
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluating Riboglow-FLIM probes for RNA sensing†
Nadia
Sarfraz
 ,
Luke K.
Shafik
,
Zachary R.
Stickelman
,
Uma
Shankar
,
Emilia
Moscoso
and
Esther
Braselmann
*
,
Luke K.
Shafik
,
Zachary R.
Stickelman
,
Uma
Shankar
,
Emilia
Moscoso
and
Esther
Braselmann
*
Department of Chemistry, Georgetown University, Washington, District of Columbia, USA. E-mail: esther.braselmann@georgetown.edu
First published on 8th January 2024
Abstract
We recently developed Riboglow-FLIM, where we genetically tag and track RNA molecules in live cells through measuring the fluorescence lifetime of a small molecule probe that binds the RNA tag. Here, we systematically and quantitatively evaluated key elements of Riboglow-FLIM that may serve as the foundation for Riboglow-FLIM applications and further tool development efforts. Our investigation focused on measuring changes in fluorescence lifetime of representative Riboglow-FLIM probes with different linkers and fluorophores in different environments. In vitro measurements revealed distinct lifetime differences among the probe variants as a result of different linker designs and fluorophore selections. To expand on the platform's versatility, probes in a wide variety of mammalian cell types were examined using fluorescence lifetime imaging microscopy (FLIM), and possible effects on cell physiology were evaluated by metabolomics. The results demonstrated that variations in lifetime were dependent on both probe and cell type. Interestingly, distinct differences in lifetime values were observed between cell lines, while no overall change in cell health was measured. These findings underscore the importance of probe selection and cellular environment when employing Riboglow-FLIM for RNA detection, serving as a foundation for future tool development and applications across diverse fields and biological systems.
Introduction
RNAs have central roles in biology across all domains of life.1 Subcellular localization and dynamics of RNAs in response to perturbations are critical to their function. Despite remarkable progress, there is still room for advancing and improving tools and approaches for visualizing RNAs in complex cellular environments. Importantly, no fluorescent RNAs have been discovered in nature, unlike fluorescent proteins,2 shifting efforts to using chemical biology to design fluorescent RNA systems for live visualization.The most common system used to visualize RNAs is the MS2/PP7 system, which allows visualization of mRNAs down to a single RNA level in complex cell systems.3,4 Complementary to MS2/PP7, much effort is focused on designing genetically encoded RNA tags that bind synthetic small molecules to yield genetically encoded fluorescent RNA tags that induce a fluorescence signal.5 Various systems exist, ranging from color-shifting ratiometric sensors to complementary RNA sensors that are spectrally distinct. The Spinach6,7 and Squash8 aptamer-fluorophore systems are examples of these, as are the Mango9 and Peach10 systems. Corn11 and, more recently, the Beetroot12 systems have further expanded RNA visualization capabilities through dimer-dependent fluorescence turn-on. A common feature of these “vegetable” RNA sensors is that the RNAs frequently adopt G-quadruplex structures,9,12,13 which may lead to cross-reactivity of RNA and probe ligand, complicating multiplexing efforts.
To complement existing tools, we developed Riboglow14,15 and recently expanded the platform to visualize model RNAs in commonly investigated U-2 OS cells using fluorescence lifetime imaging microscopy (FLIM).16,17 The components of Riboglow are a genetically encoded RNA tag and a small molecule consisting of the ligand cobalamin (Cbl) that is synthetically coupled to a fluorophore (here, ATTO 590 or Cy5).14 The RNA element in Riboglow is derived from a bacterial riboswitch,18 and as a result, the RNA tag is short and takes advantage of the selective and high-affinity binding between the riboswitch and its cognate ligand Cbl.14 The Cbl ligand is also a fluorescence quencher.19 This explains why the Cbl-fluorophore probes exhibit quenched fluorescence intensity and dramatically different fluorescence lifetime compared to their free fluorophore counterparts (Fig. 1 and 2B).14,16,17 Binding of the RNA ligand to Cbl affects this fluorescence signal, such that the intensity and lifetime readouts upon RNA tag binding change (Fig. 2).14,16,17 In our proof-of-concept study for Riboglow-FLIM in live cells, we showed compatibility with RNA multiplexing by exploiting the phylogenetic diversity of Cbl riboswitches.20 We recently expanded the system to demonstrate the potential of Riboglow-FLIM for RNA visualization in complex multicellular systems.17 Together, the Riboglow-FLIM platform includes flexibility and variability of various key elements, illustrating the capacity for expansion of the platform to physiologically relevant cell models, but systematic control evaluations are lacking.
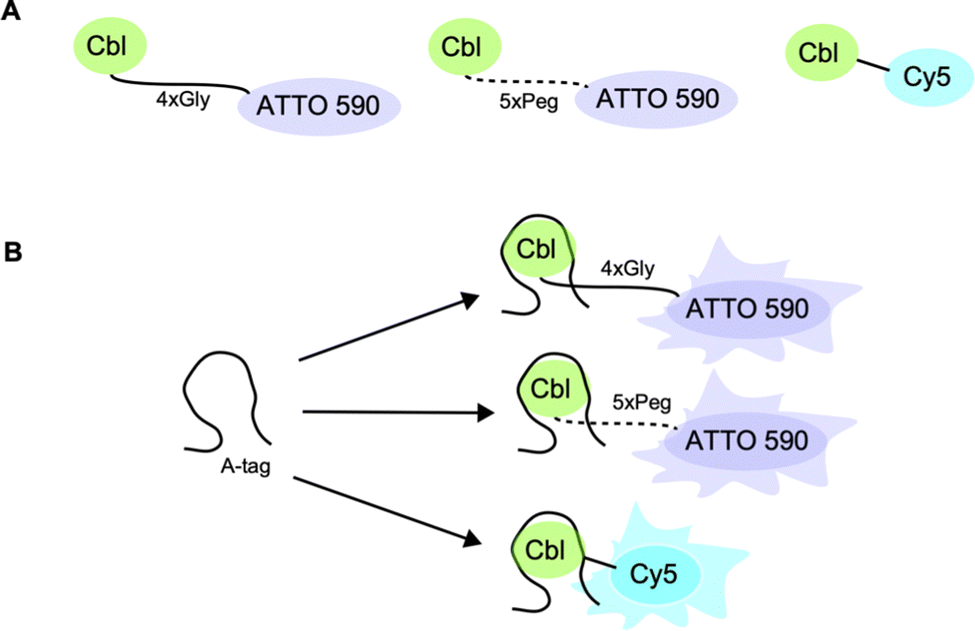 | ||
| Fig. 1 Riboglow probes used in this study. (A) Representation of Riboglow Cbl-fluorophore probe variants evaluated in this study and (B) representation of Riboglow probes binding to the RNA tag (called the A-tag throughout this study) leading to a change in fluorescence lifetime. | ||
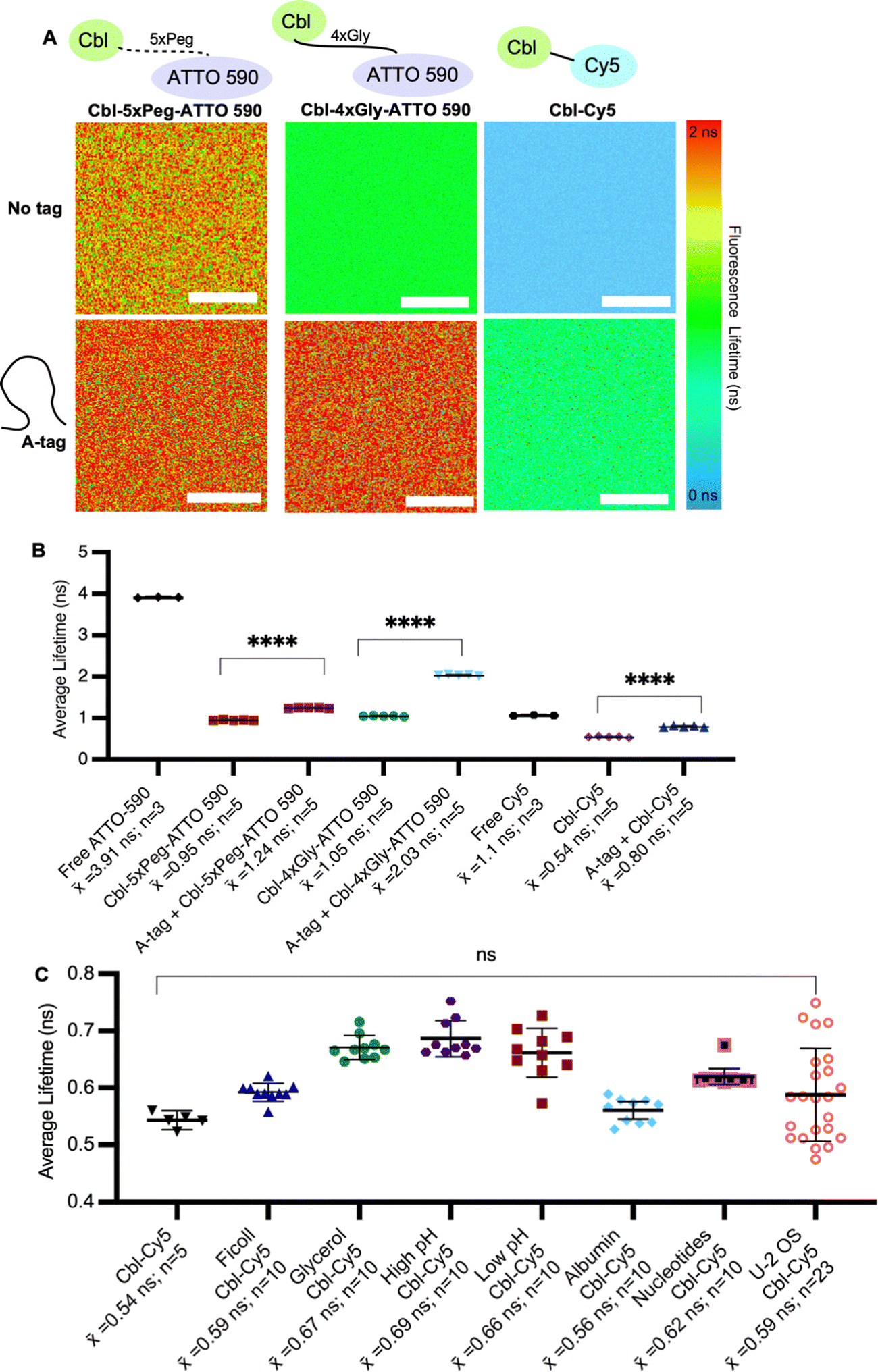 | ||
Fig. 2 Assessing fluorescence lifetimes of probe series in vitro. (A) Quantitative visualization of Riboglow probes by fluorescence lifetime imaging microscopy (FLIM) in vitro in the presence or absence of the RNA A-tag. (B) Average fluorescence lifetime values for each region of interest (ROI) (30 ROIs, 7 independent experiments) with listed probes and data processed as outlined in methods using multiexponential reconvolution fitting to n = 2 parameters (Fig. S3, ESI†). One symbol = 1 ROI, number of data points acquired are listed, p-values listed (ns: p![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ≤ 0.5; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). Error bars indicate mean and standard deviation (±SD). Scale bar = 10 μm. (C) Average fluorescence lifetime values of probe under each labelled condition for each region of interest (ROI) (68 ROIs, 17 independent experiments and data processed as outlined in methods). One symbol = 1 ROI, p-values listed (ns: p ≤ 0.5). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). ≤ 0.5; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). Error bars indicate mean and standard deviation (±SD). Scale bar = 10 μm. (C) Average fluorescence lifetime values of probe under each labelled condition for each region of interest (ROI) (68 ROIs, 17 independent experiments and data processed as outlined in methods). One symbol = 1 ROI, p-values listed (ns: p ≤ 0.5). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). | ||
Riboglow is compatible with a range of synthetic fluorophores and linkers, where the fluorescence quencher Cbl can be fused to various fluorophores using different linkers.14 Riboglow-ATTO 590 and Riboglow-Cy5 probes were designed previously and proof-of-concept studies revealed that RNAs that are genetically tagged with Riboglow can be sensed by an increase in fluorescence intensity or fluorescence lifetime in live mammalian cells.17
We recently demonstrated that Riboglow-FLIM16 offers the ability to quantitatively visualize RNAs in vitro and in live U-2 OS cells using the Cbl-4xGly-ATTO 590 probe, expanding the usability of the Riboglow platform to FLIM. However, a more comprehensive and methodical examination of probe variants and cell lines is necessary to fully understand their capabilities, applicability in diverse cellular systems, and rationally guide further tool development efforts.
Riboglow-FLIM has the advantage of concentration independence21 compared to fluorescence intensity-based measurements, but is very sensitive to the cellular context. As observed before by others22 and our own study,16 the fluorescence lifetime of a probe in different cell lines may vary, likely due to different cell characteristics and properties.23 Sampling Riboglow-FLIM in multiple cell lines is essential for a comprehensive characterization and understanding of its application, as different cell lines possess unique genetic and phenotypic features.24 We reasoned that evaluating the Riboglow's probe fluorescence lifetime in different cell lines and evaluating cell viability and effects on metabolic activity will allow us to assess performance across diverse cellular contexts and gather insights into generalizability.
Results
We began by systematically assessing the in vitro Riboglow-FLIM system, which consisted of a series of probe variants with characteristic UV Vis spectra (Fig. 1A and Fig. S1, S2A, ESI†). We confirmed binding of the RNA A-tag to Cbl (ESI,† Fig S2B and Fig. 1B), with a binding affinity (KD = 99 nM ± 30 nM) in line with the previously reported affinity (KD = 37 nM ± 1 nM).14 We quantified the fluorescence lifetime of Cbl-4xGly-ATTO 590 in the presence of our RNA A-tag (Table S1, Table S2, ESI†) by fitting the data using multiexponential reconvolution (n = 2). We have previously found that n = 2 fits the data best for this variant of Riboglow.16 For Cbl-Cy5, multiexponential reconvolution with n = 2 allows for direct comparison of lifetimes across in vitro and cell work, as this fits the data best, considering the goodness of the fit shown by overlapping decay curves of raw and fitted data and the residual maps (Fig. S3, ESI†). As seen before,14,16,17 the Riboglow probes exhibit a reduced fluorescence lifetime value compared with the free fluorophore and adding the RNA ligand induces de-quenching, leading to an increase in fluorescence lifetime (Fig. 2). We then explored whether variations in RNA/probe concentrations would impact fluorescence lifetime values. To address this question, we considered the KD value of 99 nM and compared conditions of 5 μM RNA A-tag and 5 μM probe versus 5 μM RNA A-tag and 0.5 μM probe. We do not observe changes in lifetimes, indicating that as long as sample concentrations above KD are employed, the produced lifetimes of about 2.4 ns remain similar (Table S2, ESI†). This confirms the concentration-independence of fluorescence lifetime measurements.16,21To assess Riboglow probe variants, we systematically compared fluorescence lifetimes for probes with varying linkers and fluorophores that were previously used14,16,17 in live cell proof-of-concept assays (Fig. 1A and Fig. S1, ESI†). We selected three different probe variants, Cbl-4xGly-ATTO 590, Cbl-5xPeg-ATTO 590, and Cbl-Cy5. The first two variants both contain the ATTO 590 fluorophore but have different linkers to Cbl (4xGly and 5xPeg). The third probe variant explored was a ‘linkerless’ Cy5 fluorophore probe (Fig. 1). In this direct comparison, we found that Cbl-Cy5 produced the lowest lifetimes, whereas the Cbl-4xGly-ATTO 590 and Cbl-5xPeg-ATTO 590 probes produced higher lifetimes and were around a similar range (Fig. 2A), comparable with values determined previously in isolated examples (Table S3, ESI†).14,16,17
We next tested fluorescence lifetime in the presence of the purified Riboglow RNA A-tag (Table S2, ESI†). To do this, we began by sampling the lifetimes of free fluorophores (free Cy5 and free ATTO 590) to serve as the maximum lifetime obtainable upon dequenching through RNA binding (Fig. 2). In line with previous observations,14,16,17 our side-by-side comparison revealed a significant lifetime increase across all samples in the presence of the RNA A-tag (Fig. 2B and Tables S3, S4, ESI†). Remarkably, the lifetime values are in line with previous proof-of-concept observations in live cells (Table S3, ESI†). Interestingly, we observed higher lifetimes in Cbl-4xGly-ATTO 590 samples in the presence of the A-tag than with Cbl-5xPeg-ATTO 590 in the presence of the A-tag. These differences were not observed when comparing fluorescence intensities previously,14 pointing to sensitivity of fluorescence lifetime for small chemical changes in the Riboglow linker when bound to the RNA.
Next, we complexified our conditions to bridge the gap between pristine buffer conditions and the dynamic environment of live mammalian cells. Specifically, the buffer conditions were augmented in complexity, guided by established techniques25 for emulating cellular crowding through the incorporation of protein, nucleotides, Ficoll and glycerol (Fig. 2C). Additionally, pH levels were varied within a range typically encountered in cellular investigations26 (Fig. 2C). As anticipated, these modifications did not have significant effects on fluorescence lifetimes, but a marginal increase in measurement error was observed as system complexity escalated, mirroring the increasing range observed when lifetimes are measured in live cell environments (Fig. 2C and Table S3, ESI†).
To establish the versatility of different Riboglow-FLIM probes in cells, we next asked if introducing probes into live mammalian cells affects viability. Indeed, our ‘bead loading’ process does not affect cell viability even for our broad series of cell lines tested (Fig. S4, ESI†). This observation is consistent with previous work on Riboglow14,16,17 and other studies in which bead loading was utilized to introduce proteins into live cells, and confirms that this process is not impactful to cell health.27,28
To systematically evaluate Riboglow-FLIM across different cell lines, we began by adding HOS cells to our established U-2 OS cell line model. Both HOS and U-2 OS cells originate from human osteosarcoma,29 representing a minimal and systematic change in cell model (Table S5, ESI†). Despite their shared origin, these cell lines exhibit substantial differences. U-2 OS cells have an epithelial morphology, whereas HOS cells have a more fibroblast-like morphology and exhibit tumorigenic properties.29 HOS and U-2 OS cells have been extensively characterized and widely used in research, providing a robust basis for a comparative analysis.30–32 As expected, lifetime differences were observed in both cell types depending on the probe used, with Cy5 eliciting the lowest lifetimes and ATTO 590 variants eliciting higher lifetimes (Fig. 3 and Fig. S5, ESI†). These trends mirror our observations in vitro (Fig. 2). In our investigation comparing the fluorescence lifetimes of two ATTO probes in cells (Fig. 3), namely Cbl-5xPeg-ATTO 590 and Cbl-4xGly-ATTO 590, we made an intriguing discovery. We observed that the Cbl-5xPeg-ATTO 590 probe exhibited consistently shorter fluorescence lifetimes (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) = 1.37 ns in HOS cells, = 1.20 ns in U-2 OS cells) compared to the Cbl-4xGly-ATTO 590 probe ( = 1.75 ns in HOS cells, = 1.68 ns in U-2 OS cells). This distinction is particularly noteworthy considering that the linkers in both probes are similar in length, measuring approximately 20 Å.14 These findings suggest that specific linkers and their interactions with cellular components contribute to variations in fluorescence lifetime. Furthermore, higher lifetimes were observed in HOS cells when compared to U-2 OS cells under identical conditions (Fig. 3B). We found that Cbl-Cy5 in U-2 OS cells yielded lower lifetimes, whereas Cbl-Cy5 in HOS cells yielded slightly higher lifetimes. This effect was not observed for Cbl-4xGly-ATTO 590, as the lifetimes produced were similar regardless of cell type. Together, we found that the fluorescence lifetime of Cbl-Cy5 is particularly sensitive to cell-type specific differences, an important aspect to consider for Riboglow-FLIM usage in different cell systems.
= 1.37 ns in HOS cells, = 1.20 ns in U-2 OS cells) compared to the Cbl-4xGly-ATTO 590 probe ( = 1.75 ns in HOS cells, = 1.68 ns in U-2 OS cells). This distinction is particularly noteworthy considering that the linkers in both probes are similar in length, measuring approximately 20 Å.14 These findings suggest that specific linkers and their interactions with cellular components contribute to variations in fluorescence lifetime. Furthermore, higher lifetimes were observed in HOS cells when compared to U-2 OS cells under identical conditions (Fig. 3B). We found that Cbl-Cy5 in U-2 OS cells yielded lower lifetimes, whereas Cbl-Cy5 in HOS cells yielded slightly higher lifetimes. This effect was not observed for Cbl-4xGly-ATTO 590, as the lifetimes produced were similar regardless of cell type. Together, we found that the fluorescence lifetime of Cbl-Cy5 is particularly sensitive to cell-type specific differences, an important aspect to consider for Riboglow-FLIM usage in different cell systems.
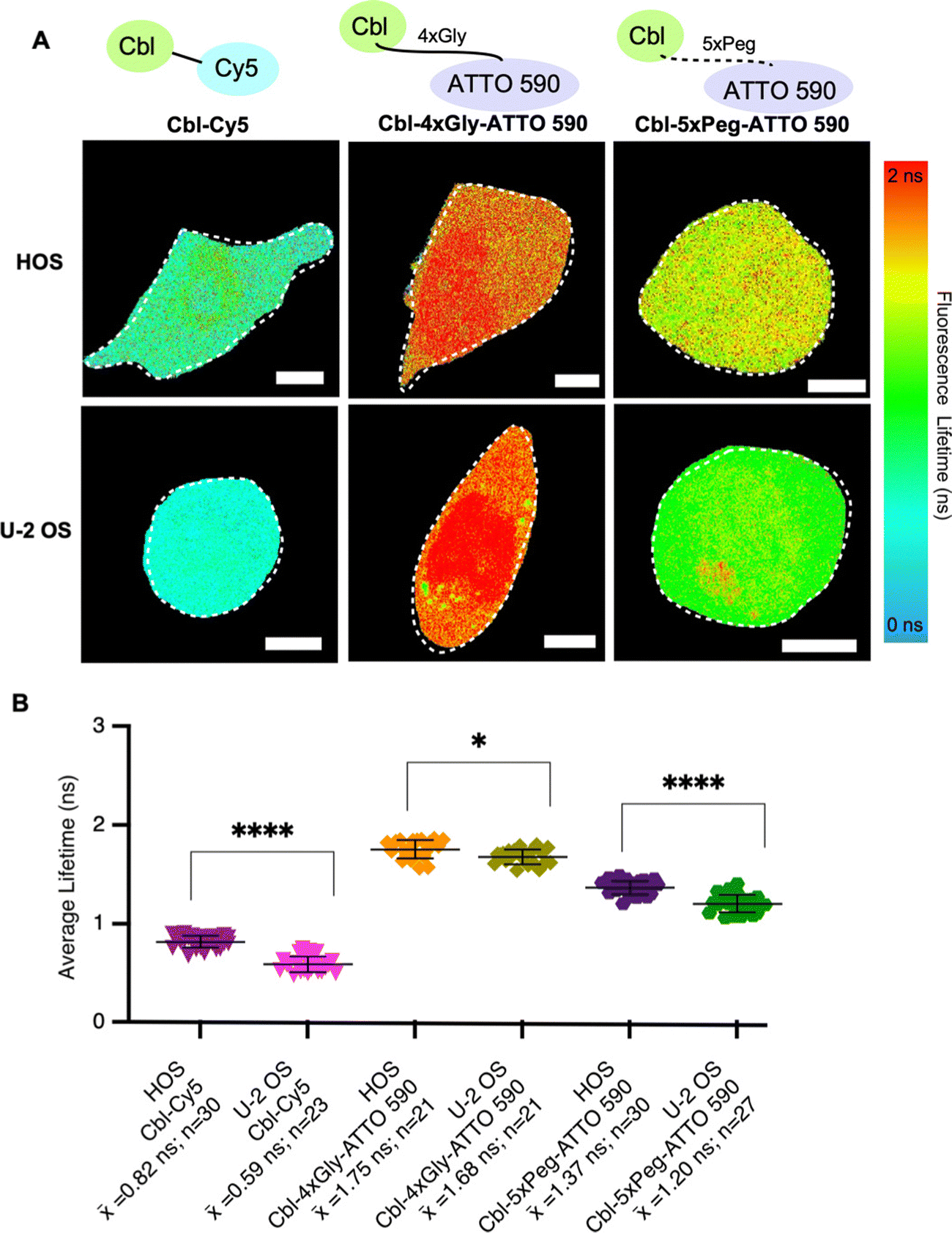 | ||
| Fig. 3 Fluorescence lifetimes of probe series in two different cell lines. (A) Quantitative visualization of Riboglow probes by fluorescence lifetime imaging microscopy (FLIM) for HOS (top) and U-2 OS (bottom) live mammalian cells. Representative cells are shown. (B) Average fluorescence lifetime values for live cells (152 cells, 8 independent experiments) loaded with listed probes and data processed as outlined in methods using multiexponential reconvolution fitting to n = 2 parameters (Fig. S3, ESI†). One symbol = 1 cell, p-values listed (ns: p ≤ 0.5; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). Error bars indicate mean and standard deviation (±SD). Scale bar = 5 μm. | ||
To explore probe lifetime differences across cell lines further, we expanded our study to more diverse cell lines (Table S5, ESI†), namely non-cancer derived fibroblasts (HFF-1) and two types of stem cells, bone marrow-derived mesenchymal and adipose-derived mesenchymal stem cells (ADM-SC and BMD-SC), while using the same probe (Cbl-Cy5). HFF-1 cells are non-cancer cells and have a more flattened and fibroblastic appearance, making them valuable for studying tissue repair.33 Stem cells (ADM-SC and BMD-SC), known for their remarkable plasticity, display a diverse range of morphologies depending on their state of differentiation.34–36 We observed significant differences in lifetime between U-2 OS cells and all other cell types used, with the same probe (Fig. 4).
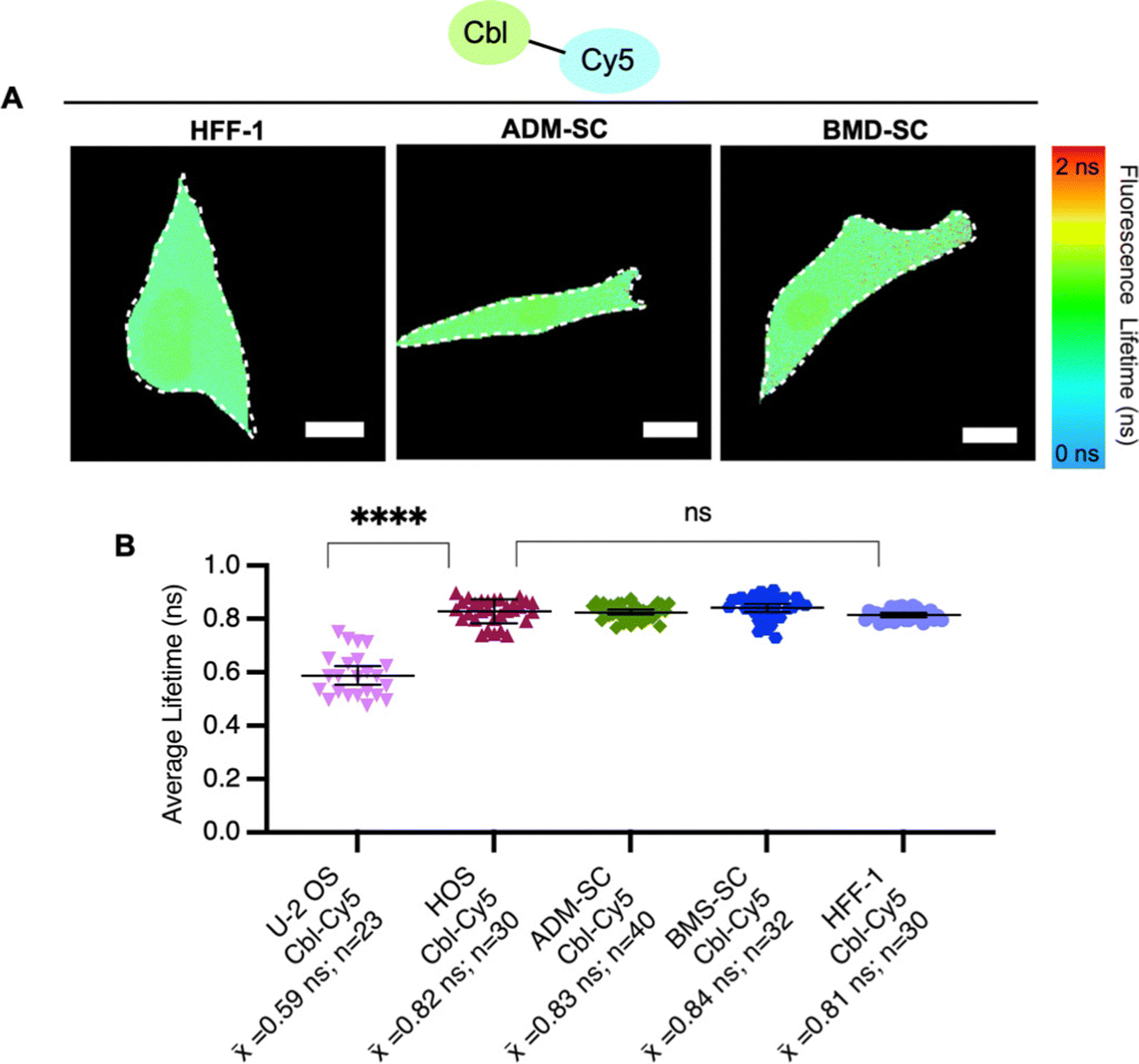 | ||
| Fig. 4 Fluorescence lifetimes of probe series in stem cell and fibroblast cell lines. (A) Quantitative visualization of Riboglow probes by fluorescence lifetime imaging microscopy (FLIM) for HFF-1 fibroblasts, adipose derived mesenchymal stem cells (ADM-SC) and bone marrow derived stem cells (BMD-SC). Representative cells are shown. (B) Average fluorescence lifetime values for live cells (155 cells, 10 independent experiments) loaded with listed probe of Cbl-Cy5 and data processed as outlined in methods using multiexponential reconvolution fitting to n = 2 parameters. One symbol = 1 cell, p-values listed (ns: p ≤ 0.5 and ****: p ≤ 0.0001). One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). Error bars indicate mean and standard deviation (±SD). Scale bar = 5 μm. | ||
To investigate factors that could contribute to lifetime variations between cell lines, we performed untargeted metabolomics for U-2 OS cells and HFF-1 cells (Fig. S7, ESI†). We chose these cell lines for this evaluation for a number of reasons. (i) They represent our ‘standard’ system (U-2 OS cells14,16) and an unrelated system (fibroblast cells, HFF-1 cells). (ii) HFF-1 and U-2 OS cells have similar probe uptake efficiency (∼60%, Fig. S6, ESI†), and so the amount of “loaded” vs. “normal” cells would be similar in this comparison. (iii) U-2 OS cells are cancer-derived epithelial cells, whereas HFF-1 are non-cancer (normal) derived fibroblasts.31,33 (iv) In comparing fluorescence lifetimes (Fig. 4), we found that U-2 OS and HFF-1 have significantly different lifetime readouts, similar to what was noted between U-2 OS cells and all other cells (Fig. 3B and 4B). We found minor metabolomic differences upon probe uptake in the HFF-1 cells compared with the U-2 OS cells where minimal changes were noted (Fig. S7, ESI†). A total of 6954 metabolites were tested, with 4 metabolites being dysregulated for U-2 OS cells and 46 being dysregulated for HFF-1, with the only commonly dysregulated metabolite being norandrosterone. We suggest that these differences could be due to cell line variability as U-2 OS cells and HFF-1 cells are derived from different tissue types (Table S5, ESI†) and so likely have different metabolic profiles and responses to treatments. Additionally, since minimal variability was noted (as seen in the volcano and scores plot in ESI† Fig. S7), the effects may be due to biological variability as individual cells can exhibit some level of heterogeneity in their metabolic responses.17 We hypothesize that the observed metabolic changes are not solely an effect of Riboglow probe treatment, but more studies would need to be done to further explore this. Overall, we conclude that metabolic stability should be generally considered when planning to use Riboglow for questions outside of epithelial adherent cells (like U-2 OS cells), as demonstrated by our comparison of U-2 OS cells vs. HFF-1 cells.
Conclusions
Our systematic characterization of Riboglow-FLIM probe variants shows that FLIM measurements are highly sensitive to the probe's environment. By exploring probe variants with different linkers and fluorophores, we observed significant differences in fluorescence lifetimes both in vitro and in live mammalian cells. The Cbl-Cy5 probe exhibited the lowest lifetime, while Cbl-4xGly-ATTO 590 and Cbl-5xPeg-ATTO 590 showed higher, similar lifetimes. This is in line with the previous establishment that Cbl functions as a quencher of a wide variation of fluorophores, leading to a marked decrease in fluorescence lifetime.14,16,17 Moreover, the presence of the Riboglow RNA A-tag resulted in a significant increase in lifetimes across all probe samples, with Cbl-4xGly-ATTO 590 displaying higher changes in lifetime compared to Cbl-5xPeg-ATTO 590. Systematically changing the buffer conditions to mimic cellular conditions resulted in more variability, in line with FLIM measurements in live cells that typically have larger error bars (Fig. 2C and 3B). It is important to note that our lifetime values were consistent and reproducible across buffer and cellular samples, pointing to using Riboglow-FLIM as a robust and sensitive platform for RNA sensing.The observed differences in fluorescence lifetimes in different environments imply that the probes exhibit distinct interactions within different cellular environments. These interactions likely involve factors such as cellular composition or molecular crowding in each cell type.37 Consequently, we conclude that the fluorescence lifetime of a probe is not solely dependent on the probe itself but also on the intricate interplay between the probe and the specific cellular context in which it is utilized. These outcomes underscore the importance of careful consideration in the selection of the Riboglow-FLIM probe and the thorough need for characterization of its behavior in the intended cellular environment to obtain results that reflect the molecular events of interest and provide meaningful insights, which goes hand in hand with ensuring accurate fitting is conducted (Fig. S3, ESI†).
Interestingly, we observed cell-type-dependent lifetime variations, first when comparing HOS cells vs. U-2 OS cells and then across a more diverse cell series (Fig. 3 and 4), further highlighting the significance of cellular context in fluorescence lifetime measurements. Overall, our study provides valuable insights into the versatility and potential applications of Riboglow-FLIM for RNA imaging, as well as the sensitivity of fluorescence lifetime. The modular nature of Riboglow-FLIM will likely inspire future applications.
Statistical methods
One-way ANOVA (95% confidence limit); post hoc test (Tukey HSD). Error bars: mean and standard deviation (±SD).Software
Data collection: microscopy images acquired on EVOS M5000 microscope (Thermo Fisher) and Abberior STEDYCON instrument with Picoquant lifetime software. Data analysis: ImageJ2 v.1.53 with Fiji plugin suite, Prism v.9.4.1(458), Canvas X draw v.2, Zotero v.6.0.17, Picoquant Symphotime 64 v.1-9, MetaboAnalystR v.4.0.Author contributions
E. B. and N. S. conceptualized and designed the study. N. S. and Z. R. S. purified riboswitch variants for in vitro work. N. S., L. K. S., Z. R. S., U. S., and E. M. performed in vitro work, designed, and performed cellular work, and analyzed data with input from all authors. N. S. and L. K. S. wrote the manuscript with edits from all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank A. Van Dyke, A. Cheema, and T. Ichiye for helpful discussions. The authors would like to acknowledge financial support from the NIH (R00GM127752 and R35GM150823 to E. B.), the Luce Foundation (to E. B.), Georgetown College, the Georgetown Department of Chemistry (Espenscheid fellowships to L. K. S. and Z. R. S.) and the American Cancer Society (ACS IRG 17-177-23, pilot award to E. B., PI: Riggins). This research was supported by Proteomics and Metabolomics Shared Resource (PMSR) and Microscopy & Imaging Shared Resource Center of the Georgetown Lombardi Comprehensive Cancer Center (P30CA051008).References
- E. J. Strobel, K. E. Watters, D. Loughrey and J. B. Lucks, RNA Systems Biology: Uniting Functional Discoveries and Structural Tools to Understand Global Roles of RNAs, Curr. Opin. Biotechnol, 2016, 39, 182–191, DOI:10.1016/j.copbio.2016.03.019.
- G.-J. Kremers, S. G. Gilbert, P. J. Cranfill, M. W. Davidson and D. W. Piston, Fluorescent Proteins at a Glance, J. Cell Sci., 2011, 124(2), 157–160 CrossRef CAS PubMed.
- E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Localization of ASH1 mRNA Particles in Living Yeast, Mol. Cell, 1998, 2(4), 437–445, DOI:10.1016/S1097-2765(00)80143-4.
- B. Wu, J. A. Chao and R. H. Singer, Fluorescence Fluctuation Spectroscopy Enables Quantitative Imaging of Single mRNAs in Living Cells, Biophys. J., 2012, 102(12), 2936–2944, DOI:10.1016/j.bpj.2012.05.017.
- E. Braselmann, C. Rathbun, E. M. Richards and A. E. Palmer, Illuminating RNA Biology: Tools for Imaging RNA in Live Mammalian Cells, Cell Chem. Biol., 2020, 27(8), 891–903, DOI:10.1016/j.chembiol.2020.06.010.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, RNA Mimics of Green Fluorescent Protein, Science, 2011, 333(6042), 642–646, DOI:10.1126/science.1207339.
- X. Li, H. Kim, J. L. Litke, J. Wu and S. R. Jaffrey, Fluorophore-Promoted RNA Folding and Photostability Enables Imaging of Single Broccoli-Tagged mRNAs in Live Mammalian Cells, Angew. Chem., Int. Ed., 2020, 59(11), 4511–4518, DOI:10.1002/anie.201914576.
- S. K. Dey, G. S. Filonov, A. O. Olarerin-George, B. T. Jackson, L. W. S. Finley and S. R. Jaffrey, Repurposing an Adenine Riboswitch into a Fluorogenic Imaging and Sensing Tag, Nat. Chem. Biol., 2022, 18(2), 180–190, DOI:10.1038/s41589-021-00925-0.
- E. V. Dolgosheina, S. C. Y. Jeng, S. S. S. Panchapakesan, R. Cojocaru, P. S. K. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, RNA Mango Aptamer-Fluorophore: A Bright, High-Affinity Complex for RNA Labeling and Tracking, ACS Chem. Biol., 2014, 9(10), 2412–2420, DOI:10.1021/cb500499x.
- K. Y. Kong, S. C. Jeng, B. Rayyan and P. J. Unrau, RNA Peach and Mango: Orthogonal Two-Color Fluorogenic Aptamers Distinguish Nearly Identical Ligands, RNA, 2021, 27(5), 604–615 CrossRef CAS PubMed.
- W. Song, G. S. Filonov, H. Kim, M. Hirsch, X. Li, J. D. Moon and S. R. Jaffrey, Imaging RNA Polymerase III Transcription Using a Photostable RNA–Fluorophore Complex, Nat. Chem. Biol., 2017, 13(11), 1187–1194, DOI:10.1038/nchembio.2477.
- L. F. M. Passalacqua, M. R. Starich, K. A. Link, J. Wu, J. R. Knutson, N. Tjandra, S. R. Jaffrey and A. R. Ferré-D’Amaré, Co-Crystal Structures of the Fluorogenic Aptamer Beetroot Show That Close Homology May Not Predict Similar RNA Architecture, Nat. Commun., 2023, 14(1), 2969, DOI:10.1038/s41467-023-38683-3.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, Broccoli: Rapid Selection of an RNA Mimic of Green Fluorescent Protein by Fluorescence-Based Selection and Directed Evolution, J. Am. Chem. Soc., 2014, 136(46), 16299–16308, DOI:10.1021/ja508478x.
- E. Braselmann, A. J. Wierzba, J. T. Polaski, M. Chromiński, Z. E. Holmes, S.-T. Hung, D. Batan, J. R. Wheeler, R. Parker, R. Jimenez, D. Gryko, R. T. Batey and A. E. Palmer, A Multicolor Riboswitch-Based Platform for Imaging of RNA in Live Mammalian Cells, Nat. Chem. Biol., 2018, 14(10), 964–971, DOI:10.1038/s41589-018-0103-7.
- E. Braselmann and A. E. Palmer, A Multicolor Riboswitch-Based Platform for Imaging of RNA in Live Mammalian Cells, Methods Enzymol., 2020, 641, 343–372, DOI:10.1016/bs.mie.2020.03.004.
- N. Sarfraz, E. Moscoso, T. Oertel, H. J. Lee, S. Ranjit and E. Braselmann, Visualizing Orthogonal RNAs Simultaneously in Live Mammalian Cells by Fluorescence Lifetime Imaging Microscopy (FLIM), Nat. Commun., 2023, 14(1), 867, DOI:10.1038/s41467-023-36531-y.
- N. Sarfraz, H. J. Lee, M. K. Rice, E. Moscoso, L. K. Shafik, E. Glasgow, S. Ranjit, B. J. Lambeck and E. Braselmann, Establishing Riboglow-FLIM to Visualize Noncoding RNAs inside Live Zebrafish Embryos, Biophys. Rep., 2023, 3(4), 100132, DOI:10.1016/j.bpr.2023.100132.
- J. T. Polaski, S. M. Webster, J. E. Johnson and R. T. Batey, Cobalamin Riboswitches Exhibit a Broad Range of Ability to Discriminate between Methylcobalamin and Adenosylcobalamin, J. Biol. Chem., 2017, 292(28), 11650–11658, DOI:10.1074/jbc.M117.787176.
- M. Lee and C. B. Grissom, Design, Synthesis, and Characterization of Fluorescent Cobalamin Analogues with High Quantum Efficiencies, Org. Lett., 2009, 11(12), 2499–2502 CrossRef CAS PubMed.
- J. T. Polaski, O. A. Kletzien, L. K. Drogalis and R. T. Batey, A Functional Genetic Screen Reveals Sequence Preferences within a Key Tertiary Interaction in Cobalamin Riboswitches Required for Ligand Selectivity, Nucleic Acids Res., 2018, 46(17), 9094–9105, DOI:10.1093/nar/gky539.
- K. Suhling, L. M. Hirvonen, J. A. Levitt, P.-H. Chung, C. Tregidgo, A. Le Marois, D. A. Rusakov, K. Zheng, S. Ameer-Beg and S. Poland, Fluorescence Lifetime Imaging (FLIM): Basic Concepts and Some Recent Developments, Med. Photonics, 2015, 27, 3–40 CrossRef.
- E. A. Shirshin, M. V. Shirmanova, A. V. Gayer, M. M. Lukina, E. E. Nikonova, B. P. Yakimov, G. S. Budylin, V. V. Dudenkova, N. I. Ignatova, D. V. Komarov, V. V. Yakovlev, W. Becker, E. V. Zagaynova, V. I. Shcheslavskiy and M. O. Scully, Label-Free Sensing of Cells with Fluorescence Lifetime Imaging: The Quest for Metabolic Heterogeneity, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(9), e2118241119, DOI:10.1073/pnas.2118241119.
- C. Allegrucci and L. E. Young, Differences between Human Embryonic Stem Cell Lines, Hum. Reprod. Update, 2007, 13(2), 103–120, DOI:10.1093/humupd/dml041.
- B. Carter and K. Zhao, The Epigenetic Basis of Cellular Heterogeneity, Nat. Rev. Genet., 2021, 22(4), 235–250, DOI:10.1038/s41576-020-00300-0.
- C. M. Davis, J. Deutsch and M. Gruebele, An in Vitro Mimic of In-Cell Solvation for Protein Folding Studies, Protein Sci., 2020, 29(4), 1046–1054, DOI:10.1002/pro.3833.
- M. Naciri, D. Kuystermans and M. Al-Rubeai, Monitoring pH and Dissolved Oxygen in Mammalian Cell Culture Using Optical Sensors, Cytotechnology, 2008, 57(3), 245–250, DOI:10.1007/s10616-008-9160-1.
- Y. Hayashi-Takanaka, K. Yamagata, T. Wakayama, T. J. Stasevich, T. Kainuma, T. Tsurimoto, M. Tachibana, Y. Shinkai, H. Kurumizaka, N. Nozaki and H. Kimura, Tracking Epigenetic Histone Modifications in Single Cells Using Fab-Based Live Endogenous Modification Labeling, Nucleic Acids Res., 2011, 39(15), 6475–6488, DOI:10.1093/nar/gkr343.
- C. A. Cialek, G. Galindo, A. L. Koch, M. N. Saxton and T. J. Stasevich, Bead Loading Proteins and Nucleic Acids into Adherent Human Cells, JoVE, 2021, 172, e62559, DOI:10.3791/62559.
- A. B. Mohseny, I. Machado, Y. Cai, K.-L. Schaefer, M. Serra, P. C. W. Hogendoorn, A. Llombart-Bosch and A.-M. Cleton-Jansen, Functional Characterization of Osteosarcoma Cell Lines Provides Representative Models to Study the Human Disease, Lab. Invest., 2011, 91(8), 1195–1205, DOI:10.1038/labinvest.2011.72.
- H. K. Brown, M. Tellez-Gabriel and D. Heymann, Cancer Stem Cells in Osteosarcoma, Cancer Lett., 2017, 386, 189–195 CrossRef CAS PubMed.
- J. Ponten and E. Saksela, Two Established in Vitro Cell Lines from Human Mesenchymal Tumours, Int. J. Cancer, 1967, 2(5), 434–447, DOI:10.1002/ijc.2910020505.
- R. M. McAllister, M. B. Gardner, A. E. Greene, C. Bradt, W. W. Nichols and B. H. Landing, Cultivation in Vitro of Cells Derived from a Human Osteosarcoma, Cancer, 1971, 27(2), 397–402, DOI:10.1002/1097-0142(197102)27:2.
- O. Hovatta, M. Mikkola, K. Gertow, A. Strömberg, J. Inzunza, J. Hreinsson, B. Rozell, E. Blennow, M. Andäng and L. Ährlund-Richter, A Culture System Using Human Foreskin Fibroblasts as Feeder Cells Allows Production of Human Embryonic Stem Cells, Hum. Reprod., 2003, 18(7), 1404–1409, DOI:10.1093/humrep/deg290.
- Q. Li, J. Xia, Y. Yao, D. Gong, H. Shi and Q. Zhou, Sulforaphane Inhibits Mammary Adipogenesis by Targeting Adipose Mesenchymal Stem Cells, Breast Cancer Res. Treat., 2013, 141(2), 317–324, DOI:10.1007/s10549-013-2672-1.
- W. Wagner and A. D. Ho, Mesenchymal Stem Cell Preparations—Comparing Apples and Oranges, Stem Cell Rev., 2007, 3(4), 239–248, DOI:10.1007/s12015-007-9001-1.
- A. Al Madhoun, S. Alkandari, H. Ali, N. Carrio, M. Atari, M. S. Bitar and F. Al-Mulla, Chemically Defined Conditions Mediate an Efficient Induction of Mesodermal Lineage from Human Umbilical Cord- and Bone Marrow- Mesenchymal Stem Cells and Dental Pulp Pluripotent-Like Stem Cells, Cell. Reprogram., 2018, 20(1), 9–16, DOI:10.1089/cell.2017.0028.
- Z. Li, Q. Xu, H. Peng, R. Cheng, Z. Sun and Z. Ye, IFN-γ Enhances HOS and U2OS Cell Lines Susceptibility to Γδ T Cell-Mediated Killing through the Fas/Fas Ligand Pathway, Int. Immunopharmacol., 2011, 11(4), 496–503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00197k |
| This journal is © The Royal Society of Chemistry 2024 |