
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTernary solvent based homogeneous liquid–liquid microextraction for the preconcentration of organochlorine pesticides from water and apple juice samples†
Kero Assefa
Ago
 *,
Shimeles Addisu
Kitte
,
Gadisa
Chirfa
and
Abera
Gure
*,
Shimeles Addisu
Kitte
,
Gadisa
Chirfa
and
Abera
Gure
Department of Chemistry, College of Natural Sciences, Jimma University, P. O. Box 378, Jimma, Ethiopia. E-mail: keassefabe16@gmail.com
First published on 21st November 2023
Abstract
In the present study, the optimal experimental conditions were determined by optimizing the effect of extraction solvent types and volume, salt types and concentration, centrifugation speed and time using one variable at a time. Under optimal experimental conditions, calibration curves were constructed separately using water and apple juice samples as representative matrices, and good linearities were achieved over a wide concentration range of 0.2–1600 ng L−1 with a coefficient of determination (r2) ≥ 0.998. The limits of detection (LOD) and limits of quantification (LOQ), determined to be 3 and 10 times the signal-to-noise ratios (S/N), were between 0.07–3.9 and 0.2–12.0 ng L−1 for water samples and 2.6–10.0 and 8.0–30.0 ng L−1 for the apple juice sample respectively. The precisions study showed %RSD values of ≤6% for both matrices, indicating satisfactory precisions. The enrichment factors and recoveries of the proposed method ranged from 41.4–74.5 and 86–109% respectively. The proposed method could be used as a simple and environmentally friendly alternative for the analysis of OCPs from environmental and food matrices. This method potentially offers a more sustainable and effective approach to monitoring OCPs in environmental and food products. Its use in the analysis of apple juice samples is particularly novel and can provide valuable insights into pesticide contamination in fruit juices.
1. Introduction
Organochlorine pesticides (OCPs) and other persistent organic pollutants (POPs) are of increasing public health concern due to their potential bioaccumulation and adverse effects on the environment. Even though the use of OCPs is banned worldwide, several developing countries still use these chemicals. Monitoring their residues in environmental and food matrices is crucial due to their persistence, bioaccumulation, and a range of negative effects on human and other animal health.1 Therefore, there is an increasing call for the development of highly sensitive and selective methods for the determination of OCP residues, which are usually present in trace amounts.Various chromatographic methodologies, including gas chromatography (GC) and high-performance liquid chromatography (HPLC), have been employed for the analysis of pesticide residues in food.2 Among these, HPLC is a type of separation technique used to separate compounds based on their interactions with the stationary phase. It separates compounds based on their differences in polarities, molecular weights, and thermal instabilities or tendency to ionize in solution. In other words, a GC equipped with diverse detectors is extensively utilized for the separation and detection of non-polar and thermally stable compounds like OCPs in environmental and/or food sample matrices. This is due to its appropriateness, sensitivity, and capability to separate compounds based on their volatility and amplified sensitivity to non-polar substances.3,4
Nevertheless, due to the complexity of food and environmental matrices and the trace concentration of OCPs, sample preparation and enrichment methods are required prior to their determination by gas chromatography-mass spectrometry (GC-MS). For the study of OCPs from such complex matrices, well-known sample preparation techniques have long been developed including liquid–liquid extraction (LLE) and solid-phase extraction (SPE).5,6 However, these methods have several inherent disadvantages, such as being time-consuming, requiring the use of hazardous chemical solvents, and being labour-intensive.7 Several microextraction techniques have been developed to alleviate these limitations, including hollow fibre liquid–liquid microextraction,8 dispersive liquid–liquid microextraction,2 and headspace solid-phase microextraction.9
At present, homogeneous liquid–liquid extraction (HLLE) is an alternative extraction method that utilizes phase separation in a homogeneous solution to produce a very small collected phase. In this method, a homogeneous phase could be formed by using pure or a mixture of water-miscible organic solvents.10 It is based on the use of water-miscible organic solvents with low dielectric constant, including acetone, acetonitrile, 2-propanol, ethanol and ethyl acetate, as extractants.11 When these solvents are added to the aqueous sample, a homogeneous solution is produced. Due to the infinite number of interfaces between the aqueous solvent and the extraction solvent, there is a rapid mass transfer of the target analytes into the organic phase. In the HLLE process, phase separation could be achieved by adding water-soluble salts or other phase separation phenomena.10,12,13
To improve the environmental sustainability of analysis, sample preparation should use lower amounts of safe organic solvents, minimize analysis time, reduce waste production, safeguard the operators' health, and maintain the greenness of the environment.14 In contrast to the traditional HLLE method, HLLME enables simpler and more environmentally friendly extraction techniques that use the lowest amount of organic solvents. Using microliters of organic solvents in HLLE to develop HLLME has many advantages, including simplicity, affordability, reduced extraction time and solvent volume, use of safer organic solvents, and lower operator exposure to chemicals than traditional HLLE.15,16
In most cases, acetonitrile is used in the HLLME process as an extraction solvent for preconcentration of various pesticides and other contaminants from different matrices. For example, triazole pesticides in water samples,17 sulfonylurea herbicides from environmental water and banana juice samples,18 and benzimidazole fungicides from high salinity samples19 were analysed using this method. The mentioned analytes are highly polar and can be easily extracted from the matrices by a binary solvent system. However, when a mixture of analytes with different volatilities exist in the sample, the use of mixed organic solvents (i.e., a mixture of cosolvent with a small amount of less polar extraction solvent) could improve the extraction efficiency of the method. This three solvent (ternary) system could facilitate phase separation more easily than traditional homogeneous binary phase liquid–liquid microextraction (HLLME).20 Solvents such as n-hexane and chloroform have been used as extractants in HLLME with cosolvents acetone or methanol.21,22 However, the stated methods require large amounts of cosolvents and chlorinated organic solvents as extractants, which requires the search for other alternative HLLME methods that use fewer cosolvents and less toxic extractants than chlorinated toxic organic solvents such as chloroform. The main disadvantage of traditional HLLE is that the extraction solvent is generally limited to solvents with higher density than water to be sedimented by centrifugation. These solvents are typically chlorinated solvents such as chlorobenzene, chloroform and carbon tetrachloride, all of which are potentially toxic to humans and the environment. Furthermore, the use of these solvents as extractants limits the broader applicability of HLLE. This is because the choice of low-density solvent is more limited compared to high-density solvents.23 To the best of researchers' knowledge, ternary solvent based HLLME (TS-HLLME) using low-density solvents as extraction and cosolvents in the preconcentration of OCPs from targeted sample matrices has not been reported.
Therefore, in this study, we developed ternary solvents (acetonitrile, ethyl acetate & sample) based HLLME (TS-HLLME) using a non-chlorinated extraction solvents for the extraction and preconcentration of 13 OCP residues from various water and apple juice samples. The use of microliter volume of solvents can reduce the amount of solvent required for extraction, resulting in less waste production and reduced environmental impact, overcoming the limitations caused by the use of high-density solvents. Furthermore, using a mixture of low-density solvents as cosolvents and extractants in TS-HLLME can improve the sensitivity, compatibility and selectivity of the method, resulting in more accurate measurement of OCPs in water and apple juice samples. This mixture can effectively extract both moderately polar and highly non-polar OCP compounds. Overall, TS-HLLME potentially offers a more sustainable and effective approach to monitoring OCPs in environmental and food matrices.
Various experimental variables that affect the extraction efficiency of the method, including the type and volume of the extraction solvent, the type and amount of salt, centrifugation speed and time, were examined and the optimal conditions were established. The analytical performances of the proposed method were validated according to the International Conference for Harmonization (ICH) guidelines.24 The performance of the proposed TS-HLLME method was compared with other reported methods for analysing OCPs from water and juice matrices. Moreover, its use in the analysis of apple juice samples is particularly novel and can provide valuable insights into pesticide contamination in fruit juices.
2. Experimental
2.1. Chemicals and reagents
All reagents and solvents used were of analytical grade. Organic solvents such as acetone were obtained from Sisco Research Laboratories Pvt. Ltd (Mumbai, India), acetonitrile (ACN) was purchased from Unichem® chemical reagent (Merck, KGaA, Darmstadt, Germany), and hexane, diethyl ether (DEE), and ethyl acetate (EA) were purchased from Loba Chemie Pvt. Ltd (Mumbai, India). Sodium chloride (NaCl), and magnesium sulfate (MgSO4, anhydrous) were supplied by Fisher Scientific (UK) and Finkem Research Chemicals Ltd (Toronto, Canada), respectively. Deionized water processed using a Millipore SAS direct-Q®-5UV water purification system (Molsheim, France) was used. Whatman Grade 1 filter paper and 3 μm nylon filters obtained from Whatman International Ltd (Maidstone, England) were used for the filtration of the water samples. Analytical standards of OCPs including benzene hexachlorides (BHC) including α-BHC (99.5%), β-BHC (99.5%), and δ-BHC (99.5%); dichlorodiphenyltrichloroethane, DDT (98.9%); dichlorodiphenyldichloroethylene, DDE (99.9%); chlorinated cyclodienes including endosulphan sulfates (ESS) (98.8%), endrin (99.3%), ɣ-chlordane (98.8%), heptachlor epoxide(HCE) (98.8%), aldrin (≥98.8%), dieldrin (97.9%), and methoxychlor (MC) (97.7%); and dibutylchlorindate (DBC) (99.5%) were obtained from Sigma Aldrich (St. Louis, MO, USA). The chemical structures, physical and chemical properties including boiling and melting points, solubility and other related properties of these analytes are presented in the cited ref. 25. Individual standard stock solutions (1000 mg L−1) of α-BHC, β-BHC, δ-BHC, and DBC; 400 mg L−1 DDT, DDE, dieldrin, ESS, HCE, ɣ-chlordane, MC, and endrin as well as 800 mg L−1 aldrin were prepared in hexane. A mixed standard solution (20 mg L−1) was prepared by diluting the stock solutions in hexane. The prepared solutions were stored below 4 °C when not used for analysis. Working standard solutions were daily prepared by diluting the mixed standard solution in n-hexane.32.2. Instrumentation
A GC (Agilent 8890) coupled with a single quadruple MS, Agilent 5977B, and an autosampler, Agilent G4513A (Agilent Technologies, USA), was used for the analysis of OCPs. An HP-5MS capillary column (30 m, 0.25 mm i. d, 0.25 μm film thickness) coated with 5% diphenyl-95% dimethylsiloxane as a stationary phase (Agilent Technologies) was used for chromatographic separations. The 15 mL falcon centrifuge tubes and medical syringes with B. Braun Sterican needle 21 G × 4¾ (0.80 × 122 mm BL/LB) obtained from B. Braun Melsungen AG (Melsungen, Germany) were used during sample preparation. High-purity (99.999%) helium was used as carrier gas at a flow rate of 1 mL min−1. The sample (1 μL) was injected in the splitless mode. The GC temperature program was: initial temperature 100 °C without holding time; and then elevated to 200 °C at a rate of 15 °C min−1, held for 5 min; ramp at 4 °C min−1 to 250 °C, held for 4 min; and finally increased to 270 °C at a rate of 10 °C min−1, held for 10 min. The injector port temperature was set at 280 °C.Mass spectrometry (MS) was operated in electron ionization (EI) mode with ionization energy of 70 eV, GC-MS transfer line temperature of 250 °C; ion source temperature of 230 °C; and quadruple temperature of 150 °C; scanning from m/z 45 to 500 at 150 s per scan; and solvent delay time of 3 min. Analysis was performed in the selected ion monitoring (SIM) mode using one quantitative and two qualifier ions as shown in ESI Table S1.† Abundances of the quantitative and qualifier ions were determined by injecting the pesticide standards in full-scan mode with the mass/charge ratio ranging from m/z 45 to 500. Quantification was done using the peak area of the quantitative ion of each analyte.
2.3. Extraction procedure
5 mL of water or apple juice sample was transferred into a 15 mL falcon tube and spiked with 5 ng mL−1 OCP mixed standard. Then, from the pre-mixed acetonitrile (ACN) and ethyl acetate (EA) solvents in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 600 μL was added. The solution was manually shaken for 30 s. Then, 1.5 g NaCl was added and manually shaken until the salt was completely dissolved. After the dissolution of the salt, the cloudy solution formed was centrifuged at 4000 rpm for 2 min and this resulted in a distinct separation of the organic and aqueous phases with the organic phase floating over the aqueous layer. Then, the bottom phase was carefully discarded and about 100 μL of the organic phase was carefully transferred into a 200 μL insert vial, which was inserted in a 1.5 mL amber autosampler vial for subsequent GC-MS analysis.
:1, 600 μL was added. The solution was manually shaken for 30 s. Then, 1.5 g NaCl was added and manually shaken until the salt was completely dissolved. After the dissolution of the salt, the cloudy solution formed was centrifuged at 4000 rpm for 2 min and this resulted in a distinct separation of the organic and aqueous phases with the organic phase floating over the aqueous layer. Then, the bottom phase was carefully discarded and about 100 μL of the organic phase was carefully transferred into a 200 μL insert vial, which was inserted in a 1.5 mL amber autosampler vial for subsequent GC-MS analysis.
3. Results and discussion
3.1. Optimization of extraction parameters
To obtain optimal conditions for HLLME to extract OCPs from the sample matrices, the effects of various parameters such as type, total volume and volume ratio of the solvents, type and amount of salts, and centrifugation rate and time on the extraction efficiency were evaluated. The effects of the parameters were examined separately rather than simultaneously in this study. This was done to better understand how each factor affects the extraction efficiency and selectivity of the method. By varying only one factor at a time, it is possible to isolate its effect and determine the optimal conditions for that particular factor. Once the optimal conditions for each factor have been identified, these can be combined to develop an optimized method that takes into account the effects of both factors simultaneously. Additionally, examining the effects of each factor separately can help identify possible interactions or trade-offs between them that may not be apparent when varying simultaneously.Parameters were optimized using a 5 mL sample with mixed standards. In this study, peak area-based numbers are used in the optimization of analytical methods to evaluate the linearity and sensitivity of the proposed method. Analysis of the linearity and slope of the line could provide valuable information about the sensitivity and reliability of the method. It is also possible to sketch numbers based on the recovery percentage that could provide a visual representation of the performance of the method at different analyte concentrations. This can help us identify any concentration-dependent biases or limitations of the method and enable a more comprehensive assessment of its accuracy and reliability.
In this study, 650 μL of an extraction solvent composed of pure (acetonitrile or acetone) and acetone mixed with diethyl ether or ethyl acetate and acetonitrile mixed with diethyl ether or ethyl acetate were initially investigated for the extraction of target analytes. For the mixed solvents, a ratio of miscible to immiscible organic solvents of 2:1 was used when selecting the extraction solvent. The results showed that the highest extraction efficiencies for all target OCPs were achieved when a mixture of acetonitrile (ACN) and ethyl acetate (EA) was used as the extraction solvent (Fig. 1). However, when using acetone, the phase separation produced was not sufficient to be sampled for subsequent analysis. The reason for this could be the different solubility of the OCPs in acetone and acetonitrile. Acetone may not be effective in extracting these analytes from the sample matrices, resulting in a lower concentration of the analytes in the extract. This can result in phase separation that is not sufficient for later analysis. On the other hand, pure acetonitrile or its mixture with ethyl acetate could have better solubility for the OCPs, resulting in a higher concentration of the analytes in the extract and more effective phase separation than acetone or its mixtures with other solvents. Furthermore, the polarity of the solvents could also play a role in their effectiveness in extracting OCPs. Therefore, a mixture of ACN and EA was selected for further studies.
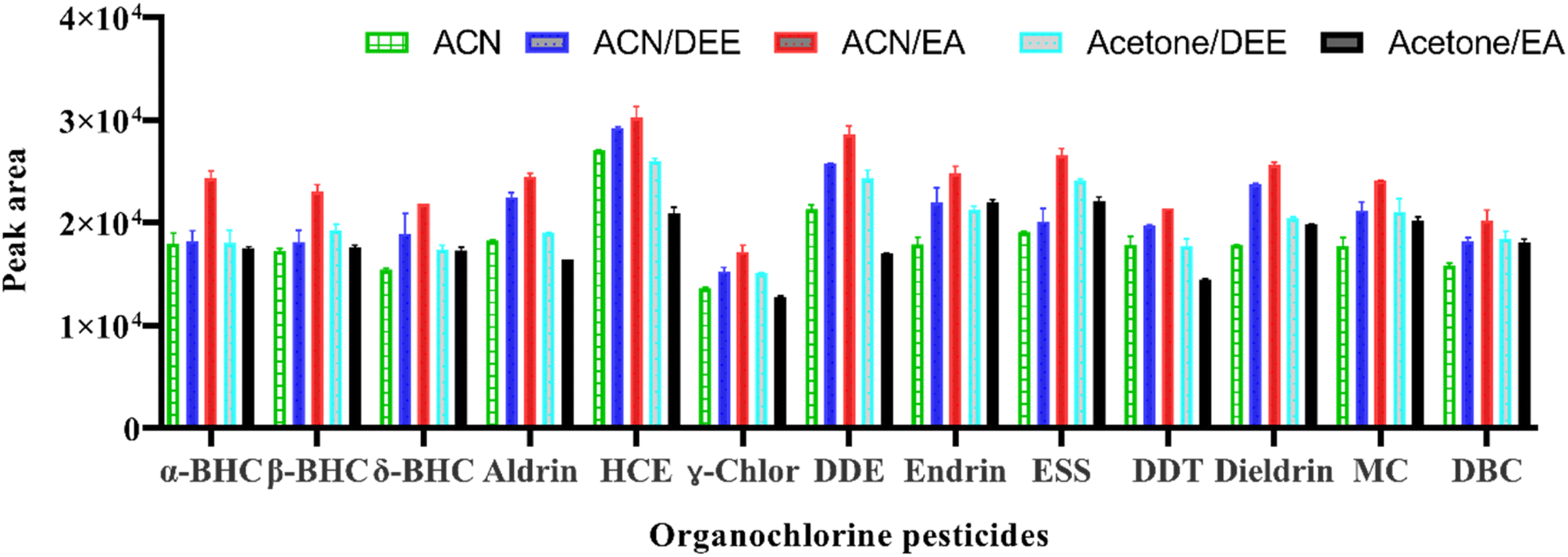 | ||
| Fig. 1 Selection of extraction solvent. Extraction conditions: sample volume 5 mL, mixed standard 5 ng mL−1, extraction solvent volume ratio 2:1, the mass of NaCl: 1.5 g, centrifugation at 4000 rpm for 2 min. | ||
:1, 3:1, 4:1, 5:1 and 6:1. The result shown in Fig. 2 indicated that a larger peak area was obtained at a ratio of 3:1 for ACN: EA. This could be due to the specific properties and interaction of both solvents with the analytes present in the sample matrices. This ratio provided the best extraction efficiency or separation ability for the target analytes in the matrices of the analyzed samples. That means, it enables more accurate and sensitive analysis of the studied contaminants.
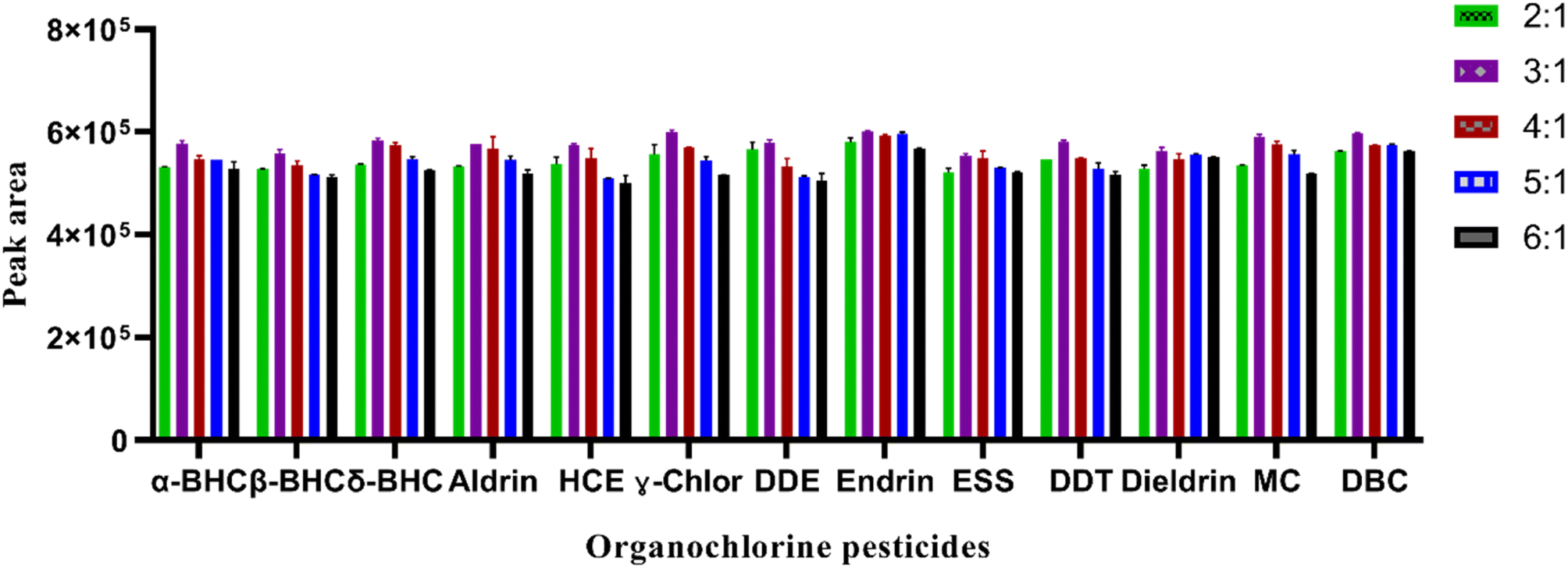 | ||
| Fig. 2 Effect of volume ratio of extraction solvents on the extraction efficiency of the proposed method. Extraction conditions: sample volume 5 mL, mixed standard 5 ng mL−1, extraction solvent (volume) ACN:EA (600 μL), the mass of NaCl: 1.5 g, centrifugation at 4000 rpm for 2 min. | ||
In this study, NaCl, anhydrous MgSO4, and a mixture of NaCl and MgSO4 in a ratio of 1:4 (w/w) were evaluated. First, 1.5 g of each salt was added to the sample and shaken manually until the salt was completely dissolved. The resulting cloudy solution was then initially centrifuged at 4000 rpm for 4 min before optimization. As can be seen from Fig. 3, both pure NaCl and MgSO4 induced comparable phase separation to their mixture. During the salting-out process in TS-HLLME, the anion of the salt is more important than the cation to achieve phase separation,28 and according to the Hofmeister series: Na+ > Mg2+ while SO42− > Cl− salting-out effects.27,29 NaCl was selected for further studies due to its high solubility in aqueous samples, low cost, availability in the laboratory, and better efficiency of the method.
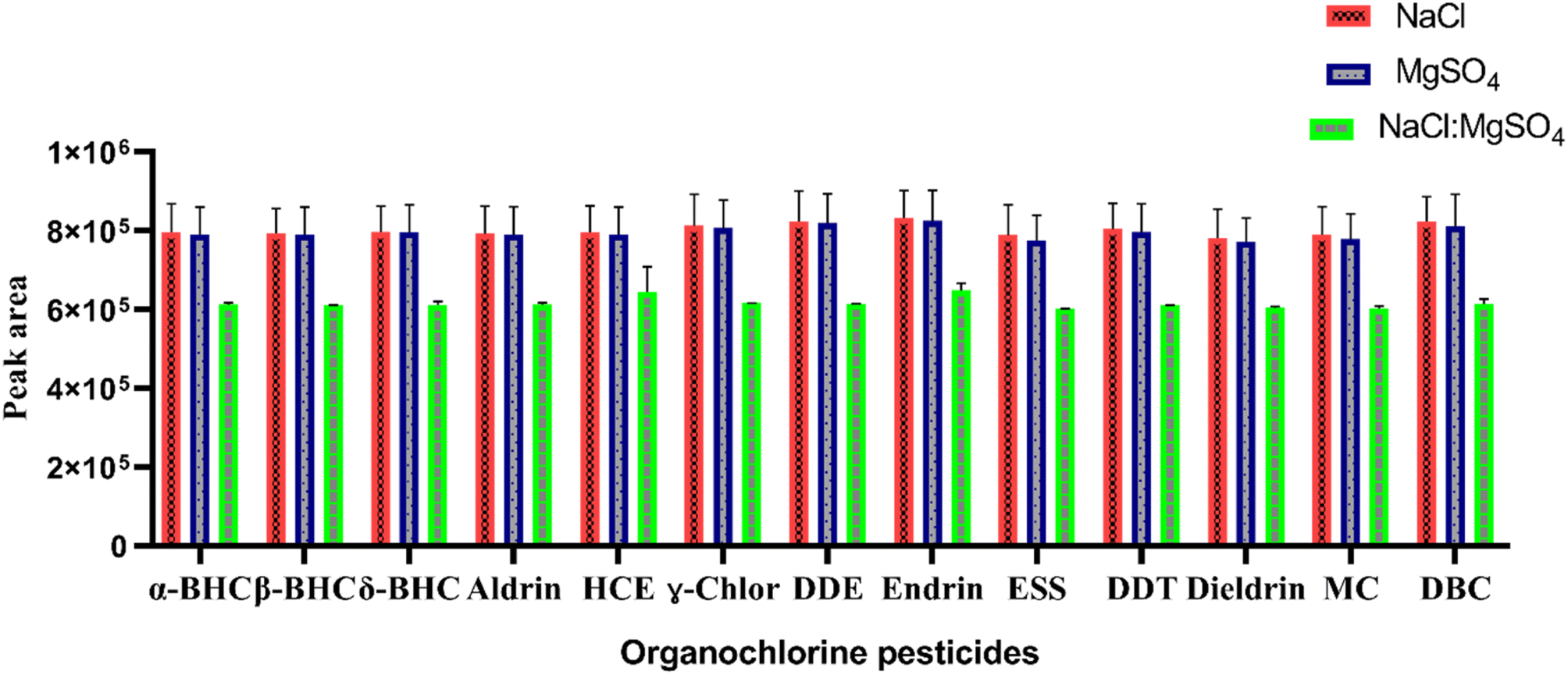 | ||
| Fig. 3 Effect of type of salt on the extraction efficiency of the proposed method. Extraction conditions: sample volume 5 mL, mixed standard 5 ng mL−1, extraction solvent (volume) ACN:EA (600 μL), extraction solvent volume ratio 3:1, the mass of salts 1.5 g, centrifugation at 4000 rpm for 2 min. | ||
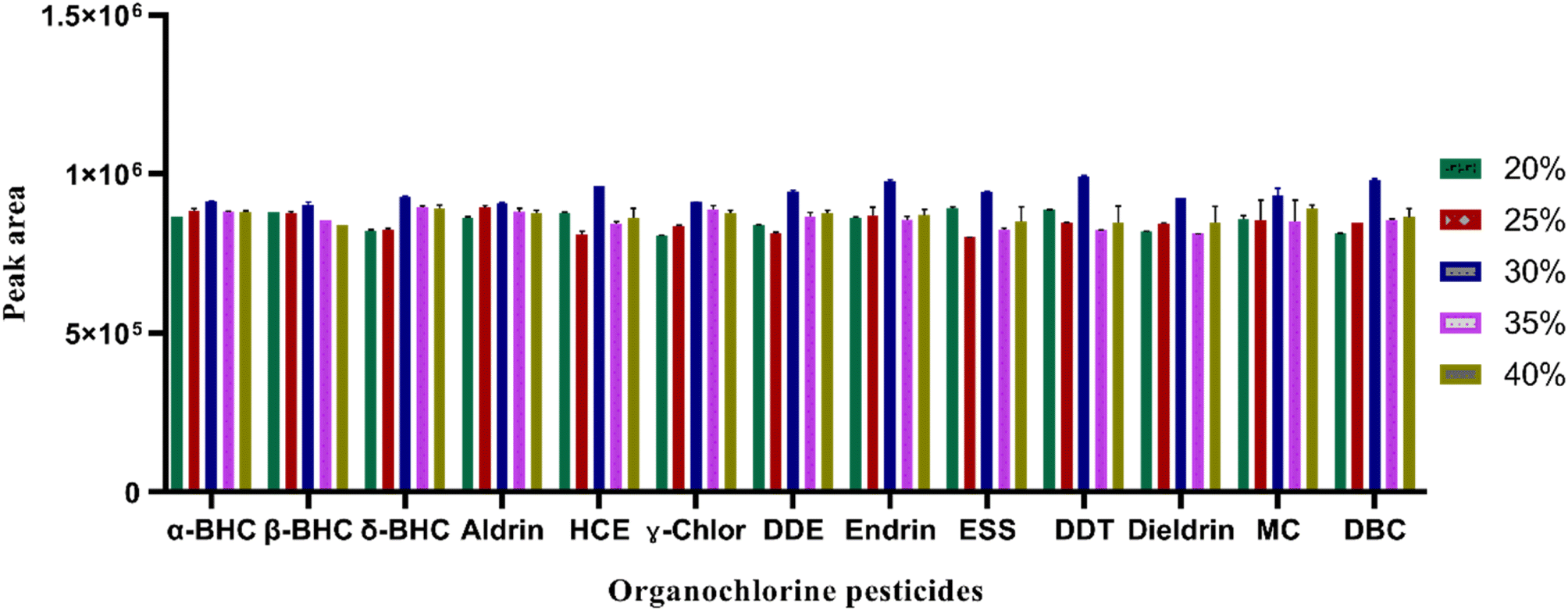 | ||
| Fig. 4 Effect of salt concentration on the extraction of OCPs: experimental conditions: sample volume: 5 mL, mixed standard 5 ng mL−1, extraction solvents and volume (ACN:EA; 600 μL in a 3:1 ratio), centrifugation at 4000 rpm for 2 min. | ||
000 rpm was selected for the subsequent studies.
3.2. Analytical performance of the proposed method
| Analytes | Water sample | Apple juice sample | This study result (ng L−1) | EU, MRL | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LRa | R 2 | LODa | LOQa | LRa | R 2 | LODa | LOQa | Apple mg kg−1 | ||
| a Unit of LR, LOD and LOQ; ng L−1, EU MRL source: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/MRLs). | ||||||||||
| δ-BHC | 0.2–1600 | 0.9999 | 0.07 | 0.2 | 9.0–1600 | 0.9998 | 3.0 | 9.0 | <LOQ | 0.01 |
| β-BHC | 12.0–1600 | 0.9999 | 3.9 | 12.0 | 30.0–1600 | 0.9990 | 10.0 | 30.0 | <LOQ | 0.01 |
| δ-BHC | 5.3–1600 | 0.9999 | 1.6 | 5.3 | 9.3–1600 | 0.9996 | 2.8 | 9.3 | <LOQ | 0.01 |
| Aldrin | 11.0–1600 | 0.9989 | 3.6 | 11.0 | 24.0–1600 | 0.9986 | 8.0 | 24.0 | <LOQ | 0.01 |
| HCE | 6.0–1600 | 0.9999 | 1.9 | 6.0 | 10.0–1600 | 0.9994 | 3.2 | 10.0 | <LOQ | 0.01 |
| γ-Chlor | 2.1–1600 | 0.9982 | 0.7 | 2.1 | 29.5–1600 | 0.9980 | 9.2 | 29.5 | <LOQ | 0.01 |
| DDE | 3–1600 | 0.9996 | 1.0 | 3.0 | 13.0–1600 | 0.9995 | 4.2 | 13.0 | <LOQ | 0.05 |
| Endrin | 5.3–1600 | 0.9997 | 1.6 | 5.3 | 11.0–1600 | 0.9997 | 3.6 | 11.0 | <LOQ | 0.01 |
| DDT | 3.0–1600 | 0.9996 | 1.0 | 3.0 | 13.0–1600 | 0.9994 | 4.2 | 13.0 | <LOQ | 0.05 |
| ESS | 2.0–1600 | 0.9995 | 2.0 | 6.0 | 14.0–1600 | 0.9992 | 4.6 | 14.0 | <LOQ | 0.05 |
| Dieldrin | 3.0–1600 | 0.9999 | 1.0 | 3.0 | 8.0–1600 | 0.9990 | 2.6 | 8.0 | <LOQ | 0.01 |
| MC | 1.8–1600 | 0.9994 | 0.6 | 1.8 | 16.0–1600 | 0.9991 | 5.2 | 16.0 | <LOQ | 0.01 |
| DBC | 2.1–1600 | 0.9984 | 0.7 | 2.1 | 16.0–1600 | 0.9994 | 5.2 | 16.0 | <LOQ | 0.01 |
The intraday precision of the proposed method was examined by extracting each added concentration level in triplicate and then injecting each extract in duplicate on the same day under the same experimental conditions. The repeatability results, expressed as relative standard deviation (RSD), ranged from 0.21 to 4.41 for water, and 0.46 to 4.58 for the apple juice samples as shown in Table 2.
| Analytes | Water sample | Apple juice sample | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-day (n = 6) | Inter-day (n = 12) | Intra-day (n = 6) | Inter-day (n = 12) | |||||||||
| 50a | 200a | 800a | 50a | 200a | 800a | 50a | 200a | 800a | 50a | 200a | 800a | |
| a Unit of each concentration level (ng L−1). | ||||||||||||
| δ-BHC | 4.41 | 1.18 | 0.39 | 5.21 | 2.07 | 0.68 | 1.54 | 1.40 | 0.59 | 4.09 | 1.71 | 0.99 |
| β-BHC | 3.22 | 1.64 | 1.84 | 3.50 | 1.29 | 1.05 | 0.50 | 1.64 | 0.68 | 2.52 | 1.16 | 0.88 |
| δ-BHC | 2.77 | 2.24 | 0.21 | 2.35 | 3.76 | 2.88 | 2.8 | 2.74 | 1.35 | 2.36 | 3.96 | 3.16 |
| Aldrin | 3.93 | 2.05 | 0.75 | 4.72 | 1.69 | 2.46 | 1.90 | 1.83 | 1.94 | 5.26 | 1.45 | 2.18 |
| HCE | 2.87 | 0.66 | 1.04 | 2.99 | 1.54 | 1.22 | 2.64 | 1.33 | 0.94 | 2.42 | 3.25 | 1.23 |
| γ-Chlor | 3.37 | 0.84 | 2.48 | 4.92 | 2.64 | 5.66 | 3.17 | 1.44 | 3.38 | 4.46 | 2.78 | 1.06 |
| DDE | 2.99 | 0.60 | 1.00 | 2.33 | 2.21 | 4.78 | 1.82 | 1.86 | 2.96 | 2.02 | 2.50 | 3.34 |
| Endrin | 2.39 | 1.32 | 0.95 | 3.19 | 1.19 | 1.69 | 1.83 | 0.65 | 1.44 | 2.60 | 1.01 | 5.95 |
| DDT | 3.04 | 1.44 | 1.76 | 2.10 | 2.69 | 3.90 | 1.17 | 1.05 | 2.98 | 4.58 | 4.20 | 2.78 |
| ESS | 3.02 | 1.24 | 0.37 | 5.91 | 3.09 | 4.92 | 1.24 | 1.77 | 1.96 | 3.82 | 2.96 | 3.28 |
| Dieldrin | 1.94 | 0.46 | 0.43 | 4.29 | 2.31 | 2.89 | 1.29 | 0.69 | 0.46 | 4.15 | 1.01 | 5.79 |
| MC | 2.72 | 1.13 | 1.29 | 5.78 | 2.29 | 1.03 | 1.20 | 0.74 | 4.38 | 4.14 | 0.97 | 5.88 |
| DBC | 2.24 | 0.73 | 1.44 | 4.8 | 2.37 | 2.75 | 4.58 | 0.47 | 1.37 | 3.42 | 1.41 | 1.11 |
The interday precision of the method was examined by extracting one sample per day for each concentration level for three consecutive days. Each extract was then injected in duplicate. The interday precision results, expressed as relative standard deviation (RSD), ranged from 0.68 to 5.78 and 0.88 to 5.88 for water and apple juice samples respectively (Table 2).
3.3. Application of the proposed method to real water and apple juice samples
In this study, two types of samples were analyzed: water and apple juice samples. Water samples, including groundwater and spring water, were collected from various locations around Jimma city in Ethiopia, while tap water was collected from the Analytical Chemistry Laboratory of Jimma University after flowing freely for about 10 minutes and the bottled water (Aqua Jimma) also sourced from the local market (wholesaler) in Jimma, Ethiopia. All water samples except tap water were then transported to the Analytical Chemistry Laboratory of the Department of Chemistry, Jimma University, where the first two water samples were filtered through 0.45 μm Whatman filter paper (quality 1 and size 8.5 cm) to remove various suspended solids before use, while others were carried out without filtration.An apple juice sample was purchased from a supermarket in Jimma town, Jimma, Ethiopia. In this study, processed apple juice was preferred over apple fruit samples. This is because processed apple juice involves various steps such as washing, peeling and pressing (juicing), which can remove or reduce the concentration of OCPs present on the surface or in the fruit. Therefore, analysis of processed apple juice provides a more accurate representation of the OCPs that consumers may be exposed to when consuming apple juice. This analysis can help ensure that the processed juice is within legal limits for OCP residues and is safe for consumption. Additionally, OCPs are fat-soluble compounds that can be concentrated in juice rather than fruit through processing, making them a better sample for analysis. Finally, it can also help evaluate the effectiveness of pesticide use and monitor apple industry compliance with pesticide use regulations. The choice of apple juice in this experiment was due to its relevance as it is a commonly consumed beverage and the presence of OCPs can be a potential concern due to their potentially harmful effects on human health. Due to its similarity to other fruit juices, apple juice is also often used as a representative sample for the analysis of pesticide residues in fruit juices. Since the processing and cultivation practices of apple juice are similar to those of other fruit juices, the results of apple juice analysis can be examined to make general conclusions about pesticide contamination in various fruit juices. By analyzing apple juice samples, it is possible to determine the level of OCP contamination in a drink that many people, including children, regularly consume.
To verify the applicability of the proposed method to juice samples, an apple juice sample was prepared based on the previously described method with some modifications.33 For instance, 5 mL of juice sample was diluted 10 times with deionized water purified using the Direct-Q 5UV water purification system to make the sample solution dilute and reduce matrix effects. Then, 5 mL of the diluted solution was added to a 15 mL conical bottom Falcon centrifuge tube. Afterwards, an appropriate concentration of OCPs was added to the solution, then sonicated for a few minutes and left for 30 minutes to equilibrate. The procedure used for water samples was then repeated in the same way. All samples were stored refrigerated at 4 °C until use.
The effectiveness of the proposed method was evaluated by determining the relative recovery (%RR) of OCPs in the water and extraction recovery (% RE)34 for apple juice samples as shown in equations eqn (1) and (2) respectively. None of the target OCPs were detected in the collected water and juice samples.
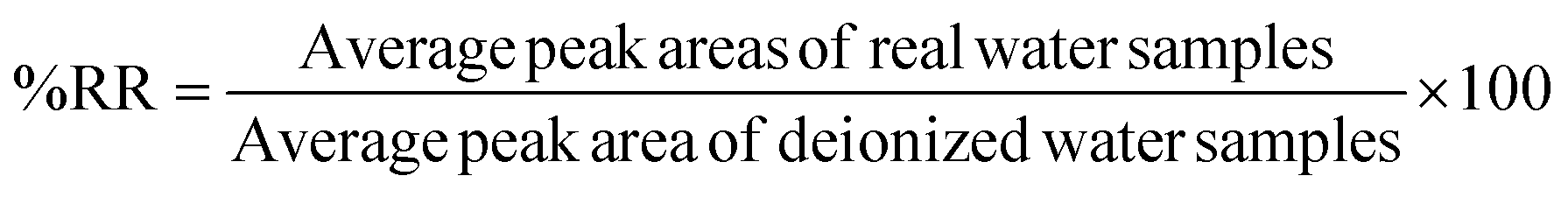 | (1) |
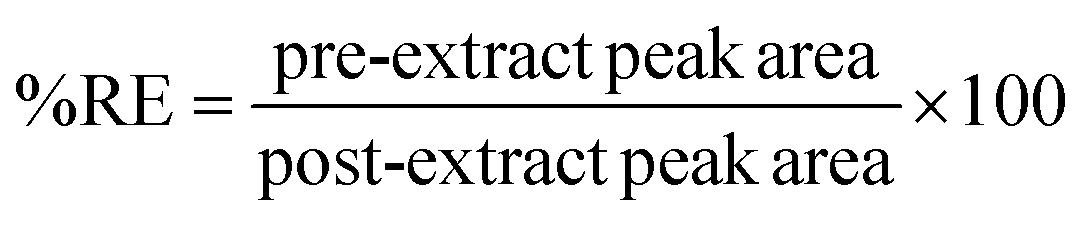 | (2) |
The percent recovery and corresponding RSD of all samples are listed in Table 3. In all samples, the percent recoveries were in the range of 86.4109.9% with RSD < 6%, showing that the proposed method has satisfactory recoveries for the analysis of OCP residues from the samples studied and other related matrices. In many references, the enrichment factor (EF) has been defined as the ratio of the final concentration of the analyte in the acceptor phase (collection phase) to the initial concentration of the analyte in the sample solution.35,36 To determine EF, a 5 mL juice sample spiked with the target analytes at an amount of 2.4 ng L−1 was extracted under the optimized conditions as described in the methods section. The extraction was performed in triplicate and the peak area of the extract was recorded and the average peak area is used for calculation. The peak area after extraction was measured at the same concentration as extraction and measured in triplicate. Finally, the average peak area was used for the EF calculation, as shown in eqn (3).
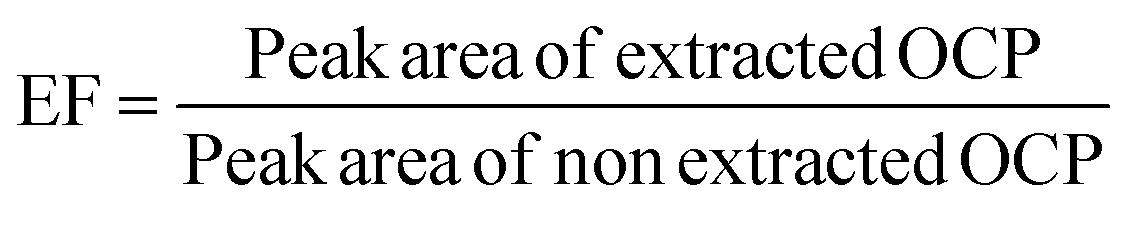 | (3) |
| Pesticides | Spiked levela | Tap water | Bottled water | Ground water | Spring water | Apple juice | EF |
|---|---|---|---|---|---|---|---|
| a Unit of spiked concentration levels (ng L−1). | |||||||
| δ-BHC | 50 | 96.8 ± 0.8 | 101.6 ± 0.3 | 103.4 ± 2.4 | 96.7 ± 1.8 | 97.1 ± 2.1 | 44.2 |
| 200 | 99.7 ± 0.5 | 103.1 ± 2.3 | 109.1 ± 2.2 | 101.2 ± 2.5 | 93.8 ± 4.0 | ||
| 800 | 98.1 ± 0.5 | 108.9 ± 0.6 | 111.2 ± 1.4 | 102.6 ± 1.3 | 87.7 ± 9.2 | ||
| β-BHC | 50 | 98.9 ± 0.9 | 102.7 ± 0.8 | 98.4 ± 2.9 | 97.1 ± 1.2 | 99.2 ± 0.5 | 41.4 |
| 200 | 99.8 ± 3.5 | 105.6 ± 1.7 | 100.1 ± 1.2 | 103.0 ± 0.6 | 94.9 ± 3.6 | ||
| 800 | 97.8 ± 1.5 | 109.7 ± 1.6 | 101.0 ± 0.4 | 101.1 ± 1.4 | 99.8 ± 0.09 | ||
| δ-BHC | 50 | 94.2 ± 4.5 | 99.8 ± 0.6 | 96.4 ± 0.9 | 93.2 ± 0.7 | 96.6 ± 2.4 | 42.8 |
| 200 | 96.3 ± 0.9 | 100.6 ± 0.3 | 96.7 ± 0.1 | 98.2 ± 1.7 | 99.9 ± 0.05 | ||
| 800 | 88.4 ± 0.4 | 100.2 ± 0.6 | 95.6 ± 0.5 | 105.4 ± 0.7 | 96.8 ± 2.3 | ||
| Aldrin | 50 | 95.1 ± 1.0 | 106.5 ± 0.2 | 98.9 ± 0.3 | 99.8 ± 0.6 | 89.9 ± 7.5 | 42.4 |
| 200 | 100.6 ± 1.2 | 108.0 ± 0.2 | 102.1 ± 1.6 | 91.4 ± 1.4 | 100.0 ± 0.1 | ||
| 800 | 88.1 ± 0.1 | 109.8 ± 2.1 | 104.2 ± 0.3 | 102.1 ± 2.0 | 94.2 ± 4.1 | ||
| HCE | 50 | 101.7 ± 4.2 | 95.6 ± 0.4 | 101.0 ± 0.4 | 98.4 ± 1.2 | 87.0 ± 9.8 | 46.7 |
| 200 | 102.9 ± 3.9 | 96.2 ± 1.4 | 99.2 ± 0.4 | 103.3 ± 2.2 | 93.7 ± 4.5 | ||
| 800 | 105.0 ± 1.9 | 101.3 ± 2.6 | 90.4 ± 2.0 | 104.1 ± 0.2 | 93.8 ± 4.4 | ||
| ɣ-chlordane | 50 | 101.2 ± 0.7 | 101.1 ± 0.1 | 99.4 ± 0.9 | 99.0 ± 4.1 | 96.4 ± 2.5 | 74.5 |
| 200 | 104.1 ± 0.8 | 95.4 ± 2.9 | 99.1 ± 1.4 | 103.4 ± 1.4 | 95.9 ± 2.9 | ||
| 800 | 105.0 ± 0.4 | 109.4 ± 0.9 | 108.3 ± 1.3 | 105 ± 0.9 | 98.4 ± 1.09 | ||
| DDE | 50 | 96.4 ± 2.8 | 106.4 ± 0.1 | 103.1 ± 0.3 | 98.5 ± 0.6 | 87.9 ± 9.03 | 51.4 |
| 200 | 105.6 ± 0.5 | 102.1 ± 1.8 | 103.3 ± 0.8 | 105.1 ± 2.5 | 98.7 ± 0.9 | ||
| 800 | 99.0 ± 1.6 | 102.4 ± 1.2 | 95.5 ± 0.8 | 103.0 ± 0.5 | 97.5 ± 1.8 | ||
| Endrin | 50 | 104.0 ± 1.4 | 99.1 ± 0.5 | 98.0 ± 0.8 | 103.1 ± 0.3 | 93.6 ± 4.6 | 63.9 |
| 200 | 94.2 ± 0.4 | 106.4 ± 3.8 | 104.5 ± 0.2 | 96.4 ± 2.9 | 95.0 ± 3.6 | ||
| 800 | 86.1 ± 6.68 | 94.2 ± 0.1 | 96.0 ± 2.2 | 97.1 ± 2.8 | 96.8 ± 2.3 | ||
| DDT | 50 | 97.0 ± 0.53 | 101.1 ± 0.1 | 91.4 ± 1.5 | 96.7 ± 0.3 | 91.7 ± 6.0 | 58.9 |
| 200 | 96.4 ± 1.60 | 100.0 ± 0.5 | 107.8 ± 2.2 | 107.8 ± 1.6 | 99.9 ± 0.5 | ||
| 800 | 91.4 ± 5.84 | 100.1 ± 0.1 | 102.0 ± 1.1 | 95.6 ± 0.2 | 96.5 ± 2.4 | ||
| ESS | 50 | 98.0 ± 0.64 | 101.4 ± 0.2 | 101.1 ± 1.5 | 92.2 ± 0.5 | 92.8 ± 1.2 | 49.6 |
| 200 | 91.2 ± 3.07 | 91.4 ± 0.9 | 102.4 ± 1.9 | 103.5 ± 3.7 | 97.3 ± 1.9 | ||
| 800 | 103.7 ± 2.1 | 99.8 ± 0.7 | 102.3 ± 1.2 | 104.4 ± 1.3 | 92.3 ± 5.6 | ||
| Dieldrin | 50 | 98.9 ± 4.6 | 107.8 ± 2.8 | 101.2 ± 0.3 | 97.4 ± 0.2 | 97.7 ± 1.6 | 65.4 |
| 200 | 97.5 ± 4.7 | 94.5 ± 4.8 | 103.1 ± 2.2 | 103.2 ± 1.2 | 90.9 ± 6.7 | ||
| 800 | 104.1 ± 2.0 | 97.8 ± 0.2 | 109.1 ± 0.6 | 106.7 ± 0.7 | 96.7 ± 2.3 | ||
| MC | 50 | 96.4 ± 0.4 | 98.9 ± 2.6 | 101.1 ± 0.8 | 102.1 ± 0.4 | 95.3 ± 3.3 | 45.2 |
| 200 | 97.4 ± 1.6 | 99.1 ± 0.6 | 102.0 ± 1.4 | 105.6 ± 1.5 | 95.9 ± 2.9 | ||
| 800 | 96.7 ± 0.9 | 104.5 ± 4.6 | 107.1 ± 1.0 | 104.5 ± 1.7 | 98.3 ± 1.1 | ||
| DBC | 50 | 99.4 ± 1.0 | 98.3 ± 1.0 | 97.4 ± 2.7 | 98.9 ± 0.2 | 91.8 ± 6.0 | 50.7 |
| 200 | 96.5 ± 4.0 | 98.9 ± 0.1 | 99.1 ± 0.09 | 99.8 ± 0.5 | 95.1 ± 3.5 | ||
| 800 | 99.8 ± 3.8 | 99.1 ± 0.2 | 97.4 ± 1.1 | 102.1 ± 3.4 | 98.4 ± 1.1 | ||
Accordingly, the EF of the method ranged from 41 to 75 (Table 3).
3.4. Selectivity of the proposed method
The selectivity of the method describes the ability of the GC process to correctly separate analytes from each other. The selectivity of the proposed method was evaluated by comparing the chromatograms of the unspiked water and apple juice samples with the corresponding spiked samples. The apple juice sample was prepared based on the previously described method with some modifications.33 Accordingly, 10-fold diluted apple juice with ultrapure water was used for sample extraction. Fig. 5a–d show representative chromatograms of the unspiked and spiked apple juice and river water samples with 50 ng L−1 of the target pesticides. As shown from the chromatograms, no interfering peaks were observed in the retention time windows of the target analytes, indicating that the proposed method has good selectivity for trace analysis of the selected pesticides in water, juice and other related matrices. Through the whole procedure, the matrix effect was minimized, because water and apple juice samples were properly prepared to remove any particulate matter or other interfering substances. This was done by filtration and centrifugation. The extraction conditions, such as the volume ratio of the solvents, the pH, and the salt concentration, were optimized to minimize the matrix effect. For instance, the addition of salts helps to reduce the solubility of some matrix components and improve the extraction efficiency. Therefore, proper sample preparation, optimization of extraction conditions, and use of matrix matched calibration was performed to control the matrix effect and to improve the performance of the method as shown in Fig. 5.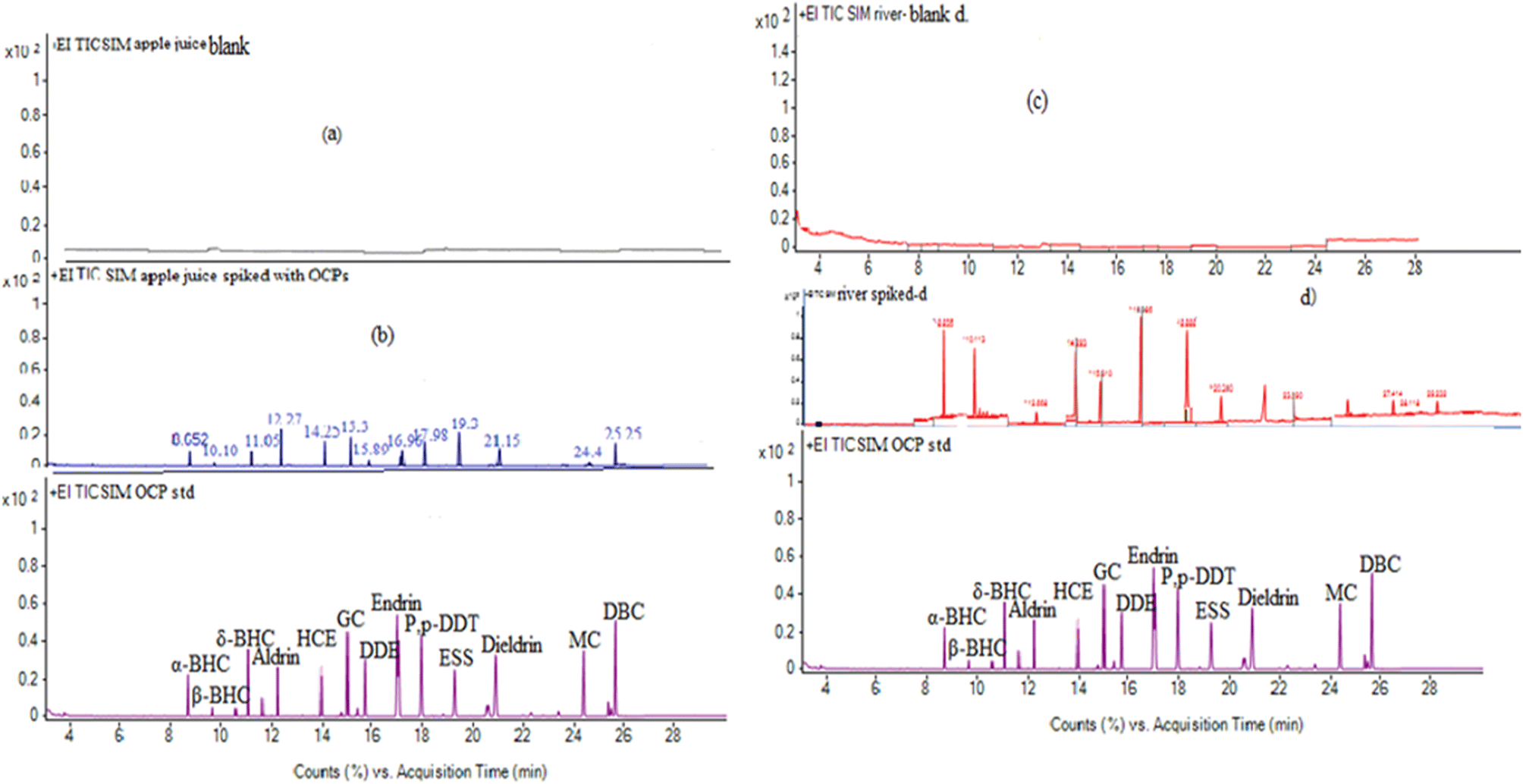 | ||
| Fig. 5 Sample chromatograms of (a) blank, and (b) spiked apple juice samples with the respective standard and (c) blank and (d) spiked river water samples with the respective OCP standard. | ||
3.5. Comparison of the proposed method with other methods
The proposed TS-HLLME combined with GC-MS analysis techniques has been compared with previously reported methods for the determination of OCPs from different matrices. On the basis of the parameters indicated in Table 4, it was noted that the technique optimized exhibited comparable analytical performance characteristics with previously reported methods. Moreover, the proposed method utilizes less toxic organic solvents (acetonitrile & ethyl acetate) which can be found in common research laboratories in developing countries. It also offered similar or better LODs and comparable linearities and recoveries with others cited for comparison. Based on the experimental findings the proposed method can be considered as one of the preferred, simple, fast, effective, and environmentally safe alternative green methods for the extraction and preconcentration of OCPs from different food, environmental, and other related matrices.| Matrices | Method | R 2 | Recovery, % | LODa | LOQa | EF | Ref. |
|---|---|---|---|---|---|---|---|
| a Unit of LOD and LOQ; ng L−1. | |||||||
| Water | GC-DLLME | ≥0.995 | 81.9–109.7 | 0.16–2.17 | 0.53–7.16 | 63–116 | 38 |
| Water | HF-SPME-UHPLC-UV | ≥0.996 | 64–113 | 0.33–0.38 | 1.00–1.25 | — | 39 |
| Water | MSPE-GC/MS/MS | ≥0.997 | 79.4–98.3 | 0.39–0.70 | 1.45–2.0 | — | 40 |
| Water | MSPE-HPLC-UV | ≥0.998 | 90.6–103.5 | 0.012–0.029 | 0.04–0.097 | — | 41 |
| Strawberry | GC-ECD-QuEChERS | ≥0.999 | 75.6–88.4 | 0.6–0.9 | — | 42 | |
| Water and apple juice | TS-SAHLLME-GC-MSD | ≥0.998 | 87.0–111.0 | 0.07–10.0 | 0.2–30 | 41–75 | This work |
The greenness of the proposed method was checked following the AGREE prep metric software. The input criteria for this metric refer to the 12 principles of green chemistry and was assigned different weights that allow for a certain flexibility. Each of the 12 input variables were transformed into a common scale in the 0–1 range, as designated in the literature.37 The final assessment result is the product of the assessment results for each principle. The output is a clock-like graph, with the overall score and color representation in the middle as depicted in ESI Fig. S4.† The performance of the procedure in each principle is reflected with the intuitive red–yellow–green color scale, while the weight of each principle is reflected with the width of its corresponding segment. The assessment was easily performed using user-friendly software available at https://agree-index.anvil.app/, with automatically generated graph and an assessment report.37 As shown in the ESI (Fig. S4†), the scores corresponding to GAC principles 1, 2, 3, 5 and 10 are low, while other principles including 4, 6, 7, 8, 9, 11 and 12 offered excellent greenness performance.
4. Conclusions
An HLLME technique in conjunction with GC-MS was developed and successfully applied for the extraction and enrichment of 13 OCPs in water and apple juice samples. The method offers a variety of advantages, including high recovery, wide linearity range, short analysis times, ease of use, and environmental friendliness. Based on the current findings, the method can be considered a promising extraction and enrichment method for trace analysis of OCPs from various environmental water samples in combination with a mixture of water-miscible solvent, ACN, and lower water-immiscible ethyl acetate as well as NaCl as a salting-out agent. In addition, it showed comparable and/or much better performances in terms of LOD and LOQ values compared to certain other reported techniques. However, the proposed method is slightly time and labour intensive since it was a manual process and uses time consuming centrifugation, which may not be feasible for large-scale applications. Therefore, for the future development, automating the entire process could significantly reduce the manual efforts required for sample preparation, extraction, and analysis.Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.Author contributions
Kero Assefa Ago: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, writing original draft, writing review & editing, Shimelis Addisu Kitte: supervision, data curation, software, writing review & editing, Gadisa Chirfa Mosisa: data curation, writing review & editing, Abera Gure Tufa: project administration, funding acquisition, resources, investigation, supervision, writing, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the College of Natural Sciences, Jimma University through the grand research project (CNS-Chem-11-2020/21-SP1); therefore it is acknowledged by all authors. Jinka University is acknowledged by Ago K. A for sponsoring the PhD study.References
- O. O. Emoyan, B. O. Peretiemo-Clarke, G. O. Tesi and E. Ohwo, Occurrence, Origin, Ecological and Human Health Risks of Organochlorine Pesticides in Soils from Selected Urban, Suburban and Rural Storm Water Reservoirs, Soil Sediment Contam., 2022, 31, 152–175, DOI:10.1080/15320383.2021.1913993.
- A. Mardani, M. Torbati, M. A. Farajzadeh and A. M. M. Mohebbi, Combination of homogeneous liquid – liquid extraction and dispersive liquid – liquid microextraction for extraction of some organochlorine pesticides from cocoa, Int. J. Environ. Anal. Chem., 2022, 102, 5092–5105, DOI:10.1080/03067319.2020.1791329.
- K. A. Ago, S. A. Kitte, G. Chirfa and A. Gure, Effervescent powder-assisted floating organic solvent-based dispersive liquid-liquid microextraction for determination of organochlorine pesticides in water by GC–MS, Heliyon, 2023, 9, e12954, DOI:10.1016/j.heliyon.2023.e12954.
- A. Mardani, M. Torbati, M. A. Farajzadeh, A. Mohebbi, A. A. Alizadeh and M. R. Afshar Mogaddam, Development of temperature-assisted solidification of floating organic droplet-based dispersive liquid–liquid microextraction performed during centrifugation for extraction of organochlorine pesticide residues in cocoa powder prior to GC-ECD, Chem. Pap., 2021, 75, 1691–1700, DOI:10.1007/s11696-020-01424-7.
- S. Bhattacharyya, R. Poi, S. Mandal, M. Baskey Sen, D. K. Hazra and S. Saha, et al., Method development, validation, monitoring, seasonal effect and risk assessment of multiclass multi pesticide residues in surface and ground water of new alluvial zone in eastern India, Environ. Sci. Pollut. Res., 2022, 29, 17174–17187, DOI:10.1007/s11356-021-16959-9.
- J. da Silva Sousa, H. O. do Nascimento, H. de Oliveira Gomes and R. F. do Nascimento, Pesticide residues in groundwater and surface water: recent advances in solid-phase extraction and solid-phase microextraction sample preparation methods for multiclass analysis by gas chromatography-mass spectrometry, Microchem J, 2021, 168, 106359 CrossRef CAS.
- N. S. Pano-Farias, S. G. Ceballos-Magana, R. Muniz-Valencia, J. M. Jurado, A. Alcazar and I. A. Aguayo-Villarreal, Direct immersion single drop micro-extraction method for multi-class pesticides analysis in mango using GC–MS, Food Chem., 2017, 237, 30–38 CrossRef CAS PubMed.
- A. Raoufi, A. M. Raoufi, A. Ismailzadeh and E. K. A. Soleimani Rad, Application of hollow fiber - protected liquid - phase microextraction combined with GC - MS in determining Endrin , Chlordane , and Dieldrin in rice samples, Environ. Geochem. Health, 2023, 1–7, DOI:10.1007/s10653-023-01570-3.
- N. Hamid, I. A. Hassan Sereshti and S. Nanthini, Superhydrophobic Nanosilica Decorated Electrospun Polyethylene Terephthalate Nanofibers for Headspace Solid Phase Microextraction of 16 Organochlorine Pesticides in Environmental Water Samples, Polymers (Basel), 2022, 14, 3682 CrossRef PubMed.
- H. Musarurwa and N. T. Tavengwa, Homogenous liquid-liquid micro-extraction of pollutants in complex matrices, Microchem. J., 2021, 170, 106750 CrossRef CAS.
- E. Teju, B. Tadesse and N. Megersa, Salting-out assisted liquid-liquid extraction for the preconcentration and quantitative determination of eight herbicide residues simultaneously in different water samples with high performance liquid chromatography, Sep. Sci. Technol., 2021, 56, 719–729 CrossRef CAS.
- A. Koltsakidou, C. K. Zacharis and K. Fytianos, A validated liquid chromatographic method for the determination of polycyclic aromatic hydrocarbons in honey after homogeneous liquid-liquid extraction using hydrophilic acetonitrile and sodium chloride as mass separating agent, J. Chromatogr. A, 2015, 1377, 46–54 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, Determination of organic compounds in water using dispersive liquid-liquid microextraction, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- S. Armenta, S. Garrigues and M. De Guardia, Green Analytical Chemistry, Trends Anal. Chem., 2008, 27, 497–511 CrossRef CAS.
- S. F. Hammad, I. A. Abdallah, A. Bedair and F. R. Mansour, Salting-out induced liquid–liquid microextraction for alogliptin benzoate determination in human plasma by HPLC/UV, BMC Chem., 2021, 15, 1–10 CrossRef PubMed.
- B. Ziban, M. Nemati and A. Shayanfar, Salting-Out Assisted Liquid-Liquid Extraction for Quantification of Ammonia Compounds in Food Samples, Anal. Bioanal. Chem. Res., 2023, 10, 425–433 CAS.
- X. Y. Xu, J. Q. Ye, J. Nie, Z. G. Li and M. R. Lee, A new liquid-liquid microextraction method by ultrasound assisted salting-out for determination of triazole pesticides in water samples coupled by gas chromatography-mass spectrometry, Anal. Methods, 2015, 7, 1194–1199 RSC.
- A. Gure, F. J. Lara, - D. Moreno, N. Megersa and A. M. García-campaña, A Salting-out assisted liquid-liquid extraction combined with capillary HPLC for the deter- mination of sulfonylurea herbicides in envir- onmental water and banana juice samples, Talanta, 2014, 127, 51–58, DOI:10.1016/j.talanta.2014.03.070.
- Y. Wen, J. Li, F. Yang, W. Zhang, W. Li and C. Liao, et al., Salting-out assisted liquid-liquid extraction with the aid of experimental design for determination of benzimidazole fungicides in high salinity samples by high-performance liquid chromatography, Talanta, 2013, 106, 119–126, DOI:10.1016/j.talanta.2012.12.011.
- W. H. Tsai, T. C. Huang, H. H. Chen, Y. W. Wu, J. J. Huang and H. Y. Chuang, Determination of sulfonamides in swine muscle after salting-out assisted liquid extraction with acetonitrile coupled with back-extraction by a water/acetonitrile/dichloromethane ternary component system prior to high-performance liquid chromatography, J. Chromatogr. A, 2010, 1217, 250–255 CrossRef CAS PubMed.
- H. Haddadi, M. Shirani, A. Semnani, M. Rezaee, H. A. Mashayekhi and A. Hosseinian, Simultaneous determination of deltamethrin and permethrin in water samples using homogeneous liquid-liquid microextraction via flotation assistance and GC-FID, Chromatographia, 2014, 77, 715–721 CrossRef CAS.
- H. Ebrahimzadeh, Y. Yamini, F. Kamarei and S. Shariati, Homogeneous liquid-liquid extraction of trace amounts of mononitrotoluenes from waste water samples, Anal. Chim. Acta, 2007, 594, 93–100 CrossRef CAS PubMed.
- M. Haji Hosseini, P. Asaadi, M. Rezaee, M. R. Rezaei, M. R. Pourjavid and M. Arabieh, et al., Homogeneous liquid-liquid microextraction via flotation assistance (HLLME-FA) method for the pretreatment of organochlorine pesticides in aqueous samples and determination by GC-MS, Chromatographia, 2013, 76, 1779–1784 CrossRef CAS.
- B. Khagga, M. V. Kaitha, R. Dammu and S. Mogili, ICH guidelines – “Q” series (quality guidelines) - A review, GSC Biol. Pharm. Sci., 2019, 6, 089–106 CrossRef.
- E. N. Tzanetou and H. Karasali, A Comprehensive Review of Organochlorine Pesticide Monitoring in Agricultural Soils: The Silent Threat of a Conventional Agricultural Past, Agric, 2022, 12, 728 CAS.
- M. M. Issa, M. S. Taha, A. M. El-Marsafy, M. M. H. Khalil and E. H. Ismail, Acetonitrile-Ethyl acetate based method for the residue analysis of 373 pesticides in beeswax using LC-MS/MS and GC–MS/MS, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1145, 122106, DOI:10.1016/j.jchromb.2020.122106.
- A. Salis and B. W. Ninham, Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y. L. Zhong, M. Shevlin and F. Peng, General Principles and Strategies for Salting-Out Informed by the Hofmeister Series, Org. Process Res. Dev., 2017, 21, 1355–1370 CrossRef CAS.
- B. Kang, H. Tang, Z. Zhao and S. Song, Hofmeister Series: Insights of Ion Specificity from Amphiphilic Assembly and Interface Property, ACS Omega, 2020, 5, 6229–6239 CrossRef CAS PubMed.
- Q. Wang, C. R. Yin and L. Xu, Optimization of hydrophilic interaction LC by univariate and multivariate methods and its combination with salting-out liquid-liquid extraction for the determination of antihypertensive drugs in the environmental waters, J. Sep. Sci., 2013, 36, 1007–1014 CrossRef CAS PubMed.
- Y. Alemayehu, T. Tolcha and N. Megersa, Salting-Out Assisted Liquid-Liquid Extraction Combined with HPLC for Quantitative Extraction of Trace Multiclass Pesticide Residues from Environmental Waters, Am. J. Anal. Chem., 2017, 8, 433–448 CrossRef CAS.
- A. Mardani, M. Torbati, M. A. Farajzadeh and A. Mohebbi, Combination of homogeneous liquid – liquid extraction and dispersive liquid – liquid microextraction for extraction of some organochlorine pesticides from cocoa, Int. J. Environ. Anal. Chem., 2020, 102, 5092–5105 CrossRef.
- A. Tighrine, Y. Amir, P. Alfaro, M. Mamou and C. Nerín, Simultaneous extraction and analysis of preservatives and artificial sweeteners in juices by salting out liquid-liquid extraction method prior to ultra-high per- formance liquid chromatography, Food Chem., 2018, 1–30 Search PubMed.
- H. Trufelli, P. Palma and A. C. Giorgio Famiglini, An overview of Matrix Effects in Liquid chromatography-mass spectrometry, Mass Spectrom. Rev., 2011, 30, 491–509 CrossRef CAS PubMed.
- E. Yildiz and H. Cabuk, A new solidified effervescent tablet-assisted dispersive liquid-liquid microextraction for the analysis of fungicides in fruit juice samples, Anal. Methods, 2018, 10, 330–337 RSC.
- M. Ghambarian, N. Yazdanfar and Y. Yamini, Homogeneous Liquid – Liquid Microextraction for Determination of Organochlorine Pesticides in Water and Fruit Samples, Chromatographia, 2014, 77, 329–336 CrossRef.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, AGREE - Analytical GREEnness Metric Approach and Software, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
- D. Hou, O. Khureldavaa, F. Zhang, J. He and B. Amarsanaa, Determination of OCPs and PCBs in environmental water samples by GC-DLLME optimized by response surface methodology, Mong. J. Chem., 2019, 20, 13–23 CrossRef CAS.
- L. Pang, P. Yang, R. Pang and S. Li, Bis(trifluoromethylsulfonyl)imide-based frozen ionic liquid for the hollow-fiber solid-phase microextraction of dichlorodiphenyltrichloroethane and its main metabolites, J. Sep. Sci., 2017, 40, 3311–3317 CrossRef CAS PubMed.
- Y. Liu, Z. Gao, R. Wu, Z. Wang, X. Chen and T. W. D. Chan, Magnetic porous carbon derived from a bimetallic metal–organic framework for magnetic solid-phase extraction of organochlorine pesticides from drinking and environmental water samples, J. Chromatogr. A, 2017, 1479, 55–61 CrossRef CAS PubMed.
- Q. Zhoua, Y. Wu, Y. Sun, X. Sheng, Y. Tong and J. Guo, et al., Magnetic polyamidoamine dendrimers for magnetic separation and sensitive determination of organochlorine pesticides from water samples by high-performance liquid chromatography, J. Environ. Sci., 2021, 102, 64–73 CrossRef PubMed.
- S.-J. Lim, Y.-T. Oh, Y.-S. Jo, J.-H. Ro, G.-H. Choi, J.-Y. Yang and B. J. Park, Persistent Organic Pollutants (POPs) Residues in Greenhouse Soil and Strawberry Organochlorine Pesticides, Korean J. Environ. Agric., 2016, 35, 6–14 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay01751f |
| This journal is © The Royal Society of Chemistry 2024 |