
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peroxi-electrocoagulation for treatment of trace organic compounds and natural organic matter at neutral pH†
Donald R.
Ryan
a,
Patrick J.
McNamara
 a,
Claire K.
Baldus
a,
Yin
Wang
b and
Brooke K.
Mayer
*a
a,
Claire K.
Baldus
a,
Yin
Wang
b and
Brooke K.
Mayer
*a
aDepartment of Civil, Construction and Environmental Engineering, Marquette University, Milwaukee, WI 53233, USA. Tel: +414-288-2161E-mail: Brooke.Mayer@marquette.edu
bDepartment of Civil and Environmental Engineering, University of Wisconsin-Milwaukee, Milwaukee, WI 53201, USA
First published on 4th October 2023
Abstract
Iron-based oxidation technologies can be advantageous for mitigating trace organic compounds (TOrCs) during water and wastewater treatment due to their production of hydroxyl radicals. However, iron-based oxidation often occurs at acidic pH to promote Fenton's reaction, which limits the processes' feasibility for treatment applications. This study focused on utilizing iron-electrocoagulation (EC) paired with ex situ H2O2 addition (peroxi-electrocoagulation [EC:H2O2]) to promote oxidative reactions at neutral pH conditions. The hydroxyl radical probe para-chlorobenzoic acid (pCBA) was used to gauge oxidant activity and serve as a representative TOrC. The impact of water pH, current density, iron dose, H2O2 dose (i.e., [H2O2]initial/[Fe2+]generated ratio), and the presence of natural organic matter (NOM) were evaluated. Multivariable regressions showed that high levels of H2O2 relative to iron (i.e., [H2O2]initial/[Fe2+]generated ratio >0.7) inhibited the rate of pCBA oxidation, likely due to additional radical quenching from extra H2O2. Oxidation of pCBA was confirmed at neutral pH conditions, indicating that EC:H2O2 may potentially serve as a multi-mechanistic treatment technology capable of oxidation. Experiments were also conducted in real-world water samples to gauge EC:H2O2 applications for treating groundwater, river water, and primary treated wastewater. Overall, H2O2 addition enhanced the oxidative degradation of TOrCs while still removing NOM. The one exception was the primary effluent sample, which had the highest degree of oxidant scavenging of all matrices tested. The electrical energy per order (EEO) metric demonstrated that EC:H2O2 is competitive with other TOrC oxidation technologies, with the added benefit of NOM mitigation in the same unit process.
Environmental significanceAs trace organic compounds are increasingly being monitored in drinking water, technologies are needed that can mitigate their risks. Iron-electrocoagulation paired with hydrogen peroxide can potentially serve as an oxidative technology for these contaminants, and can simultaneously remove trace organics and bulk organics such as natural organic matter within the same unit process. This combined process can be particularly advantageous for rural and decentralized systems due to multiple treatment processes occurring within the same reactor and favorable energy requirements as compared to oxidation technologies such as UV-H2O2 and ozonation. |
1. Introduction
Iron has expansive applications for water and wastewater treatment. Different iron-based treatment pathways proceed depending on the valence state of the iron (e.g., ferrous [Fe2+] or ferric [Fe3+]). Iron speciation varies as a function of pH and the presence of dissolved oxygen in water. Ferric iron predominates in the oxygen-rich neutral and basic pH conditions that are typical for water and wastewater treatment. During coagulation, iron is dosed as ferric chloride or ferric sulfate targeting removal of turbidity and natural organic matter (NOM).1 Alternately, Fe2+ predominantly exists in acidic conditions, and can mediate oxidative treatment via Fenton's reaction, which produces hydroxyl radicals (HO˙) that can oxidize trace organic compounds (TOrCs).2–4 Accordingly, iron-based treatments typically feature either non-destructive removal or oxidative destruction due to dominant pathways under different pH conditions.5 Research is needed to simultaneously promote both non-destructive and oxidative destructive pathways through Fenton's reaction at circumneutral pH for treating multiple classes of contaminants such as bulk organics (i.e., NOM) and TOrCs in a single unit process, which can be beneficial for water and wastewater treatment facilities.Fenton's reaction relies on non-complexed Fe2+ and H2O2 as reagents to form HO˙ (Reaction 1, Table 1). Hydroxyl radicals are highly reactive (2.8 V vs. standard hydrogen electrode) and can react with Fenton's reagents (Reactions 4 and 5) at faster rates (108 M−1 s−1) than the radicals are generated (40–80 M−1 s−1), which terminates Fenton's reaction due to oxidant and reagent depletion and hinders treatment effectiveness. However, in acidic conditions (pH 2–4), soluble Fe3+ can be recycled into Fe2+ (Reaction 2), thereby continuing HO˙ generation without reagent depletion.
 and
and  a
a

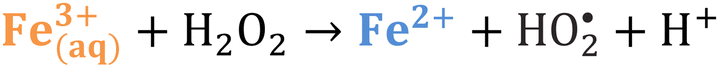
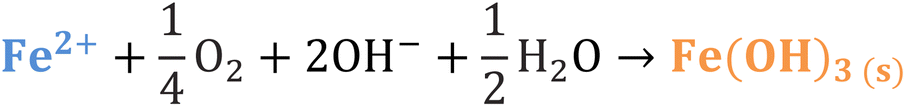

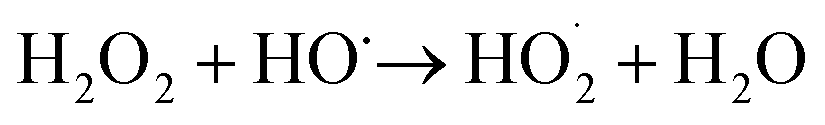
At neutral pH conditions in water and wastewater treatment, the feasibility of Fenton's reaction is limited for several reasons:
1. Iron speciation shifts toward Fe3+, which is less soluble and more prone to floc formation compared to Fe2+, resulting in termination of the Fenton's reaction cycle by inhibiting regeneration of Fe2+ required for oxidant generation.
2. Dissolved oxygen readily oxidizes Fe2+ in neutral and basic pH conditions (Reaction 3). Each increase in pH unit increases the oxidation rate of Fe2+ 100-fold, leading to less available Fenton's reagents.9,10
3. Anionic ligands in natural waters (e.g., OH− and CO32−) form complexes with Fe2+, which decreases the amount of non-complexed Fe2+ available to react with H2O2 to generate oxidants.6
Accordingly, pH limitations restrict Fenton applications to a narrow pH range (pH 2–4), which impedes implementation in water and wastewater treatment due to the intensive pH adjustments to acidify and neutralize waters before and after treatment. Additionally, acidic waters can enhance corrosion of infrastructure and shift the pH of natural waters following discharge.6
To facilitate Fenton oxidation at neutral pH, the key premise relies on generating or stabilizing the Fe2+ needed to react with H2O2 to form HO˙. Accordingly, electrochemical water treatment processes, such as electrocoagulation (EC), may be used for Fenton oxidation at neutral pH by generating non-complexed Fe2+via anodic dissolution of iron electrodes.11 Continuous generation of Fe2+ can be advantageous for Fenton oxidation at neutral pH by minimizing the need for Fe3+ reduction to Fe2+via H2O2 (Reaction 2). Prior research has also shown that EC alone can generate HO˙ to treat TOrCs through the in situ generation of Fe2+ at the anode and H2O2 production at the cathode.12–14 During electrolysis, the microenvironment near the anode surface is acidic.15 Consequently, Fenton reactions may occur at the vicinity of the anode surface even if the bulk solution pH is circumneutral, potentially leading to oxidative conditions at neutral pH between H2O2 and the iron anode surface. Supplemental addition of H2O2 as a radical promotor, known as peroxi-electrocoagulation (EC:H2O2), can further enhance EC's oxidizing capacity and serve as a multi-mechanistic process. During EC:H2O2, Fe2+ is continually generated at low concentrations (nM s−1 based on Faraday's law) over the course of electrolysis, such that non-complexed Fe2+ is available for oxidation by H2O2. As a result, less Fe2+ is “wasted” as a Fenton's reagent by non-radical generating side reactions such as ligand complexation or oxygenation.11 This combination of Fe2+ reagent generation and minimal reliance on Fe3+ reduction to Fe2+ can make EC:H2O2 an advantageous dosing method compared to ex situ reagent dosing in Fenton applications. Of note, while in situ Fe2+ dosing can be advantageous, ex situ H2O2 dosing may still be needed, depending on EC:H2O2 reactor design.
Pratap and Lemley (1998, 1994)8,16 demonstrated point-of-concept use of EC:H2O2 for remediation of the herbicides atrazine and metalochlor at neutral pH conditions. Since the inception of EC:H2O2, research has primarily focused on coagulation/flocculation during industrial wastewater treatment for removing bulk organic pollutants (such as chemical oxygen demand) at high concentrations (mg L−1 levels).5,17–22 However, these high-strength wastewater studies do not translate well to municipal wastewater and drinking water treatment applications. For example, environmental waters have lower conductivity, fewer oxidant scavengers, higher dissolved oxygen, and neutral pH conditions, all of which impact the oxidative efficiency of EC:H2O2 and speciation of iron in water. Considering iron's treatment capabilities, EC:H2O2 may also offer an opportunity for simultaneous treatment of TOrCs and bulk organics (e.g., NOM and chemical oxygen demand) in a single unit process as the Fe3+ produced following Fenton's reaction can subsequently contribute to physical removal (i.e., non-destructive removal) of contaminants through coagulation, flocculation, and sedimentation processes.
The goal of this research was to evaluate EC:H2O2 for simultaneous treatment of both TOrCs and NOM at neutral pH conditions. To vet oxidation, para-chlorobenzoic acid (pCBA) was selected as the representative TOrC, and also served as a HO˙ probe for advanced oxidation process (AOP) effectiveness.23 The relative impacts of current density (i.e., iron dosing rate), H2O2 dose, and the corresponding [H2O2]initial/[Fe2+]generated ratio were tested in synthetic matrices. Experiments were conducted to differentiate non-destructive removal via EC-only from oxidative destructive removal and to assess the contribution of potential oxidants generated in EC:H2O2 such as HO˙ and H2O2. Experiments were also conducted using surface water, groundwater, and wastewater sources to evaluate the influence of water quality parameters (i.e., dissolved organic carbon [DOC], pH, conductivity, and ions) and the feasibility of EC:H2O2 for different treatment applications. Finally, electrical energy per order of magnitude reduction (EEO) was calculated for all matrices to provide a means of comparing EC:H2O2 energy requirements relative to other advanced oxidation processes.
2. Materials and methods
2.1. Experimental protocols for EC:H2O2 tests of pCBA removal
The EC:H2O2 batch experiments were conducted for 15 minutes of electrolysis with 150 rpm mixing (G = 180 s−1) in 4 mM HCO3− buffer solutions containing 400 μg L−1pCBA. The pCBA concentration of 400 μg L−1 was selected based on reliable analytical quantification of >90% removal at a target pCBA concentration below that of mg L−1-level background oxidant scavengers (i.e., NOM). Electrolysis was performed in 200 mL polypropylene beakers using 1020 steel iron electrodes (VMetals, Milwaukee, WI), which were sanded and wet polished with 320 grit silicon carbide sandpaper prior to experiments. An XPH 75-2D Dual DC power supply (Sorenson Electronics, Cedar City, UT) was used to carry out electrolysis at currents ranging from 40 mA to 200 mA through a submerged electroactive surface area of 13.5 cm2, as described in Ryan et al. (2020).24 The power supply was equipped with a polarity reversal device to alternate the anode and cathode every 30 seconds based on prior works.25 For evaluating oxidative treatment, pCBA (99%, Sigma Aldrich, St. Louis, MO) was selected as the HO˙ probe due to its resistance to sorption on iron flocs and frequent use as a radical probe to demonstrate the treatability of TOrCs by HO˙ exposure.23,26–28 Compared to other TOrCs, pCBA is classified as having “moderate reactivity” with HO˙, as reported by Gerrity et al. (2012),23 which is similar to TOrCs of concern such as atrazine and 1,4-dioxane.23Three reactor inputs – current density, H2O2 dose, and the corresponding [H2O2]initial/[Fe2+]generated ratio – were evaluated to gauge their relative influence on treatment. For EC experiments, the current density was synonymous with the iron loading rate. The iron applied in each test was varied by adjusting the current density (and consequently the iron loading rate). For these experiments, the current density ranged from 3 to 15 mA cm−2 (charge loading rate = 12–60 Coulomb L−1 min−1, iron loading rate = 3.5–17.3 mg-Fe2+ per L-min). The H2O2 stock (ACS reagent grade, Sigma Aldrich, St. Louis, MO) was added at the beginning of EC:H2O2 experiments at levels ranging from 10 to 200 mg H2O2 per L to assess pCBA treatment resulting from a fixed amount of H2O2 available for Fe2+ to generate radicals. The corresponding [H2O2]initial/[Fe2+]generated was 0.3–1.6 based on current density and H2O2 inputs. Notably, the [H2O2]initial/[Fe2+]generated ratio reflects the total H2O2 added at the beginning of the reaction, divided by the amount of Fe2+ generated by EC (estimated by Faraday's Law) by the end timepoint when pCBA removal ceased due to H2O2 depletion. The end timepoint of the pCBA degradation reaction was determined as the time point at which less than 10% difference in pCBA removal compared to the preceding time point was observed, likely indicating depletion of H2O2. Samples were collected every 2.5 minutes for 10 minutes, with a final sample at 15 minutes for kinetic analyses. Kinetic curves were fit to at least four data points (R2 > 0.95 for all) to calculate first order rate constants for pCBA degradation for samples collected prior to H2O2 depletion, assessed as noted above.
2.2. Removal pathway control experiments
Experiments were conducted under the same electrolysis and current density conditions described in Section 2.1 to isolate the impact of different system inputs and delineate the potential treatment pathways in EC:H2O2, including oxidation by HO˙ and H2O2 as well as physical removal by sorption to iron flocs. For HO˙ oxidation controls, methanol was spiked in stoichiometric excess (12 mM MeOH) of pCBA and H2O2 to quench HO˙ that would otherwise react with pCBA. In this case, H2O2 is reactive with electron-dense compounds and unlikely to react with unsaturated alcohols such as methanol. For no electricity controls (e.g., H2O2 controls), 30 mg-H2O2 per L was spiked into the reactors containing the iron electrodes and stirred for 15 minutes to assess the potential pCBA removal due to H2O2 under treatment conditions without electricity in addition to potential losses via sorption to the electrode surface.Kinetic analyses were conducted to estimate the competition between H2O2 and O2 as a function of H2O2 inputs and water chemistry conditions (H2O2 dose, O2, and pH). These tests assessed the feasibility of HO˙ generation under neutral pH conditions and informed mechanistic analyses (oxidation by O2 limits HO˙ production by generating Fe3+). The relative rates of oxidation and associated rate constants are provided in the ESI S4.†
2.3. Water quality conditions
All EC:H2O2 experiments were conducted in 4 mM bicarbonate solution (with the exception of the environmental waters) to simulate buffered conditions for neutral pH environmental waters containing alkalinity, and to supply an electrolyte for electrochemical reactions (Table 2). Environmental waters were sampled to assess the impact of water quality and treatment performance in real waters relative to synthetic waters containing different NOM sources (Table 2). These analyses are important for informing the role of other environmentally relevant water quality parameters such as NOM characteristics and concentration, conductivity, and divalent cations, all of which can impact treatment efficacy. A sample from the Milwaukee River (Milwaukee, WI) was used to test the impact of NOM and mid-range conductivity water. Groundwater from a drinking water well in West Bend, WI, was tested to reflect low dissolved organic carbon (DOC) and high conductivity conditions. Finally, primary effluent from an urban water reclamation facility in Milwaukee, WI, was tested for the impact of high DOC due to anthropogenic NOM and other oxidant scavengers (such as bulk chemical oxygen demand). The wastewater also served as a point of comparison to previous EC:H2O2 wastewater studies. For DOC quantification experiments, a sedimentation phase was required after EC:H2O2 to allow the flocs to settle prior to DOC analysis. Batch tests were performed as described in Section 2.1 followed by an additional tapered flocculation phase (10 minutes at 40 rpm [G = 25 s−1] and 10 minutes at 20 rpm [G = 9 s−1]) and a 20 minute sedimentation period to remove flocs (method adapted from Ryan et al. (2020)).24| Water matrix | Initial pH | H2O2 demand,a mg L−1 (% H2O2 removal) | DOC, mg-C per L | Alkalinity, mg L−1 as CaCO3 | Conductivity, μS cm−1 | Ca2+, mg L−1 | Mg2+, mg L−1 |
|---|---|---|---|---|---|---|---|
| a H2O2 demand is reported as the decrease in H2O2 concentration after 15 minutes (the length of batch experiments), where the initial concentration was 30 mg L−1 H2O2. b All model waters were prepared in Milli-Q water with 4 mM HCO3−. c pH varied from 3 to 10.3 depending on the experiment. The unadjusted pH was 8.3. d For NOM tests, NOM was added as International Humic Substance Society Suwannee River NOM. e Conductivity varied for pH tests due to the addition of acid (HCl) or base (NaOH) for pH adjustment. At pH 3, conductivity = 920 μS cm−1. At pH 6.3, conductivity = 450 μS cm−1. At pH 10.3, conductivity = 750 μS cm−1. For unadjusted pH, conductivity = 370 μS cm−1. | |||||||
| Bicarbonate bufferb | 8.3c | 0 (0%) | 0, 7.5d | 210 | 370e | 0 | 0 |
| Groundwater | 7.30 | 10 (33%) | 3.3 | 400 | 1430 | 70 | 40 |
| River water | 8.4 | 5 (15%) | 7.0 | 240 | 755 | 30 | 20 |
| Primary wastewater effluent | 7.1 | 23 (75%) | 55 | 280 | 1400 | 40 | 15 |
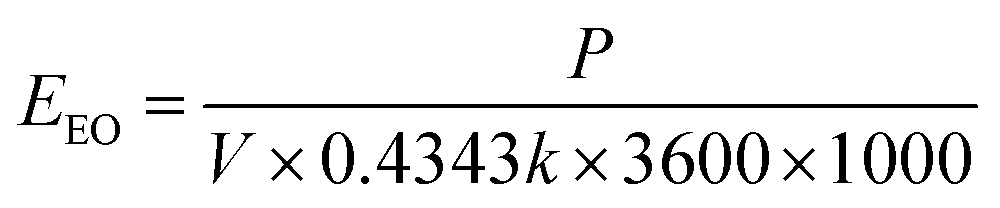 | (1) |
3. Results and discussion
3.1. Para-chlorobenzoic acid removal for hydroxyl radical validation
Removal of pCBA during EC:H2O2 primarily proceeded via oxidation at neutral pH conditions due to the system's combination of iron and H2O2 (Fig. 1). EC-only controls yielded an average pCBA removal of approximately 15%, presumably due to the low levels of HO˙ that can be generated during EC alone.13,14 For EC:H2O2 + MeOH experiments, the high MeOH concentration (12 mM) scavenged the oxidants and resulted in negligible pCBA degradation. This scavenging indirectly underscores the role of homogeneous oxidants (such as HO˙). Negligible pCBA removal in the EC:H2O2 + MeOH test further indicates that pCBA does not sorb to iron flocs. The ‘No Electricity Control’ experiments demonstrated that potential reactions between H2O2 and the iron electrode surface had minimal removal relative to the EC:H2O2 conditions with electricity (p < 0.0001, one-way ANOVA) at circumneutral pH conditions. Overall, these data demonstrate that the addition of H2O2 can enhance oxidant production in EC:H2O2 relative to EC alone and induce oxidative processes at neutral pH conditions.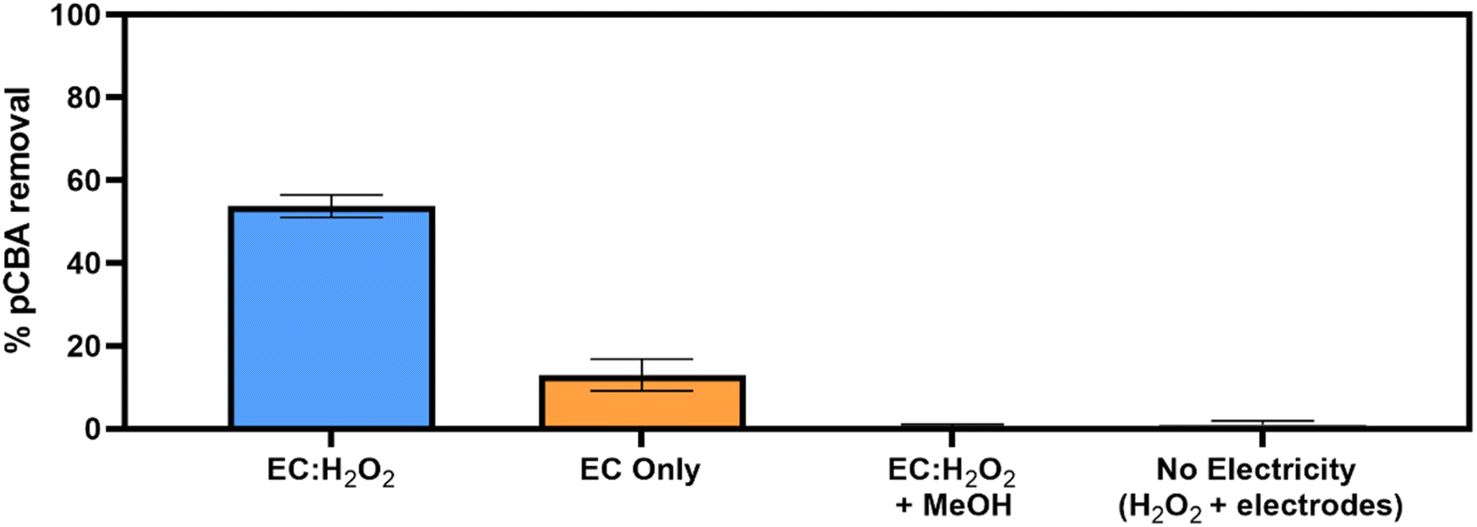 | ||
| Fig. 1 Mechanisms for pCBA removal during EC:H2O2 at 7.40 mA cm−2. A series of controlled batch experiments were run in 4 mM bicarbonate buffer at pH 8.3 for 15 minutes. In “EC only,” electrolysis was conducted using iron electrodes with no peroxide addition. For “EC:H2O2 + MeOH”, methanol was spiked in stoichiometric excess of pCBA (12 mM MeOH:2.5 μM pCBA) to quench oxidants that would otherwise degrade pCBA. In “No Electricity (H2O2 + electrodes)”, 30 mg L−1 H2O2 was spiked into the solution with the iron electrodes and mixed for 15 minutes. All experiments were conducted in duplicate and error bars indicate ± 1 standard deviation. EC-only results are the average of all duplicate experiments for each EC-only control, including current densities of 3.5 mA cm−2, 5.5 mA cm−2, 11.1 mA cm−2, and 15 mA cm−2, where n = 8. | ||
The occurrence of oxidation at neutral pH conditions during EC:H2O2 is important in the context of Fenton literature since traditional Fenton oxidation proceeds at highly acidic pH 3 conditions. These conventional Fenton conditions limit the feasibility of EC:H2O2 applications as the high acidity can damage infrastructure, enhance corrosion, and incur chemical costs for acidifying and neutralizing water during treatment.
3.2. The impact of reactor inputs on pCBA degradation during EC:H2O2: removal and kinetics
Following oxidant verification, the impact of EC:H2O2 reactor inputs and water quality were assessed. The discussion centers on the role of [H2O2]initial/[Fe2+]generated ratios, current density, and pH. Multivariable linear regressions were used to parameterize the contribution of all inputs. | ||
| Fig. 2 pCBA removal after 15 minute EC:H2O2 batch experiments as a function of (a) H2O2 initially dosed into the system, (b) total ferrous iron generated (estimated by Faraday's law) over the course of the EC:H2O2 experiment, and (c) current density, which is proportional to the iron loading rate (mg-Fe per L). Iron loading rates were estimated using Faraday's law, where 3 mA cm−2 = 3.5 mg-Fe per L-min, 5.5 mA cm−2 = 6.5 mg-Fe per L-min, 7.4 mA cm−2 = 8.6 mg-Fe per L-min, 11.1 mA cm−2 = 13 mg-Fe per L-min, and 15 mA cm−2 = 17.4 mg-Fe per L-min. All experiments were conducted in duplicate and error bars indicate ± 1 standard deviation. | ||
The key role of current density in this study was to adjust the iron loading rate to add Fe2+ as Fenton's reagent (Fig. 2B). When no H2O2 was present (i.e., EC-only), pCBA removal was consistently less than 20% regardless of current density (Fig. 2C). Hence, EC offered effective pCBA removal only when H2O2 was present, indicating that the [H2O2]initial/[Fe2+]generated ratio was the key driver of treatment efficacy. During EC:H2O2, pCBA removal improved with increases in current density up to 7.4 mA cm−2 (Rcurrent density=3 to 7.4 mA cm−22 = 0.63, p = 0.008, Pearson correlation, Table S16†) and plateaued after 7.4 mA cm−2 (Rcurrent density>7.4 mA cm−22 = 0.141, p = 0.6, Pearson correlation). As treatment inputs increased, the higher ratio of [H2O2]initial/[Fe2+]generated = 1.6 had the least pCBA removal for EC:H2O2 regardless of current density. The plateau in pCBA removal for higher current density may suggest that a minimum level of iron is needed for this system, and beyond that level, additional iron no longer improves treatment. Here, the lowest Fe2+ loading rate was 3.5 mg Fe per L-min (resulting from 3 mA cm−2 current density).
In summary, [H2O2]initial/[Fe2+]generated ratios were the key driver for pCBA removal where lower ratios (0.33–0.7) had higher removal (βH2O2/Fe=0–0.7 = 0.77, p < 0.0001, multivariable linear regression: “% R, low ratio, neutral pH”) from minimal HO˙ scavenging, and higher ratios (1.6) decreased removal (βH2O2/Fe=0.3–1.6 = −0.42, p = 0.0008, multivariable linear regression: “% R EC: H2O2 neutral pH”). This finding is important when considering material requirements including the ex situ H2O2 additions and the power demands associated with iron generation. For this system, H2O2 levels determined the treatment capacity because pCBA removal ceased after depletion of the one-time dose of H2O2 at the start of the test, whereas the Fe2+ was continually generated via electrolysis.
It is important to note that [H2O2]initial/[Fe2+]generated ratios do not translate to the actual ratio of H2O2 relative to Fe2+ at any timepoint during the test. During EC:H2O2, H2O2 is initially in large excess to Fe2+ as Fe2+ is formed during EC, which may drive the rate of oxidant formation resulting from interactions between Fe2+ and H2O2. This excess is a result of Fe2+ being generated at nM levels (e.g., 2500 nM s−1 for 7.4 mA cm−2 based on Faradays law) during electrolysis, which highlights the benefits of using iron electrolysis for Fe2+ dosing to avoid side reactions and encourage efficient Fe2+ utilization by H2O2.
 | ||
| Fig. 3 Summary of pseudo-first order rate constants for degradation of para-chlorobenzoic acid (pCBA) following 15 minute EC:H2O2 batch experiments. All experiments were conducted in 4 mM HCO3− buffer at pH = 8.3. Samples were taken every 2.5 minutes for a total of 15 minutes. (a) Pseudo-first order kinetic curves showing the impact of [H2O2]initial/[Fe2+]generated ratios for a fixed current density of 5.5 mA cm−2. (b) Pseudo-first order kinetic curves showing the impact of current density for a fixed [H2O2]initial/[Fe2+]generated of 0.5. (c) Summary of pseudo-first order rate constants for all EC:H2O2 batch experiments for all current density and [H2O2]initial/[Fe2+]generated ratios. Pseudo-first order constants were determined and verified based on R2 ≥ 0.95 over the course of treatment to capture the linear range prior to H2O2 depletion. The end timepoint of the pCBA degradation reaction used in kinetic modeling was determined as the time point at which pCBA removal was less than 10% different than the preceding time point. Error bars show ± 1 standard deviation of duplicate experiments. | ||
As shown in Fig. 3, for a fixed [H2O2]initial/[Fe2+]generated ratio of 0.5, the rate constants were comparable for current densities of 5.5, 7.4, and 11.1 mA cm−2 (1.3 to 1.6 × 10−3 s−1, Table S5†). However, 3 mA cm−2 had the lowest rate of removal (8.4 × 10−4 s−1, Table S5†). This trend aligns with the removal data, in which the removal plateaued as current (i.e., iron loading rate) increased, indicating that additional iron after a threshold level no longer improved treatment.
Overall, the ratio had the highest influence on rate of removal based on multivariable linear regressions (β[H2O2]/[Fe2+]=0–0.77 = 0.76 ± 0.11, p < 0.0001, multivariable linear regression: “k, EC:H2O2 low ratio, neutral pH”) (Fig. 3C). Considering both major inputs in terms of [H2O2]initial/[Fe2+]generated ratios, pseudo-first order rate constants were grouped into three clusters for a range of current densities at neutral pH conditions (Fig. 3C) to understand the general impact of different ratio levels. The clusters were EC-only conditions (i.e., no H2O2), [H2O2]initial/[Fe2+]generated ratios = 0.3–0.7, and [H2O2]initial/[Fe2+]generated ratios greater than 0.7. The lower [H2O2]initial/[Fe2+]generated ratios of 0.3–0.7 had the highest rate constants, ranging from 1.1 × 10−3 to 1.6 × 10−3 s−1. The higher ratios of >0.7 to 1.6 resulted in lower rate constants, ranging from 5.8 × 10−4 to 7.7 × 10−4 s−1 (β[H2O2]/[Fe2+]=0.3 to 1.6 = −0.59 ± 0.11, p < 0.0001, multivariable linear regression: “k, EC:H2O2 only, neutral pH”). For all cases, experiments containing H2O2 had faster rates of pCBA removal compared EC-only controls (1.4 × 10−4 to 2.7 × 10−4 s−1). However, higher levels of H2O2 (greater than 40 mg L−1) did not increase the rate of pCBA removal, likely due to radical quenching by H2O2. This is notable given that the effective H2O2 doses found in this study are less than reagent demands in other EC:H2O2 studies for industrial treatment applications.5,17–22 Overall, these findings indicate that less H2O2 may be required than previously thought for effective oxidation during EC:H2O2.
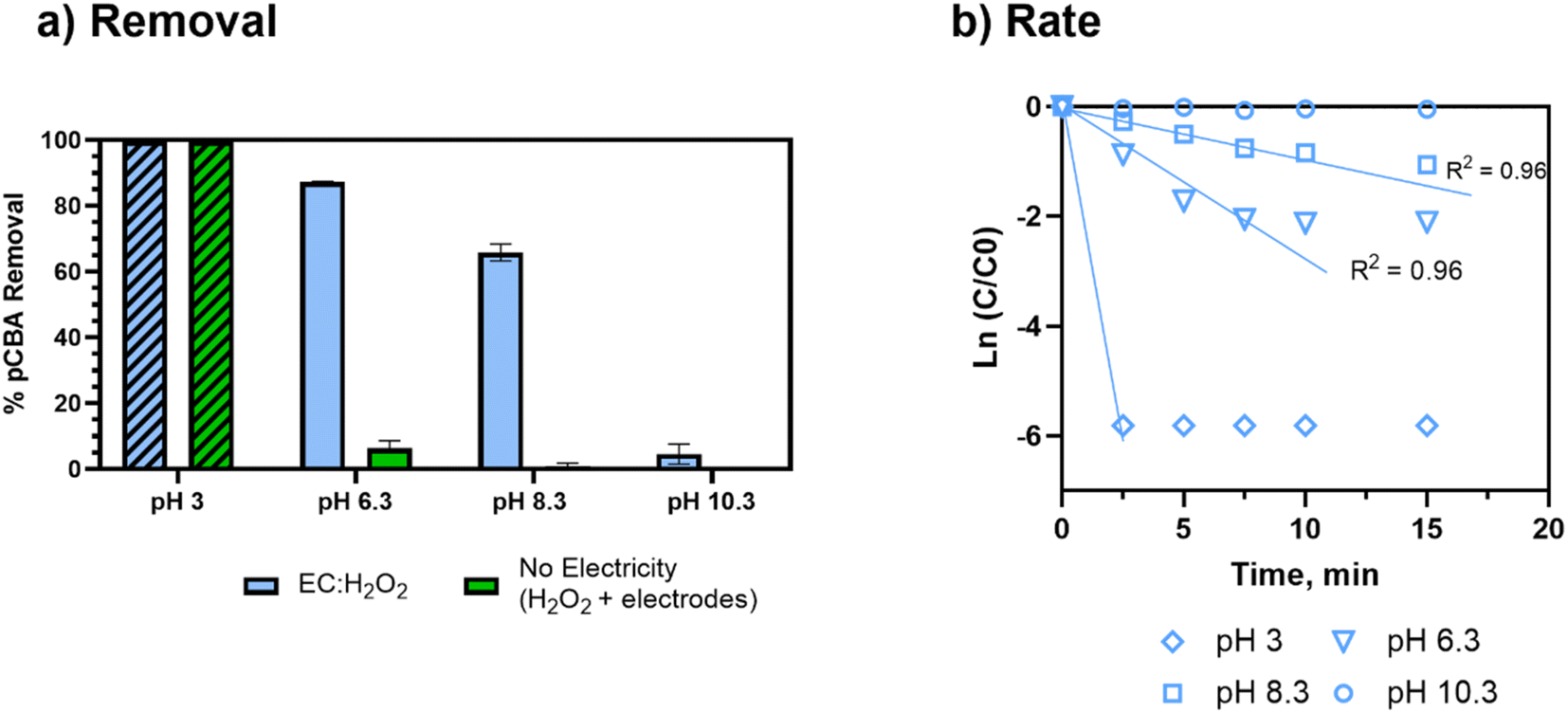 | ||
| Fig. 4 Degradation of pCBA at different pH conditions prepared in 4 mM bicarbonate buffer. During EC:H2O2, current density = 7.4 mA cm−2, H2O2 = 30 mg L−1, 15 minutes of treatment time. (a) Removal of pCBA as a function of pH. The no electricity control experiment shows removal due only to iron electrodes and the addition of 30 mg L−1 H2O2. Striped bars indicate concentrations below the limit of detection (4 μg L−1). (b) pCBA removal rate as a function of pH. R2 correlations are not shown for pH 3 due to insufficient points above the limit of detection after treatment (removal at all pH 3 treatments was calculated using the limit of detection as the final concentration). R2 is not shown for pH 10.3 due to no removal. The end timepoint of the pCBA degradation reaction used in kinetic modeling was determined as the time point at which pCBA removal was less than 10% different than the preceding time point. Error bars show ± 1 standard deviation of triplicate experiments. Of note, the error bars for b are not visible due to low standard deviation. | ||
The acidic conditions (pH 3, encouraging Fenton's reactions) resulted in the greatest and fastest pCBA removal (>99% removal, to below the detectable limit). However, at pH 3, enhanced corrosivity led to >99% pCBA removal even without electricity, wherein increased iron dissolution was visually observed. For no electricity controls, pCBA removal was likely due to non-faradaic iron dissolving from the electrodes (30 mg Fe per L) and reacting with ex situ H2O2 (98% H2O2 removal; ESI S7†) to generate HO˙. The no electricity control resulted in a [H2O2]initial/[Fe2+]generated ratio of 1.5. Although this ratio was toward the upper end of ratios tested, the pH 3 conditions were expected to enhance the rate of reaction. For circumneutral conditions, the no-electricity controls had minimal pCBA removal, indicating no oxidant generation in the absence of electricity at the conditions tested.
Overall, the pCBA removal trends agree with the kinetic modeling performed to estimate the competition between H2O2 and O2 for oxidizing Fe2+ (ESI S4†). The modeling scenarios included 0 to 200 mg L−1 H2O2 concentrations. At pH 6.3, the rate of Fe2+ oxidation by H2O2 was up to 10 orders of magnitude higher than Fe2+ oxidation by O2, suggesting that there was minimal competition for ferrous oxidation between H2O2 and O2. Accordingly, these kinetic analyses support that HO˙ generation was driven by Fe2+ oxidation via H2O2 (not O2). Removal was minimal at pH 10.3, likely due to enhanced O2 activity (Fig. 4). As pH increases, the inhibition of HO˙ generation due to O2 becomes more apparent given that the oxidation of Fe2+ by O2 is second order with respect to [OH−] (based on Stumm and Lee, 19619) and increases 100-fold for each pH unit increase (ESI S4†).
The pseudo-first order rate constants were used to estimate the HO˙ concentration (ESI S2†). For pH 8.3, [HO˙] ranged from 2–4.1 × 10−13 M for the [H2O2]initial/[Fe2+]generated ratios of 0.33 to 0.7. At pH 6.3, when pCBA treatment was more effective, [HO˙] was approximately 9 × 10−13 M. These estimates of radical concentrations can be applied to future studies to compare the [HO˙] yield for a range of TOrC oxidation technologies such as UV/H2O2.
Overall, [H2O2]initial/[Fe2+]generated ratios, pH, and current density were significantly correlated to pCBA removal (p = 0.028, 0.008, and <0.0001, respectively, multivariable regression “% R all”). The most influential parameter for pCBA removal was pH (βpH = −0.91 ± 0.15, multivariable linear regression: “% R, all”), where lower pH led to higher pCBA removal. The ratio of [H2O2]initial/[Fe2+]generated and current density had smaller impacts relative to pH, but similar magnitude of contributions to one another (β[H2O2]/[Fe2+] = 0.22 ± 0.09, βcurrent density = 0.36 ± 0.09, multivariable linear regression: “% R, all”).
A separate regression was performed for experiments with [H2O2]initial/[Fe2+]generated ratios = 0–0.77 to rank the inputs that yielded higher rate constants and higher pCBA removal. For these tests, pH still had the greatest influence (βpH = −0.79 ± 0.08, p < 0.0001, multivariable linear regression: “k, all”) followed by [H2O2]initial/[Fe2+]generated ratios (β[H2O2]/[Fe2+] = 0.38 ± 0.05, p < 0.0001, multivariable linear regression: “k, all”). However, variations in current density alone had an insignificant influence on the rate of pCBA removal (βcurrent density = 0.09 ± 0.05, p = 0.073, multivariable linear regression: “k, all”), implying that pH and [H2O2]initial/[Fe2+]generated ratios are the key parameters influencing oxidant production.
3.3. Co-treatment of pCBA and NOM using EC:H2O2 to treat environmental waters and synthetic matrices
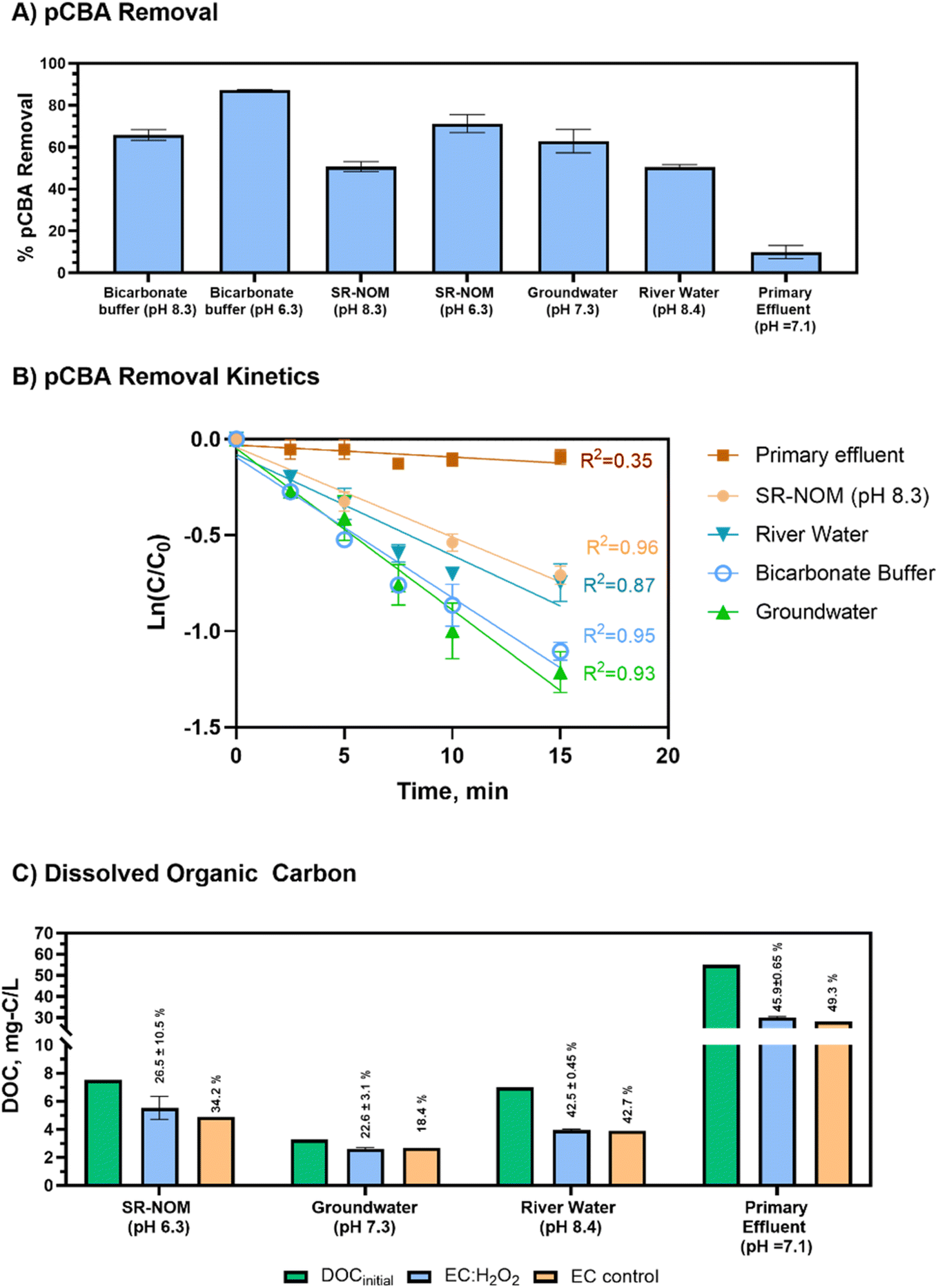 | ||
| Fig. 5 Removal of pCBA and dissolved organic carbon (DOC) in synthetic and real-world waters following 15 minutes of EC:H2O2 (7.40 mA cm−2, H2O2 = 30 mg L−1). (A) pCBA removal following EC:H2O2 (Ci = 400 μg L−1 for all matrices). (B) Pseudo-first order degradation of pCBA in EC:H2O2 at [H2O2]initial/[Fe2+]generated ratio = 0.55 (7.4 mA cm−2, 30 mg H2O2 per L) in synthetic and real-world waters. (C) DOC in the waters initially and remaining following EC:H2O2 or EC-only treatment. A pH 6.3 test was conducted for SR-NOM to reflect enhanced coagulation conditions. Bicarbonate buffer is not included in panel (C) as there was no DOC in the synthetic matrix. The % DOC removal by EC:H2O2 is shown in vertical text above each bar. All DOC and pCBA data are from the same batch experiments for the respective water matrix. Error bars show ± 1 standard deviation of triplicate experiments, with the exception of the “EC control” values in panel C, which are single replicates. | ||
The initial concentration of DOC had a small impact on pCBA removal (βDOC = −0.07, p = 0.34, not including primary effluent) for matrices containing low-to mid-range DOC levels (<10 mg-C per L) that reflect drinking water source matrices. This trend implies that the presence of NOM may not heavily impede pCBA oxidation when treating typical environmental source waters.
Compared to real-world waters and synthetic matrices, the primary effluent had the least pCBA removal. Decreased removal was likely due to high H2O2 demand (Table 2) and high DOC levels. In this case, the high H2O2 demand rapidly depleted the H2O2 that was initially dosed into the reactor, which hindered HO˙ production. Thus, for EC:H2O2 applications, the water's H2O2 demand should be accounted for to gauge potential negative impacts on process performance. For example, Serra-Clusellas et al. (2021)32 demonstrated TOrC mitigation via EC:H2O2 at pH 3 in municipal tertiary treated wastewater containing ng L−1 TOrCs by using elevated 220–440 H2O2 mg L−1 doses (resulting in [H2O2]initial/[Fe2+]generated ratios of 1.7 to 2 during treatment), which offset oxidant scavenging by wastewater constituents.
A multivariable regression of all test matrices showed that DOCinitial and pH were the key water quality parameters that impacted pCBA removal (βDOC = −0.72 ± 0.18, p = 0.008 and βpH −0.81 ± 0.08, p=<0.0001, respectively). Pearson correlations showed that DOCinitial and H2O2 demand were multicollinear (RDOC vs. H2O2 demand2 = 0.893, p < 0.05, Pearson correlations). As anticipated, higher DOC levels typical of wastewater impeded treatment efficacy, whereas lower DOC conditions improved radical yield and offered less competition for oxidants. However, it is important to note that other water matrix constituents beyond DOC (including chemical oxygen demand, reduced metals, and sulfides, which were not assessed in this study) also likely contributed to H2O2 depletion and impeded DOC removal.
The river water and SR-NOM matrices are of particular interest for DOC removal given that they are representative of surface waters that could be treated for drinking water. Using EC:H2O2, DOC removal for the river water complied with recommendations in the US Environmental Protection Agency's Enhanced Coagulation Guidance manual (>30% DOC removal for matrices containing >120 mg L−1 as CaCO3 alkalinity).33 The synthetic SR-NOM matrix only had effective DOC removal when pH was 6.3. At pH 8.3, EC:H2O2 formed no flocs or precipitates in SR-NOM, indicating unsuccessful coagulation, precipitation, and subsequent sedimentation of flocs (ESI S9†). This difference between real and synthetic waters suggests that other constituents in environmental waters (such as divalent cations, i.e., calcium and magnesium) may improve coagulation processes in real waters by promoting ionic interactions between NOM and ions that promote co-sorption to flocs, as shown for a calcium-fulvic acid-goethite iron mineral system.34 Overall, the addition of H2O2 during EC:H2O2 can enhance treatment applications by simultaneously treating TOrCs such as pCBA as well as bulk organics such as DOC in a single unit process in lieu of a multi-stage treatment train such as coagulation/flocculation/sedimentation followed by filtration and oxidation to achieve both non-destructive removal and oxidative destruction of contaminants.
3.4. Engineering implications: rate constants and electrical energy per order
| [H2O2]initial/[Fe2+]generated | Current density, mA cm−2 | ||||
|---|---|---|---|---|---|
| 3 | 5.5 | 7.4 | 11.1 | 15 | |
| 0.35 | 0.74 ± 0.04 | 3.13 ± 0.13 | 6.10 ± 0.21 | 12.54 ± 2.12 | |
| 0.5 | 0.76 ± 0.03 | 0.62 ± 0.02 | 3.15 ± 0.39 | 5.89 ± 0.06 | |
| 0.7 | 0.74 ± 0.03 | 7.45 ± 0.84 | |||
| 1.6 | 1.11 ± 0.08 | 1.22 ± 0.05 | 12.56 ± 2.01 | ||
| Water matrix | k, s−1 | E EO, kW h m−3 |
|---|---|---|
| Bicarbonate buffer (pH 8.3) | 1.2 × 10−3 | 2.86 ± 0.20 |
| Bicarbonate buffer (pH 6.3) | 4.7 × 10−3 | 0.68 ± 0.005 |
| Bicarbonate buffer + NOM | 6.3 × 10−3 | 7.57 ± 0.20 |
| Bicarbonate buffer + NOM (pH 6.3) | 2.9 × 10−3 | 1.13 ± 0.08 |
| Groundwater | 1.6 × 10−3 | 1.00 ± 0.13 |
| River water | 1.3 × 10−3 | 1.91 ± 0.21 |
| Primary effluent | 2.5 × 10−4 | 6.49 ± 1.34 |
At circumneutral pH, current densities above 3 mA cm−2 thus exceeded the recommended 1 kW h m−3EEO threshold to be competitive with conventional HO˙-mediated advanced oxidation processes (AOPs) such as UV/H2O2 and ozone-based AOPs.36 Accordingly, EC:H2O2 may be operated at lower current densities for more favorable energy demands. However, the benchmark EEO values for conventional AOPs rely on preliminary treatment technologies such as coagulation and membrane filtration to remove oxidant scavengers, primarily DOC. Additional DOC removal technologies add materials and energy demands to overall treatment of TOrCs that are not accounted for in standalone EEO values for conventional AOPs. Alternately, EC:H2O2 offers the benefit of simultaneous TOrC and DOC treatment, which can minimize preliminary treatment needs and decrease overall energy inputs compared to conventional AOP treatment trains.
Although EC:H2O2 was higher than the EEO benchmark for conventional AOPs under high current conditions, EC:H2O2 generally resulted in a lower range of EEO values (0.6 to 12.5 kW h kW h m−3 [Table 3]) compared to pCBA mitigation using other electrochemical technologies such as boron-doped diamond electrooxidation. For example, Lanzarini-Lopes et al. (2017)37 reported EEO values for pCBA mitigation ranging from 39.3 kW h m−3 to 332 kW h m−3 for electrooxidation current densities from 16.6 to 100 mA cm−2. Consistent with this study, increasing current density in the electrochemical treatment process yielded higher, less favorable EEO values.
Multivariable regressions were used to assess how water quality in environmental and synthetic water matrices influenced EEO. The EEO trends followed the removal trends, where DOC concentration and alkalinity increased EEO by decreasing pCBA removal and increasing treatment inputs (βDOC = 1.2 ± 0.15, p < 0.0001; βalkalinity = 0.45 ± 0.11, p = 0.0013, multivariable regression: “EEO, water quality”). In terms of water quality parameters, DOCinitial had the largest negative influence on EEO. For parameters that improved EEO, higher water matrix conductivity improved EEO by reducing the electrochemical cell's power demands (βconductivity = −0.29 ± 0.1, p < 0.0001, multivariable regression: “EEO, water quality” ESI S10†). Accordingly, groundwater required the lowest EEO of the environmental waters likely due to the low DOC concentration and high conductivity. The energy demands of the EC:H2O2 system operated at 7.4 mA cm−2 were in the range of competitive performance (e.g., 1 kW h m−3 according to Miklos et al. (2018)36) for several water matrices (Table 4), making EC:H2O2 a promising option for scaled applications for treating TOrCs in environmental waters such as groundwater and river water, with the added benefit of DOC removal in the same reactor.
3.5. Conclusions
The goal of this research was to evaluate EC:H2O2 as a combined destructive and non-destructive treatment technology at neutral pH. This performance was assessed as a function of reactor inputs and solution pH. The treatment efficacy of environmental source waters containing varying levels of NOM, scavengers, and ionic constituents was also evaluated. Neither current density nor H2O2 alone promoted pCBA oxidation, although the combination of these parameters heavily influence performance. At neutral pH conditions, [H2O2]initial/[Fe2+]generated ratio was the key driver of oxidative performance, where ratios <0.7 had higher pCBA removal and higher ratios (0.7–1.6) decreased pCBA removal, likely due to H2O2 scavenging.For water quality, pH was the key driver of improved removal, where lower pH conditions minimized the competition between H2O2 and O2 for oxidation of Fe2+ to better encourage radical generation. When treating groundwater and river water, EC:H2O2 had both oxidative treatment of TOrCs and non-destructive treatment of DOC. The pseudo-first order rate constants and EEO values demonstrated that EC:H2O2 can be competitive with other AOPs for TOrC treatment based on energy requirements and treatment performance (depending on current density and water quality, e.g., low DOC, high conductivity waters are easier to treat), with an added benefit of DOC removal due to coagulation and flocculation in the same reactor.
In real world treatment trains, EC:H2O2 could be operated to promote both oxidative and non-destructive treatments in a single process, which could replace multiple conventional unit processes. However, post-EC:H2O2 particle separation via sedimentation or rapid sand filtration would be needed to separate the iron flocs from solution. Additionally, a final disinfection step would likely be needed to ensure sufficient pathogen inactivation and maintain disinfectant residual. Future assessment of the performance of the full treatment train and the related energy efficiency can help to inform treatment train comparisons.
Future work is needed to evaluate EC:H2O2 treatment trains from a systems-engineering perspective wherein the additional benefits such as DOC removal and in situ chemical generation are parameterized to compare against the treatment costs associated with conventional treatment trains. These findings are needed to quantify the benefits of utilizing EC:H2O2 for combined treatment and provide a more comprehensive comparison of EC:H2O2 to current AOP technologies as well as conventional treatment trains. Additionally, the byproducts generated during EC:H2O2 should be evaluated. For example, the co-dissolution of regulated metals from iron electrodes (e.g., manganese) could add secondary contamination. Ex situ H2O2 addition is another challenge for decentralized EC:H2O2 treatment. Accordingly, research is needed to inform reactor setups for EC:H2O2 and to explore H2O2 dosing technologies such as air-diffusion cathodes that can promote in situ H2O2 generation, thereby decreasing ex situ chemical additions and enhancing the process' potential as a small footprint decentralized treatment technology.
Author contributions
DRR: conceptualization, data curation, methodology, laboratory analyses, investigation, visualization, writing – original draft; writing – review & editing. PJM: methodology, funding acquisition, project administration, supervision, resources, validation, visualization, writing – review & editing. CKB: investigation, laboratory analysis, writing – original outline, writing – review & editing. YW: funding acquisition, resources, writing – review &editing. BKM: methodology, funding acquisition, project administration, supervision, resources, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by the Water Equipment and Policy Research Center (WEP) located at the University of Wisconsin-Milwaukee (IIP-1540032) and Marquette University (IIP-1540010). WEP operates under the auspices of the National Science Foundation Industry/University Cooperative Research Center Program.References
- J. C. Crittenden, R. R. Trussell, D. W. Hand, K. J. Howe and G. Tchobanoglous, MWH's Water Treatment: Principles and Design, 3rd edn, 2012 Search PubMed.
- J. J. Pignatello, E. Oliveros and A. Mackay, Advanced Oxidation Processes for Organic Contaminant Destruction Based on the Fenton Reaction and Related Chemistry, Crit. Rev. Environ. Sci. Technol., 2006, 36(1), 1–84 CrossRef CAS.
- H. J. H. Fenton, Oxidation of tartaric acid in presence of iron, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- S. J. Hug and O. Leupin, Iron-catalyzed oxidation of Arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the Fenton reaction, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS PubMed.
- S. Garcia-Segura, M. M. S. G. Eiband, J. V. de Melo and C. A. Martínez-Huitle, Electrocoagulation and advanced electrocoagulation processes: A general review about the fundamentals, emerging applications and its association with other technologies, J. Electroanal. Chem., 2017, 801, 267–299 CrossRef CAS.
- A. Fischbacher, C. von Sonntag and T. C. Schmidt, Hydroxyl radical yields in the Fenton process under various pH, ligand concentrations and hydrogen peroxide/Fe(II) ratios, Chemosphere, 2017, 182, 738–744 CrossRef CAS PubMed.
- F. Haber and J. Weiss, The catalytic decomposition of hydrogen peroxide Iron Salts, Proc. R. Soc. London, 1934, 147, 1070–1091 Search PubMed.
- K. Pratap and A. T. Lemley, Fenton Electrochemical Treatment of Aqueous Atrazine and Metolachlor, J. Agric. Food Chem., 1998, 46, 3285–3291 CrossRef CAS.
- W. Stumm and G. F. Lee, Oxygenation of Ferrous Iron, Ind. Eng. Chem., 1961, 53, 143–146 CrossRef CAS.
- J. Heffron, B. McDermid and B. K. Mayer, Bacteriophage inactivation as a function of ferrous iron oxidation, Environ. Sci., 2019, 5, 1309–1317 CAS.
- D. Lakshmanan, D. A. Clifford and G. Samanta, Ferrous and ferric ion generation during iron electrocoagulation, Environ. Sci. Technol., 2009, 43, 3853–3859 CrossRef CAS PubMed.
- K. Govindan, A. Angelin, M. Kalpana, M. Rangarajan, P. Shankar and A. Jang, Electrocoagulants Characteristics and Application of Electrocoagulation for Micropollutant Removal and Transformation Mechanism, ACS Appl. Mater. Interfaces, 2020, 12, 1775–1788 CrossRef CAS PubMed.
- E. K. Maher, K. N. O'Malley, J. Heffron, J. Huo, Y. Wang, B. K. Mayer and P. J. McNamara, Removal of estrogenic compounds: Via iron electrocoagulation: Impact of water quality and assessment of removal mechanisms, Environ. Sci., 2019, 5, 956–966 CAS.
- A. Qian, S. Yuan, S. Xie, M. Tong, P. Zhang and Y. Zheng, Oxidizing Capacity of Iron Electrocoagulation Systems for Refractory Organic Contaminant Transformation, Environ. Sci. Technol., 2019, 53, 12629–12638 CrossRef CAS PubMed.
- B. P. Chaplin, Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 RSC.
- K. Pratap and A. T. Lemley, Electrochemical Peroxide Treatment of Aqueous Herbicide Solutions, J. Agric. Food Chem., 1994, 42, 209–215 CrossRef CAS.
- S. Vasudevan, An efficient removal of phenol from water by peroxi-electrocoagulation processes, J. Water Process. Eng., 2014, 2, 53–57 CrossRef.
- A. R. Yazdanbakhsh, M. Kermani, S. Komasi, E. Aghayani and A. Sheikhmohammadi, Humic acid removal from aqueous solutions by peroxi- electrocoagulation process, Environ. Health Eng. Manage. J., 2015, 2, 53–58 CAS.
- J. Behin, N. Farhadian, M. Ahmadi and M. Parvizi, Ozone assisted electrocoagulation in a rectangular internal-loop airlift reactor: Application to decolorization of acid dye, J. Water Process. Eng., 2015, 8, 171–178 CrossRef.
- A. Kumar, P. V. Nidheesh and M. Suresh Kumar, Composite wastewater treatment by aerated electrocoagulation and modified peroxi-coagulation processes, Chemosphere, 2018, 205, 587–593 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, A comparative study of electrocoagulation, electrochemical Fenton, electro-Fenton and peroxi-coagulation for decolorization of real textile wastewater: Electrical energy consumption and biodegradability improvement, Biochem. Pharmacol., 2015, 3, 499–506 CAS.
- E. Yüksel, I. A. Şengil and M. Özacar, The removal of sodium dodecyl sulfate in synthetic wastewater by peroxi-electrocoagulation method, Chem. Eng. J., 2009, 152, 347–353 CrossRef.
- D. Gerrity, S. Gamage, D. Jones, G. V. Korshin, Y. Lee, A. Pisarenko, R. A. Trenholm, U. von Gunten, E. C. Wert and S. A. Snyder, Development of surrogate correlation models to predict trace organic contaminant oxidation and microbial inactivation during ozonation, Water Res., 2012, 46, 6257–6272 CrossRef CAS PubMed.
- D. R. Ryan, P. J. McNamara and B. K. Mayer, Iron-electrocoagulation as a disinfection byproduct control strategy for drinking water treatment, Environ. Sci., 2020, 6, 1116–1124 CAS.
- E. K. Maher, K. N. O'Malley, J. Heffron, J. Huo, B. K. Mayer, Y. Wang and P. J. McNamara, Analysis of Operational Parameters, Reactor Kinetics, and Floc Characterization for the Removal of Estrogens via Electrocoagulation, Chemosphere, 2018, 220, 1141–1149 CrossRef PubMed.
- Y. Pi, J. Schumacher and M. Jekel, The use of para-chlorobenzoic acid (pCBA) as an ozone/hydroxyl radical probe compound, Ozone: Sci. Eng., 2006, 27(6), 431–436 CrossRef.
- E. J. Rosenfeldt, K. G. Linden, S. Canonica and U. Von Gunten, OH radical formation during ozonation and the advanced oxidation processes O3/H2 O2 and UV/H2O2, Water Res., 2006, 40, 3695–3704 CrossRef CAS PubMed.
- B. J. Vanderford, F. L. Rosario-Ortiz and S. A. Snyder, Analysis of p-chlorobenzoic acid in water by liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 2007, 1164, 219–223 CrossRef CAS PubMed.
- J. R. Bolton, K. G. Bircher, W. Tumas and C. A. Tolman, Figures-of-merit for the technical development and application of advanced oxidation technologies for both electric- and solar-driven systems (IUPAC Technical Report), Pure Appl. Chem., 2009, 73(4), 627–637 CrossRef.
- D. Borkin, A. Némethová, G. Michaľčonok and K. Maiorov, Impact of Data Normalization on Classification Model Accuracy, Res. Pap. – Fac. Mater. Sci. Technol., Slovak Univ. Technol., 2019, 27, 79–84 Search PubMed.
- M. Olusegun Akinwande, H. Garba Dikko and A. Samson, Variance Inflation Factor: As a Condition for the Inclusion of Suppressor Variable(s) in Regression Analysis, Open J. Stat., 2015, 5, 754–767 CrossRef.
- A. Serra-Clusellas, L. Sbardella, P. Herrero, A. Delpino-Rius, M. Riu, M. de L. Correa, A. Casadellà, N. Canela and X. Martínez-Lladó, Erythromycin abatement from water by electro-fenton and peroxyelectrocoagulation treatments, Water, 2021, 13(8), 1129 CrossRef CAS.
- Environmental Protection Agency (EPA), Enhanced Coagulation and Enhanced Precipitative Softening Guidance Manual, 1999, EPA Office of Water, EPA 815-R-99-012 Search PubMed.
- L. P. Weng, L. K. Koopal, T. Hiemstra, J. C. L. Meeussen and W. H. Van Riemsdijk, Interactions of calcium and fulvic acid at the goethite-water interface, Geochim. Cosmochim. Acta, 2005, 69, 325–339 CrossRef CAS.
- R. Stirling, W. S. Walker, P. Westerhoff and S. Garcia-Segura, Techno-economic analysis to identify key innovations required for electrochemical oxidation as point-of-use treatment systems, Electrochim. Acta, 2020, 338(1), 135874 CrossRef CAS.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Evaluation of advanced oxidation processes for water and wastewater treatment – A critical review, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- M. Lanzarini-Lopes, S. Garcia-Segura, K. Hristovski and P. Westerhoff, Electrical energy per order and current efficiency for electrochemical oxidation of p-chlorobenzoic acid with boron-doped diamond anode, Chemosphere, 2017, 188, 304–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00138e |
| This journal is © The Royal Society of Chemistry 2023 |