break, and (D) ultimate tensile strength. Data are presented as mean ± SD of three independent experiments (n = 3, per condition) performed in triplicates. Significant differences were found for ** p ≤ 0.01. | ||
3.5 FITC-BSA loading capacity and in vitro release profile
The use of the HTCC/ALG FS membranes as platforms for drug delivery was studied by employing FITC-BSA as a model drug, placed in two different positions of the multilayered assemblies. The in vitro release profile of FITC-BSA from (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes was assessed by the dialysis method and the amounts of FITC-BSA loaded and released were determined from a standard calibration curve of FITC-BSA. The FITC-BSA loading capacity of the (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes was estimated to be 292.2 ± 10.2 and 52.0 ± 5.6 μg mL−1 mm−2, respectively. Although both membranes showed an intrinsic ability to release FITC-BSA, the release of FITC-BSA from the (HTCC/ALG)200/HTCC/FITC-BSA membranes exhibited a burst release profile (Fig. 9A and B, orange line). On the other hand, the (HTCC/ALG/HTCC/FITC-BSA)100 membranes prevented the initial burst release and promoted a much more sustained release of FITC-BSA over time, suggesting a quasi-zero-order release kinetics (Fig. 9A, blue line), thus enabling drug release at a constant rate for longer periods of time. The (HTCC/ALG/HTCC/FITC-BSA)100 and (HTCC/ALG)200/HTCC/FITC-BSA FS membranes released ∼39.8 ± 4.1 and 8.1 ± 0.6 μg mL−1 mm−2 of the loaded FITC-BSA, respectively, within 4 days (Fig. 9A). We hypothesize that the distinct behaviour observed in Fig. 9 could be assigned to the difference in the film structure, indicating that FITC-BSA molecules are more complexed with HTCC layers when assembled as an intrinsic building block of the multilayered film rather than when adsorbed as an outer layer, thus corroborating the more sustained release profile obtained for the (HTCC/ALG/HTCC/FITC-BSA)100 FS membranes. Several publications have described the preparation of fully natural-origin polymer- or hybrid synthetic/natural polymer-based FS multilayered membranes as reservoirs of model drugs or proteins for controlled drug/protein release. In particular, the incorporation of the bone morphogenetic protein 2 (BMP-2) as an intrinsic LbL ingredient of [poly(β-aminoester)/chondroitin sulfate (CS)/BMP-2/CS]100 multilayered films coated 3D printed scaffolds conducted to a very small burst release in the first few hours followed by a sustained linear release profile over a period of about 3 days,91 corroborating the release profile of our (HTCC/ALG/HTCC/FITC-BSA)100 FS membranes. Besides, other studies have reported the production of either (CHT/ALG)200 or (CHT/ALG)100 FS multilayered membranes post-loaded with BMP-2 or FGF-2,23,92 showcasing an initial burst release followed by a sustained and prolonged release along time until a plateau was reached, similar to the release kinetics observed for the (HTCC/ALG)200/HTCC/FITC-BSA FS membranes.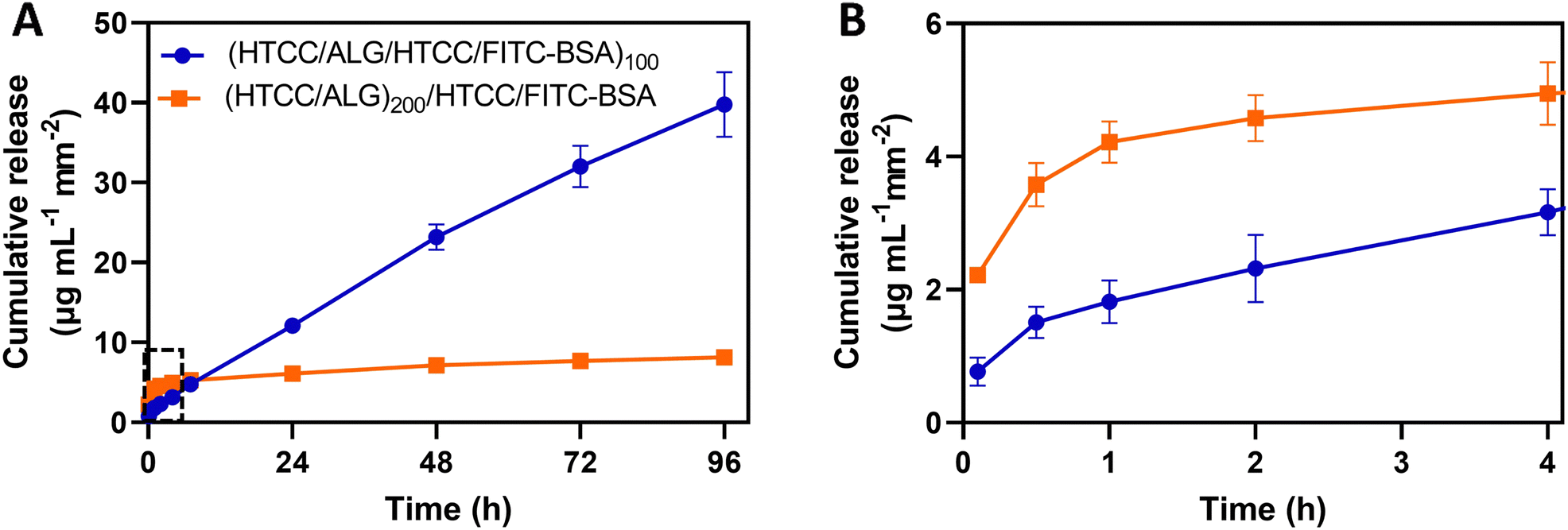 | ||
| Fig. 9 (A) Cumulative mass release profile of FITC-BSA in PBS at pH 7.4 from (HTCC/ALG/HTCC/FITC-BSA)100 (blue line and circles) and (HTCC/ALG)200/HTCC/FITC-BSA (orange line and squares) FS membranes over 96 h. (B) Zoom-in on the cumulative release of FITC-BSA for the first 4 h (represented with a dotted line in A). Data are presented as mean ± SD of three independent experiments (n = 3, per condition) performed in triplicates. | ||
3.6 In vitro biological performance
The viability of hUC-MSCs for the different FS membranes was assessed by fluorescence microscopy after 1 and 3 days of in vitro culture by performing a live/dead assay, and the cellular metabolic activity was investigated via an MTS assay. The live/dead images showed that, similarly to the positive control condition (cells cultured with normal medium), the cells cultured with the culture medium containing the extracts released from the FS membranes remained viable after 1 and 3 days of culture (Fig. 10A), confirming the cytocompatibility of all FS membranes for this cell line. Moreover, the metabolic activity of hUC-MSCs was similar for either the FITC-BSA-free or FITC-BSA loaded membranes, and in the range of the control (Fig. 10B). Besides, the metabolic activity of hUC-MSCs increased along the culture time, demonstrating the cytocompatibility of all the studied FS membranes, thus being very appealing for biomedical applications.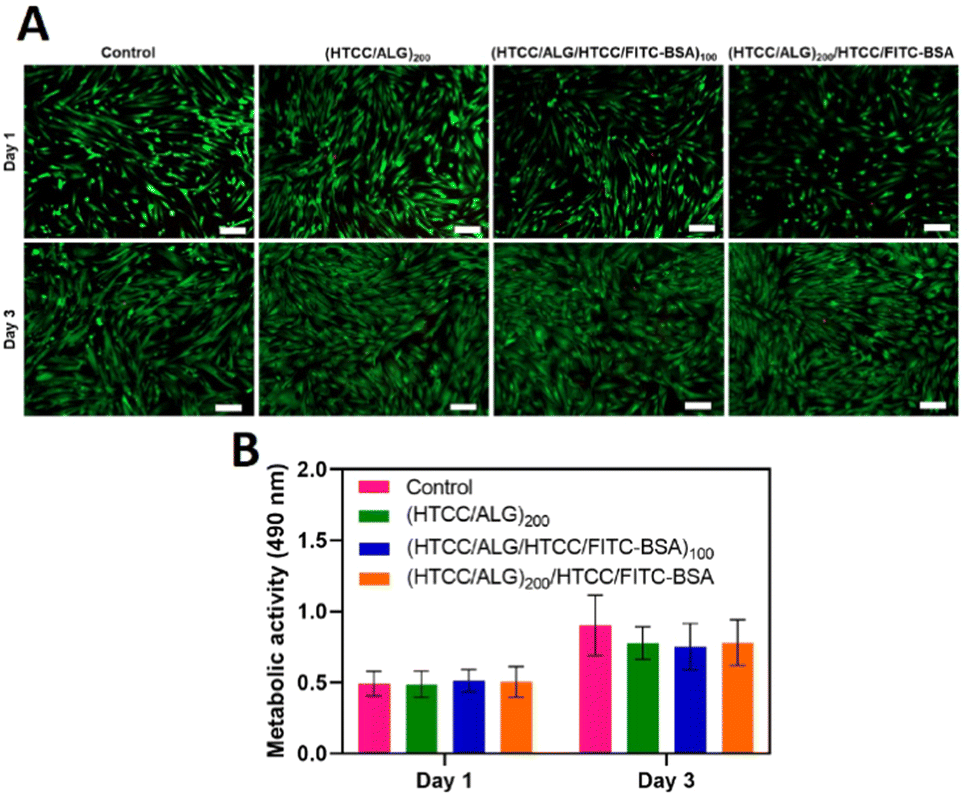 | ||
| Fig. 10 (A) Representative fluorescence microscopy images of live (green) and dead (red) hUC-MSCs and (B) metabolic activity of hUC-MSCs at 1 and 3 days of in vitro culture with the medium containing the extracts released from the FS membranes. Scale bars represent 200 μm. Data are presented as mean ± SD of three independent experiments (n = 3, per condition) performed in triplicates. | ||
3.7 Cell adhesion and morphology
The cellular adhesion and morphology of hUC-MSCs were evaluated by DAPI-phalloidin assay after 1 and 3 days of in vitro culture (Fig. 11). The cells adhered, denoted a stretched morphology, and were well spread in all the studied conditions, and exhibited a similar shape to the one observed on the control (cells cultured with normal medium). Moreover, the percentage of area covered by the cells increased over time.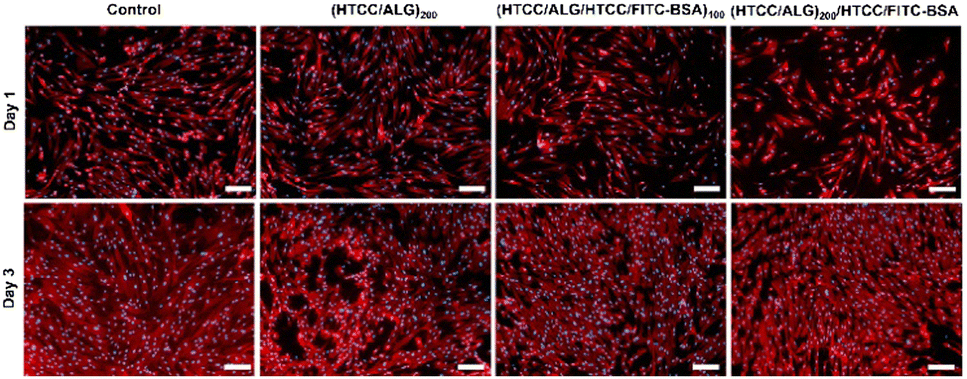 | ||
| Fig. 11 Representative fluorescence microscopy images of hUC-MSCs with F-actin filaments in the cytoskeleton stained with phalloidin (red) and nuclei stained with DAPI (blue) at 1 and 3 days of in vitro culture with the medium containing the extracts released from the FS membranes. Scale bars represent 200 μm. | ||
4. Conclusions
In this study, we have successfully developed thicker and robust FS membranes encompassing water-soluble HTCC and ALG biopolymers, under physiological conditions, for controlled drug delivery, aiming for biomedical applications. The FITC-BSA was incorporated either as an intrinsic building block or added as the outer layer on the FS membranes to demonstrate the influence of the film structure in the drug release profile. While the (HTCC/ALG)200/HTCC/FITC-BSA membranes exhibited a burst release of FITC-BSA within the first 4 h, the (HTCC/ALG/HTCC/FITC-BSA)100 FS membranes triggered a controlled release over time, following a quasi-zero-order release kinetics, thereby enabling a sustained drug delivery for extended periods of time. The HTCC-derived FS membranes denoted enhanced mechanical properties when assembled at physiological pH when compared to the native CHT-derived FS membrane counterparts, assembled under acidic pH environments. All the HTCC-derived FS membranes demonstrated to be biocompatible for hUC-MSCs, as confirmed by the live/dead and MTS assays, holding great promise for biomedical applications. We foresee that the HTCC-derived multilayered biomaterials together with the high versatility imparted by the LbL assembly technology could open new perspectives in the development of a wide repertoire of smart, sustainable, size and shape tunable implantable and injectable biomaterials for sustained drug/gene/therapeutics delivery, within the desired therapeutic window, in multiple tissue engineering and regenerative medicine strategies. Moreover, the drug nanoreservoirs herein proposed are expected to increase the use and application of the CHT biopolymer in biologically relevant scenarios, potentially reducing the dose frequency and adverse side effects, and improving drug efficacy.Author contributions
J. B. and J. F. M. conceived the original idea and research direction, provided the resources, supervised the project and acquired funding. C. F. V. S., J. B. and J. F. M. designed the experiments and methodology. C. F. V. S and L. P. G. M. performed the experiments. J. M. M. R. provided experimental advice regarding the NMR experiments. C. F. V. S. wrote the original draft of the manuscript. All authors analysed the data, discussed the results, and reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union's Horizon Europe research and innovation programme under the grant agreement No. 101079482 (“SUPRALIFE”). The financial support by the Portuguese Foundation for Science and Technology (FCT) through the individual PhD grants (2020.04408.BD, C. F. V. S.; 2020.06767.BD, L. P. G. M.), and individual Junior Researcher (CEECIND/01363/2018, J. M. M. R.) and Assistant Researcher (2020.00758.CEECIND, J. B.) contracts under the Scientific Employment Stimulus – Individual Call is gratefully acknowledged. This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MCTES (PIDDAC). The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project No. 022161 (co-financed by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC). The authors gratefully acknowledge Dr Hélène L. Lauzon from Primex ehf (Siglufjordur, Iceland) for kindly providing the chitosan batch used in this work, and Prof. Rute Ferreira and Dr Sandra Correia from CICECO-Aveiro Institute of Materials, Department of Physics, University of Aveiro (Aveiro, Portugal) for the kind assistance with the WCA measurements.References
- J. W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- Y. Ping, J. Guo, H. Ejima, X. Chen, J. J. Richardson, H. Sun and F. Caruso, Small, 2015, 11, 2032–2036 CrossRef CAS PubMed.
- H. Acar, J. M. Ting, S. Srivastava, J. L. LaBelle and M. V. Tirrell, Chem. Soc. Rev., 2017, 46, 6553–6569 RSC.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Adv. Mater., 2018, 30, 1705328 CrossRef PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- A. Bochot and E. Fattal, J. Controlled Release, 2012, 161, 628–634 CrossRef CAS PubMed.
- S. D. Steichen, M. Caldorera-Moore and N. A. Peppas, Eur. J. Pharm. Sci., 2013, 48, 416–427 CrossRef CAS PubMed.
- K. Ariga, Y. M. Lvov, K. Kawakami, Q. Ji and J. P. Hill, Adv. Drug Delivery Rev., 2011, 63, 762–771 CrossRef CAS PubMed.
- A. C. Lima, P. Sher and J. F. Mano, Expert Opin. Drug Delivery, 2012, 9, 231–248 CrossRef CAS PubMed.
- R. R. Costa, M. Alatorre-Meda and J. F. Mano, Biotechnol. Adv., 2015, 33, 1310–1326 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- C. Sanson, O. Diou, J. Thévenot, E. Ibarboure, A. Soum, A. Brûlet, S. Miraux, E. Thiaudière, S. Tan, A. Brisson, V. Dupuis, O. Sandre and S. Lecommandoux, ACS Nano, 2011, 5, 1122–1140 CrossRef CAS PubMed.
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- Y. Song, Y. Chen, P. Li and C. M. Dong, Biomacromolecules, 2020, 21, 5345–5357 CrossRef CAS PubMed.
- M. G. Gouveia, J. P. Wesseler, J. Ramaekers, C. Weder, P. B. V. Scholten and N. Bruns, Chem. Soc. Rev., 2023, 52, 728–778 RSC.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- V. Mohanta, G. Madras and S. Patil, J. Mater. Chem. B, 2013, 1, 4819–4827 RSC.
- C. D. Easton, A. J. Bullock, G. Gigliobianco, S. L. McArthur and S. Macneil, J. Mater. Chem. B, 2014, 2, 5558–5568 RSC.
- R. R. Costa and J. F. Mano, Chem. Soc. Rev., 2014, 43, 3453–3479 RSC.
- D. Choi and J. Hong, Arch. Pharmacal Res., 2014, 37, 79–87 CrossRef CAS PubMed.
- S. G. Caridade, C. Monge, J. Almodóvar, R. Guillot, J. Lavaud, V. Josserand, J. L. Coll, J. F. Mano and C. Picart, Acta Biomater., 2015, 15, 139–149 CrossRef CAS PubMed.
- M. Criado-Gonzalez, M. Fernandez-Gutierrez, J. San Roman, C. Mijangos and R. Hernández, Carbohydr. Polym., 2019, 206, 428–434 CrossRef CAS PubMed.
- C. Husteden, Y. A. Brito Barrera, S. Tegtmeyer, J. Borges, J. Giselbrecht, M. Menzel, A. Langner, J. F. Mano, C. E. H. Schmelzer, C. Wölk and T. Groth, Adv. Healthcare Mater., 2023, 12, 2201978 CrossRef CAS PubMed.
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS.
- J. Borges, L. C. Rodrigues, R. L. Reis and J. F. Mano, Adv. Funct. Mater., 2014, 24, 5624–5648 CrossRef CAS.
- M. J. Cardoso, S. G. Caridade, R. R. Costa and J. F. Mano, Biomacromolecules, 2016, 17, 1347–1357 CrossRef CAS PubMed.
- K. Park, D. Choi and J. Hong, Sci. Rep., 2018, 8, 3365 CrossRef PubMed.
- J. Borges, M. P. Sousa, G. Cinar, S. G. Caridade, M. O. Guler and J. F. Mano, Adv. Funct. Mater., 2017, 27, 1605122 CrossRef.
- X. Zhang, Y. Xu, X. Zhang, H. Wu, J. Shen, R. Chen, Y. Xiong, J. Li and S. Guo, Prog. Polym. Sci., 2019, 89, 76–107 CrossRef CAS.
- S. Zhao, F. Caruso, L. Dähne, G. Decher, B. G. De Geest, J. Fan, N. Feliu, Y. Gogotsi, P. T. Hammond, M. C. Hersam, A. Khademhosseini, N. Kotov, S. Leporatti, Y. Li, F. Lisdat, L. M. Liz-Marzan, S. Moya, P. Mulvaney, A. L. Rogach, S. Roy, D. G. Shchukin, A. G. Skirtach, M. M. Stevens, G. B. Sukhorukov, P. S. Weiss, Z. Yue, D. Zhu and W. J. Parak, ACS Nano, 2019, 13, 6151–6169 CrossRef CAS PubMed.
- J. L. Lutkenhaus, K. D. Hrabak, K. McEnnis and P. T. Hammond, J. Am. Chem. Soc., 2005, 127, 17228–17234 CrossRef CAS PubMed.
- S. S. Ono and G. Decher, Nano Lett., 2006, 6, 592–598 CrossRef CAS PubMed.
- R. Polak, T. Crouzier, R. M. Lim, K. Ribbeck, M. M. Beppu, R. N. M. Pitombo, R. E. Cohen and M. F. Rubner, Biomacromolecules, 2014, 15, 3093–3098 CrossRef CAS PubMed.
- S. G. Caridade, C. Monge, F. Gilde, T. Boudou, J. F. Mano and C. Picart, Biomacromolecules, 2013, 14, 1653–1660 CrossRef CAS PubMed.
- J. M. Silva, A. R. C. Duarte, S. G. Caridade, C. Picart, R. L. Reis and J. F. Mano, Biomacromolecules, 2014, 15, 3817–3826 CrossRef CAS PubMed.
- J. Borges, S. G. Caridade, J. M. Silva and J. F. Mano, Macromol. Rapid Commun., 2015, 36, 405–412 CrossRef CAS PubMed.
- J. M. Silva, S. G. Caridade, R. L. Reis and J. F. Mano, Soft Matter, 2016, 12, 1200–1209 RSC.
- N. I. Martins, M. P. Sousa, C. A. Custódio, V. C. Pinto, P. J. Sousa, G. Minas, F. Cleymand and J. F. Mano, Acta Biomater., 2017, 57, 313–323 CrossRef CAS PubMed.
- Z. Liang, A. S. Susha, A. Yu and F. Caruso, Adv. Mater., 2003, 15, 1849–1853 CrossRef CAS.
- S. Hou, J. Wang and C. R. Martin, Nano Lett., 2005, 5, 231–234 CrossRef CAS PubMed.
- T. Komatsu, H. Terada and N. Kobayashi, Chem. – Eur. J., 2011, 17, 1849–1854 CrossRef CAS PubMed.
- J. M. Silva, C. A. Custódio, R. L. Reis and J. F. Mano, ACS Biomater. Sci. Eng., 2016, 2, 2304–2314 CrossRef CAS PubMed.
- B. G. De Geest, S. De Koker, K. Immesoete, J. Demeester, S. C. De Smedt and W. E. Hennink, Adv. Mater., 2008, 20, 3687–3691 CrossRef CAS.
- A. Elbakry, A. Zaky, R. Liebl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- Z. Poon, J. B. Lee, S. W. Morton and P. T. Hammond, Nano Lett., 2011, 11, 2096–2103 CrossRef CAS PubMed.
- J. Huang, Q. Shu, L. Wang, H. Wu, A. Y. Wang and H. Mao, Biomaterials, 2015, 39, 105–113 CrossRef CAS PubMed.
- F. Huang, W. C. Liao, Y. S. Sohn, R. Nechushtai, C. H. Lu and I. Willner, J. Am. Chem. Soc., 2016, 138, 8936–8945 CrossRef CAS PubMed.
- C. Ribeiro, J. Borges, A. M. S. Costa, V. M. Gaspar, V. de Zea Bermudez and J. F. Mano, Molecules, 2018, 23, 625 CrossRef PubMed.
- C. R. Correia, I. M. Bjørge, J. Zeng, M. Matsusaki and J. F. Mano, Adv. Healthcare Mater., 2019, 8, 1901221 CrossRef CAS PubMed.
- S. Nadine, S. G. Patrício, C. C. Barrias, I. S. Choi, M. Matsusaki, C. R. Correia and J. F. Mano, Adv. Biosyst., 2020, 4, 2000127 CrossRef CAS PubMed.
- O. Kulygin, A. D. Price, S. F. Chong, B. Städler, A. N. Zelikin and F. Caruso, Small, 2010, 6, 1558–1564 CrossRef CAS PubMed.
- R. R. Costa, E. Castro, F. J. Arias, J. C. Rodríguez-Cabello and J. F. Mano, Biomacromolecules, 2013, 14, 2403–2410 CrossRef CAS PubMed.
- S. Nadine, I. Fernandes, S. G. Patrício, C. R. Correia and J. F. Mano, Adv. Healthcare Mater., 2022, 11, 2200651 CrossRef CAS PubMed.
- P. Sher, C. A. Custódio and J. F. Mano, Small, 2010, 6, 2644–2648 CrossRef CAS PubMed.
- P. Sher, S. M. Oliveira, J. Borges and J. F. Mano, Biofabrication, 2015, 7, 011001 CrossRef PubMed.
- C. F. V. Sousa, C. A. Saraiva, T. R. Correia, T. Pesqueira, S. G. Patrício, M. I. Rial-Hermida, J. Borges and J. F. Mano, Biomolecules, 2021, 11, 863 CrossRef CAS PubMed.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- R. R. Costa, A. I. Neto, I. Calgeris, C. R. Correia, A. C. M. Pinho, J. Fonseca, E. T. Öner and J. F. Mano, J. Mater. Chem. B, 2013, 1, 2367–2374 RSC.
- K. f Ren, M. Hu, H. Zhang, B. c Li, W. x Lei, J. y Chen, H. Chang, L. m Wang and J. Ji, Prog. Polym. Sci., 2019, 92, 1–34 CrossRef CAS.
- J. M. Silva, R. L. Reis and J. F. Mano, Small, 2016, 12, 4308–4342 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- M. T. Cook, G. Tzortzis, V. V. Khutoryanskiy and D. Charalampopoulos, J. Mater. Chem. B, 2013, 1, 52–60 RSC.
- J. M. Silva, S. G. Caridade, N. M. Oliveira, R. L. Reis and J. F. Mano, J. Mater. Chem. B, 2015, 3, 4555–4568 RSC.
- D. Silva, L. F. V. Pinto, D. Bozukova, L. F. Santos, A. P. Serro and B. Saramago, Colloids Surf., B, 2016, 147, 81–89 CrossRef CAS PubMed.
- S. Petroni, I. Tagliaro, C. Antonini, M. D’Arienzo, S. F. Orsini, J. F. Mano, V. Brancato, J. Borges and L. Cipolla, Mar. Drugs, 2023, 21, 147 CrossRef CAS PubMed.
- M. A. Pujana, L. Pérez-Álvarez, L. C. C. Iturbe and I. Katime, Polymer, 2012, 53, 3107–3116 CrossRef CAS.
- B. I. Andreica, X. Cheng and L. Marin, Eur. Polym. J., 2020, 139, 110016 CrossRef CAS.
- K. Kamiński, K. Szczubiałka, K. Zazakowny, R. Lach and M. Nowakowska, J. Med. Chem., 2010, 53, 4141–4147 CrossRef PubMed.
- L. Wang, C. Qin, W. Wang and W. Li, Carbohydr. Polym., 2011, 84, 1289–1292 CrossRef CAS.
- B. Lorkowska-Zawicka, K. Kamiński, J. Ciejka, K. Szczubiałka, M. Białas, K. Okoń, D. Adamek, M. Nowakowska, J. Jawień, R. Olszanecki and R. Korbut, Mar. Drugs, 2014, 12, 3953–3969 CrossRef PubMed.
- E. D. Freitas, C. F. Moura, J. Kerwald and M. M. Beppu, Polymer, 2020, 12, 2878 CAS.
- M. Kumorek, D. Kubies and T. Riedel, Physiol. Res., 2016, 65, S253–S261 CAS.
- T. Urbaniak, G. S. García-Briones, A. Zhigunov, S. Hladysh, E. Adrian, V. Lobaz, T. Krunclová, O. Janoušková, O. Pop-Georgievski and D. Kubies, Biomacromolecules, 2022, 23, 4734–4748 CrossRef CAS PubMed.
- W. Huang, X. Li, Y. Xue, R. Huang, H. Deng and Z. Ma, Int. J. Biol. Macromol., 2013, 53, 26–31 CrossRef CAS PubMed.
- R. Yuan, N. Yang, S. Fan, Y. Huang, D. You, J. Wang, Q. Zhang, C. Chu, Z. Chen, L. Liu and L. Ge, Small, 2021, 17, 2103997 CrossRef CAS PubMed.
- Z. Cele, L. Ndlandla, A. Somboro, D. Gyamfi and M. Balogun, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 430, 012048 Search PubMed.
- X. Yang, C. Zhang, C. Qiao, X. Mu, T. Li, J. Xu, L. Shi and D. Zhang, Carbohydr. Polym., 2015, 130, 325–332 CrossRef CAS PubMed.
- R. J. Hunter, Zeta Potential in Colloid Science: Principle and Applications, Academic Press, London, UK, 1988 Search PubMed.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- J. Hatami, S. G. Silva, M. B. Oliveira, R. R. Costa, R. L. Reis and J. F. Mano, Polymers, 2017, 9, 440 CrossRef PubMed.
- M. Montazer and M. G. Afjeh, J. Appl. Polym. Sci., 2007, 103, 178–185 CrossRef CAS.
- Y. Wen, Z. Tan, F. Sun, L. Sheng, X. Zhang and F. Yao, Mater. Sci. Eng., C, 2012, 32, 2026–2036 CrossRef CAS PubMed.
- C. Picart, P. Lavalle, P. Hubert, F. J. G. Cuisinier, G. Decher, P. Schaaf and J. C. Voegel, Langmuir, 2001, 17, 7414–7424 CrossRef CAS.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099–1120 CrossRef CAS PubMed.
- C. Picart, J. Mutterer, L. Richert, Y. Luo, G. D. Prestwich, P. Schaaf, J. C. Voegel and P. Lavalle, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12531–12535 CrossRef CAS PubMed.
- P. Lavalle, C. Picart, J. Mutterer, C. Gergely, H. Reiss, J. C. Voegel, B. Senger and P. Schaaf, J. Phys. Chem. B, 2004, 108, 635–648 CrossRef CAS.
- A. I. Neto, A. C. Cibrão, C. R. Correia, R. R. Carvalho, G. M. Luz, G. G. Ferrer, G. Botelho, C. Picart, N. M. Alves and J. F. Mano, Small, 2014, 10, 2459–2469 CrossRef CAS PubMed.
- M. L. Macdonald, R. E. Samuel, N. J. Shah, R. F. Padera, Y. M. Beben and P. T. Hammond, Biomaterials, 2011, 32, 1446–1453 CrossRef CAS PubMed.
- A. Hautmann, D. Kedilaya, S. Stojanović, M. Radenković, C. K. Marx, S. Najman, M. Pietzsch, J. F. Mano and T. Groth, Biomater. Adv., 2022, 14, 213166 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |