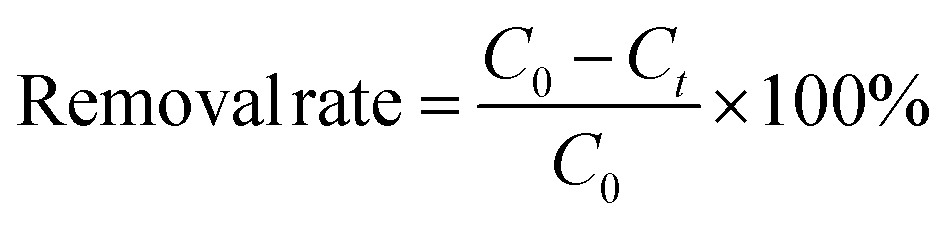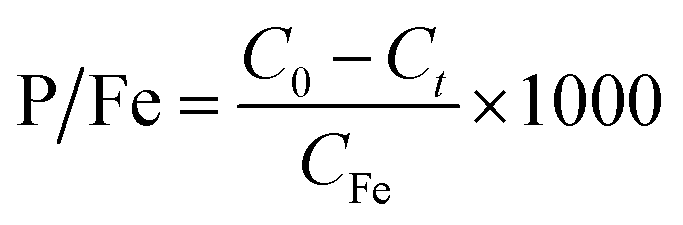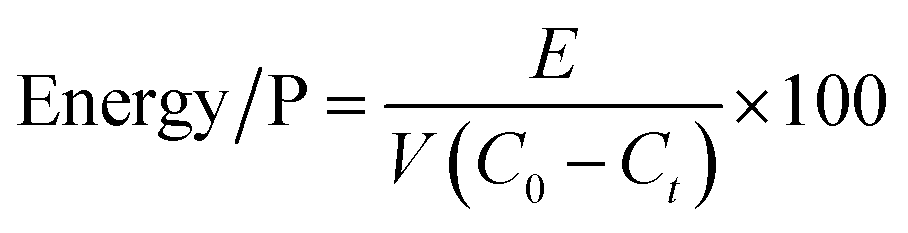Phosphate removal by ex situ generated Fe (hydr)oxides from scrap iron electrocoagulation: the critical role of coprecipitation†
Received
4th February 2023
, Accepted 24th April 2023
First published on 24th April 2023
Abstract
The immobilization of phosphate by Fe (hydr)oxides is a promising method with low cost and high efficiency. However, the complicated synthesis process and particle aggregation limited their application. In this study, we utilized electrocoagulation with a scrap iron anode to generate Fe (hydr)oxides in the prepared NaCl electrolyte, which are ready to use for phosphate removal on-site. The Fe (hydr)oxides generated via scrap iron electrocoagulation without aeration removed 86.7% of phosphate and showed an energy cost of 35.9 W h g−1 P. The structure and morphology of Fe precipitates containing phosphate were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). The results showed that vivianite, strengite, and amorphous Fe–P precipitates were formed immediately after the mixing of Fe (hydr)oxides with phosphate. Compared to coprecipitation, adsorption by Fe (hydr)oxides (e.g., lepidocrocite) plays a minor role in phosphate removal. The findings gained from this study shed light on the feasibility and mechanism of scrap iron materials in the immobilization of phosphate through on-site electrocoagulation.
Environmental significance
The looming P crisis makes the removal of P from wastewater the main challenges of the twenty-first century. We report a new strategy of ex situ production of Fe (hydr)oxides by scrap iron electrocoagulation in a prepared NaCl electrolyte that is facile and low cost for the sequestration of phosphate, expanding the scope of application of Fe-based materials for phosphate sequestration in wastewater.
|
1. Introduction
Inefficient use and loss of phosphorus pose increasing risk to food and water security, freshwater biodiversity, and human health.1,2 For example, P fertilizer manufacturing emits around 200–280 million tons of phosphogypsum waste per year, which leads to serious water and soil contamination during the storage.3,4 Increasing P load possibly leads to the eutrophication of freshwater bodies as well as harmful algal blooms. These phenomena are especially serious in closed waterbodies, such as lakes and ponds, as revealed by exhaustion of dissolved oxygen, death of fish, and bad smell.5 The looming P crisis makes the removal of P from wastewater the main challenges of the twenty-first century.6–8
In both China and the United states, the discharge of phosphate is limited to 0.5–1.0 mg L−1.5 To meet the strict discharge requirements on P content, technologies including chemical precipitation,9,10 adsorption,11–13 and enhanced biological P removal14 have been developed. Among the methods, adsorption by a suite of Fe (hydr)oxides (e.g., ferrihydrite, magnetite, and goethite) has shown promise for phosphate removal owing to the advantages of environmental friendliness, low cost, and easy regeneration.11,15 The adsorption of PO43− on Fe (hydr)oxides was found to be endothermic and spontaneous, possibly via electrostatic attraction and surface precipitation.11,15–17 The adsorption capacities of Fe (hydr)oxides largely depend on the synthesis methods, (hydr)oxide structure, solution pH, and competitive anions.12 For example, Fe hydroxides (e.g., ferrihydrite and green rust) showed higher adsorption capacity in comparison to oxides (e.g., magnetite and goethite).11,15 Moreover, freshly prepared Fe (hydr)oxides were found to have a higher adsorption capacity than aged ones.18 Fe (hydr)oxides are usually synthesized by co-precipitation or hydrothermal treatment.15,19 However, these methods are either time consuming or difficult to control, limiting the application of Fe (hydr)oxides as a P sequestrator.
Electrocoagulation is an efficient method for the production of a range of high-quality Fe (hydr)oxides that are potential sequestrators of phosphate. In the electrocoagulation process, Fe ions are delivered in situ by the electrochemical dissolution of Fe(0) anodes, which react with the hydroxyl ions generated on the cathode to form Fe (hydr)oxides.20,21 This process could produce various Fe (hydr)oxide nanoparticles including green rust, magnetite, lepidocrocite, and goethite, depending on the current density, dissolved oxygen, and electrolyte.22,23 For example, an electrocoagulation process using steel electrodes produced pure magnetite particles of 100 nm diameter, which showed significant magnetic susceptibility.24 Similarly, uniform magnetite nanorods were prepared by a pulsed current electrocoagulation method.25 Ying et al.26 found that magnetite particles could form in NaCl solution regardless of the presence of Mg2+ and Ca2+ ions by electrocoagulation with carbon steel electrodes. Although some studies reported that the electrocoagulation process was efficient for in situ treatment of excess P in wastewater,27–29 complexed wastewater components would lead to electrode passivation and energy increase.30 Besides, wastewater of low electricity conductivity would lead to high energy consumption in the electrocoagulation process due to the high ohmic drop. Thus, dosing Fe (hydr)oxides generated by electrocoagulation in well-controlled electrolytes such as prepared NaCl solution would be more attractive for the removal of phosphate. However, the knowledge on this aspect is still limited.
In this study, we utilized scrap iron as a sacrificial anode to generate Fe (hydr)oxides to reduce cost and promote waste recycle. The performance of P removal by ex situ generated Fe (hydr)oxides was evaluated by using the removal rate, P/Fe ratios, and energy consumption. Specifically, we first compared the performance of different types of scrap iron materials. Then, we investigated the impact of aerations and current intensity on the performance. Ultimately, we characterized the Fe precipitations containing Fe–P minerals by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). The results obtained from this study shed light on the feasibility of scrap iron materials in the treatment of P wastewater and recycling P to achieve the goal of turning waste into treasure.
2. Materials and methods
2.1. Experimental operation
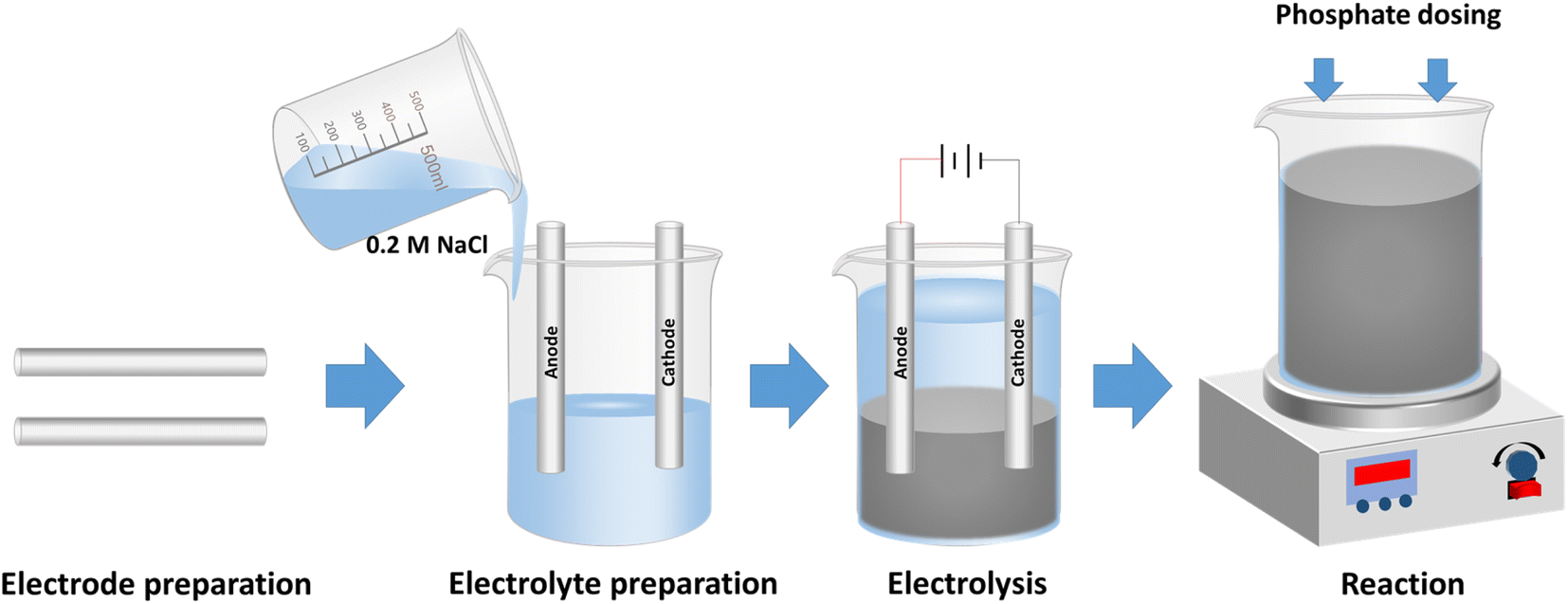
The description and physical properties of waste materials used as electrodes are presented in Table 1. Electrocoagulation experiments were carried out in a glass cell of 600 mL at room temperature (Fig. S1 in the ESI†). Electrodes made of scrap iron were installed in parallel with a distance of 2.0 cm. Prior to each experiment, the iron electrodes were polished mechanically, and cleaned with deionized (DI) water. 500 mL electrolyte was prepared with 0.2 M NaCl. NaCl was chosen as it is cheap and widely available, and retards the passivation of electrodes.31 After 25 min electrolysis, suspensions containing Fe (hydr)oxides were generated. Then, a stock solution of KH2PO4 was added into the suspensions to initiate the experiments of phosphate removal. The solution was thoroughly mixed with a magnetic stirrer during the electrocoagulation and phosphate removal tests. A schematic diagram of the experimental procedure is shown in Scheme 1. The aeration was supplied by an air pump (SB748, Zhongshan Songbao Electric Group) at a flow rate recorded by a gas flow meter (LZB-3WB, Shuanghuan). Constant currents were supplied by a DC power supply (GPS-3303C, Gw-INSTEK) during the electrocoagulation. The energy consumption during the processes was monitored by using a wattmeter (DPT1502, JUWEI). The initial pH values of the suspensions were adjusted with diluted hydrochloric acid (HCl) and sodium hydroxide (NaOH) solution during the phosphate removal tests.
Table 1 Description and physical properties of the waste materials used as electrodes in this study
| Material used |
Scrap cast iron |
Scrap mild steel |
| Alloy type |
Cast iron |
Mild steel |
| Composition |
Fe, C, Si |
Fe, Mn, C |
| Density (g cm−3) |
7.35 |
7.85 |
| Electric conductivity (S m−1) at 20 °C |
2.00 × 106 |
6.21 × 106 |
| Shape |
Cylinder |
Cylinder |
| Diameter of material (mm) |
9.863 |
7.006 |
| Length (mm) |
82.22 |
60.70 |
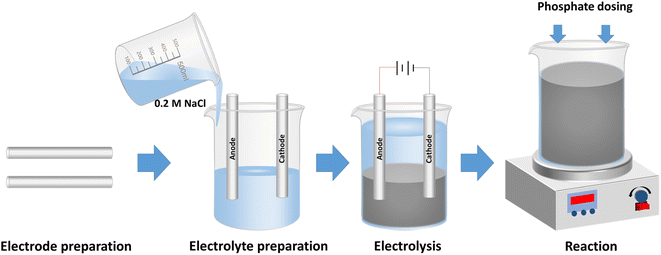 |
| | Scheme 1 Schematic diagram of the experimental procedure in this study. Electrodes made of scrap iron were polished and cleaned before usage. Electrolyte was prepared with 0.2 M NaCl solution. After 25 min electrolysis, suspensions containing Fe (hydr)oxides were generated. Then, a stock solution of KH2PO4 was added into the suspensions to initiate the experiments of phosphate removal. | |
2.2. Calculations
The removal rate (%) was calculated using eqn (1):| | 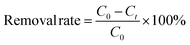 | (1) |
where C0 and Ct are the initial and final concentrations of phosphorus (mg L−1).
The phosphate removal capacity of Fe (hydr)oxides (P/Fe, mg g−1) was calculated using eqn (2):
| | 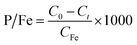 | (2) |
where
CFe is the final concentration of total Fe generated by electrocoagulation (mg L
−1).
The mean energy consumption for phosphate removal (Energy/P, W h g−1) was calculated using eqn (3):
| | 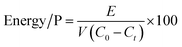 | (3) |
where
E is the power consumption recorded by the wattmeter (W h), and
V is the volume of the phosphate solution (L).
2.3. Measurement and analysis
Two samples of the solution were taken at predetermined times during the phosphate removal experiments with one of these samples filtered immediately through a 0.22 μm nylon membrane. The concentrations of phosphate in the filtered samples were determined by the molybdenum blue spectrophotometric method at 700 nm using an AOE UV-1800 spectrophotometer (Ao Yi Instrument, Shanghai). The solution pH values and the dissolved oxygen (DO) concentrations were measured by using a multifunctional water quality analyzer (Multi-350i, WTW Group, Germany). Dissolved Fe(II) was measured at a wavelength of 562 nm by a ferrozine method.32 Another set of samples acquired from the suspensions were digested in 3 M HCl for the determination of total Fe through the reduction of hydroxylamine hydrochloride.
The solid samples were collected during the phosphate removal tests for X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), and scanning TEM (STEM) combined with energy-dispersive X-ray detection (EDX). The suspensions generated after 25 min electrocoagulation were centrifugated and freeze-dried under anoxic conditions filled with N2 gas (99.999%). XPS was collected on an AXIS SUPRA+ Spectrometer (SHIMADZU, Japan) equipped with a monochromatic Al Kα source at 58.7 eV. The binding energy was calibrated using the C 1s XPS peak at 284.8 eV. XRD was carried out on an X-ray diffractometer (SmartLab SE, Rigaku, Japan) with Cu K radiation at a scanning speed of 1° per min and a step size of 0.02°. To characterize the morphology of precipitates, a field-emission gun TEM (Tecnai G2 F20, FEI, USA) was operated at 200 kV. The elemental distribution was obtained on the same machine equipped with an EDX detector.
3. Results and discussion
3.1. Impact of aeration
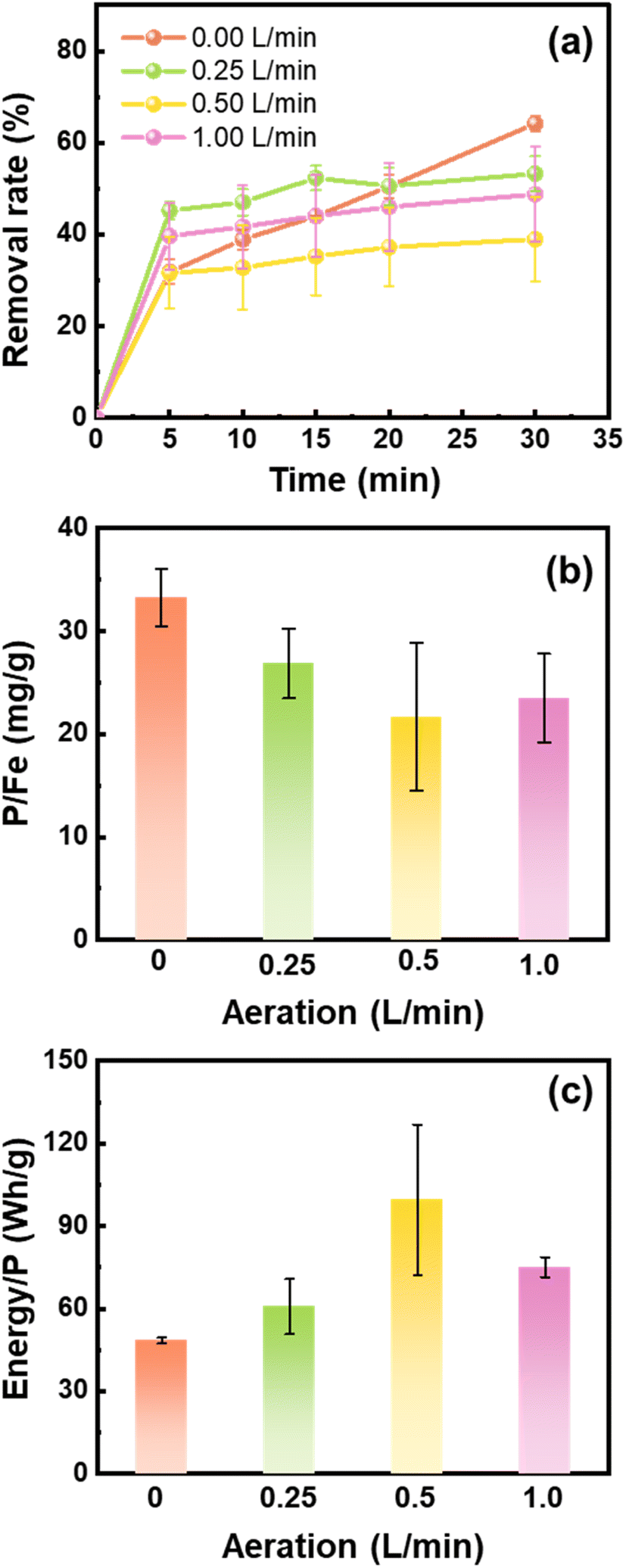
The Fe (hydr)oxides generated during iron electrocoagulation depend on the DO content that is controlled by aeration. We compared the performance of Fe (hydr)oxides generated under different flow rates of aeration for phosphate removal. As shown in Fig. 1a, phosphate was largely removed in the first 5 min, with a small fraction being removed in the following 25 min. The phosphate removal rates approached 64.16%, 53.26%, 38.94%, and 48.76% at 30 min when 0, 0.25, 0.5, and 1.0 L min−1 air was supplied to the electrocoagulation processes, respectively. In line with the results of removal rates, the phosphate removal capacity of Fe (hydr)oxides decreased with the air flow rate from 0 to 0.5 L min−1, but increased slightly from 0.5 to 1.0 L min−1 (Fig. 1b). The energy consumption for phosphate removal increased from 48.4 W h g−1 to 99.5 W h g−1 as the air flow rate increased from 0 to 0.5 L min−1, but decreased slightly to 75.1 W h g−1 at 1.0 L min−1 (Fig. 1c).
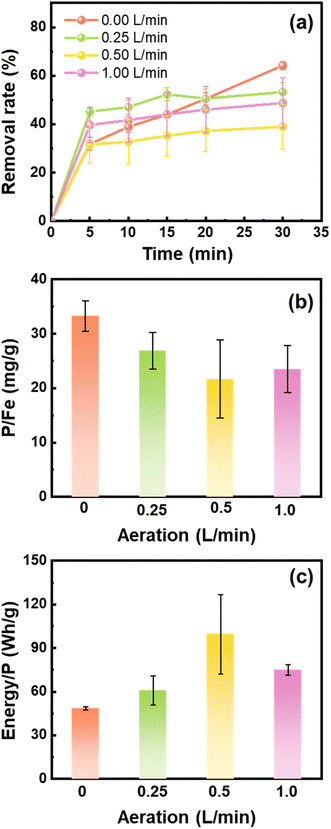 |
| | Fig. 1 (a) Variations of the phosphate removal rate, (b) removal capacity of phosphate, and (c) electricity energy input for the removal of 1 g phosphate by the Fe (hydr)oxides generated from scrap iron electrocoagulation under different air flow rates. Experiment conditions: [electrolyte] = 0.2 M NaCl, 25 min electrocoagulation using the cast iron anode at 0.2 A, and [phosphate]0 = 10 mg L−1. | |
After 25 min of electrocoagulation, the majority of Fe ions dissolved from the anode were precipitated (Fig. 2a). Especially for 0 L min−1, there was only 0.1 mg L−1 of dissolved Fe detected (Fig. 2a). The low content of dissolved Fe at 0 L min−1 might be due to the rise in solution pH to 10.4 (Fig. 2b), under which conditions both Fe2+ and Fe3+ were hydrolyzed and precipitated to Fe (hydr)oxides. However, as the solution pH decreased to about 7.3 during the phosphate removal experiments (Fig. 2b), Fe2+ would be dissolved and contributed to the increase of the phosphate removal rate after 5 min. Thermodynamic calculation by using Visual MINTEQ v3.1 showed that phosphate competed with hydroxyl ions for Fe2+ to form vivianite in the pH range of 4.5–8.0 (Fig. S2a†). However, the formation of vivianite would be unfavorable at pH > 9.33 Therefore, coprecipitation might be a contributor to the phosphate removal under anoxic conditions.
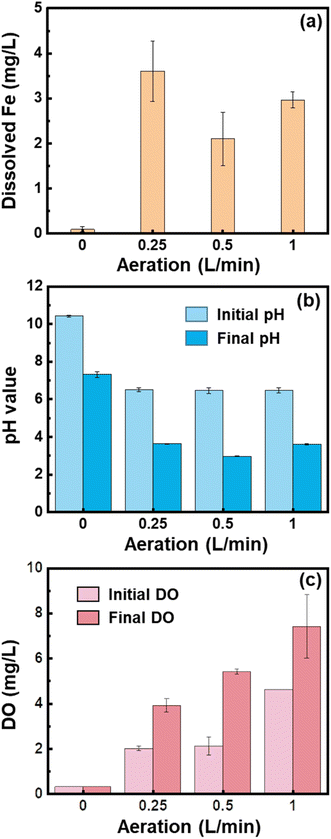 |
| | Fig. 2 (a) Dissolved Fe content after 25 min electrocoagulation, change of (b) pH, and (c) DO during the phosphate removal processes by the Fe (hydr)oxides generated from scrap iron electrocoagulation under different air flow rates. Experiment conditions: [electrolyte] = 0.2 M NaCl, electrolysis at 0.2 A for 25 min, and [phosphate]0 = 10 mg L−1. | |
In contrast, when 0.25–1.0 L min−1 air was supplied to the electrolytes, the solution pH value was kept at about 6.5 and dissolved Fe could be observed (Fig. 2a and b). The concentrations of DO increase with increasing the air flow rate (Fig. 2c). Under oxic conditions, Fe(II) hydroxides were oxidized quickly, leading to Fe(III) oxides or hydroxides being the main components of precipitates. It has been found that lepidocrocite is the main product of precipitation in the aerobic electrocoagulation system.23,32 The initial quick removal of phosphate might be induced by the dissolved Fe (mainly as Fe2+) that resulted in the formation of vivianite and strengite (Fig. S2†). Consistent with the concentrations of dissolved Fe, the phosphate removal rate at 5 min was in an order of 0.25 L min−1 >1.0 L min−1 >0.5 L min−1. In the subsequent processes, the phosphate removal rate was slow under all conditions. The adsorption of phosphate by the Fe(III) hydroxides was facilitated by the presence of hydroxyl groups (–OH and –OH2).34 However, the fraction of these hydroxyl groups decreased with the aggregation process of nanoprecipitates, and the formation of large flocs reduced the surface area available for phosphate adsorption.35 Therefore, the removal of phosphate slowed down with increasing reaction time, which was in line with previous observations.36
3.2. Impact of scrap iron materials
To study the effects of different iron materials on the Fe (hydr)oxide production and phosphate removal, scrap cast iron and scrap mild steel were selected for the experiments. As shown in Fig. 3a, the phosphate removal rates were 64.16% and 56.99% in 30 min adsorption by the Fe (hydr)oxides generated from the electrocoagulation process using the cast iron electrodes and the mild steel electrodes, respectively. The phosphate removal capacity of the Fe (hydr)oxides was comparable between the two materials, both about 33.0 mg g−1 (Fig. 3b). However, the energy consumption using cast iron (48.4 W h g−1) was lower than that using mild steel (65.8 W h g−1) (Fig. 3c).
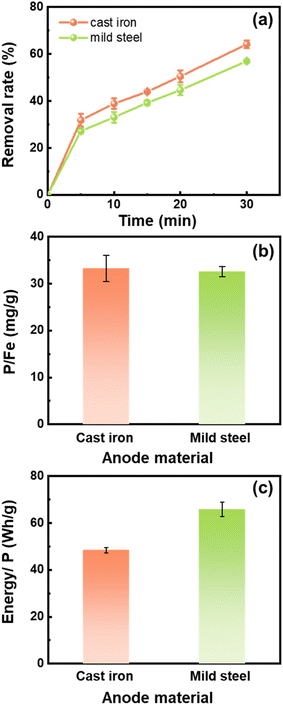 |
| | Fig. 3 (a) The phosphate removal rate during the experiments by the Fe (hydr)oxides generated from the electrocoagulation processes with different scrap iron materials. (b) P/Fe ratios and (c) energy consumption of the experiments. Experimental conditions: 0.2 M NaCl, 0.2 A for 25 min electrocoagulation, no aeration, and [phosphate]0 = 10 mg L−1. | |
The production of dissolved Fe ions from the anode is crucial for the phosphate removal by the electrocoagulation process since the Fe ions are the source of Fe (hydr)oxides. The Fe concentration generated by the cast iron electrodes was 181 ± 20 mg L−1 at 30 min, higher than that by the mild steel electrodes (163 ± 4 mg L−1). The different Fe production might result from the different C contents in the materials. The C content in the cast iron is 5.0–7.9%, which is higher than that in the mild steel (0.6–1.9%). The carbon is mainly present as graphite in the cast iron. The release of Fe on the surface leaves behind a skeleton of graphite flakes, which would contribute to the electrode corrosion.37 Previous work also presented a higher production of Fe ions in the first 25 min using the cast iron anode than that in the case of the mild steel anode.28 Altogether, the high removal rate and low energy consumption made the cast iron outperform mild steel when serving as the anode of the electrocoagulation process.
3.3. Impact of current intensity
The phosphate removal and energy cost performance under different current intensities differed remarkedly. Since high concentrations of Fe (hydr)oxides were generated under high current intensity, the reaction time with phosphate was prolonged to 300 min. The phosphate removal rate substantially increased from 28.0% at 100 mA to 86.7–99.3% when the applied current was ≥200 mA (Fig. 4a). Among the experiments, the removal capacity of the Fe (hydr)oxides generated at 200 mA was highest being 44.5 mg g−1 (Fig. 4b). Although a high removal rate was reached at 400 and 600 mA, surplus Fe (hydr)oxides generated did not contribute to the phosphate removal. Moreover, the energy consumption increased with the current intensity. For example, the energy consumption at 200 mA (35.9 W h g−1) was less than a third of that at 400 mA (112.2 W h g−1) (Fig. 4c).
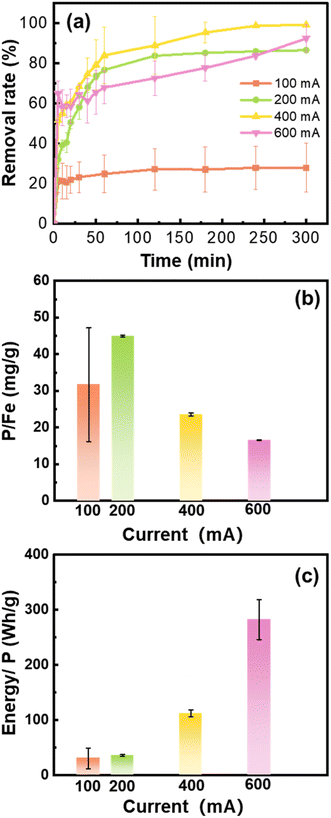 |
| | Fig. 4 (a) The phosphate removal rate during the adsorption experiments by the Fe (hydr)oxides generated from the electrocoagulation processes at different current intensities. (b) P/Fe ratios and (c) energy consumption of the experiments. Experimental conditions: 0.2 M NaCl, 25 min electrocoagulation using the cast iron anode, no aeration, and [phosphate]0 = 10 mg L−1. | |
Compared to the quick equilibrium of phosphate removal under aeration, the phosphate removal approached the steady stage until 180 min of reactions under no aeration. The DO concentration gradually increased from 0.3 mg L−1 to 6.1 mg L−1, and the solution pH decreased from 10.4 to 5.5 at 200 mA (Fig. S3†). During the phosphate removal process, the Fe(II) (hydr)oxides were gradually oxidized by the DO that diffused naturally, which resulted in the release of protons. Then, the weakly acidic conditions were favorable for PO4-P precipitation as vivianite and strengite as we have calculated in Fig. S2.† However, the solution pH increased to 11.0 after 25 min of electrocoagulation at 600 mA, which only decreased to be pH 9.1 after 300 min reaction. The abundant hydroxyl ions generated during electrocoagulation at high current would not be neutralized during the oxidation. Thus, considering the phosphate removal and energy cost, 200 mA is the optimal current for Fe (hydr)oxide generation by electrocoagulation.
The phosphate removal capacity achieved by the Fe (hydr)oxides at 200 mA was comparable to those reported in the literature. For example, Ajmal et al. obtained the maximum adsorption capacity of 57.8 mg g−1 for phosphate using well-prepared magnetite at pH 7.0.11 The removal of phosphate by the in situ electrocoagulation is also a promising process, although it depends on wastewater compositions. For the treatment of simulated wastewater containing P of 1–3 mg L−1, the removal capacity of electrocoagulation approached 50 mg P g−1 Fe.38 However, the removal capacities of electrocoagulation were found to be 21.7–93.5 mg P g−1 Fe for the treatment of wastewater containing P of 1.3 mg L−1.36
3.4. Characterization of Fe precipitates
To reveal the phosphate removal mechanism by the Fe (hydr)oxides, XRD, XPS, and TEM were performed to characterize the Fe precipitates. As shown in Fig. 5, vivianite and strengite, formed by the reactions of PO43− with ferrous and ferric ions (eqn (4) and (5)), were produced immediately after the reaction. However, the intensity of vivianite gradually disappeared at 120 min, which might result from the oxidation of vivianite.| | | 3Fe2+ + 2PO43− → Fe3(PO4)2 | (4) |
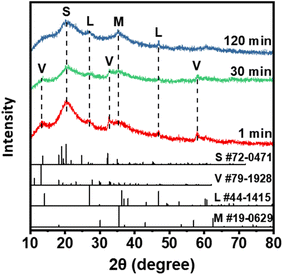 |
| | Fig. 5 XRD pattern of solid samples collected at 0, 30, and 120 min after phosphate dosing into the Fe (hydr)oxide suspensions generated under 200 mA without aeration. In the figure, S is the abbreviation of strengite, V is vivianite, L is lepidocrocite, and M is magnetite. | |
In contrast, the intensity of magnetite and lepidocrocite becomes strong with time, which provides further evidence of the oxidation of Fe hydroxides. The transformation of vivianite to strengite solids might improve the phosphate removal efficiencies, since strengite has a low solubility (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ksp = −26) and is easy to settle.39,40 These observations confirmed that coprecipitation contributed to the initial removal of phosphate, and the oxidation of Fe(II) minerals promoted the removal efficiency in the later stage.
Ksp = −26) and is easy to settle.39,40 These observations confirmed that coprecipitation contributed to the initial removal of phosphate, and the oxidation of Fe(II) minerals promoted the removal efficiency in the later stage.
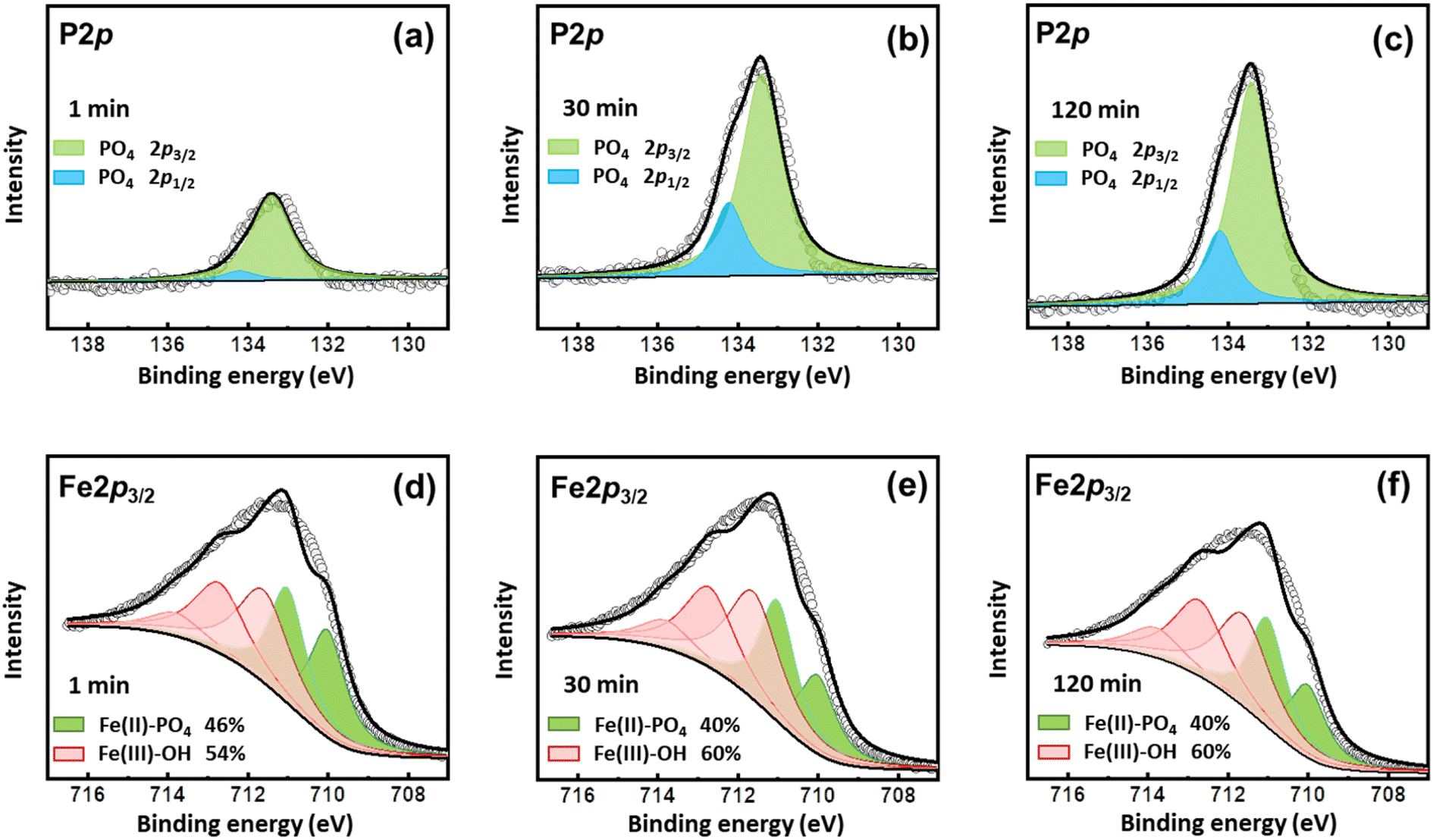
The coprecipitation of Fe–P and the transformation of Fe3(PO4)2 to FePO4 were also evidenced by XPS analyses (Fig. 6). The P 2p3/2 at 133.4 eV and 2p1/2 at 134.2 eV were ascribed to the peaks of PO4.41,42 As shown in the P 2p spectra, PO4 appeared on the surface once the reaction started, which increased to a high level at 30 min and kept stable during the subsequent processes (Fig. 6a–c). The Fe 2p3/2 data were successfully modeled using a combination of ferrous and ferric multiplet patterns (Fig. 6d–f and Table S1†). The multiplet peaks at 710.0 and 711.0 eV were ascribed to Fe(II)-PO4, and multiplet peaks at 711.6, 712.7, 713.8, and 715.1 eV belong to Fe(III)–OH.42 Vivianite (Fe(II)-PO4) accounted for 46% of the surface Fe precipitates in the beginning. However, at 30 min, the content of Fe(II)-PO4 decreased to 40%, suggesting that oxidation occurred during the process, in line with the findings of XRD results. The oxidation might be partial, since the content of Fe(II)-PO4 still accounted for 40% at 120 min, due to the stability of the electronic configuration of a Fe(II)B–Fe(III)B pair relative to a Fe(III)B–Fe(III)B pair.42 Thus, the vivianite on the surface was partly oxidized during the reactions.
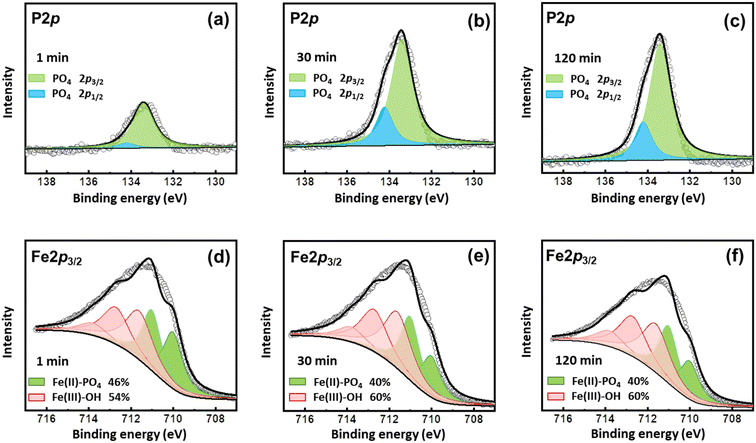 |
| | Fig. 6 XPS spectra of solid samples collected at (a and d) 1 min, (b and e) 30 min, and (c and f) 120 min after phosphate addition to the Fe (hydr)oxide suspensions generated under 200 mA without aeration. | |
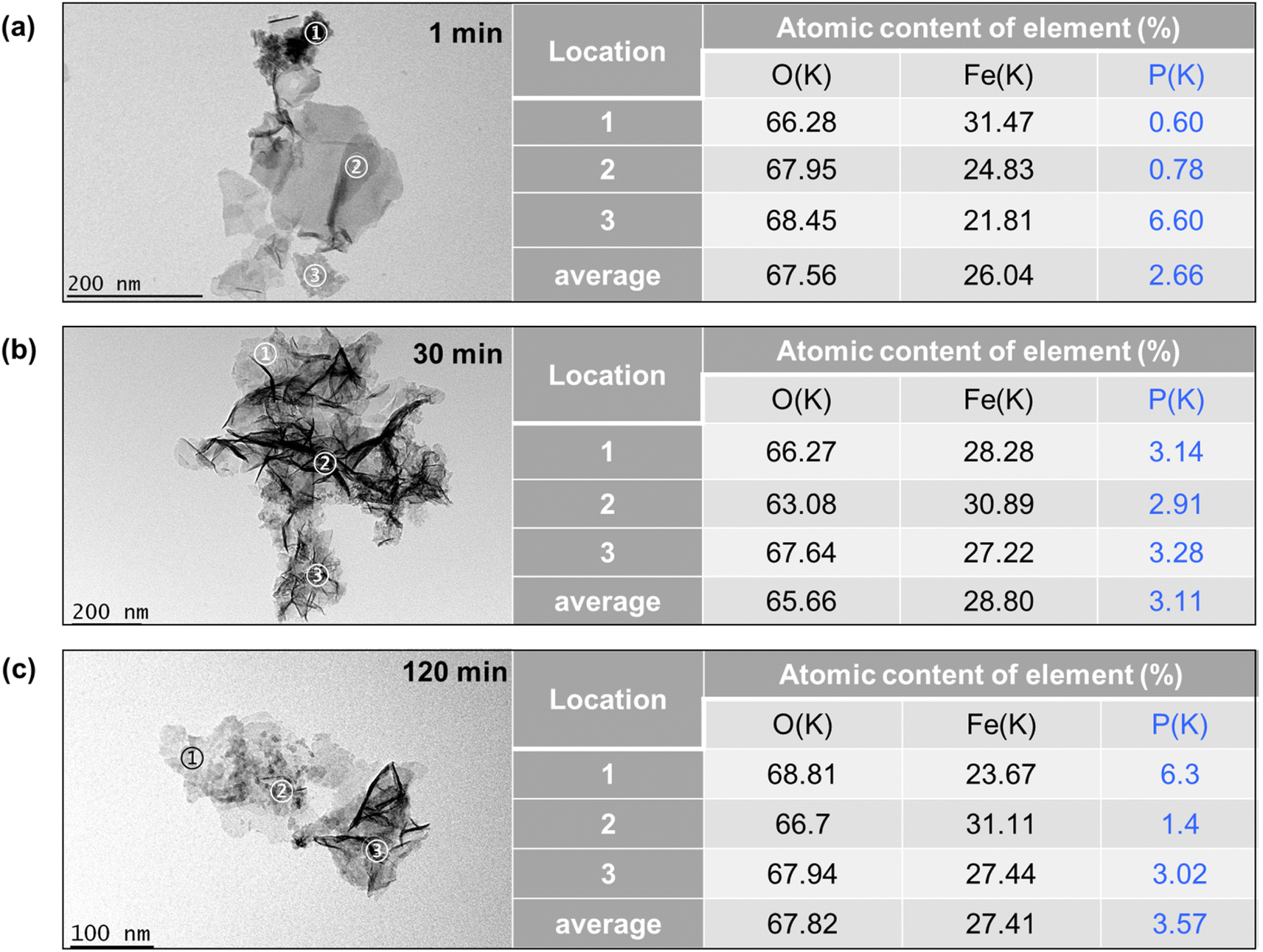
The TEM images of Fe precipitates collected at 1 min, 30 min, and 120 min of the experiments are shown in Fig. 7a–c and S4.† At 1 min, the Fe precipitates showed spherical-shaped substances at locations 1 and 3, planar-shaped substances with smooth surface textures at location 2, and individual lath-like particles with lengths of 20–50 nm. These lath-like particles are lepidocrocite that forms under conditions of rapid oxidation of Fe2+.43 The element composition of the Fe precipitates was further examined by STEM-EDX, with 3 locations reported on the right of Fig. 7. The atomic contents of phosphate are less than 0.8% at locations 1 and 2, while it is up to 6.6% at location 3. This finding suggests that the planar-shaped substances and spherical-shaped substances with crystal edges are Fe precipitates with low phosphate adsorption capacity. The spherical-shaped substances devoid of crystal edges and lattice fringes at location 3 might be amorphous Fe-phosphate precipitates, since the Fe polymerization is largely limited to the stage of monomers or oligomers by the presence of phosphate.43,44 At 30 min, the Fe precipitates exhibited more lath-like particles with lengths of 100–200 nm, which could be ascribed to the oxidation of Fe(II) precipitates to lepidocrocite.34 Furthermore, the STEM-EDX results showed a phosphate content of about 3% at all locations examined, which suggested adsorption of phosphate on these lepidocrocite nanoparticles and formation of Fe-phosphate polymers. At 120 min, beyond individual lath-like particles, there are spherical-shaped substances containing high contents of P (6.3% at location 1). Since the oxidation of Fe(II) precipitates decreased the solution pH to about 5.5, the dissolution and reformation of Fe precipitates might result in the reoccurrence of spherical-shaped substances. The average contents of P increased from 2.66% to 3.57% as the reaction time increased from 1 min to 120 min, which is consistent with the decrease of phosphate concentrations in the solution. The phosphate content in the precipitate is much higher than the reported sorption capacities of lepidocrocite at near neutral pH (1.5%).44 Thus, phosphate removal could not be explained by merely adsorption. In contrast, the coprecipitation of phosphate and Fe would not be limited by the surface area. As a result, the high content of P might mainly come from coprecipitation of Fe–P.
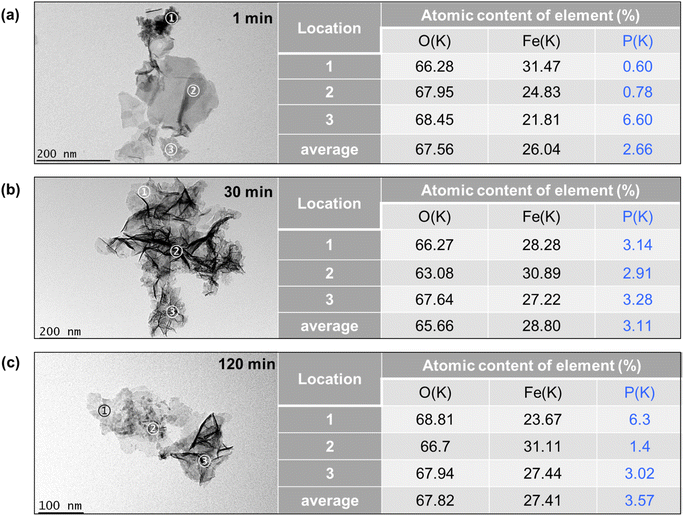 |
| | Fig. 7 Bright field TEM images of the Fe–P solid samples obtained at (a) 1 min, (b) 30 min, and (c) 120 min. The element contents of the locations 1–3 identified by EDX are presented on the right. | |
3.5. Environmental implications
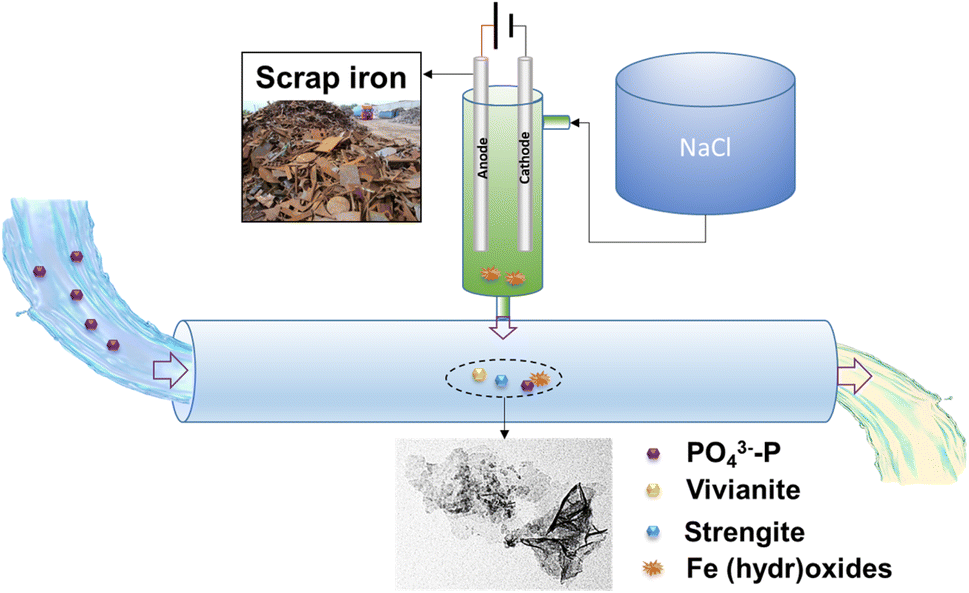
A conceptual model of phosphate removal by electrocoagulation using scrap iron electrodes is described in Scheme 2. As a low-cost and easy-access material, scrap iron (e.g., cast iron stick) could be used for the electrode after a simple treatment, such as cutting and mechanical clean. Electrode passivation is a major challenge of electrocoagulation. To solve this issue, we proposed to use the prepared NaCl solution as the electrolyte, instead of electrocoagulation performed directly in wastewater. As evidenced in this work, we found that the current efficiencies were always close to 100% independent of aeration, anode material, and current intensity of 100–600 mA (Fig. S5†). The electrocoagulation in NaCl solution could be installed on-site for Fe (hydr)oxide dosing in P-burden wastewater treatment facilities. The Fe (hydr)oxides generated by this method exhibited high efficiency (86.7–99.3%) for phosphate removal. The normalized specific energy consumption is 35.9–112.2 kW h per kg P, or 18.0–56.1 CNY per kg P assuming that the price of electricity is 0.5 CNY per kW h. The phosphate coprecipitated or adsorbed with Fe (hydr)oxides could be separated from the wastewater by sedimentation or filtration. The strategy proposed in this work utilizing available and low-cost materials in daily life provides a promising solution for phosphate removal from wastewater.
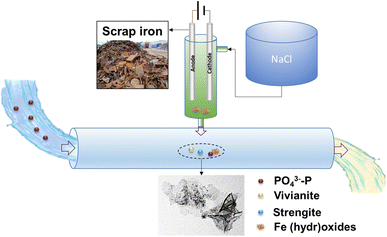 |
| | Scheme 2 A conceptual model of on-site dosing of Fe (hydr)oxides for phosphate removal by electrocoagulation with scrap iron electrodes. | |
4. Conclusions
This study evaluated the feasibility of an ex situ electrocoagulation utilizing scrap iron electrode for phosphate removal over a range of conditions representative of wastewater. The Fe precipitates generated at 25 min of electrocoagulation with cast iron electrodes could remove 86.7% of phosphate (10 mg L−1), which showed a phosphate removal capacity of 44.5 mg g−1 Fe and energy cost of 35.9 W h g−1 P. In a 0.2 M NaCl electrolyte, the current efficiency of electrocoagulation could approach 100%, which avoids electrode passivation. Compared to the electrocoagulation under air aeration, the Fe precipitates generated by electrocoagulation without aeration showed better performance, which also saved the energy consumption. The high removal rate and low energy consumption for phosphate removal made the cast iron outperform mild steel when serving as the anode of the electrocoagulation process. The phosphate retained in the Fe precipitates was mainly incorporated into the structural as vivianite, strengite, and amorphous Fe–P precipitates. We acknowledge that the efficiency of this method for the treatment of real wastewater with varied concentration of phosphate and other contaminants is still to be explored. Ongoing research is required to apply this new strategy for phosphate removal from domestic wastewater on different scales.
Author contributions
Shiwei Xie: conceptualization, writing-original draft, writing-reviewing and editing, funding acquisition, and supervision. Zhengkang Bai and Wei Shao: writing-original draft, investigation, and visualization. Chen Wang, Jianglong Qin and Ze Liu: methodology and investigation. Peng Liao: writing-reviewing and editing, funding acquisition, and supervision.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51808415 and 42177237) and Hubei Provincial Undergraduate Innovation and Entrepreneurship Training Program (S202110488025). We would like to thank Mr Zhang and Mr Wang at the Analytical & Testing Center of Wuhan University of Science and Technology for their help with XPS and XRD analysis.
References
-
W. Brownlie, M. A. Sutton, K. Heal, D. Reay and B. Spears, The Our Phosphorus Future, UK Centre for Ecology & Hydrology, Edinburgh, 2022 Search PubMed.
- F. Yang, L. Sui, C. Tang, J. Li, K. Cheng and Q. Xue, Sustainable advances on phosphorus utilization in soil via addition of biochar and humic substances, Sci. Total Environ., 2021, 768, 145106 CrossRef CAS PubMed.
- Y. Jia, S. Sun, S. Wang, X. Yan, J. Qian and B. Pan, Phosphorus in water: A review on the speciation analysis and species specific removal strategies, Crit. Rev. Environ. Sci. Technol., 2022, 1–22 CrossRef.
- J. Xu, L. Fan, Y. Xie and G. Wu, Recycling-equilibrium strategy for phosphogypsum pollution control in phosphate fertilizer plants, J. Cleaner Prod., 2019, 215, 175–197 CrossRef CAS.
- A. Bakshi, A. K. Verma and A. K. Dash, Electrocoagulation for removal of phosphate from aqueous solution: Statistical modeling and techno-economic study, J. Cleaner Prod., 2020, 246, 118988 CrossRef CAS.
- G.-J. Jiao, J. Ma, Y. Li, D. Jin, Z. Ali, J. Zhou and R. Sun, Recent advances and challenges on removal and recycling of phosphate from wastewater using biomass-derived adsorbents, Chemosphere, 2021, 278, 130377 CrossRef CAS PubMed.
- Y. Ye, H. H. Ngo, W. Guo, Y. Liu, J. Li, Y. Liu, X. Zhang and H. Jia, Insight into chemical phosphate recovery from municipal wastewater, Sci. Total Environ., 2017, 576, 159–171 CrossRef CAS PubMed.
- Y. Wang, P. Kuntke, M. Saakes, R. D. van der Weijden, C. J. N. Buisman and Y. Lei, Electrochemically mediated precipitation of phosphate minerals for phosphorus removal and recovery: Progress and perspective, Water Res., 2022, 209, 117891 CrossRef CAS PubMed.
- Y. Lei, M. Saakes, R. D. van der Weijden and C. J. N. Buisman, Electrochemically mediated calcium phosphate precipitation from phosphonates: Implications on phosphorus recovery from non-orthophosphate, Water Res., 2020, 169, 115206 CrossRef CAS PubMed.
- N. Martin, V. Ya, N. Leewiboonsilp, K.-H. Choo, P. Noophan and C.-W. Li, Electrochemical crystallization for phosphate recovery from an electronic industry wastewater effluent using sacrificial iron anodes, J. Cleaner Prod., 2020, 276, 124234 CrossRef CAS.
- Z. Ajmal, A. Muhmood, M. Usman, S. Kizito, J. Lu, R. Dong and S. Wu, Phosphate removal from aqueous solution using iron oxides: Adsorption, desorption and regeneration characteristics, J. Colloid Interface Sci., 2018, 528, 145–155 CrossRef CAS PubMed.
- R. Liu, L. Chi, X. Wang, Y. Sui, Y. Wang and H. Arandiyan, Review of metal (hydr)oxide and other adsorptive materials for phosphate removal from water, J. Environ. Chem. Eng., 2018, 6, 5269–5286 CrossRef CAS.
- Q. He, H. Zhao, Z. Teng, Y. Wang, M. Li and M. R. Hoffmann, Phosphate removal and recovery by lanthanum-based adsorbents: A review for current advances, Chemosphere, 2022, 303, 134987 CrossRef CAS PubMed.
- C. Zhang, A. Guisasola and J. A. Baeza, A review on the integration of mainstream P-recovery strategies with enhanced biological phosphorus removal, Water Res., 2022, 212, 118102 CrossRef CAS PubMed.
- M. Li, J. Liu, Y. Xu and G. Qian, Phosphate adsorption on metal oxides and metal hydroxides: A comparative review, Environ. Rev., 2016, 24, 319–332 CrossRef CAS.
- P. Chen, D. Song, X. Zhang, Q. Xie, Y. Zhou, H. Liu, L. Xu, T. Chen and K. M. Rosso, Understanding Competitive Phosphate and Silicate Adsorption on Goethite by Connecting Batch Experiments with Density Functional Theory Calculations, Environ.
Sci. Technol., 2022, 56, 823–834 CrossRef CAS PubMed.
- A. Ler and R. Stanforth, Evidence for Surface Precipitation of Phosphate on Goethite, Environ. Sci. Technol., 2003, 37, 2694–2700 CrossRef CAS PubMed.
- D. Conidi and W. J. Parker, The effect of solids residence time on phosphorus adsorption to hydrous ferric oxide floc, Water Res., 2015, 84, 323–332 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterizations, and Biological Applications, Chem. Rev., 2010, 110, 2574 CrossRef CAS.
- M. Y. A. Mollah, R. Schennach, J. R. Parga and D. L. Cocke, Electrocoagulation (EC)—science and applications, J. Hazard. Mater., 2001, 84, 29–41 CrossRef CAS.
- I. D. Tegladza, Q. Xu, K. Xu, G. Lv and J. Lu, Electrocoagulation processes: A general review about role of electro-generated flocs in pollutant removal, Process Saf. Environ. Prot., 2021, 146, 169–189 CrossRef CAS.
- K. L. Dubrawski and M. Mohseni,
In situ identification of iron electrocoagulation speciation and application for natural organic matter (NOM) removal, Water Res., 2013, 47, 5371–5380 CrossRef CAS PubMed.
- K. L. Dubrawski, C. M. van Genuchten, C. Delaire, S. E. Amrose, A. J. Gadgil and M. Mohseni, Production and transformation of mixed-valent nanoparticles generated by Fe(0) electrocoagulation, Environ. Sci. Technol., 2015, 49, 2171–2179 CrossRef CAS PubMed.
- C. Tsouris, D. W. DePaoli, J. T. Shor, M. Z. C. Hu and T. Y. Ying, Electrocoagulation for magnetic seeding of colloidal particles, Colloids Surf., A, 2001, 177, 223–233 CrossRef CAS.
- H. Karami, Heavy metal removal from water by magnetite nanorods, Chem. Eng. J., 2013, 219, 209–216 CrossRef CAS.
- T.-Y. Ying, S. Yiacoumi and C. Tsouris, An electrochemical method for the formation of magnetite particles, J. Dispersion Sci. Technol., 2002, 23, 569–576 CrossRef CAS.
- D. D. Nguyen, H. H. Ngo, W. Guo, T. T. Nguyen, S. W. Chang, A. Jang and Y. S. Yoon, Can electrocoagulation process be an appropriate technology for phosphorus removal from municipal wastewater?, Sci. Total Environ., 2016, 563–564, 549–556 CrossRef CAS PubMed.
- X. Zhang, H. Lin and B. Hu, Phosphorus removal and recovery from dairy manure by electrocoagulation, RSC Adv., 2016, 6, 57960–57968 RSC.
- I. Mishima, M. Hama, Y. Tabata and J. Nakajima, Long-term investigation of phosphorus removal by iron electrocoagulation in small-scale wastewater treatment plants, Water Sci. Technol., 2018, 78, 1304–1311 CrossRef CAS PubMed.
- M. Ingelsson, N. Yasri and E. P. L. Roberts, Electrode passivation, faradaic efficiency, and performance enhancement strategies in electrocoagulation—a review, Water Res., 2020, 187, 116433 CrossRef CAS PubMed.
- S. Müller, T. Behrends and C. M. van Genuchten, Sustaining efficient production of aqueous iron during repeated operation of Fe(0)-electrocoagulation, Water Res., 2019, 155, 455–464 CrossRef PubMed.
- A. Qian, S. Yuan, S. Xie, M. Tong, P. Zhang and Y. Zheng, Oxidizing capacity of iron electrocoagulation systems for refractory organic contaminant transformation, Environ. Sci. Technol., 2019, 53, 12629–12638 CrossRef CAS PubMed.
- J. Zhang, M. W. Bligh, P. Liang, T. D. Waite and X. Huang, Phosphorus removal by in situ generated Fe(II): Efficacy, kinetics and mechanism, Water Res., 2018, 136, 120–130 CrossRef CAS PubMed.
- X. Li, N. J. D. Graham, W. Deng, M. Liu, T. Liu and W. Yu, Structural variation of precipitates formed by Fe(II) oxidation and impact on the retention of phosphate, Environ. Sci. Technol., 2022, 56, 4345–4355 CrossRef CAS PubMed.
- R. P. Kralchevska, R. Prucek, J. Kolařík, J. Tuček, L. Machala, J. Filip, V. K. Sharma and R. Zbořil, Remarkable efficiency of phosphate removal: Ferrate(VI)-induced in situ sorption on core-shell nanoparticles, Water Res., 2016, 103, 83–91 CrossRef CAS PubMed.
- Y. Yang, Y. Li, R. Mao, Y. Shi, S. Lin, M. Qiao and X. Zhao, Removal of phosphate in secondary effluent from municipal wastewater treatment plant by iron and aluminum electrocoagulation: Efficiency and mechanism, Sep. Purif. Technol., 2022, 286, 120439 CrossRef CAS.
- R. E. Melchers, Long-term corrosion of cast irons and steel in marine and atmospheric environments, Corros. Sci., 2013, 68, 186–194 CrossRef CAS.
- D. D. Nguyen, Y. S. Yoon, X. T. Bui, S. S. Kim, S. W. Chang, W. Guo and H. H. Ngo, Influences of operational parameters on phosphorus removal in batch and continuous electrocoagulation process performance, Environ. Sci. Pollut. Res., 2017, 24, 25441–25451 CrossRef CAS PubMed.
- M. Ueshima, M. Kalin and D. Fortin, Microbial effects of natural phosphate rock (NPR) addition to mining wastes, JASMR, 2003, 2003 Search PubMed.
- T. Li, W. Dong, Q. Zhang, D. Xing, W. Ai and T. Liu, Phosphate removal from industrial wastewater through in situ Fe2+ oxidation induced homogenous precipitation: Different oxidation approaches at wide-ranged pH, J. Environ. Manag., 2020, 255, 109849 CrossRef CAS.
- M. Wu, J. Liu, B. Gao and M. Sillanpää, Phosphate substances transformation and vivianite formation in P-Fe containing sludge during the transition process of aerobic and anaerobic conditions, Bioresour. Technol., 2021, 319, 124259 CrossRef CAS PubMed.
- A. R. Pratt, Vivianite auto-oxidation, Phys. Chem. Miner., 1997, 25, 24–27 CrossRef CAS.
- R. Kaegi, A. Voegelin, D. Folini and S. J. Hug, Effect of phosphate, silicate, and Ca on the morphology, structure and elemental composition of Fe(III)-precipitates formed in aerated Fe(II) and As(III) containing water, Geochim. Cosmochim. Acta, 2010, 74, 5798–5816 CrossRef CAS.
- A. Voegelin, A.-C. Senn, R. Kaegi, S. J. Hug and S. Mangold, Dynamic Fe-precipitate formation induced by Fe(II) oxidation in aerated phosphate-containing water, Geochim. Cosmochim. Acta, 2013, 117, 216–231 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Zhengkang
Bai
a,
Wei
Shao
a,
Chen
Wang
a,
Jianglong
Qin
a,
Ze
Liu
a and
Peng
Liao
ab,
Zhengkang
Bai
a,
Wei
Shao
a,
Chen
Wang
a,
Jianglong
Qin
a,
Ze
Liu
a and
Peng
Liao