
The role of solvation on the conformational landscape of α-synuclein†
Melanie
Cheung See Kit
a,
Tyler C.
Cropley
b,
Christian
Bleiholder
bc,
Christopher D.
Chouinard
 d,
Frank
Sobott
e and
Ian K.
Webb
*af
d,
Frank
Sobott
e and
Ian K.
Webb
*af
aDepartment of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, USA. E-mail: ikwebb@iu.edu
bDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, USA
cInstitute of Molecular Biophysics, Florida State University, Tallahassee, Florida 32306, USA
dDepartment of Chemistry, Clemson University, Clemson, South Carolina 29625, USA
eAstbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK
fCenter for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA
First published on 17th November 2023
Abstract
Native ion mobility mass spectrometry has been used extensively to characterize ensembles of intrinsically disordered protein (IDP) conformers, but the extent to which the gaseous measurements provide realistic pictures of the solution conformations for such flexible proteins remains unclear. Therefore, we systematically studied the relationship between the solution and gaseous structural ensembles by measuring electrospray charge state and collision cross section (CCS) distributions for cationic and anionic forms of α-synuclein (αSN), an anionic protein in solution, as well as directly probed gas phase residue to residue distances via ion/ion reactions between gaseous α-synuclein cations and disulfonic acid linkers that form strong electrostatic bonds. We also combined results from in-solution protein crosslinking identified from native tandem mass spectrometry (MS/MS) with an initial αSN ensemble generated computationally by IDPConformerGenerator to generate an experimentally restrained solution ensemble of αSN. CCS distributions were directly calculated for the solution ensembles determined by NMR and compared to predicted gaseous conformers. While charge state and collision cross section distributions are useful for qualitatively describing the relative structural dynamics of proteins and major conformational changes induced by changes to solution states, the predicted and measured gas phase conformers include subpopulations that are significantly different than those expected from completely “freezing” solution conformations and preserving them in the gas phase. However, insights were gained on the various roles of solvent in stabilizing various conformers for extremely dynamic proteins like α-synuclein.
Introduction
An overwhelming amount of evidence in the literature exists for the retention of major aspects of solution structure of proteins when analyzed in the gas phase under nondenaturing solution conditions while avoiding collisional activation during the transfer of gaseous ions through ion mobility/mass spectrometry (IM/MS) instruments.1 Charge state distributions (CSD) of proteins electrosprayed (ESI) from nondenaturing conditions have been used as a measurement of the extent proteins are unfolded or folded in solution2,3 since nearly the advent of electrospray.4,5 Collision cross section (CCS) distributions measured by IM/MS have shown that for low charge states of ions analyzed under nondenaturing, non-activating conditions, many aspects of the overall solution structure are maintained,6–9 making so-called “native”10 IM/MS (n-IM/MS) an important technique in structural biology, especially due to the exquisite ability of n-IM/MS to rapidly measure proteins with structural11 and proteoform12 heterogeneity from very small samples13 (picomoles or less of total protein). However, these relationships become much more tenuous at higher charge states due to gas phase extension of structures by coulombic repulsion.14,15Intrinsically disordered proteins (IDPs),16,17 proteins that lack a single well-defined native structure, are important in many cellular processes and diseases.18,19 The ability of n-IM/MS to characterize their many functional/structural states that are “averaged out” in bulk solution methods makes the development and application of n-IM/MS towards characterizing IDPs an important research area. For instance, IDPs exhibit high charge states,20–22 broad CSDs, and broad CCS distributions, facilitating the identification of intrinsically disordered regions (IDRs).11 n-IM/MS has been successfully applied to characterizing α-synuclein (αSN), which is a disordered monomer in solution at pH 7 and low concentrations.23 For instance, the effects of pH,24 small molecule binding,25 metal binding,26–28 and the presence of membrane mimics29 on the α-synuclein conformational ensemble have all been characterized by n-IM/MS.
The ability of n-IM/MS to directly probe kinetically-trapped solution-like conformers30 of IDPs has been evaluated by comparing small-angle X-ray scattering (SAXS) ensembles of a set of IDPs against measured and calculated CCS distributions.31 This work revealed that in the absence of solvent, IDPs can explore a much broader conformational space than solution, which was attributed to differences in electrospray mechanisms between more compact and extended conformations, while the authors did note that the ESI-generated structures have to be related in some form to the solution structures. Herein, we have sought to systematically characterize changes in conformations and long-range interactions experienced by αSN in n-IM/MS experiments using CCS as well as solution structure molecular descriptors calculated from the solution ensembles and predicted gas phase ensembles. The solution phase and gas phase data are compared, and the implications of the similarities and differences are discussed.
Materials and methods
Details on the materials used, including various chemicals and reagents, the expression and purification of αSN, in-solution crosslinking (XL), gas-phase XL and ion/ion chemistry, ion mobility measurements, data analysis, and computational methods can be found in the ESI.† Briefly, dilute concentrations of αSN, produced by recombinant expression in E. coli, were reacted in buffered solution with either lysine/amine or aspartic acid/glutamic acid XLs. The solutions were electrosprayed and the CSD, CCS, and fragmentation spectra were determined by n-IM/MS. Additionally, XL in the gas phase was performed with electrosprayed disulfonate dianionic reagents to link positively charged sites of αSN. These data, obtained from three method replicates each, were integrated with computational approaches to determine solution and gaseous ensembles of αSN.Results
Charge state specific analyses of αSN
αSN was first reacted with 2.5 molar excess XL reagent for 15 minutes at room temperature in PBS, pH 7.4, and then buffer exchanged into 50 mM ammonium acetate pH 6.85 to quench the reaction and ionize from a more MS-compatible solution. Products of the αSN XL reaction were first assessed by native ESI-MS through static nanoelectrospray (Fig. 1). The sulfo-NHS ester reagents BS2G, BS3 and sulfo-EGS were chosen to target primary amines such as the N-terminus amine group on methionine at position 1 and side chains of lysine residues. The resulting linker regions span a range of XL distances highlighted in gray (Fig. 1A). The CSD of the unmodified protein was broad (+7 ≤ z ≤ +14) as expected for an IDP such as αSN monomer11 and was centered around z = +10. Similar trends were observed for the CSDs of XL αSN, suggesting that the presence of the XL did not significantly affect the αSN structural ensemble in solution being sampled by ESI-MS (Fig. 1B). We also measured CSDs of unmodified αSN in negative mode as α-synuclein is anionic in pH 7 solutions (Fig. S1B and C†).24 Our observations of high charge states over a broad distribution for αSN compared to narrower, lower charged distributions for cooperatively folded proteins of similar mass in both positive and negative mode matches previous studies, strong evidence that the monomeric state of αSN explores many conformations in solution at pH 7.11,24,26,32 Higher charge states on average were observed in the anion distributions measured by TWIMS/MS than by DTIMS/MS, but this can perhaps be explained by differences in ESI emitter size and flow rate (standard electrospray for Agilent DTIMS and static nanoelectrospray for Waters TWIMS) as well as significant differences in the source regions. We also note that the CSD of αSN can be highly variable, and thus caution against the overinterpretation of the CSD and its direct correlation to solution structures.33 | ||
| Fig. 1 XL-native MS/MS approach using sulfo-NHS ester reagents and native ESI mass spectra (MS) of αSN products at different charge states (z = +8, +9, +11 and +13). (A) Structures of XL reagents BS2G, BS3 and sulfo-EGS, with linker regions of various lengths (highlighted in gray). (B) Full range MS1 showing the CSDs of unmodified protein (U) as well as XL products, which are all centered around z = +10. (C)–(F) Zoomed in views of charge states after XL, labeled with the number of XL (0–4XL) observed per αSN monomer. | ||
Our modified Synapt G2-Si mass spectrometer (Q-TWIMS-TOF instrument) allows for selection of individual charge states of XL αSN via the quadrupole, and the electromagnetostatic cell, positioned between the ion mobility and transfer cells, enables fragmentation by ECD34 and identification of the XL sites on αSN. Products with multiple XL were observed, with up to 4 covalent XL in the case of the +8 charge state XL with BS3 (Fig. 1C). The reaction conditions were optimized to predominantly form the single XL (1XL) product labeled ‘1’ (Fig. 1C–F) and no dead-end XL (where only one of the sulfo-NHS ester groups reacts) were observed. The additional peaks clustered around the major product peaks were due to Na+ adducts despite multiple buffer exchange steps prior to ESI-MS in ammonium acetate. Finally, no evidence of intermolecular XL was observed in Fig. 1B (no observed oligomers or XL oligomers).
In addition, the +8 and +9 αSN charge states showed more additions of XL compared to +11 and +13. For example, the major XL BS3 product observed for the +8 and +9 was the 2XL while the 1XL product was the main peak for the higher charge states (Fig. 1C–F). The compactness of protein conformers in solution strongly affects their ionization by electrospray. Less compact conformers produce ions with higher net charges as more charges can be accommodated on the protein surface.35 The lower charge states likely emerge from more compact solution conformers, resulting in more primary amines being within the XL range. Also, increased XL can potentially make structures artificially compact, which would decrease the charge of the resulting electrospray charge state. Hence, more XL events per αSN monomer were observed for αSN +8 and +9 compared to +11 and +13 charge states. However, the presence of only a monomodal CSD was observed, which may suggest that the increase in XL was not due to structural differences.
Intrinsically disordered proteins such as αSN are not only characterized by their wide CSDs observed by ESI-MS, but also show a diverse conformational ensemble when studied by IM-MS. IM-MS has previously been used to characterize the effects of pH and ligand binding on the conformational ensemble of αSN from solution.26,33,36 The following charge states, z = +8, +9, +11 and +13 were chosen to broadly represent the conformational heterogeneity of αSN as demonstrated by the different arrival time distributions (ATDs) of the unreacted protein (bottom plots labeled ‘U’) (Fig. S2†). The ATDs for the lower charge states +8 and +9 showed more conformer heterogeneity since the drift time ranges were broader and at least 3 distinct IM peaks were observed, suggesting at least 3 co-existing conformer subpopulations. The +11 and +13 charge states had narrower ATDs indicating less gas phase conformational dynamics. The ion mobility data was assessed qualitatively to determine the effects of the different XL on conformational distribution. Overall, a single XL did not perturb the ATDs of αSN at different charges states as the drift time range and IM peak features remained mostly unchanged. In contrast, the presence of two and three XL per αSN monomer decreased the relative abundance of the more extended conformers at later drift times, especially for the lower charge states z = +8 and +9, suggesting that the multiple XL are restricting the conformer population to become more compact with an earlier drift time (Fig. S2A and B†). Despite containing more extended conformer populations, the higher charge states z = +11 and +13 showed less drastic changes after multiple XL, with the appearance of relatively low abundance peaks at earlier drift times (Fig. S2C and D†). Among the three XL reagents, sulfo-EGS XL caused the most pronounced ATD shifts to more compact conformer subpopulations, especially with the multiply XL +8 charge state, possibly due to its different linker group chemistry (ethylene glycol vs. hydrocarbon). Also, although sulfo-EGS is more extended relative to BS2G and BS3, MD simulations using Amber15 have shown that it often adopts a more compact conformation, making its spacer arm shorter than BS337 and perhaps driving reduced reactivity relative to BS3. The tightening of the αSN conformational space after multiple BS3 XL has been observed before by IM-MS using a drift tube instrument,33 which suggests that the extent of XL should be monitored by, e.g., n-IM/MS, analogous to covalent labeling “dosimetry”.38,39
The XL samples were also analyzed by a bottom-up approach using Glu-C digestion and pLink software to search for XL peptides, with the ones identified in at least two method replicates listed in Table S3.† Due to the nature of bottom-up proteomics, information on the total number of XL, specific proteoform, protein–ligand interaction, or protein–protein interaction is lost during digestion. Though these assignments include digestion and thus a mixture of the doubly, triply, and quadruply linked protein (Fig. 1), they do provide a definitive list of all the possibilities for XL that could be encountered in our XL/n-MS/MS singly linked data. Sulfo-EGS data were omitted due to the lack of identified XL.
In contrast, owing to the restraining effects observed from multiple XL per αSN monomer, only the 1XL products were selected for fragmentation by ECD to identify the XL pairs for each charge state conformer population. ECD fragments corresponding to the unmodified ions and modified ions (with the added mass of covalent adduct) were mapped onto the primary sequence of αSN to visualize the sequence coverage for individual charge states and their respective 1XL products, with the assigned XL shown in gray (Fig. S3 and S4†). Individual charge states of the non-XL (NC) protein were also analyzed using the same instrument conditions to obtain NC fragment maps, which showed efficient ECD fragmentation for all the charge states in the absence of XL (Fig. S5†). Details on the identification of XL with n-MS/MS are provided in the ESI.†
Table 1 summarizes the XL that could unambiguously be assigned to each charge state. The XL pairs N-term–K12 and K43–K96 were observed with BS2G and BS3. K96–K102 was unique to BS2G while K-60–K102 was unique to BS3. Sulfo-EGS data were omitted due to the lack of coverage from both bottom-up and top-down methods. We also note that the identified XL seem to have no trend based upon charge state, and the C-terminal acidic region was not probed. Therefore, we used acidic reactive linkers to probe the C-terminal tail (Fig. S6 and S7†). Again, with the acidic linkers, there was also no trend based on charge state and linker location. This supports that each charge state for αSN, observed in a monomodal distribution, is not itself representative of a particular solution conformational family. This also supports that the gaseous structures measured by ion mobility are thus largely governed by gas-phase electrostatics, not kinetically trapped solution states. However, though cases where the CSD of αSN was observed as a bimodal or multimodal distribution were indicative of the presence of a more compact conformational subfamily;26,28,33 we observed no evidence of a second distribution under our experimental conditions.
| Reagent | z = +8 | z = +9 | z = +11 | z = +13 |
|---|---|---|---|---|
| BS2G | N-term/K12 | N-term/K12 | N-term/K12 | N-term/K12 |
| K96/K102 | K43/K96 | K96/K102 | K43/K96 | |
| BS3 | N-term/K12 | N-term/K12 | N-term/K12 | N-term/K12 |
| K60/K102 | K60/K102 | K43/K96 | K60/K102 | |
| MDH | E130/E137 | D121/E139 | D121/E137 | E130/E137 |
| ADH | D121/E137 | D121/E137 | E130/E137 | E130/E137 |
The utility of XL/n-MS/MS for defining the solution αSN conformational space
IDPs form highly dynamic structural ensembles in solution in contrast to folded globular proteins, which cooperatively form more closely related structures. Solution experiments and computational techniques such as NMR40–42 and MD43,44 respectively are often combined to address the challenges of analyzing interconverting IDP conformers. For instance, an implementation of paramagnetic relaxation enhancement (PRE) NMR has utilized cysteine mutations at Q24, S42, Q62, S87, N103 and N122 for labeling with a nitroxide spin label. The PRE-derived distance restraints (12–20 Å range) together with MD simulations then produced an ensemble consisting of structures collected every 5 ps for 1.2 ns per replica. The ensemble was biased to obtain a hydrodynamic radius of 32.0 Å, measured from pulsed-field gradient NMR and SAXS,45 with every 10th structure uploaded on Protein Ensemble Database (PED00024).46 This ensemble (n = 576) was used as reference for benchmarking the XL/n-MS/MS approach. Here, IDPConformerGenerator (IDPConf) was used to produce the initial ensemble of conformations for αSN. This open-source software platform generates IDP conformer ensembles by constructing side chains from Monte Carlo algorithms and backbone angles for various secondary structural elements from secondary structure and loops in the Protein Data Bank and producing coordinates using a machine learning approach.47 Finally, the XL identified experimentally by native MS/MS were used as distance restraints when evaluating the initial αSN ensemble obtained from IDPConf. For XL pairs identified for two reagents, the shorter XL distance was used as the restraint distance, such as the BS2G XL distance used for the N-term–K12 pair (Table 1).The following descriptors were used to characterize the structures populating the conformational spaces occupied by the reference PRE NMR- (again, those measured by Allison et al.46) and XL-filtered IDPConf ensembles. The radius of gyration (Rg) and end to end distance (Ree), i.e., the Cα–Cα distance measured from M1 to A140, for αSN provided a measure of the overall size and shape of protein structure. Rg has previously been used as an indicator of protein compactness48 as proteins undergo conformational transitions upon ligand binding.49 The distributions of the spin label NMR reference ensemble (NMR, n = 576) and the XL/n-MS/MS filtered IDPConf ensemble (IDPConf, n = 621) were plotted against Rg (Fig. 2). The XL/n-MS/MS filtered structures were biased to an experimentally determined Rg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 by the removal of random structures with Rg < 30 Å, until the average Rg was between 30 and 34 Å (XL, Rg-Biased, Fig. 2). The unbiased and biased Rg ranges are very similar. The calculated average Rg from the deposited NMR ensemble using GROMACS is 31.3 Å, while the XL IDPConf unbiased and biased data gave an average of 27.9 and 30.0 Å, respectively. However, our data did not show evidence of the highest Rg structures identified in the NMR data. The Ree data agree strongly with the NMR data, though the longest outliers were not supported by the XL data. We note that these differences can be related to the means of generating a starting ensemble and side chain arrangements. Additionally, single molecule FRET (smFRET) experiments performed on αSN monomers reported a mean distance between dyes attached to cysteines (inserted by G to C mutation at residues 7 and 84) of roughly 50 Å (ref. 50) which agrees quite well with the mean Cα to Cα distance from residue 7 to 84 from the unbiased XL-IDPConf ensemble, 52.8 ± 1.79 Å (99% confidence interval).
45 by the removal of random structures with Rg < 30 Å, until the average Rg was between 30 and 34 Å (XL, Rg-Biased, Fig. 2). The unbiased and biased Rg ranges are very similar. The calculated average Rg from the deposited NMR ensemble using GROMACS is 31.3 Å, while the XL IDPConf unbiased and biased data gave an average of 27.9 and 30.0 Å, respectively. However, our data did not show evidence of the highest Rg structures identified in the NMR data. The Ree data agree strongly with the NMR data, though the longest outliers were not supported by the XL data. We note that these differences can be related to the means of generating a starting ensemble and side chain arrangements. Additionally, single molecule FRET (smFRET) experiments performed on αSN monomers reported a mean distance between dyes attached to cysteines (inserted by G to C mutation at residues 7 and 84) of roughly 50 Å (ref. 50) which agrees quite well with the mean Cα to Cα distance from residue 7 to 84 from the unbiased XL-IDPConf ensemble, 52.8 ± 1.79 Å (99% confidence interval).
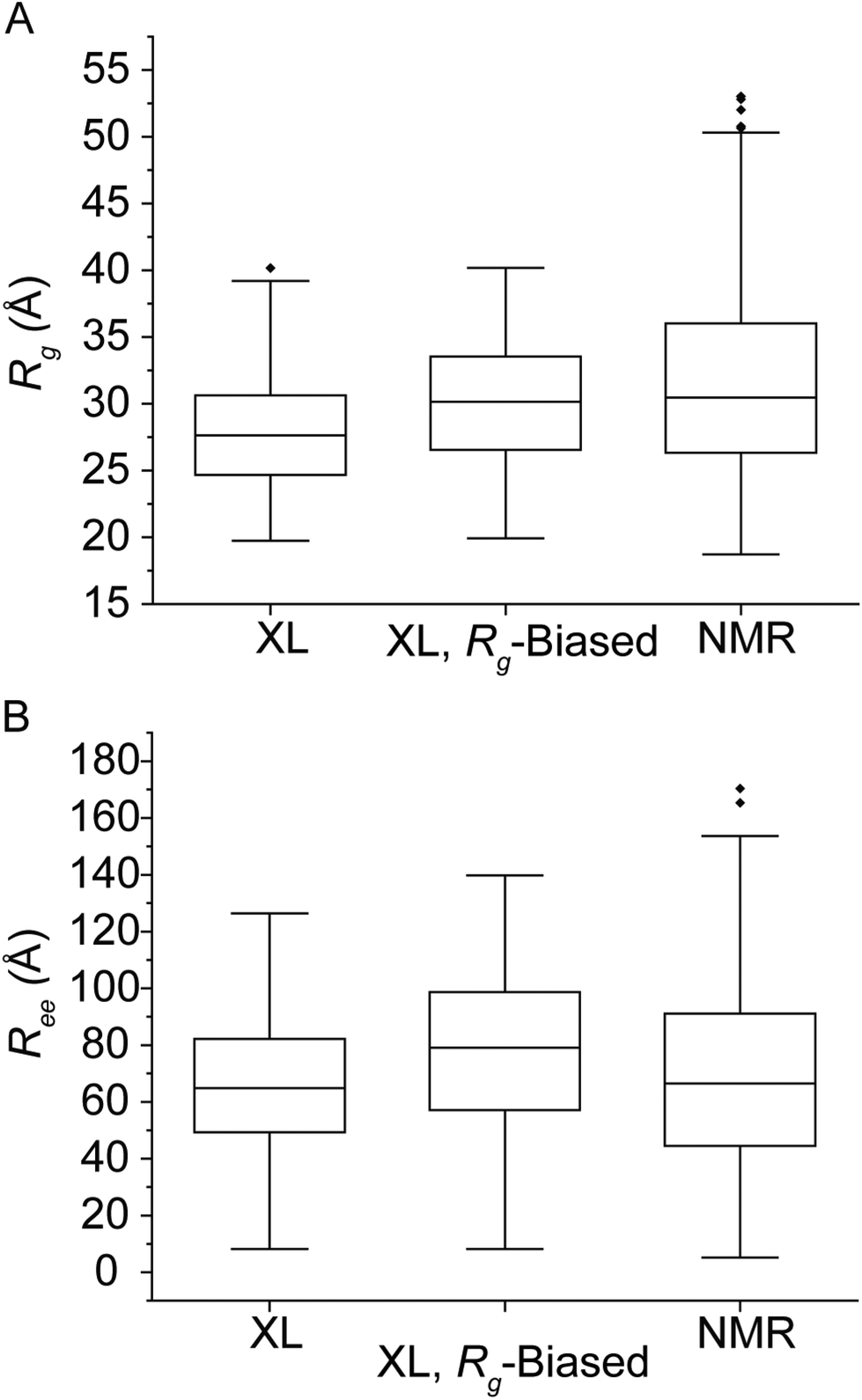 | ||
| Fig. 2 Box and whisker plots of the NMR ensemble calculated in ref. 48 compared to XL-native MS/MS and Rg-biased XL-native MS/MS results for calculated Rg (A) and Ree (B). | ||
Short-distance XL data have also been used as constraints in discrete molecular dynamics to characterize the conformational landscape of αSN.51 An all-atom Replica Exchange (REX) simulation was applied to a completely unfolded starting αSN structure and the resulting trajectories were ranked so that the lowest 10% energy structures were selected, as determined by a DMD Medusa force field. αSN was XL using various short-distance XLs and combined with a bottom-up proteomics approach to identify XL peptides, resulting in experimental restraints in the 0–7 Å range, which fit much more compact structures compared to the structures populating the PRE NMR and IDPConf ensembles, with their observed Rg ranging from 5–21 Å and a major structural subpopulation Rg around 16 Å. The authors noted that the bias to more compact conformers could be due to the short-range XL reagents and the Medusa force field used in their study. Also, differences in Rg could be explained by the fact that PRE NMR data sampled an equilibrium of monomeric and multimeric states under the experimental conditions used. However, using our XL/n-MS/MS approach allows for m/z specific XL site identification so that only XL monomers are analyzed, and the agreement in Rg between the unbiased XL-IDPConf data, SAXS, and PRE NMR suggests that this situation did not occur frequently under the low αSN concentration, salty conditions used in these three measurements.
The conformers populating the PRE NMR and unbiased IDPConf ensembles were clustered based on structural similarity, with one representative structure shown per cluster (C1–C6) as well as the calculated Rg (Fig. S8† and Fig. 3, respectively). C3 represented 32% of the IDPConf conformational space, with the 2nd highest Rg, where the more extended conformers clustered to C6, representing 18% of the conformational space. The other half of the represented conformational space ranged Rg values 19.9 to 26.0 Å. While the N- and C-termini (blue and red) were modeled to be folded onto themselves to different extents in all 6 representative structures, lower Rg, i.e. structure compactness, resulted from the folding of the NAC region (gray) as well as close interactions between the N-terminal portion and C-terminal tail and the NAC region (C5) and between the N- and C-terminal portions (C2, C1, C4). The solvent accessible surface area (SASA) ranged from 129 nm2 for C2 to 144 nm2 for C1 (lowest and highest SASA for the six representative structures, respectively), in agreement with recent MD studies.52 Together, C1 and C3 (58%) represented the most compact conformers with the lowest Rg for the PRE NMR ensemble while C2 (7%) represented the most extended conformer with Rg = 52.0 Å (Fig. S8†). A more compact NAC region and closer interactions between the C-terminus and NAC region and between the N- and C-terminal portions resulted in the more compact representative structures from the PRE NMR ensemble, similar trends to the unbiased XL-IDPConf ensemble.
 | ||
| Fig. 3 Summary of clustered filtered IDPConf structural ensemble (n = 377, C1–C6). (A) Cluster dendrogram showing distribution of conformational space into specific clusters. (B) Table summarizing the number of structures per cluster in order of decreasing Rg of the representative structures per cluster. (C) Representative structures for each cluster, rotated 90° along the x-axis. | ||
CCS distributions of αSN monomers
Insights into gas phase ion structure can be made by measuring CCS distributions. Due to the lack of nitrogen CCS values available for protein anions, αSN anions were measured with DTIMS to improve CCS accuracy. Fig. 4 shows a comparison of the estimated CCS distributions calculated from the PRE NMR ensemble to the measured values from positive and negative mode. The distributions are all centered near the 3000–4000 Å2 range indicating a large overall gas phase shape/size11,24 compared to similar mass proteins (for example, the average nitrogen CCS of native-like cytochrome C in positive mode electrosprayed from aqueous 20 mM ammonium acetate is 1536 Å2).53 The overall features are quite broad, showing that the gas phase conformers populate extremely diverse states, either through gas-phase processes or during the electrospray process itself. However, the positive mode CCS distribution was more compact than negative mode distribution at similar but opposite polarity charge states. We investigated this phenomenon as a function of charge state (Fig. S9†), with similar CCS distributions for positive and negative polarities with nine and ten net charges, but substantially larger CCS for anions at charge state 11 and above. These results showed that the anionic gas phase structural ensemble more closely matched the overall sizes/shapes of the solution ensemble while the cation gas phase ensemble was significantly more compact. This illustrates the importance of examining proteins with ESI/MS conducted in the same polarity as their solution net charge, in agreement with a recent study showing significantly different gas phase behavior for avidin and β-lactoglobulin ions with the same magnitude total charge but opposite overall polarity.54 Therefore, and since positive ion mode is the typical choice for n-MS, we sought to investigate deviations of the positive mode CCS from the other distributions by predicting the positive mode gas phase conformers with the structure relaxation approximation (SRA, see ESI† methods for description) which has been applied to and recapitulates the average cross sections and widths of cross section distributions from monomeric proteins55 and protein complexes.56 | ||
| Fig. 4 CCS distributions plotted for gas phase ensembles measured experimentally using positive (z = +8 to +15) and negative (z = −9 to −15) ESI and the CCS distribution calculated from the NMR ensemble. | ||
Comparison of structural descriptors (Rg, Ree)
The CCS distribution of α-synuclein cations predicted by the SRA is shown as a function of charge state in Fig. S9.† Though the relative intensities and shapes of features do show differences, the overall range of CCS for each charge state agrees with the measured cation cross sections. Therefore, structural descriptors from the final SRA ensembles were used to compare the similarity between gas phase and solution ensembles beyond using CCS alone. CCS measurements include the interaction potentials between gaseous ions and drift gas, while Ree and Rg are calculated directly from atomic coordinates. The structural descriptors for the SRA, XL-native MS/MS, and NMR distributions are illustrated in Fig. 5. Fig. 5A shows the Rg distribution as a function of charge state for the SRA ensembles. First, the Rg for the SRA ensembles are extremely dependent on charge state, with a portion of the +8 distribution being more compact than the solution distribution, and the +13 distribution being significantly larger than the solution Rg distributions. This observation agrees with previous observations of the ability of gaseous disordered proteins to sample a larger conformational space in the gas phase than in solution.11,31 | ||
| Fig. 5 Comparison of gas phase structural ensembles for s = +8, +11 and +13 calculated using the SRA and solution ensembles with distance restraints obtained by spin label NMR and the unbiased XL-native MS/MS data. (A) Box and whisker plots of the calculated Rg distributions for SRA +8, +11, and +13 ensembles, (B) Reevs. Rg for SRA +8, +11 and +13 ensembles (labeled in black, red, and blue respectively). (C) Reevs. Rg for NMR (grey) and XL-native MS/MS (orange) ensembles. | ||
Strikingly, the Ree distribution plotted as a function of Rg in Fig. 5B for the SRA conformers versus the solution conformers in Fig. 5C highlights not only that a portion of the +13 charge state conformers is extremely extended (Ree ≫ 175 Å) but also shows a strange feature with similar Rg to the solution ensembles but extremely small Ree. This shows that a significant subpopulation has mass distributed away from the center of mass, but the N and C termini are positioned very close to one another, indicative of head-to-tail cyclization which was not observed for the solution ensemble.32 Therefore, while some of the SRA conformers are in fact similar to the solution distributions, there are new subpopulations at the extremes that are only populated in the gas phase.
Comparison of SRA GP representative conformers to NMR and IDPConf
Next, the gas phase conformers from the SRA performed on each charge state were clustered by backbone RMSD, with the results and representative conformers from each cluster shown in Fig. 6 and Fig. S11.† Representative conformers C1 and C3 from +8 (Fig. S11C†) are quite compacted, while the rest, as well as +11 and +13, (e.g., C6 in Fig. 6C and C6 in Fig. S11D†) show virtually no “empty space” in the structure in stark contrast to the clustered NMR and XL-native MS/MS ensembles. The structural compaction for the +8 conformers is similar to what is observed for well-folded proteins where cavities collapse due to the absence of water.57–59 The representative solution conformers are characterized by longer range contacts (e.g., NMR C5 in Fig. S10C† and XL C1 in Fig. 3C) and show substantial space in the structure which is inhabited by solvent molecules and formed by interaction with the solvent (see NMR C5, XL C1, C4, C6). Long range contacts for solution conformers are between the acidic C-terminal region or amphipathic N-terminal region and non-amyloid component (NAC) (e.g., NMR C1, XL C3, C6), and between the N- and C-terminal regions (e.g., NMR C5, XL C4). In contrast, several of the +11 and +13 SRA representative conformers show cyclization between the very beginning of the N-terminal region (which contains 3 lysine residues in the first 12) and the acidic C-terminal tail. This cyclization is likely driven by long-range electrostatic forces, including the formation of new salt bridges, which dominate in the absence of solvent.60 This cyclization takes two forms, an open ring type conformation (e.g., +11 C4, +13 C4) or a closed type conformation (e.g., +13 C5) that is likely stabilized by additional electrostatic and van der Waals forces. Though the +8 ions follow well-understood “compaction” mechanisms, it is clear that some of the highly charged ions explore a huge range of conformations in the gas phase, including the formation of new intramolecular interactions that have not been identified for αSN with solution measurements.32,46,61 Therefore, while the CCS distributions are accurate descriptors of the relative order or disorder of proteins11 and reflect changes in solution conformational space upon change in solution conditions24 or ligand/metal binding,25–28 the structural features of the conformers themselves may not reflect the solution conformers. | ||
| Fig. 6 Clustered SRA +11 conformational ensembles prior to (n = 724) and post (n = 126) filtering using gas phase XL (GP-XL) restraints. Cluster dendrograms show distribution of conformational space into specific clusters (A) before and (B) after GP-XL filtering, respectively. Representative conformers for each cluster, rotated 90° along the x-axis and the αSN structure is colored as blue (N-terminal region, residues 1–60), gray (nonamyloid-β component, residues 61–95) and red (C-terminal tail, residues 96–140) before (C) and after (D) GP-XL filtering respectively. | ||
Validation of the SRA ensemble with gas phase XL
We sought to validate the SRA predictions of αSN with gas phase XL via ion/ion reactions of αSN with various length disulfonate salts.62,63 Cα–Cα maximum distances included the lengths of the linkers and the sulfur to lysine side chain/N-terminus salt bridge lengths calculated previously62 as well as the length of lysine side chains or the N-terminus to residue M1 Cα distance depending on the residues involved in the XL. Also, we added an additional 15 Å “padding” to the length of the linker and used Cα–Cα distances. Previous work has suggested that a cutoff distance of >14 Å is suitable for gaseous electrostatics in molecular dynamics of proteins.64 Therefore, the total maximum lysine to lysine Cα–Cα distances were 37.7, 40.0, 41.2, 49.8, 54.0, and 58.3 Å for 1,2-BDSA, 1,3-BDSA, 1,5-NDSA, BS2G, BS3 and sulfo-EGS, respectively, and the total maximum lysine to N-terminus Cα–Cα distances were 32.8, 35.1, 36.3, 44.9, 49.1, and 53.4 Å for 1,2-BDSA, 1,3-BDSA, 1,5-NDSA, BS2G, BS3 and sulfo-EGS, respectively.The singly XL +11 and +13 charge states resulted in net charges of +9 and +11, respectively, after ion/ion reactions with disulfonate dianions. As a control, the unmodified forms of the +9 and +11 charge states were produced by proton transfer charge reduction reactions with two deprotonated PFO anions.65–67 IM allowed for the separation of the XL and charge reduced ions from the precursors as a function of net charge (Fig. S12†). ECD of the +9 and +11 charge reduced ions gave extensive coverage over all the positively charged residues (Fig. S13†). Again, fragment assignments were validated by three replicates and used only in cases where the signal to noise of the precursor and fragment ions for the XL (Fig. S14 and S15† for the +11 and +13 charge states, respectively) and proton transferred precursors were similar. Use of the 1,5-NDSA XL badly over restrained the data (for example, only 6 of the 724 conformers from SRA of the +11 charge state could be explained using the 1,5-NDSA links), so the 1,5-NDSA identifications were not used to filter the SRA conformers. This is likely due to the rigidity of the XL inducing structural artifacts.
The SRA ensembles for each charge state were filtered by whether the predicted distances from the atomic coordinates of the conformers were less than the maximum allowed distance between the XL residues, given in ESI Table 5.† 21% of the +11 SRA conformers and 87% of the +13 SRA conformers were supported by the XL results (only the N terminus to K21 XL was used for +13). The XL-selected +11 conformers had an average Rg of 31.4 Å, versus the entire +11 SRA ensemble having an average Rg of 37.4 Å, while the XL-selected +13 had an average Rg of 42.3 Å and the entire +13 ensemble from SRA had an average Rg of 42.8 Å. Comparing representative conformers from the entire +11 SRA ensemble and the XL distance filtered +11 ensemble reveals that the gas phase XL conformers were structurally similar to the representative conformers from the overall SRA ensemble (Fig. 6). Specifically, representative conformers for both the overall ensemble (Fig. 6C) and the XL-satisfying subpopulation (Fig. 6D) both show evidence of the gas phase-specific structural motifs of compaction and head to tail cyclization. Overall, these data support structural patterns and CCS distributions predicted by the SRA for IDP cations.
Recently, solution molecular dynamics has been combined with native IM/MS and atomic force microscopy for αSN.52 The predicted structures were clustered into four clusters, based on observed CCS measurements. These clusters, named C0, C1, C2, and C3, respectively, had reported CCS of 2188, 2388, 2722, and 2944 Å2, roughly matching our observations for positive mode analysis. The Rg of these structures were similar to +11 from our SRA data for C3 and the C1 state close to the +8 from the SRA. Our solution data in Fig. 2A best supports structures resembling the C2 and C3 states, and the smallest representative structure from Fig. 3, representative structure C2, with Rg of 19.9 Å, is in good agreement with the C0 cluster.52 Although we do expect compaction in the gas-phase, it is of interest to note that using CCS as a constraint for molecular dynamics appears to be, overall, a useful way to measure IDP conformational families, even though the gas-phase structures of IDPs likely do not represent the solution structures.31 Therefore, further research on comparing XL, CCS, and molecular modeling is warranted to gain insight into IDP conformational states, since there is a clear correlation between solution order/disorder and gaseous order/disorder, and the ability of CCS distributions, in general, to guide accurate solution state modeling.
Conclusions
In this study, a systematic comparison of solution and gaseous αSN conformers was conducted, utilizing solution XL determined by native MS/MS, CCS measurements and predictions alongside additional information from calculated molecular descriptors, and gas phase XL, which served as an orthogonal approach to CCS measurements. The solution XL studies illustrated the importance of knowing the reaction stoichiometry of XL to protein for measuring tertiary structure and the ability of XL coupled to emerging computational tools to provide insight into protein conformational dynamics. Good agreement with SM-FRET and PRE-NMR was realized with this method.Charge state and CCS distributions are highly effective in qualitatively describing the disordered nature of αSN monomers, reinforcing these tools as valuable for gaining insights into how changes in solution conditions and binding to other molecules impact the protein's structure. However, it was observed that non-CCS descriptors and visual examination of representative gaseous conformers revealed significant differences when compared to solution ensembles. This finding provided crucial insights into the role of the solvent in the system. In solution, water and the high concentration of electrolytes in biochemical buffers and inside the cell screen the head-to-tail electrostatic interactions and mediate the protection of the most hydrophobic part of the protein through long-range interactions of the NAC with the C-terminal and N-terminal regions. Although interactions between the C-terminal and N-terminal regions were observed, solution long-range interactions were not proximal (i.e., ring like) to the N-terminus itself and did not show collapse, unlike the characteristic gas phase cyclization. This indicates that while electrostatic interactions remain vital in solution, the strengths and localization of these forces differ for αSN in solution versus in vacuo, highlighting the significant role of solvation in mediating protein folding and dynamics, even for IDPs. Finally, the ESI process (from droplet to bare ion) is not modeled by the SRA, but there was still excellent agreement between the SRA, CCS, and gas phase XL results. This perhaps indicates that differences between the observed gas phase ensemble and solution ensemble for αSN monomers are driven by changing the strengths of electrostatic and other intramolecular interactions in the presence and absence of water and electrolytes, not by different ESI mechanisms.
The knowledge that can be gained from systematic solution versus gas phase comparisons will be instrumental in further investigations into the structural roles of the solvent and intrinsic interactions, both for cooperatively folded and unfolded proteins. Understanding these aspects will not only enhance our understanding of protein behavior but also shed light on how the solvent environment influences the conformational dynamics of biomolecules.
Author contributions
MCSK: Investigation, formal analysis, methodology, writing – original draft; TCC: Formal analysis, investigation, methodology, software; CB: Conceptualization, formal analysis, funding acquisition, methodology, software, supervision; CDC: Investigation, formal analysis; FS: Conceptualization, IKW: Conceptualization, formal analysis, funding acquisition, methodology, software, supervision, writing – original draft, writing – review and editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Portions of this work were funded by the National Institute of General Medical Sciences of the National Institutes of Health under R21GM134408 (IKW), R35GM151251 (IKW), and R01GM135682 (CB), and by the National Science Foundation under CHE-1654608 (CB). This research was also supported in part by Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute. IKW thanks Professor Jared Shaw, University of Nebraska Lincoln, and Professor Nicholas Borotto, University of Nevada Reno for helpful discussions.References
- S.-H. Chen and D. H. Russell, How Closely Related Are Conformations of Protein Ions Sampled by IM-MS to Native Solution Structures?, J. Am. Soc. Mass Spectrom., 2015, 26, 1433–1443 CrossRef CAS.
- L. Konermann and D. J. Douglas, Acid-Induced Unfolding of Cytochrome c at Different Methanol Concentrations: Electrospray Ionization Mass Spectrometry Specifically Monitors Changes in the Tertiary Structure, Biochemistry, 1997, 36, 12296–12302 CrossRef CAS PubMed.
- L. Konermann and D. J. Douglas, Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes, J. Am. Soc. Mass Spectrom., 1998, 9, 1248–1254 CrossRef CAS.
- S. K. Chowdhury, V. Katta and B. T. Chait, Probing conformational changes in proteins by mass spectrometry, J. Am. Chem. Soc., 1990, 112, 9012–9013 CrossRef CAS.
- J. A. Loo, C. G. Edmonds, H. R. Udseth and R. D. Smith, Effect of reducing disulfide-containing proteins on electrospray ionization mass spectra, Anal. Chem., 1990, 62, 693–698 CrossRef CAS PubMed.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Evidence for macromolecular protein rings in the absence of bulk water, Science, 2005, 310, 1658–1661 CrossRef CAS.
- T. Wyttenbach and M. T. Bowers, Structural stability from solution to the gas phase: native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment, J. Phys. Chem. B, 2011, 115, 12266–12275 CrossRef CAS PubMed.
- K. Breuker, S. Bruschweiler and M. Tollinger, Electrostatic stabilization of a native protein structure in the gas phase, Angew. Chem., Int. Ed., 2011, 50, 873–877 CrossRef CAS PubMed.
- Y. Zhong, L. Han and B. T. Ruotolo, Collisional and Coulombic unfolding of gas-phase proteins: high correlation to their domain structures in solution, Angew. Chem., Int. Ed., 2014, 53, 9209–9212 CrossRef CAS.
- S. Tamara, M. A. den Boer and A. J. R. Heck, High-Resolution Native Mass Spectrometry, Chem. Rev., 2022, 122, 7269–7326 CrossRef CAS PubMed.
- D. Stuchfield and P. Barran, Unique insights to intrinsically disordered proteins provided by ion mobility mass spectrometry, Curr. Opin. Chem. Biol., 2018, 42, 177–185 CrossRef CAS PubMed.
- L. M. Smith and N. L. Kelleher, Consortium for Top Down, P. Proteoform: a single term describing protein complexity, Nat. Methods, 2013, 10, 186–187 CrossRef CAS.
- Y. Zhong, S.-J. Hyung and B. T. Ruotolo, Ion mobility–mass spectrometry for structural proteomics, Expert Rev. Proteomics, 2012, 9, 47–58 CrossRef CAS.
- D. E. Clemmer and M. F. Jarrold, Ion Mobility Measurements and their Applications to Clusters and Biomolecules, J. Mass Spectrom., 1997, 32, 577–592 CrossRef CAS.
- S. Vahidi, B. B. Stocks and L. Konermann, Partially Disordered Proteins Studied by Ion Mobility-Mass Spectrometry: Implications for the Preservation of Solution Phase Structure in the Gas Phase, Anal. Chem., 2013, 85, 10471–10478 CrossRef CAS.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff and K. W. Hipps, et al., Intrinsically disordered protein, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS PubMed.
- P. Tompa, Intrinsically unstructured proteins, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS PubMed.
- P. E. Wright and H. J. Dyson, Intrinsically disordered proteins in cellular signalling and regulation, Nat. Rev. Mol. Cell Biol., 2015, 16, 18–29 CrossRef CAS PubMed.
- B. K. Maity, A. K. Das, S. Dey, U. K. Moorthi, A. Kaur, A. Dey, D. Surendran, R. Pandit, M. Kallianpur and B. Chandra, et al., Ordered and Disordered Segments of Amyloid-beta Drive Sequential Steps of the Toxic Pathway, ACS Chem. Neurosci., 2019, 10, 2498–2509 CrossRef CAS PubMed.
- L. Testa, S. Brocca and R. Grandori, Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins, Anal. Chem., 2011, 83, 6459–6463 CrossRef CAS.
- L. Testa, S. Brocca, C. Santambrogio, A. D'Urzo, J. Habchi, S. Longhi, V. N. Uversky and R. Grandori, Extracting structural information from charge-state distributions of intrinsically disordered proteins by non-denaturing electrospray-ionization mass spectrometry, Intrinsically Disord. Proteins, 2013, 1, e25068 CrossRef.
- A. Natalello, C. Santambrogio and R. Grandori, Are Charge-State Distributions a Reliable Tool Describing Molecular Ensembles of Intrinsically Disordered Proteins by Native MS?, J. Am. Soc. Mass Spectrom., 2017, 28, 21–28 CrossRef CAS.
- W. Wang, I. Perovic, J. Chittuluru, A. Kaganovich, L. T. Nguyen, J. Liao, J. R. Auclair, D. Johnson, A. Landeru and A. K. Simorellis, et al., A soluble alpha-synuclein construct forms a dynamic tetramer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17797–17802 CrossRef CAS.
- S. L. Bernstein, D. Liu, T. Wyttenbach, M. T. Bowers, J. C. Lee, H. B. Gray and J. R. Winkler, α-Synuclein: Stable compact and extended monomeric structures and pH dependence of dimer formation, J. Am. Soc. Mass Spectrom., 2004, 15, 1435–1443 CrossRef CAS.
- M. Grabenauer, S. L. Bernstein, J. C. Lee, T. Wyttenbach, N. F. Dupuis, H. B. Gray, J. R. Winkler and M. T. Bowers, Spermine binding to Parkinson's protein alpha-synuclein and its disease-related A30P and A53T mutants, J. Phys. Chem. B, 2008, 112, 11147–11154 CrossRef CAS PubMed.
- R. Moons, A. Konijnenberg, C. Mensch, R. Van Elzen, C. Johannessen, S. Maudsley, A. M. Lambeir and F. Sobott, Metal ions shape alpha-synuclein, Sci. Rep., 2020, 10, 16293 CrossRef CAS PubMed.
- A. D. Stephens, M. Zacharopoulou, R. Moons, G. Fusco, N. Seetaloo, A. Chiki, P. J. Woodhams, I. Mela, H. A. Lashuel and J. J. Phillips, et al., Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity, Nat. Commun., 2020, 11, 2820 CrossRef CAS PubMed.
- E. J. Byrd, M. Wilkinson, S. E. Radford and F. Sobott, Taking Charge: Metal Ions Accelerate Amyloid Aggregation in Sequence Variants of α-Synuclein, J. Am. Soc. Mass Spectrom., 2023, 34, 493–504 CrossRef CAS.
- R. Moons, R. van der Wekken-de Bruijne, S. Maudsley, F. Lemière, A.-M. Lambeir and F. Sobott, Effects of Detergent on α-Synuclein Structure: A Native MS-Ion Mobility Study, Int. J. Mol. Sci., 2020, 21, 7884 CrossRef CAS PubMed.
- D. E. Clemmer, D. H. Russell and E. R. Williams, Characterizing the Conformationome: Toward a Structural Understanding of the Proteome, Acc. Chem. Res., 2017, 50, 556–560 CrossRef CAS PubMed.
- A. J. Borysik, D. Kovacs, M. Guharoy and P. Tompa, Ensemble Methods Enable a New Definition for the Solution to Gas-Phase Transfer of Intrinsically Disordered Proteins, J. Am. Chem. Soc., 2015, 137, 13807–13817 CrossRef CAS PubMed.
- M. Cheung See Kit and I. K. Webb, Surveying the Conformational Landscape of α-Synuclein Using Native Crosslinking, Ion Mobility-Mass Spectrometry and Ensemble Modeling, ChemRxiv, 2023, This content is a preprint and has not been peer-reviewed.
- A. S. Phillips, A. F. Gomes, J. M. D. Kalapothakis, J. E. Gillam, J. Gasparavicius, F. C. Gozzo, T. Kunath, C. MacPhee and P. E. Barran, Conformational dynamics of α-synuclein: insights from mass spectrometry, Analyst, 2015, 140, 3070–3081 RSC.
- J. P. Williams, L. J. Morrison, J. M. Brown, J. S. Beckman, V. G. Voinov and F. Lermyte, Top-Down Characterization of Denatured Proteins and Native Protein Complexes Using Electron Capture Dissociation Implemented within a Modified Ion Mobility-Mass Spectrometer, Anal. Chem., 2020, 92, 3674–3681 CrossRef CAS PubMed.
- I. A. Kaltashov, C. E. Bobst and R. R. Abzalimov, Mass spectrometry–based methods to study protein architecture and dynamics, Protein Sci., 2013, 22, 530–544 CrossRef CAS PubMed.
- R. Beveridge, A. S. Phillips, L. Denbigh, H. M. Saleem, C. E. MacPhee and P. E. Barran, Relating gas phase to solution conformations: Lessons from disordered proteins, Proteomics, 2015, 15, 2872–2883 CrossRef CAS.
- Y.-H. Ding, S.-B. Fan, S. Li, B.-Y. Feng, N. Gao, K. Ye, S.-M. He and M.-Q. Dong, Increasing the Depth of Mass-Spectrometry-Based Structural Analysis of Protein Complexes through the Use of Multiple Cross-Linkers, Anal. Chem., 2016, 88, 4461–4469 CrossRef CAS.
- B. Xie and J. S. Sharp, Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method, Anal. Chem., 2015, 87, 10719–10723 CrossRef CAS PubMed.
- P. K. Limpikirati, B. Zhao, X. Pan, S. J. Eyles and R. W. Vachet, Covalent labeling/mass spectrometry of monoclonal antibodies with diethylpyrocarbonate: reaction kinetics for ensuring protein structural integrity, J. Am. Soc. Mass Spectrom., 2020, 31, 1223–1232 CrossRef CAS PubMed.
- M. R. Jensen, M. Zweckstetter, J. R. Huang and M. Blackledge, Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy, Chem. Rev., 2014, 114, 6632–6660 CrossRef CAS PubMed.
- D. H. Kim, J. Lee, K. H. Mok, J. H. Lee and K. H. Han, Salient Features of Monomeric Alpha-Synuclein Revealed by NMR Spectroscopy, Biomolecules, 2020, 10, 428 CrossRef CAS.
- E. C. Cook, G. A. Usher and S. A. Showalter, The Use of (13)C Direct-Detect NMR to Characterize Flexible and Disordered Proteins, Methods Enzymol., 2018, 611, 81–100 CAS.
- U. R. Shrestha, P. Juneja, Q. Zhang, V. Gurumoorthy, J. M. Borreguero, V. Urban, X. Cheng, S. V. Pingali, J. C. Smith and H. M. O'Neill, et al., Generation of the configurational ensemble of an intrinsically disordered protein from unbiased molecular dynamics simulation, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20446–20452 CrossRef CAS.
- U. R. Shrestha, J. C. Smith and L. Petridis, Full structural ensembles of intrinsically disordered proteins from unbiased molecular dynamics simulations, Commun. Biol., 2021, 4, 243 CrossRef CAS PubMed.
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernández, Interaction of α-Synuclein with Divalent Metal Ions Reveals Key Differences: A Link between Structure, Binding Specificity and Fibrillation Enhancement, J. Am. Chem. Soc., 2006, 128, 9893–9901 CrossRef CAS PubMed.
- J. R. Allison, P. Varnai, C. M. Dobson and M. Vendruscolo, Determination of the Free Energy Landscape of α-Synuclein Using Spin Label Nuclear Magnetic Resonance Measurements, J. Am. Chem. Soc., 2009, 131, 18314–18326 CrossRef CAS PubMed.
- J. M. C. Teixeira, Z. H. Liu, A. Namini, J. Li, R. M. Vernon, M. Krzeminski, A. A. Shamandy, O. Zhang, M. Haghighatlari and L. Yu, et al., IDPConformerGenerator: A Flexible Software Suite for Sampling the Conformational Space of Disordered Protein States, J. Phys. Chem. A, 2022, 126, 5985–6003 CrossRef CAS.
- M. Y. Lobanov, N. Bogatyreva and O. Galzitskaya, Radius of gyration as an indicator of protein structure compactness, Mol. Biol., 2008, 42, 623–628 CrossRef CAS.
- D. Seeliger and B. L. de Groot, Conformational transitions upon ligand binding: Holo structure prediction from apo conformations, Biophys. J., 2010, 98, 428a CrossRef.
- A. C. Ferreon, Y. Gambin, E. A. Lemke and A. A. Deniz, Interplayof alpha-synuclein binding and conformational switching probed by single-molecule fluorescence, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5645–5650 CrossRef CAS PubMed.
- N. I. Brodie, K. I. Popov, E. V. Petrotchenko, N. V. Dokholyan and C. H. Borchers, Conformational ensemble of native α-synuclein in solution as determined by short-distance crosslinking constraint-guided discrete molecular dynamics simulations, PLoS Comput. Biol., 2019, 15, e1006859 CrossRef PubMed.
- O. Palomino-Hernandez, C. Santambrogio, G. Rossetti, C. O. Fernandez, R. Grandori and P. Carloni, Molecular Dynamics-Assisted Interpretation of Experimentally Determined Intrinsically Disordered Protein Conformational Components: The Case of Human α-Synuclein, J. Phys. Chem. B, 2022, 126, 3632–3639 CrossRef CAS PubMed.
- J. C. May, E. Jurneczko, S. M. Stow, I. Kratochvil, S. Kalkhof and J. A. McLean, Conformational Landscapes of Ubiquitin, Cytochrome c, and Myoglobin: Uniform Field Ion Mobility Measurements in Helium and Nitrogen Drift Gas, Int. J. Mass Spectrom., 2018, 427, 79–90 CrossRef CAS PubMed.
- S. Hong and M. F. Bush, Collision-Induced Unfolding Is Sensitive to the Polarity of Proteins and Protein Complexes, J. Am. Soc. Mass Spectrom., 2019, 30, 2430–2437 CrossRef CAS.
- C. Bleiholder and F. C. Liu, Structure Relaxation Approximation (SRA) for Elucidation of Protein Structures from Ion Mobility Measurements, J. Phys. Chem. B, 2019, 123, 2756–2769 CrossRef CAS PubMed.
- T. C. Cropley, F. C. Liu, T. Pedrete, M. A. Hossain, J. N. Agar and C. Bleiholder, Structure Relaxation Approximation (SRA) for Elucidation of Protein Structures from Ion Mobility Measurements (II). Protein Complexes, J. Phys. Chem. B, 2023, 127, 5553–5565 CrossRef CAS PubMed.
- P. W. A. Devine, H. C. Fisher, A. N. Calabrese, F. Whelan, D. R. Higazi, J. R. Potts, D. C. Lowe, S. E. Radford and A. E. Ashcroft, Investigating the Structural Compaction of Biomolecules Upon Transition to the Gas-Phase Using ESI-TWIMS-MS, J. Am. Soc. Mass Spectrom., 2017, 28, 1855–1862 CrossRef CAS.
- C. J. Hogan Jr., B. T. Ruotolo, C. V. Robinson and J. Fernandez de la Mora, Tandem differential mobility analysis-mass spectrometry reveals partial gas-phase collapse of the GroEL complex, J. Phys. Chem. B, 2011, 115, 3614–3621 CrossRef PubMed.
- A. D. Rolland and J. S. Prell, Computational Insights into Compaction of Gas-Phase Protein and Protein Complex Ions in Native Ion Mobility-Mass Spectrometry, TrAC, Trends Anal. Chem., 2019, 116, 282–291 CrossRef CAS PubMed.
- K. Breuker and F. W. McLafferty, Stepwise evolution of protein native structure with electrospray into the gas phase, 10(-12) to 10(2) s, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18145–18152 CrossRef CAS.
- N. Bisi, L. Feni, K. Peqini, H. Perez-Pena, S. Ongeri, S. Pieraccini and S. Pellegrino, alpha-Synuclein: An All-Inclusive Trip Around its Structure, Influencing Factors and Applied Techniques, Front. Chem., 2021, 9, 666585 CrossRef CAS.
- M. Cheung See Kit and I. K. Webb, Application of Multiple Length Cross-linkers to the Characterization of Gaseous Protein Structure, Anal. Chem., 2022, 94(39), 13301–13310 CrossRef CAS PubMed.
- M. Cheung See Kit, V. V. Carvalho, J. Z. Vilseck and I. K. Webb, Gas-Phase Ion/Ion Chemistry for Structurally Sensitive Probes of Gaseous Protein Ion Structure: Electrostatic and Electrostatic to Covalent Cross-Linking, Int. J. Mass Spectrom., 2021, 463, 116549–116559 CrossRef CAS PubMed.
- R. J. Loncharich and B. R. Brooks, The effects of truncating long–range forces on protein dynamics, Proteins: Struct., Funct., Bioinf., 1989, 6, 32–45 CrossRef CAS.
- K. J. Laszlo and M. F. Bush, Analysis of Native-Like Proteins and Protein Complexes Using Cation to Anion Proton Transfer Reactions (CAPTR), J. Am. Soc. Mass Spectrom., 2015, 26, 2152–2161 CrossRef CAS.
- S. A. McLuckey and J. L. Stephenson Jr., Ion/ion chemistry of high-mass multiply charged ions, Mass Spectrom. Rev., 1998, 17, 369–407 CrossRef CAS PubMed.
- J. T. Kline, C. Mullen, K. R. Durbin, R. N. Oates, R. Huguet, J. E. P. Syka and L. Fornelli, Sequential Ion–Ion Reactions for Enhanced Gas-Phase Sequencing of Large Intact Proteins in a Tribrid Orbitrap Mass Spectrometer, J. Am. Soc. Mass Spectrom., 2021, 32, 2334–2345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting methods, crosslink scheme, CSDs, ATDs, fragmentation maps, representative clusters of ensembles, tuning parameters, ion/ion products, and crosslinking site IDs. See DOI: https://doi.org/10.1039/d3an01680c |
| This journal is © The Royal Society of Chemistry 2024 |