
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTracking the cellular uptake and phototoxicity of Ru(II)-polypyridyl-1,8-naphthalimide Tröger's base conjugates†
Sandra A.
Bright‡
ab,
MariaLuisa
Erby‡
a,
Fergus E.
Poynton
b,
Daniel
Monteyne
c,
David
Pérez-Morga
cd,
Thorfinnur
Gunnlaugsson
 *be,
D. Clive
Williams
*a and
Robert B. P.
Elmes
*efg
*be,
D. Clive
Williams
*a and
Robert B. P.
Elmes
*efg
aSchool of Biochemistry and Immunology, Biomedical Sciences Institute, Trinity College, Dublin 2, Ireland. E-mail: clive.williams@tcd.ie; Tel: +353 1 8962596
bSchool of Chemistry, Centre for Synthesis and Chemical Biology, Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie; Tel: +353 1 8963459
cLaboratoire de Parasitologie Moléculaire, IBMM-DBM Université Libre de Bruxelles, Gosselies, Belgium
dCenter for Microscopy and Molecular Imaging CMMI Université Libre de Bruxelles, Gosselies, Belgium
eSynthesis and Solid State Pharmaceutical Centre (SSPC), University of Limerick, Ireland
fDepartment of Chemistry, Maynooth University, National University of Ireland, Maynooth, Co. Kildare, Ireland. E-mail: robert.elmes@mu.ie; Tel: +353 1708 4615
gKathleen Lonsdale Institute for Human Health Research, Maynooth University, Maynooth, Co. Kildare, Ireland
First published on 21st February 2024
Abstract
Ruthenium(II) complexes are attracting significant research attention as a promising class of photosensitizers (PSs) in photodynamic therapy (PDT). Having previously reported the synthesis of two novel Ru(II)-polypyridyl-1,8-naphthalimide Tröger's base compounds 1 and 2 with interesting photophysical properties, where the emission from either the Ru(II) polypyridyl centres or the naphthalimide moieties could be used to monitor binding to nucleic acids, we sought to use these compounds to investigate further and in more detail their biological profiling, which included unravelling their mechanism of cellular uptake, cellular trafficking and cellular responses to photoexcitation. Here we demonstrate that these compounds undergo rapid time dependent uptake in HeLa cells that involved energy dependent, caveolae and lipid raft-dependent mediated endocytosis, as demonstrated by confocal imaging, and transmission and scanning electron microscopy. Following endocytosis, both compounds were shown to localise to mostly lysosomal and Golgi apparatus compartments with some accumulation in mitochondria but no localisation was found to the nucleus. Upon photoactivation, the compounds increased ROS production and induced ROS-dependent apoptotic cell death. The photo-activated compounds subsequently induced DNA damage and altered tubulin, but not actin structures, which was likely to be an indirect effect of ROS production and induced apoptosis. Furthermore, by changing the concentration of the compounds or the laser used to illuminate the cells, the mechanism of cell death could be changed from apoptosis to necrosis. This is the first detailed biological study of Ru(II)-polypyridyl Tröger's bases and clearly suggests caveolae-dependent endocytosis is responsible for cell uptake – this may also explain the lack of nuclear uptake for these compounds and similar results observed for other Ru(II)-polypyridyl complexes. These conjugates are potential candidates for further development as PDT agents and may also be useful in mechanistic studies on cell uptake and trafficking.
Introduction
1,8-Naphthalimide Tröger's base (TB) structures are unique organic and orthogonal shaped molecular scaffolds, where two naphthalimide units are connected together via a diazocine moiety, which is chiral.1,2 In the past, we have shown the versatile application of such structures as DNA binders, therapeutic agents, sensors and imaging agents, coordination compounds, and polymers, where various functionalities have been incorporated at the imide side (e.g. amines, amino acids, peptides, sugars, coordination moieties, etc.) that dictate the nature of the application.3–13 Likewise, Shanmugaraju and co-workers have recently developed several excellent examples of naphthalimide Tröger's bases and demonstrated their application in a range of supramolecular chemistry applications.14–18 Ru(II) polypyridyl complexes are known DNA and RNA targeting agents, but biological profiling of such complexes has only recently been appreciated and studied in some detail.19–28 Hence, the pursuit of Ru(II) polypyridyl complexes as cellular imaging agents and potential theranostics is gathering pace with the recent progression of the thiophane containing TLD1433 to a phase II study for treating non-muscle invasive bladder cancer – the first ruthenium polypridyl based PDT agent to progress to clinical trials.29 Ru(II) polypyridyl complexes exhibit many advantageous biological properties including DNA binding, ligand exchange potential, solubility, lipophilicity, charge, and importantly, useful photophysical properties that have all driven an explosion of interest in this class of compounds.30,31 In particular, their potential as photo-activated chemotherapeutics and antimicrobial agents32–39 has been driven by low dark toxicity, exceptional photostability and their aforementioned spectroscopic characteristics. Recent reviews have extensively highlighted their in vitro and in vivo investigations.29,40–46Our interest in Ru(II) polypyridyls has focussed on novel Ru(II) conjugates where tethering the metal centre to various other functional subunits such as gold nanoparticles,47,48 or appended organic chromophores20,49–52 has yielded various modified designs that display high affinity DNA targeting, enhanced photophysical properties, and effective DNA photocleavage that we have shown can affect numerous biological pathways and initiate apoptosis. We have also investigated the various binding modes of metal ion complexes with oligonucleotides using ultrafast spectroscopy such as transient IR and transient absorption.53–55 Conjugation of Ru(II) centres to Tröger's bases has led to systems displaying interesting photophysical and DNA binding behaviour.56,57 Previously, we reported the synthesis and photophysical investigation into dinuclear Ru(II) polypyridyl complexes, with each metal core linked by 1,8-naphthalimide Tröger's base units, 1 and 2 (Fig. 1), and we demonstrated that they possess desirable photophysical properties, where energy transfer from the naphthalimide moiety can be used to populate the metal-to-ligand charge transfer (MLCT) excited state of the polypyridyl complexes. Furthermore, preliminary investigation showed that these complexes accumulate intracellularly in HeLa cells, most likely in organelles within the cytoplasm. However, it was clear to us that if we were to fully exploit the potential of this class of compounds for medicinal chemistry applications, a more in-depth knowledge of their underlying chemical biology was desirable. Particularly, regarding their cellular uptake and localisation, which has a direct effect on their downstream biological properties. There are numerous possible routes of entry into cancer cells from passive diffusion to various forms of active transport.58–61 For example, pinocytosis is commonly used to take up small samples of extracellular fluid through invaginations in the plasma membrane and it can occur in any cell type and is non-specific. On the other hand, phagocytosis is reserved for specialised cells, is specific, and uses pseudopodia for the uptake of particles greater than 0.75 μm in diameter such as cellular debris or micro-organisms.62 Receptor-dependent endocytosis such as clathrin-dependent endocytosis; caveolae-dependent endocytosis63 and the more recently described clathrin/caveolae-independent (flotillin, CLIC/GEEC, arf6) forms of endocytosis64 are also highly specific. Lipid rafts have been associated with both caveolae-dependent and clathrin/caveolae independent endocytosis mechanisms.65
 | ||
| Fig. 1 Structures of the binuclear Ru(II)-1,8-naphthalimide containing Tröger's bases, 1 and 2. | ||
There have been conflicting reports in the literature as to the localisation of Ru(II) polypyridyl complexes with some examples selectively localising to the nucleus of cells,66 while structurally analogous examples (with just a structural tweak) can lead to localisation in the endoplasmic reticulum.67 Moreover, while the desired target for the majority of Ru(II) complexes is to nuclear DNA (due to their interacting photophysical behaviour and photoreactivity with DNA) it is currently unclear if nuclear localisation is required to illicit any desired biological effect.68
From a photodynamic therapy (PDT) perspective, the therapeutic effect is mediated by the generation of reactive oxygen species (ROS), normally produced as a by-product of oxidative phosphorylation, and/or the formation of singlet oxygen (1O2).69–71 Tumour destruction is most efficient using compounds with a long lived triplet excited state and a high quantum yield for the generation of 1O2.72 Unlike conventional chemotherapy, photosensitisers outside the tumour region being illuminated are not photoactivated and are easily excreted over time with minimal side-effects. This suggests that nuclear targeting is not necessarily required for optimal activity. Any number of subcellular targets can be attacked during PDT including mitochondria (including mitochondrial DNA), lysosomes, plasma membranes and nuclei.73–76 However, a knowledge of the location of the photosensitizer following uptake into the cell is important due to the transient nature of 1O2.72 Such photo-induced damage leads to cell death and can induce inflammation and other tumour-specific immune reactions.77 Additionally, unlike classical PDT agents, Ru(II) polypyridyl complexes can also initiate apoptosis in cells by other means than through the singlet oxygen activation, as recently outlined by us and others in the field.19,78–82 In fact, often more than one activation pathway is available to such potential therapeutics, which makes Ru(II) polypyridyl complexes highly versatile and exciting therapeutic candidates for cancer and other diseases. The study of such agents in vitro and in vivo has been extensively featured in the review by Poynton and co-workers amongst others.41,83–86
Herein, we report a detailed biological investigation of the Ru(II) polypyridyl compounds 1 and 2 where we seek to evaluate their imaging potential, their mechanism of uptake into cells and subcellular localisation, together with their ability to act as PDT photosensitisers in a cancer cell model.56 This work provides a valuable insight in to the biological fate of this class of Ru(II)-NapTBs and may provide valuable information that aids in the design of further candidates with therapeutic potential.
Results
Cellular uptake of 1 and 2 in HeLa and peripheral blood mononuclear (PBMN) cells
Cell uptake experiments were performed to determine if the compounds were taken up by cancer cells and to investigate any effects of the compounds on cell morphology. HeLa is a human adherent cervix epithelial adenocarcinoma cell line, and HeLa cells were incubated with either 5 μM 1 or 2 for 2, 4, 8 or 24 h (Fig. 2(A)). During one photon confocal microscopy, the complexes were excited by a 488 nm argon laser while DAPI was used as a nuclear stain. Results from the 2 h incubation demonstrated 1 and 2 to be rapidly taken up into HeLa cells. After longer incubation times both compounds were increasingly observed at a peri-nuclear location within the cells. In most cases, the cells tolerated the compounds well and appeared healthy. In comparison, uptake of 1 or 2 into peripheral blood mononuclear cells (PBMCs) from healthy individuals (n = 3) was to a much lower level than for HeLa cells, with only a minority of cells exhibiting compound luminescence. While this result suggests that the compounds are preferentially taken up by cancer cells over non-cancer cells (Fig. 2(B)), cell uptake was also assessed and quantified by using flow cytometry in order to confirm this observation. Results showed luminescence within treated HeLa cells within 5 min, and the luminescence intensity increased in a time-dependent manner with the mean luminescence of compound 1 being higher than that observed for compound 2 at the same concentration (Fig. 2(C)). The emission spectra of the compounds within cells was also quantified by using confocal fluorescence microscopy and showed characteristic Ru(II) MLCT emission ranging from 540 nm to >750 nm, with a maximum at 620 nm (Fig. 2(D)). These emission spectra are consistent with the emission observed from the free complexes in solution.56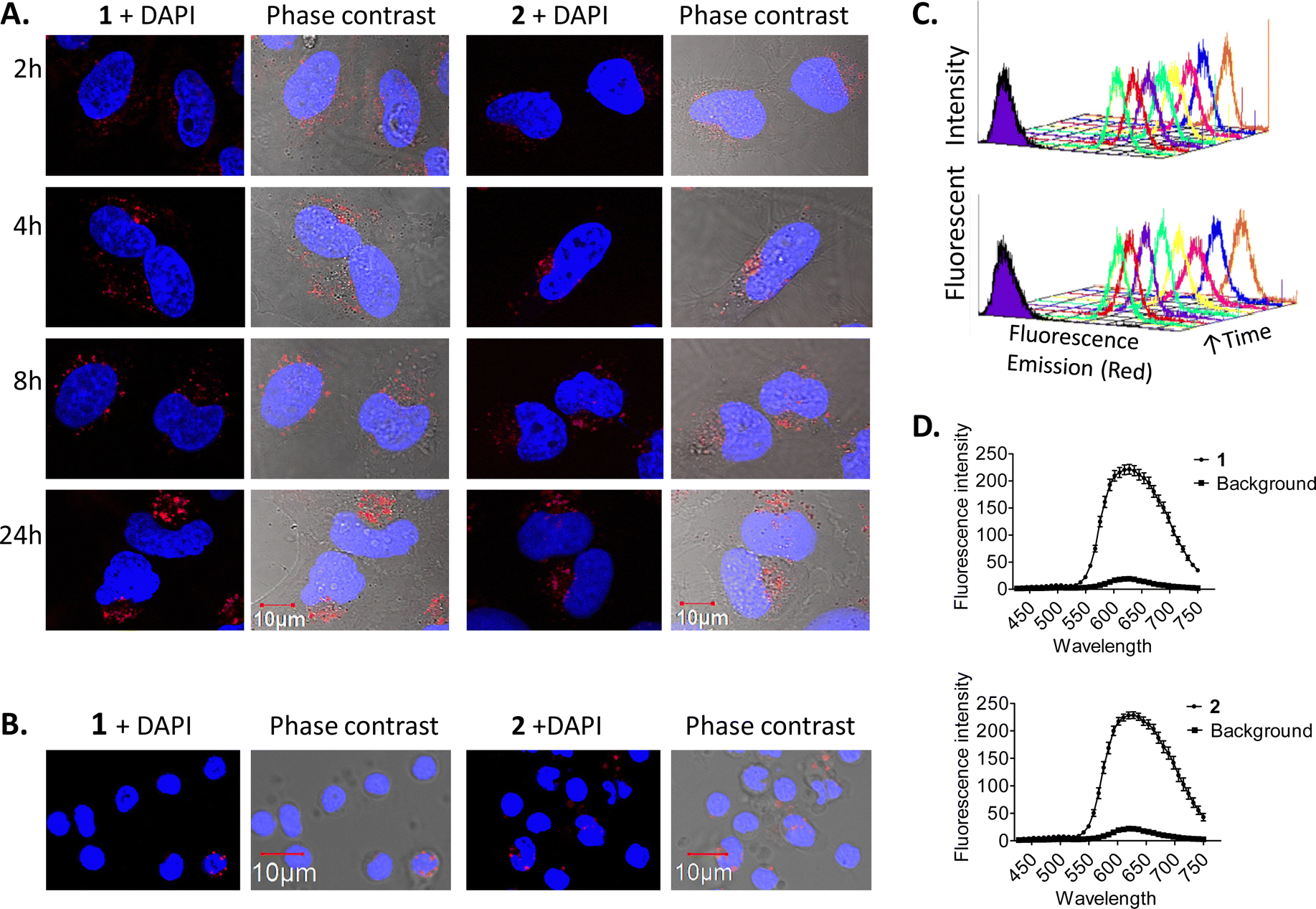 | ||
| Fig. 2 Time dependent uptake of 1 and 2 in HeLa cells. HeLa cells were incubated overnight before treatment with 5 μM of 1 or 2 for the indicated time-periods. (A) HeLa cells and (B) PBMCs were imaged by confocal microscopy, compounds were excited by a 488 nm argon laser, emission 620 nm and DAPI was excited by a 405 diode laser, emission 461 nm. (C) HeLa cells were analysed by flow cytometry. The compounds were excited by a 488 nm argon laser, with emission observed at 630 nm. Representative histograms for compounds 1 (top) and 2 (bottom) are displayed showing their uptake. (D) Emission spectra of the compounds in HeLa cells. Cells were imaged by confocal microscopy, compounds were excited by a 405 nm laser, fluorescence emission was quantified over a range of 410–780 nm. Results are representative of three independent experiments. | ||
Binding of ruthenium complexes to serum proteins has been shown to reduce the amount of free circulating Ru and can have a significant impact on the cellular uptake and localisation of complexes within cells.87–90 Indeed, most serum proteins cannot efficiently enter into cells, unless denatured or aggregated.91,92 We wished to ascertain whether 1 or 2 may bind to serum proteins and that the protein bound complexes may be responsible for the mechanism of uptake as opposed to the free complexes being uptaken. In order to investigate this theory, HeLa cells were cultured in normal serum-containing medium, rinsed and incubated with serum-free medium, followed by incubation with 1 or 2 for 4 h. Confocal fluorescence microscopy imaging confirmed that both compounds were successfully taken up into cells under serum-free conditions (Fig. 3(A)). After serum protein was ruled out as a possible mechanism for compound uptake, we next sought to investigate whether passive diffusion could play a significant role in their uptake. However, passive diffusion was eliminated as an uptake route for both compounds as no cellular internalisation was observed following a 4 h incubation at 4 °C as observed with confocal fluorescence microscopy (Fig. 3(B)) for either of these complexes.
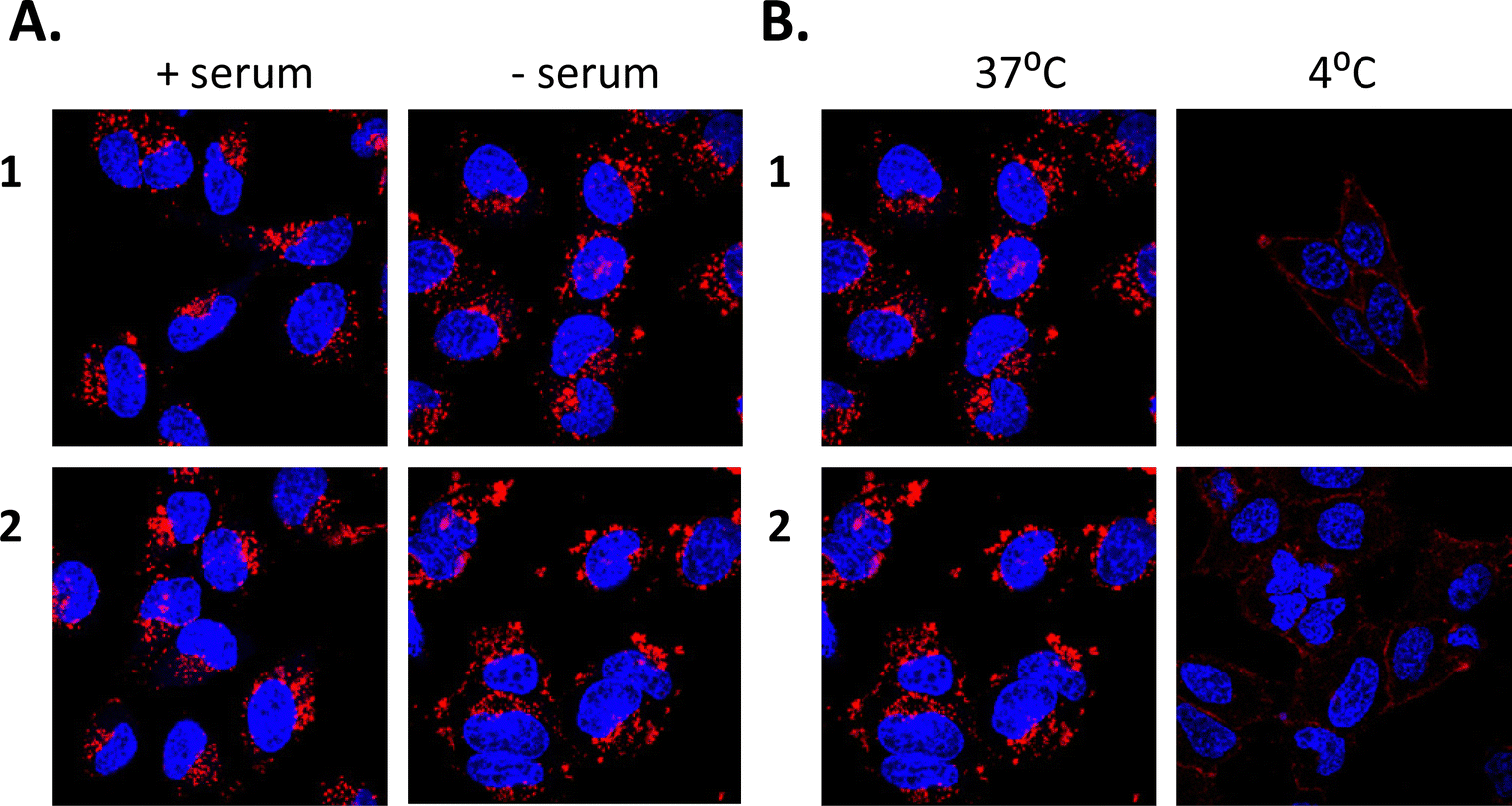 | ||
| Fig. 3 Uptake of 1 and 2 into HeLa cells is not due to passive diffusion or by binding to serum. HeLa cells were incubated overnight before treatment with 5 μM of 1 or 2 for the indicated time-periods. HeLa cells (A) with and without serum and (B) at different temperatures. Cells were imaged by confocal microscopy, compounds were excited by a 488 nm argon laser, emission 620 nm and DAPI was excited by a 405 diode laser, emission 461 nm. Results are representative of three independent experiments. | ||
Conversely, Dynasore, a general endocytosis inhibitor, effectively blocked uptake of compounds 1 and 2 into HeLa cells (Fig. 4(A)) after a 3 h pre-treatment with no obvious intracellular luminescence being observed by confocal fluorescence microscopy. To study this further, we used a range of inhibitors to determine which form of endocytosis could be operating; these included the use of chlorpromazine (which inhibits clathrin dependent endocytosis), genistein (which inhibits caveolae dependent endocytosis) and methyl-beta-cyclodextrin (MβCD) (which inhibits lipid raft endocytosis). Results from these confocal fluorescence microscopy experiments suggested that both genistein and MβCD blocked the uptake of 1 and 2 with the luminescence arising from the compounds only being observed in, or around, the edges of the plasma membrane. This would suggest that the compounds are taken up into cells via caveolae- and lipid raft-dependent endocytosis where chlorpromazine did not prevent any endocytosis of the compounds (Fig. 4(A)). These experiments were also repeated and quantified with flow cytometry and the results from these measurements showed a 9-fold reduction in uptake following pre-treatment with either genistein or dynasore (Fig. 4(B)). Interestingly, MβCD did not reduce the intracellular fluorescence of 1 or 2 as measured by flow cytometric analysis.
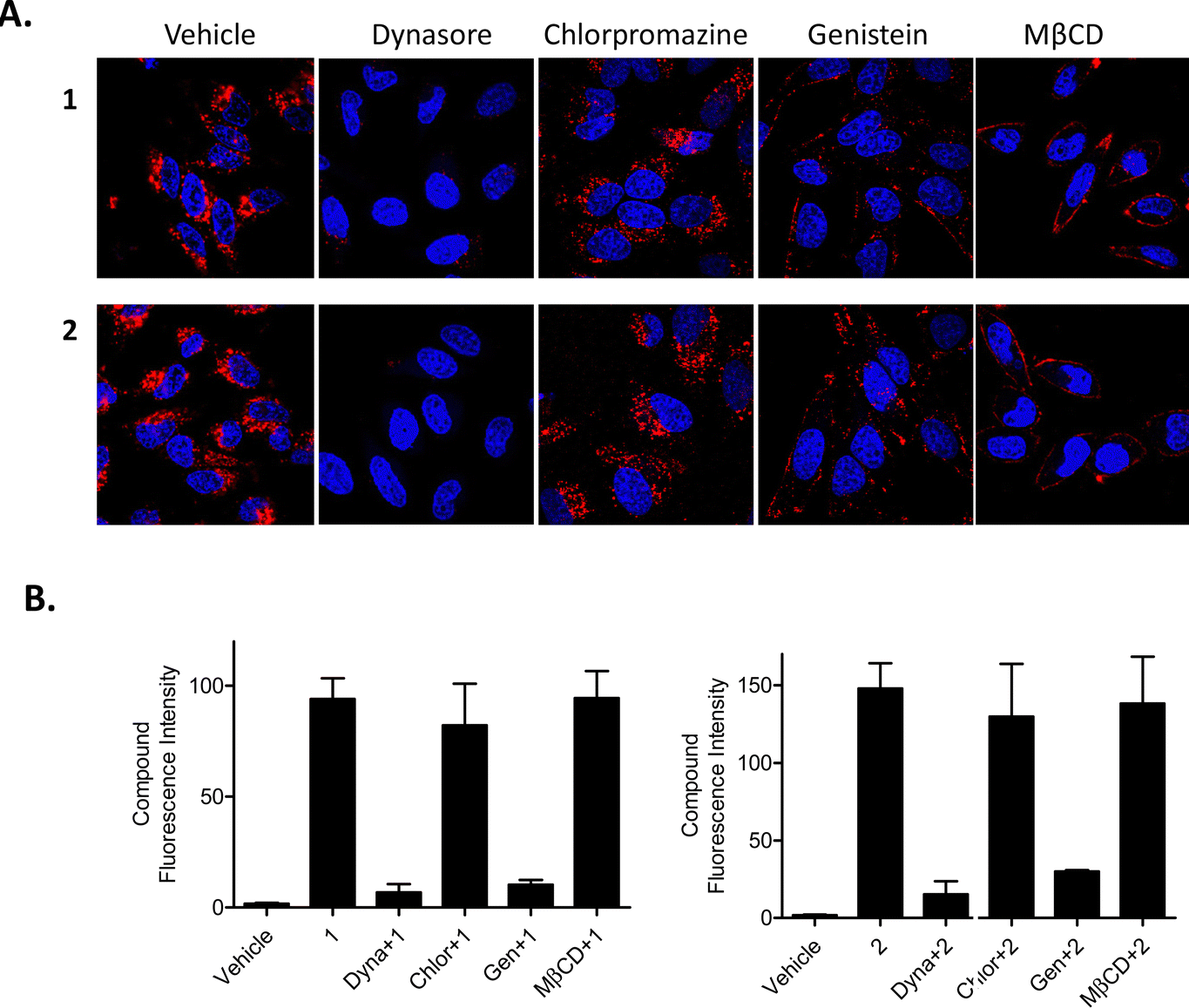 | ||
| Fig. 4 Compounds are taken up into cells via caveolae-dependent endocytosis. HeLa cells were incubated overnight before being subjected to the indicated treatment conditions. General, clathrin, caveolae and lipid raft-dependent endocytosis as assessed by (A) confocal microscopy and (B) flow cytometry. Compounds 1 and 2 were excited by a 488 nm argon laser, emission 620 nm, DAPI was excited by a 405 diode laser, emission 461 nm. Results are representative of three independent experiments. | ||
To further confirm the mechanism of endocytosis, the cells were subjected to TEM and SEM analysis following pre-treatment with either genistein or MβCD, and incubation with and without compound 1. In the absence of inhibitors, cells incubated with compound 1 displayed a typical morphology, similar to that of the control cells; cells were polymorphic containing a mix of lamellipodia and microvilli at the plasma membrane as indicated by SEM. All organelles looked healthy in cells incubated with or without compound 1 as indicated by TEM, confirming a general lack of toxicity with compound 1. A small amount of compound 1 could be observed as an electron dense ‘haze’ outside of the cells but darker contrast staining inside cells prevented observation of intracellular compound 1.
When cells were pre-treated with genistein, with or without compound 1, invaginations or open endocytotic vesicles were visible at the plasma membrane, where there was an inhibition of ‘pinching-off’ of these invaginations which were all in an omega-like state. Lipid droplets, swollen ER, mitophagy and peroxyphagy were also observed (Fig. 5(A)–(D)). Cells pretreated with genistein, and then incubated with compound 1 also showed the above-described morphologies, but now compound 1 could be observed as an electron-dense material, that was accumulated at the surface of the cells and at open invaginations. When cells were pre-treated with MβCD, some toxicity to the cells was observed and it was clear that MβCD induced some degree of cell death even in the absence of these complexes. In live cells, protrusions were smaller, fused together and of variable sizes compared to untreated cells. Microvilli were longer and more abundant. Multiple polymorphic vesicles of variable sizes were also observed at the plasma membranes and, along with autophagy and swollen ER particles were again associated with inhibitor treatment. As observed for genistein inhibition, 1 was again shown to accumulate on the outside of the cells (Fig. 5(E)–(H)).
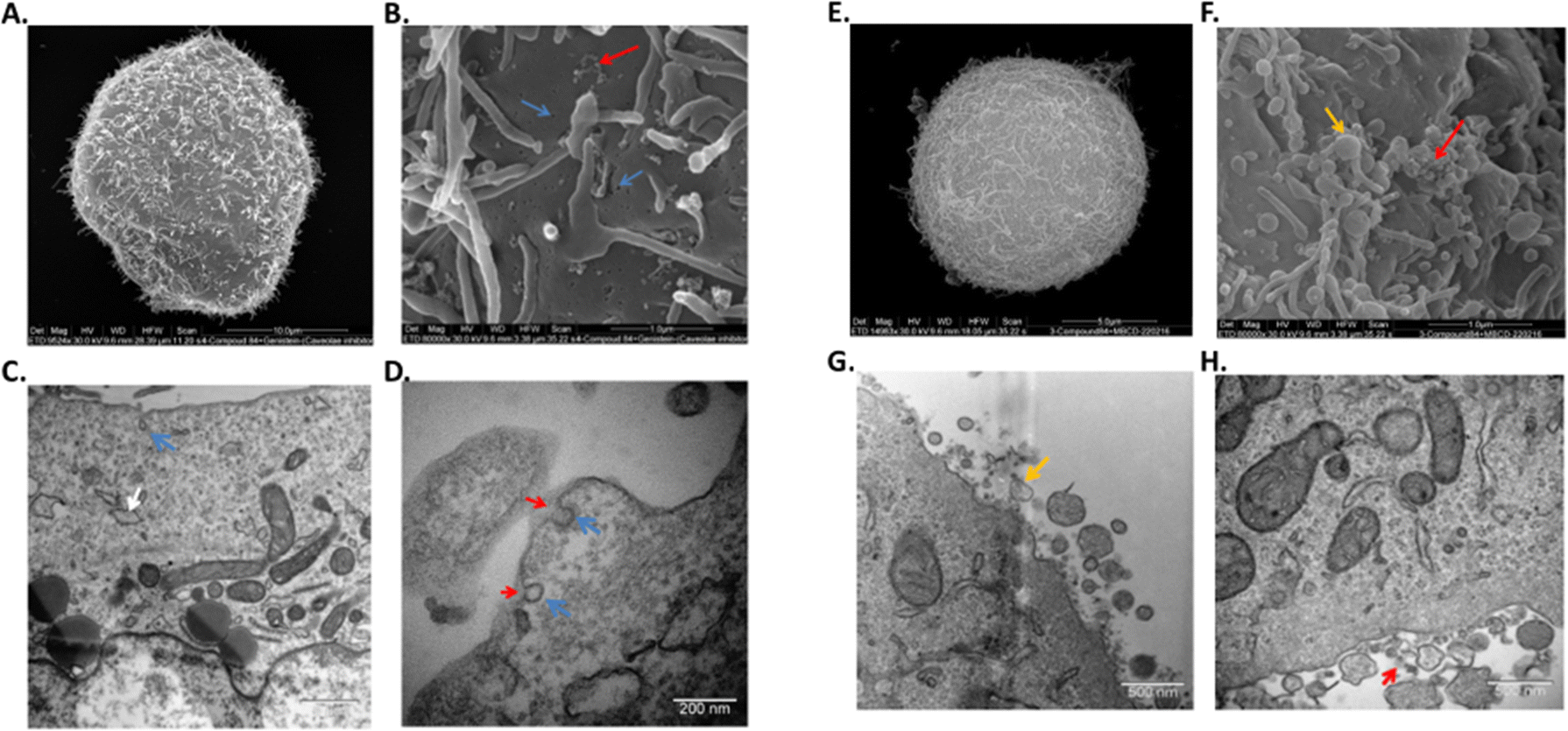 | ||
| Fig. 5 Endocytosis of compound 1 is caveoloe- and lipid raft-dependent. HeLa cells were incubated overnight before being subjected to pretreatment for 3 h with genistein (left) or MβCD (right) followed by 4 h incubation with compound 1. Images of (A), (E) a whole cell and (B), (F) plasma membrane by SEM. Images of (C) (G), (D) (H) cell and plasma membrane by TEM. Results are representative of two independent experiments. Invaginations – blue arrow, Compound 1 – red arrow, lipid droplets – white arrow, extracellular vesicles – yellow arrow. | ||
These results, taken together with the confocal fluorescence microscopy studies and the flow cytometry results, suggest the complexes prior to endocytosis, may be incorporated into vesicles at the plasma membrane with MβCD but inhibited from entering the cytoplasm or other organelles. With genistein, the complexes were also inhibited from entering the cytoplasm or other organelles, but additionally, they do not appear to be physically tethered to the plasma membrane. In this case, wash steps in sample preparation appear to eliminate the compound from being detected via flow cytometry. These results also suggest caution while interpreting confocal or electron microscopy results, without additional flow cytometry techniques.
Although after endocytosis, one might expect 1 and 2 to be routed to endosomes, lysosomes and/or to the Golgi apparatus/complex, previous results from our group suggested that some localised to and had a profound effect on mitochondria.93 We therefore wished to determine the subcellular location(s) of compounds 1 and 2 after uptake. Confocal microscopy of HeLa cells tagged with mitochondrial, Golgi apparatus and lysosomal markers, demonstrated the compounds to localise mostly to lysosomes (Fig. 6(B) and (D)) and the Golgi complex (Fig. 6(A) and (D)), with a small degree of co-localisation to mitochondria (Fig. 6(C) and (D)) following 24 h of uptake. These results suggest that after transport across the cellular plasma membrane via caveosomes, 1 and 2 localise mostly to Golgi complex and lysosomes, with a smaller amount present in mitochondria. This result is not entirely surprising as it is known that caveosomes fuse with the Golgi complex. Similarly, there are a number of reports demonstrating evidence of caveosomes fusing with lysosomes.91,94,95 Other reports have also demonstrated mitochondrial96 and lysosomal localisation97 or a mixture of both mitochondrial and lysosomal localization.93,98,99 In addition, Ru(II) polypyridyl compounds complexed to a porphyrin have been shown to mainly localise to lysosomes (in human nasopharyngeal carcinoma HK-1 cells100 and also to the cytoplasm of HeLa and HK-1 cells),101 whilst other Ru(II) polypyridyl complexes have demonstrated nuclear localisation.87,102–105
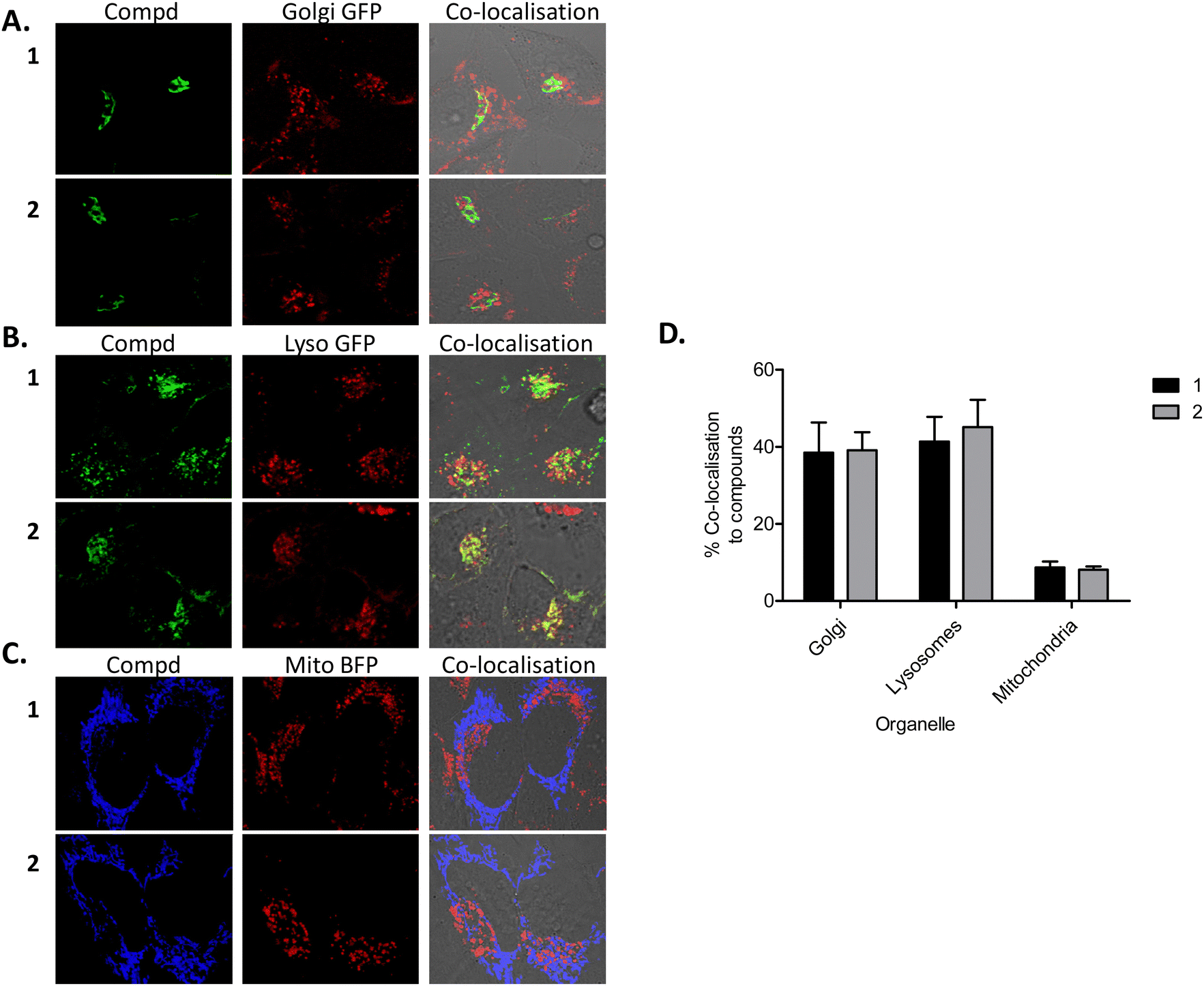 | ||
| Fig. 6 Compounds co-localise with lysosomes and the Golgi apparatus. Transfected HeLa cells were incubated for 24 h with 1 or 2, stained with DAPI and assessed for co-localisation. Compounds 1 and 2 were excited by a 488 nm argon laser, emission 620 nm and DAPI was excited by a 405 diode laser, emission 461 nm. Results are representative of three independent experiments. Confocal microscopy showing compounds (red), (A) Golgi apparatus (green) (B) lysosomes (green), (C) mitochondria (blue) and co-localisation (yellow). (D) Co-localisation quantified using Amaris software. Results are representative of three independent experiments. | ||
From the combination of experiments reported above, it is clear that both 1 and 2 enter HeLa cells predominantly by an endocytotic route. Serum protein binding of compounds was found not to be involved in endocytosis. However, further dissections of the uptake mechanism showed that caveolae and lipid raft-dependent endocytosis were the predominant routes of uptake. Little or no contribution to uptake was found from passive diffusion or active transport. Caveolae-dependent endocytosis has been associated with the uptake of a wide range of molecules including nanoparticles,106,107 polyplexes,108 liposomes109 and albumin,91,92 and as demonstrated by Cao et al., also associated with some ruthenium complexes, where a partial inhibition of uptake of a Ru complex with a caveolae inhibitor was recorded.95
The endocytotic route of entry into cells leading to accumulation into endosomes, lysosomes and Golgi apparatus, explains why these compounds and possibly other similar ones, cannot enter the nucleus as they are trapped in these organelles and thus are not free in the cytoplasmic milieu to diffuse to, and enter the nucleus via the nuclear pores. It could thus be anticipated that if these compound-loaded cells were fixed using alcohol or formaldehydes for microscopy studies, this would result in organelle membrane permeabilisation allowing diffusion of compound to the nuclear compartment, as found in studies by Svensson et al.110 and Ye et al.111 The location of 1 and 2 to the mitochondrial compartment is less pronounced than with previous compounds from our group93 and may be due to plasma-membrane potentially-driven uptake of the compounds across the plasma membrane or some release from the organelles. Either way, it is clear that the positively-charged compounds accumulate in various organelles inside the cells, are well tolerated, and display useful photophysics that allows their tracking through the various cellular compartments. Moreover, their rapid uptake and lack of toxicity in the dark provides the potential that 1 and 2 may be useful photoactivated therapeutic agents, as we demonstrate in the next section.
Light dependent cellular toxicity of 1 and 2
As many Ru(II) polypyridyl complexes are capable of efficiently generating singlet oxygen upon light activation,112,113 as well as partake in (photoinduced) electron or energy transfer mechanisms, compounds 1 and 2 were also expected to function as possible light activated therapeutic agents, where either of these mechanisms could operate either independently, or in synergy. In order to explore this, an AlamarBlue cell viability assay was undertaken with 1 and 2. The results confirmed that neither compounds reduced cell viability in the dark at concentrations below 10 μM. However, upon photo-activation (using 18 J cm−2) for 1 h followed by a further 23 h incubation, a significant concentration-dependent reduction in cell viability was observed with 1 and 2 showing IC50 values of 1.8 ± 0.3 and 4.6 ± 0.7 μM, respectively, compared to IC50 values of >10 μM for the non-photo-activated conditions (Fig. 7).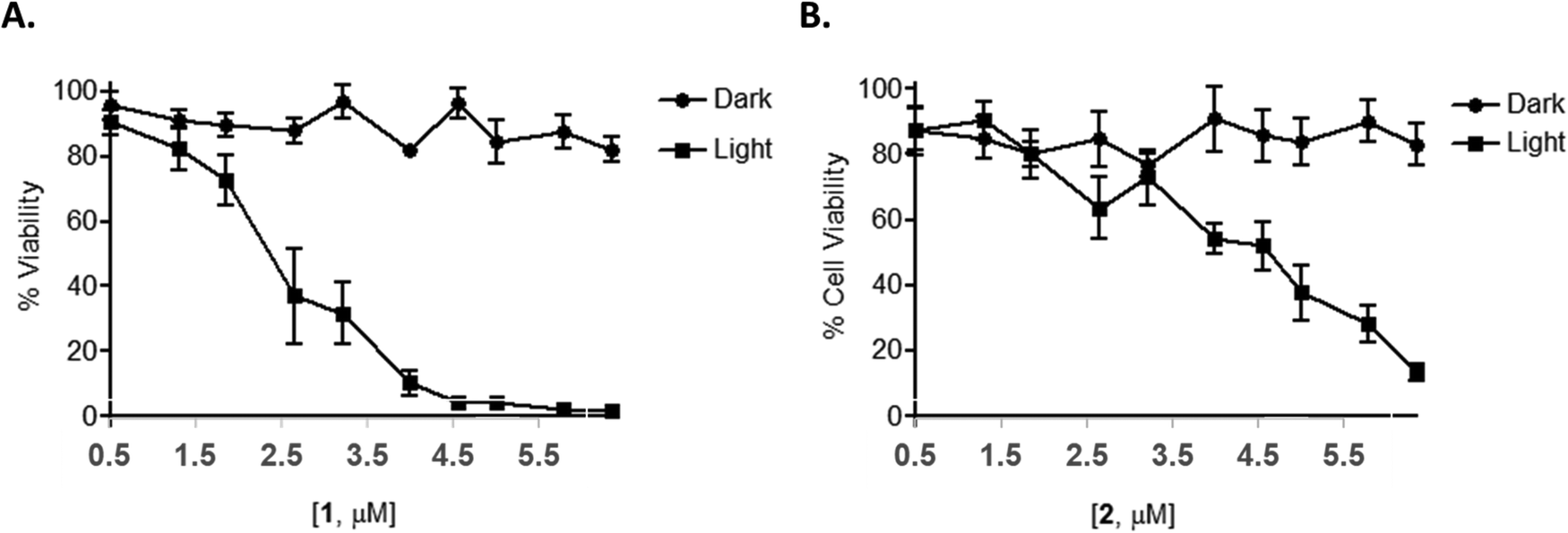 | ||
| Fig. 7 Light activated-1/2 potently reduce cellular viability in HeLa cells. HeLa cells were treated with a range of concentrations of 1 or 2. Cells were then either exposed to light for 1 h to give a light dose of 18 J cm−2 and further incubated overnight or maintained in the dark. Cells were subsequently assessed for cellular viability via an Alamar blue assay. (A) Compound 1 and (B) compound 2. | ||
To determine whether this photo-activated reduction of cellular viability also induced cell death, cells were subjected to flow cytometric Annexin V/propidium iodide (AnV/PI) analysis. The results showed a small increase in the number of cells undergoing early-stage apoptosis (AnV+/PI−) and a much larger number of cells undergoing late-stage apoptosis (AnV+/PI+) upon light activation at concentrations of 1 μM or higher for 1 and 2 (Fig. 8(A)). In order to determine if there was any necrotic cytotoxicity associated with photo-activated 1/2-induced apoptosis, a lactate dehydrogenase (LDH) assay was performed showing only a minimal increase in the release of LDH with increasing concentration of the compounds (Fig. 8(B)). Furthermore, PI cell cycle analysis also showed a significant increase in cell death as indicated by a hypodiploid (pre-G1) peak (and a corresponding decrease in G1 phase, between photo-activated and non photo-activated compounds) (Fig. 8(C)). There was no change in the number of cells in the G2/M phase of the cell cycle, regardless of treatment regime.
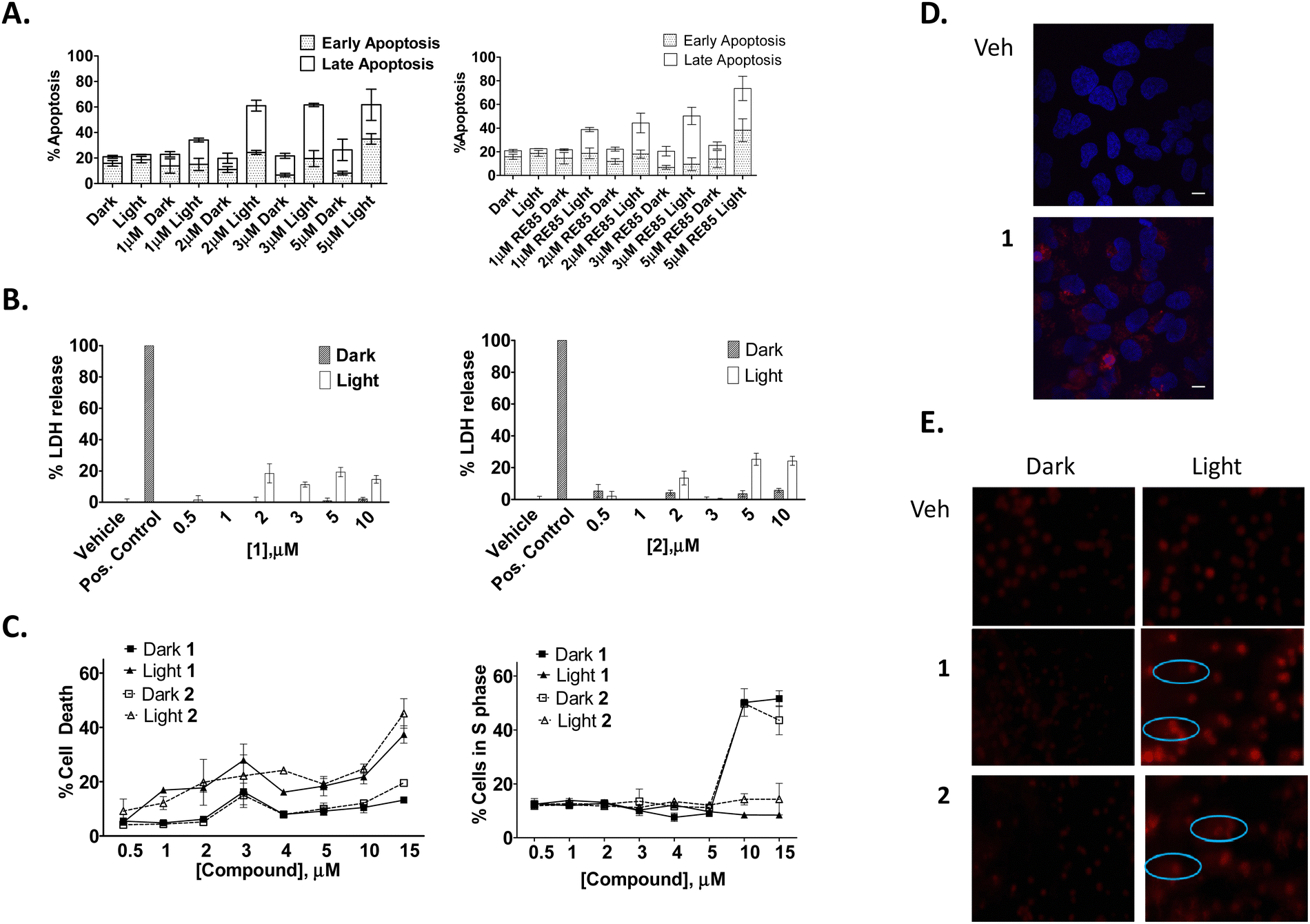 | ||
| Fig. 8 Compounds 1 and 2 potently induce photo activatable-apoptotic cell death and DNA damage. HeLa cells were seeded and treated with the indicated concentrations of 1 or 2. Where appropriate, cells were either exposed to light for 1 h to give a light dose of 18 J cm−2 and assessed for (A) apoptotic cell death via Annexin V/PI staining, (B) necrosis via an LDH assay, (C) flow cytometric cell cycle analysis, (D) fluorescent emission via two photon microscopy and (E) DNA damage via a comet assay. All data points were analysed using GRAPH PAD Prism software. Results are representative of three independent experiments. | ||
Given the known propensity of Ru(II) polypyridyl complexes to initiate DNA cleavage, a Comet assay was also performed in order to investigate intracellular nuclear DNA fragmentation with and without light treatment. This experiment showed that HeLa cells treated with a range of concentrations of 1 or 2 and incubated for 24 h before being exposed to light or maintained in the dark (Fig. 8(D)) showed some comet tails at 1 μM. However, at higher concentrations the DNA was not visible. We suspect that this is due to the efficiency of cell death where DNA strand cleavage is so advanced that fragments are too small to be retained in the gel.
Next, a second set of phototoxicity experiments were conducted using 2-photon excitation. The range of light used in PDT is most often between 600–900 nm due to strong absorption of endogenous molecules, such as haemoglobin and cytochromes that capture most of the incoming photons. Longer wavelengths of light can penetrate deeper into the tissue,114 however above 900 nm, water can absorb infrared light leading to high scattering in tissues. It is therefore important to develop photosensitizers which absorb light within this 600–900 nm near-IR region of the spectrum. As compounds 1 and 2 are activated below these wavelengths (488 nm), we used two photon microscopy to determine if the compounds could similarly be activated at a higher wavelength. Two photon microscopy results showed both compounds to be easily visualised in cells when activated at 790 nm using this technique (Fig. 8(E)). The luminescence from the compounds was observed, as with one photon microscopy, at 630 nm suggesting any further mechanistic experiments would yield the same results with either technique. Other studies have also demonstrated the two-photon induced emission spectrum of Ru(II) polypyridyl complexes in solution,100 in cells101,115 and in vivo.87,116,117 However, this is the first time that we have successfully used two-photon excitation to visualize a Ru(II) polypyridyl based naphthalimide conjugates in vitro.
As the predominant therapeutic effect in PDT is mediated by the generation of ROS, it was of interest to see if 1 and 2 illicit their cytotoxic effects in a similar manner. A ROS indicator, 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (10 μM), was used to quantify any increase in the production of ROS following photo-activation of 1 and 2 treated HeLa cells. This experiment showed an increase in ROS production in a concentration- and light-dependent manner (effects were more pronounced with compound 1) (Fig. 9(A)), suggesting ROS production as one of the causes of cell death. Moreover, the addition of NAC (N-acetyl cysteine), a ROS scavenger, reduced the light-dependent ROS production, but only in cells incubated with compound 1 (Fig. 9(B)).
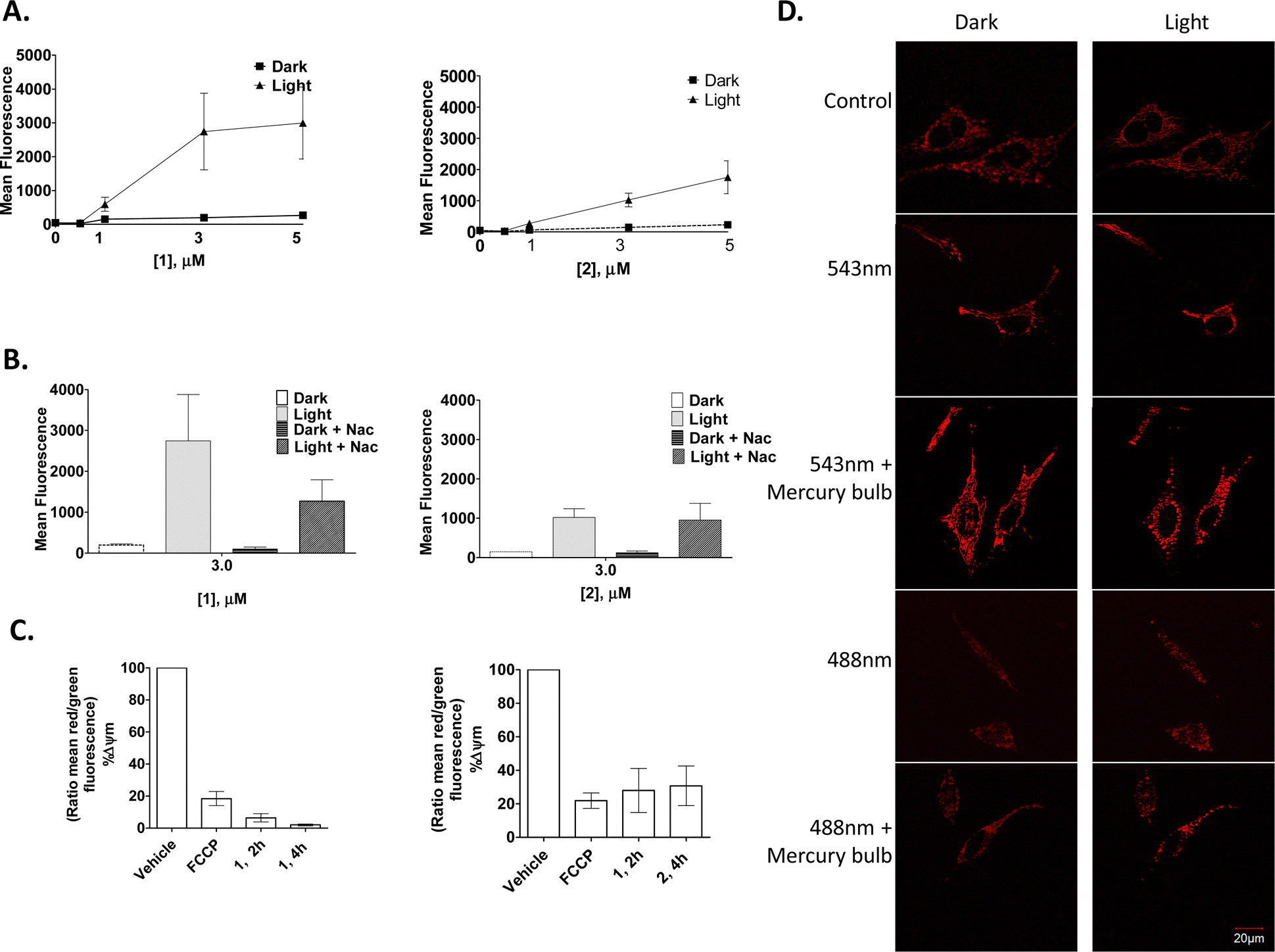 | ||
| Fig. 9 1 and 2 induce ROS production, decrease mitochondrial membrane potential and distort mitochondrial morphology in HeLa cells. | ||
To investigate if the compounds by themselves, without any photo-activation, could affect mitochondrial membrane potential (MMP), the JC-1 MMP-sensing dye was utilised. Here, a large and rapid decrease in the mitochondrial membrane potential occurred after only 2 h of treatment, and again, as seen above, complex 1 decreased the mitochondrial membrane potential to a greater extent than that seen for 2 (Fig. 9(C)).
Confocal fluorescence microscopy was used to investigate any effect of the compounds on mitochondrial morphology. For this, stably transfected HeLa DsRed were used to visualise the mitochondria. Results showed 1 to have a dramatic effect on the mitochondria within a few minutes after a short laser irradiation, which consisted of rounding and/or swelling of the mitochondria. It was observed that cells irradiated with a 488 nm laser together with the 543 nm laser accelerated this mitochondrial rounding. Moreover, the use of the mercury bulb (white-green region) in the confocal fluorescence microscopy made this process even more dramatic. These drastic changes in the mitochondrial-morphology suggest that mitochondrial fission occurred after light exposure in the presence of 1, as the mitochondrial morphology of the untreated cells did not change after light exposure with any laser combination or exposure time (Fig. 9(D)).
HeLa cells were seeded and treated with 1 or 2 as indicate and assessed for (A) ROS production using the indicator DCFH-DA or (B) ROS production following pretreatment (1 h) with the ROS scavenger NAC, (C) mitochondrial membrane potential using JC-1 or (D) mitochondrial morphology with either 4% 543 nm laser, 10% 488 nm laser and/or mercury bulb, left – cells in the dark; right – light activation. Results are representative of three independent experiments.
The above results show that, although only accumulating in the mitochondria to a smaller extent than other organelles (see discussion above), both complexes 1 and 2 are capable of inducing rapid effects on the mitochondrial morphology, and that they reduce the mitochondrial membrane potential as well as inducing a large amount of ROS production following light activation with PDT. Additionally, it is likely that both 1 and 2 also exert their effects on mitochondria indirectly through activation of signalling pathways, through release from both the Golgi apparatus and lysosome vesicles, or possibly via transport directly from the plasma membrane into the cytoplasm. These results are in good agreement with results published for several Ru(II) polypyridyl complexes that have also been reported to reduce the mitochondrial membrane potential upon light activation.87,90,118,119 In fact, through some of our own work, we have demonstrated reduced mitochondrial membrane potential with other Ru(II) polypyridyl compounds upon light activation, increasing intracellular ROS production.93 The Warburg effect, where cancer cells produce most of their ATP through glycolysis even under aerobic conditions, may explain why non-photoactivated cells can remain healthy and viable despite such a great reduction in the mitochondrial membrane potential, indicative of mitochondrial damage.
Importantly, given the observed lack of nuclear penetration, and hence DNA binding, of either 1 or 2, it might have been expected that they would not show such light-dependent cell death. However, potent light and ROS-dependent cell death was found, suggesting a different mode of action to what might have been expected. When compared to structurally simpler Ru(II) complexes, 1 and 2 are more potent with lower IC50 values and wider therapeutic intervals.93 IC50 values were comparable with literature reports of other Ru compounds.120–123 This data together with an absence of toxicity in cells without light treatment, suggest 1 and 2 as potentially suitable PDT agents.
The effect of 1 and 2 on F-actin and α-Tubulin
Finally, to probe the effects of 1 and 2 on the cytoskeleton structure, their binding to, actin and tubulin filaments were next investigated through the use of cellular imaging. For this, fixed samples were used where HeLa cells were incubated with 1 or 2 (5 μM) for 24 h and either incubated in the dark or illuminated for 1 h, followed by a further 23 h incubation. Samples were then stained for either F-actin or tubulin with DAPI used to visualise the nucleus. The confocal fluorescence microscopy images showed that there was no obvious change in the actin structure after light treatment (Fig. 10(A)), however, cell shrinkage was evident in these samples. On the other hand, a drastic tubulin disruption was shown to occur after light treatment with compounds 1 and 2, with evidence of depolymerised-microtubules fragments around the nucleus after light exposure. This was supported by the fact that cells that were not subjected to light activation stayed healthy, showing no morphological changes indicating that cells can tolerate either complex extremely well in the absence of light activation (Fig. 10(B)).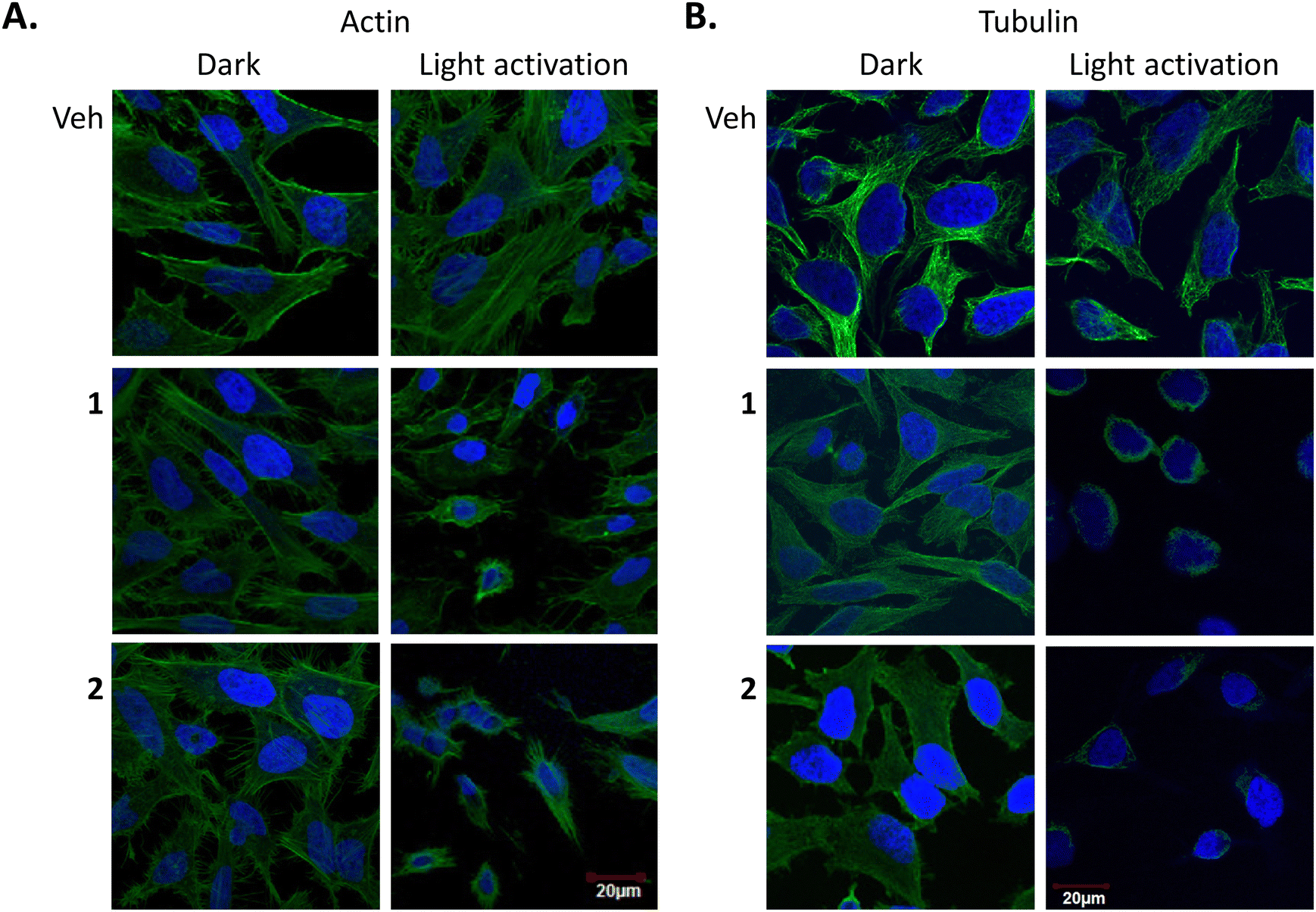 | ||
| Fig. 10 Effect of 1 and 2 on actin and α-tubulin in HeLa cells. | ||
These results confirm that compounds 1 and 2 have a profound effect on microtubules. Tubulin appeared depolymerised and was most likely a late downstream effect of apoptosis. Previous studies have shown other PDT agents cause damage to the cytoskeleton, however to the best of our knowledge, this is the first study to demonstrate the effect of Ru(II) polypyridyl PDT compounds on actin and tubulin. For example, studies on the photosensitizing effects of zinc(II)-phthalocyanine (ZnPc) on the cytoskeleton of HeLa cells showed photodamage to microtubules, actin microfilaments and intermediate filaments of keratin, as well as on α-actinin.124
HeLa cells were seeded and treated with the indicated concentrations of 1 or 2 for 48 h. Where appropriate, cells were either exposed to light for 1 h to give a light dose of 18 J cm−2. Cells were then fixed in paraformaldehyde and stained for DAPI and actin/tubulin. Image viewing using Olympus FV1000 confocal microscopy with a 60× oil immersion lens and analysed using FluoView Version 7.1 Software. DAPI was excited by a 405 laser, emission 461 nm (A) Actin: phalloidin 488 (ex 488 nm, em 518 nm). (B) Tubulin: anti-α-tubulin (ex 633 nm, em 647 nm). Results are representative of three independent experiments.
Conclusion
Given the reported applications of Ru(II) polypyridyl complexes as DNA binding and photoreactive agents, it is true that many of the reported examples either haven’t been assessed in vitro, or have been shown not to enter the nucleus of cells in live cell imaging studies.87 Thus, a study of the detailed mechanism of uptake and cell trafficking of this important group of compounds, which have such potential for cellular imaging applications or photoactivated light therapy, is needed to facilitate further use within such settings and with the view of developing more targeting and potent analogues. Here, we sought to use the two Ru(II)-polypyridyl-1,8-naphthalimide Tröger's base complexes 1 and 2, which are rapidly uptaken by HeLa cells (and do not show any degree of ‘dark toxicity’ up to a concentration of 10 μM), to investigate the detailed mechanism of the cellular uptake, cellular trafficking and cellular responses. We have confirmed that both 1 and 2 are rapidly taken up into HeLa cells within 2 h while uptake into non-cancerous PBMCs under the same conditions was minimal. The mechanism of uptake into cancer cells was determined to be caveolae and lipid raft-dependent endocytosis where 1 and 2 were efficiently endocytosed even under serum free conditions. Following endocytosis, 1 and 2 accumulated in several organelles including lysosomes, the Golgi apparatus, and to a lesser extent, the mitochondria.Upon photoactivation in the visible region, both compounds 1 and 2 induce a large amount of ROS that causes rapid distortion of mitochondrial morphology by inhibiting fusion, destruction of mitochondrial function and loss of mitochondrial proteins into the cytoplasm. These events trigger the initiation of signal-transduction pathways and the induction of apoptosis. Finally, cytoskeleton and DNA damage was also observed, and thought to be an indirect result of ROS formation that may diffuse through the cell or a downstream effect of apoptosis.
The results here further demonstrate the utility of Ru(II) polypyridyl complexes as strong candidates for development as a new molecular systems for light activated therapeutics in the treatment of cancer. While complexes such as those studied here require activation with light in the visible region, using a two-photon excitation strategy these complexes could be activated with light at 790 nm, thereby enabling their activation deeper in tissues. Moreover, the ability to understand the underlying mechanism of action will allow for rational design of future candidates. We are in the progress of developing alternative systems based on these results with the view of enhancing both their targeting and therapeutic nature.
Materials and methods
Compounds 1 and 2 were synthesised and described as previously reported.56Cell culture
HeLa cell line was cultured in DMEM + Glutamax medium supplemented with 10% (v/v) FBS, pen-strep (50 μg ml−1). For phototoxicity studies, cells were treated with the indicated concentration of the compounds, or a control, for 24 h, before being washed with fresh media and irradiated using a Hamamatsu L2570 200 W HgXe arc lamp equipped with a NaNO2 filter to give light doses of ∼18 J cm−2, followed by incubation for a further 24 h before being analysed. The radiation spectrum of the light source is shown in Fig. S1 (ESI†). A sodium nitrate liquid filter was used to exclude light at wavelengths lower than 390 nm.HeLaDsRed cells were stably transfected with pDsRed2-mito plasmid (Clontech, CA, USA) which encodes a fusion fluorescent protein and mitochondrial targeting sequence from subunit VIII of human cytocrome c oxidase.
Peripheral blood mononuclear cells (PBMCs)
Fresh peripheral blood (10 ml) was collected with written informed consent from healthy volunteers (n = 3) in EDTA-anticoagulant tubes. Peripheral blood was diluted with an equal volume of RPMI medium and PBMCs were isolated via lymphoprep Ficoll gradient centrifugation. PBMCs were resuspended in DMEM (Glutamax) medium supplemented with 10% (v/v) FBS, 50 μg ml−1 penicillin/streptomycin and seeded and treated as required.Live subject statement
Written informed consent was obtained from all subjects according to the guidelines specified in the Declaration of Helsinki. The study was approved by the St James's Hospital and Federated Dublin Voluntary Hospitals’ Joint Ethics Committee, Dublin, Ireland.Confocal fluorescence microscopy
Electron microscopy
For scanning electron microscopy, cells were fixed with 2.5% glutaraldehyde overnight and rinsed in cacodylate buffer 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M, pH 7.0. After serial dehydration in increasing concentrations of ethanol and finally acetone, samples were dried at CO2 critical point, then opened and mounted on scanning electron microscopy stubs. Observations were performed with an ESEM Quanta F200 (FEI-Thermo Fisher) microscope and secondary electron images captured with an Everhart-Thornley detector. Images were analyzed and processed by iTEM software. For transmission electron microscopy analyses of thin sections, cells were fixed with 2.5% glutaraldehyde (EM grade, electron microscopy sciences) and post-fixed twice in 1% OsO4 (with 1.5% ferrocyanide). Samples were stained with uranyl acetate (UA), serially dehydrated in increasing ethanol concentrations, and embedded in epoxy resin (Agar 100 resin, Agar Scientific Ltd, UK). Sectioning was done on a Leica EM UC7 ultramicrotome and ultrathin (50–70nm thick) sections were further stained with UA and lead citrate by electron microscopy standard procedures. Observations were made on a Tecnai 10 electron microscope (FEI-Thermo Fisher) and images were captured with a Veleta CCD camera and processed with SIS iTem software (Olympus).
000 cells and the CELLQUEST software package. The compounds were excited by a 488 nm argon laser, with emission observed between 593 and 633 nm. The percentage of luminescence in the cells was analysed and graphed as the mean ± S.E.M in GRAPHPAD Prism software.
M, pH 7.0. After serial dehydration in increasing concentrations of ethanol and finally acetone, samples were dried at CO2 critical point, then opened and mounted on scanning electron microscopy stubs. Observations were performed with an ESEM Quanta F200 (FEI-Thermo Fisher) microscope and secondary electron images captured with an Everhart-Thornley detector. Images were analyzed and processed by iTEM software. For transmission electron microscopy analyses of thin sections, cells were fixed with 2.5% glutaraldehyde (EM grade, electron microscopy sciences) and post-fixed twice in 1% OsO4 (with 1.5% ferrocyanide). Samples were stained with uranyl acetate (UA), serially dehydrated in increasing ethanol concentrations, and embedded in epoxy resin (Agar 100 resin, Agar Scientific Ltd, UK). Sectioning was done on a Leica EM UC7 ultramicrotome and ultrathin (50–70nm thick) sections were further stained with UA and lead citrate by electron microscopy standard procedures. Observations were made on a Tecnai 10 electron microscope (FEI-Thermo Fisher) and images were captured with a Veleta CCD camera and processed with SIS iTem software (Olympus).
000 cells and the CELLQUEST software package. The compounds were excited by a 488 nm argon laser, with emission observed between 593 and 633 nm. The percentage of luminescence in the cells was analysed and graphed as the mean ± S.E.M in GRAPHPAD Prism software.
Cell death assays
000 gated cells once cell debris had been excluded.
000 cells and using Summit software package. The standard compensation was performed using the untreated control, cells stained only with Annexin V or PI, cells stained with both AnnexinV/PI and by excluding the fluorescence of the compounds. The percentage of fluorescence into the cells was analysed using Flow Jo software.
Mitochondrial assays
000 cells and using Summit software package. The percentage of fluorescence within the cells was analysed using Flow Jo software. The ratio of red/green mean fluorescence values represent the mitochondrial ΔΨ and were expressed as a percentage of the untreated control which is taken as 100%.
Abbreviations
| ER | Endoplasmic reticulum |
| FACS | Fluorescent activated cell sorter |
| MβCD | Methyl-β-cyclodextrin |
| MLCT | Metal-to-ligand charge-transfer |
| MMP | Mitochondrial membrane potential |
| NAC | N-acetyl cysteine |
| PDT | Photodynamic therapy |
| PI | Propidium iodide |
| ROS | Reactive oxygen species |
| PBMCs | Peripheral blood mononuclear cells |
| SEM | Scanning electron microscopy |
| TEM | Transmission electron microscopy |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Science Foundation Ireland (SFI RFP 2009, SFI 2010 PI and 2013 PI grants), HEA PRTLI Cycle 4, The Irish Research Council (IRC Postgraduate Studentship to RBPE and FEP), Master and Back Altaformazione Programma, 2009, Autonomous Region of Sardinia and TCD for financial support. RBPE and TG acknowledge funding from Science Foundation Ireland (SFI, SSPC), grant number 12/RC/2275/P2, which is co-funded under the European Regional Development Fund. SFI are also acknowledged for the funding of the NMR facility (12/RI/2346/SOF) at Maynooth University through the Research Infrastructure Programme and the Advion Compact Mass Spec through the Opportunistic Infrastructure Fund (16/RI/3399). The CMMI is supported by the European Regional Development Fund and the Walloon Region. We would also like to thank Brenton Cavanagh, RCSI, Ireland, for assisting with the two-photon studies.References
- N. R. Deprez, K. A. McNitt, M. E. Petersen, R. G. Brown and D. E. Lewis, Tetrahedron Lett., 2005, 46, 2149–2153 CrossRef CAS.
- E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513–5525 CrossRef CAS PubMed.
- S. Murphy, S. A. Bright, F. E. Poynton, T. McCabe, J. A. Kitchen, E. B. Veale, D. C. Williams and T. Gunnlaugsson, Org. Biomol. Chem., 2014, 12, 6610–6623 RSC.
- S. Shanmugaraju, C. Dabadie, K. Byrne, A. J. Savyasachi, D. Umadevi, W. Schmitt, J. A. Kitchen and T. Gunnlaugsson, Chem. Sci., 2017, 8, 1535–1546 RSC.
- S. Shanmugaraju, C. S. Hawes, A. J. Savyasachi, S. Blasco, J. A. Kitchen and T. Gunnlaugsson, Chem. Commun., 2017, 53, 12512–12515 RSC.
- S. Shanmugaraju, D. Umadevi, L. M. González-Barcia, J. M. Delente, K. Byrne, W. Schmitt, G. W. Watson and T. Gunnlaugsson, Chem. Commun., 2019, 55, 12140–12143 RSC.
- J. M. Delente, D. Umadevi, K. Byrne, W. Schmitt, G. W. Watson, T. Gunnlaugsson and S. Shanmugaraju, Supramol. Chem., 2020, 32, 508–517 CrossRef CAS.
- J. I. Lovitt, D. Umadevi, P. Raja Lakshmi, B. Twamley, T. Gunnlaugsson and S. Shanmugaraju, Supramol. Chem., 2020, 32, 620–633 CrossRef CAS.
- S. A. Murphy, O. Kotova, S. Comby and T. Gunnlaugsson, Results Chem., 2021, 3, 100128 CrossRef CAS.
- T. Gorai, J. I. Lovitt, D. Umadevi, G. McManus and T. Gunnlaugsson, Chem. Sci., 2022, 13, 7805–7813 RSC.
- B. Mohan, S. Estalayo-Adrián, D. Umadevi, B. La Cour Poulsen, S. Blasco, G. J. McManus, T. Gunnlaugsson and S. Shanmugaraju, Inorg. Chem., 2022, 61, 11592–11599 CrossRef CAS PubMed.
- E. B. Veale, D. O. Frimannsson, M. Lawler and T. Gunnlaugsson, Org. Lett., 2009, 11, 4040–4043 CrossRef CAS PubMed.
- E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513–5525 CrossRef CAS PubMed.
- B. Mohan and S. Shanmugaraju, Dalton Trans., 2023, 52, 2566–2570 RSC.
- S. Kannan, R. Maayuri and S. Shanmugaraju, Inorg. Chim. Acta, 2023, 550, 121432 CrossRef CAS.
- B. Mohan, D. Sarkar, P. Raja Lakshmi, D. Umadevi and S. Shanmugaraju, J. Photochem. Photobiol., A, 2023, 441, 114727 CrossRef CAS.
- B. Mohan, D. Umadevi and S. Shanmugaraju, Sens. Diagn., 2023, 2, 262–267 RSC.
- A. Shanmughan, M. A. Nithasha, B. Mohan, D. Umadevi and S. Shanmugaraju, Polym. Chem., 2023, 14, 4153–4159 RSC.
- S. M. Cloonan, R. B. P. Elmes, M. Erby, S. A. Bright, F. E. Poynton, D. E. Nolan, S. J. Quinn, T. Gunnlaugsson and D. C. Williams, J. Med. Chem., 2015, 58, 4494–4505 CrossRef CAS PubMed.
- G. J. Ryan, F. E. Poynton, R. B. P. Elmes, M. Erby, D. C. Williams, S. J. Quinn and T. Gunnlaugsson, Dalton Trans., 2015, 44, 16332–16344 RSC.
- B. C. Poulsen, S. Estalayo-Adrián, S. Blasco, S. A. Bright, J. M. Kelly, D. C. Williams and T. Gunnlaugsson, Dalton Trans., 2016, 45, 18208–18220 RSC.
- K. L. Smitten, H. M. Southam, J. B. De La Serna, M. R. Gill, P. J. Jarman, C. G. W. Smythe, R. K. Poole and J. A. Thomas, ACS Nano, 2019, 13, 5133–5146 CrossRef CAS PubMed.
- N. A. Yusoh, S. W. Leong, S. L. Chia, S. N. Harun, M. B. A. Rahman, K. A. Vallis, M. R. Gill and H. Ahmad, ACS Chem. Biol., 2020, 15, 378–387 CrossRef CAS PubMed.
- S. N. Harun, H. Ahmad, H. N. Lim, S. L. Chia and M. R. Gill, Pharmaceutics, 2021, 13, 150 CrossRef CAS PubMed.
- M. R. Gill, P. J. Jarman, V. Hearnden, S. D. Fairbanks, M. Bassetto, H. Maib, J. Palmer, K. R. Ayscough, J. A. Thomas and C. Smythe, Angew. Chem., Int. Ed., 2022, 61, e202117449 CrossRef CAS PubMed.
- N. A. Yusoh, S. L. Chia, N. Saad, H. Ahmad and M. R. Gill, Sci. Rep., 2023, 13, 1456 CrossRef CAS PubMed.
- N. A. Yusoh, P. R. Tiley, S. D. James, S. N. Harun, J. A. Thomas, N. Saad, L. W. Hii, S. L. Chia, M. R. Gill and H. Ahmad, J. Med. Chem., 2023, 66, 6922–6937 CrossRef CAS PubMed.
- K. Smitten, H. M. Southam, S. Fairbanks, A. Graf, A. Chauvet and J. A. Thomas, Chem. Eur. J., 2023, 29, e202203555 CrossRef CAS PubMed.
- S. Monro, K. L. Colon, H. Yin, J. Roque, 3rd, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, A. Leonidova, S. Ferrari and G. Gasser, Eur. J. Inorg. Chem., 2015, 3879–3891 CrossRef CAS.
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Chem. Sci., 2016, 7, 6115–6124 RSC.
- A. Notaro and G. Gasser, Chem. Soc. Rev., 2017, 46, 7317–7337 RSC.
- M. Flamme, E. Clarke, G. Gasser and M. Hollenstein, Molecules, 2018, 23, 1515 CrossRef PubMed.
- E. Villemin, Y. C. Ong, C. M. Thomas and G. Gasser, Nat. Rev. Chem., 2019, 3, 261–282 CrossRef CAS.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed.
- K. L. Smitten, E. J. Thick, H. M. Southam, J. Bernardino de la Serna, S. J. Foster and J. A. Thomas, Chem. Sci., 2020, 11, 8828–8838 RSC.
- F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- M. Jakubaszek, B. Goud, S. Ferrari and G. Gasser, Chem. Commun., 2018, 54, 13040–13059 RSC.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC.
- L. Holden, C. S. Burke, D. Cullinane and T. E. Keyes, RSC Chem. Biol., 2021, 2, 1021–1049 RSC.
- J. Mo, N. P. Mai Le and R. Priefer, Eur. J. Med. Chem., 2021, 225, 113770 CrossRef CAS PubMed.
- Pragti, B. K. Kundu and S. Mukhopadhyay, Coord. Chem. Rev., 2021, 448, 214169 CrossRef CAS.
- J. Jiang, T. Teunens, J. Tisaun, L. Denuit and C. Moucheron, Molecules, 2022, 27, 1541 CrossRef CAS PubMed.
- R. B. P. Elmes, K. N. Orange, S. M. Cloonan, D. C. Williams and T. Gunnlaugsson, J. Am. Chem. Soc., 2011, 133, 15862–15865 CrossRef CAS PubMed.
- M. Martinez-Calvo, K. N. Orange, R. B. P. Elmes, B. la Cour Poulsen, D. C. Williams and T. Gunnlaugsson, Nanoscale, 2016, 8, 563–574 RSC.
- B. C. Poulsen, S. Estalayo-Adrián, S. Blasco, S. A. Bright, J. M. Kelly, D. C. Williams and T. Gunnlaugsson, Dalton Trans., 2016, 45, 18208–18220 RSC.
- G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401–403 CrossRef CAS PubMed.
- R. B. P. Elmes, G. J. Ryan, M. L. Erby, D. O. Frimannsson, J. A. Kitchen, M. Lawler, D. C. Williams, S. J. Quinn and T. Gunnlaugsson, Inorg. Chem., 2020, 59, 10874–10893 CrossRef CAS PubMed.
- G. J. Ryan, T. Gunnlaugsson and S. J. Quinn, Inorg. Chem., 2022, 61, 12073–12086 CrossRef CAS PubMed.
- F. R. Baptista, S. J. Devereux, S. P. Gurung, J. P. Hall, I. V. Sazanovich, M. Towrie, C. J. Cardin, J. A. Brazier, J. M. Kelly and S. J. Quinn, Chem. Commun., 2020, 56, 9703–9706 RSC.
- S. J. Devereux, F. E. Poynton, F. R. Baptista, T. Gunnlaugsson, C. J. Cardin, I. V. Sazanovich, M. Towrie, J. M. Kelly and S. J. Quinn, Chem. – Eur. J., 2020, 26, 17103–17109 CrossRef CAS PubMed.
- P. M. Keane, K. O’Sullivan, F. E. Poynton, B. C. Poulsen, I. V. Sazanovich, M. Towrie, C. J. Cardin, X. Z. Sun, M. W. George, T. Gunnlaugsson, S. J. Quinn and J. M. Kelly, Chem. Sci., 2020, 11, 8600–8609 RSC.
- R. B. P. Elmes, M. Erby, S. A. Bright, D. C. Williams and T. Gunnlaugsson, Chem. Commun., 2012, 48, 2588–2590 RSC.
- S. Shanmugaraju, B. la Cour Poulsen, T. Arisa, D. Umadevi, H. L. Dalton, C. S. Hawes, S. Estalayo-Adrián, A. J. Savyasachi, G. W. Watson, D. C. Williams and T. Gunnlaugsson, Chem. Commun., 2018, 54, 4120–4123 RSC.
- M. Groessl, O. Zava and P. J. Dyson, Metallomics, 2011, 3, 591–599 CrossRef CAS PubMed.
- K. Lin, Z. Z. Zhao, H. B. Bo, X. J. Hao and J. Q. Wang, Front. Pharmacol., 2018, 9, 1323 CrossRef CAS PubMed.
- S. Y. Lee, C. Y. Kim and T. G. Nam, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- Y. Lu, D. Zhu, Q. Le, Y. Wang and W. Wang, Nanoscale, 2022, 14, 16339–16375 RSC.
- M. Bohdanowicz and S. Grinstein, Physiol. Rev., 2013, 93, 69–106 CrossRef CAS PubMed.
- M. Kirkham and R. G. Parton, Biochim. Biophys. Acta., 2005, 1745, 273–286 CrossRef CAS PubMed.
- C. M. Blouin and C. Lamaze, Front. Immunol., 2013, 4, 267 Search PubMed.
- H. Ewers and A. Helenius, Cold Spring Harbor Perspect. Biol., 2011, 3, a004721 Search PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- W. M. Sharman, Drug Discovery Today, 1999, 507–517 CrossRef CAS PubMed.
- N. L. Oleinick and H. H. Evans, Radiat. Res., 1998, 150, S146–156 CrossRef CAS PubMed.
- P. Calin, J. Optoelectron. Adv. Mater., 2006, 8, 1173–1179 Search PubMed.
- Z. Luksiene, Medicina, 2003, 39, 1137–1150 Search PubMed.
- T. J. Dougherty, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- J. V. Moore, Phys. Med. Biol., 1997, 42, 913–935 CrossRef CAS PubMed.
- M. Ochsner, J. Photochem. Photobiol., B, 1997, 39, 1–18 CrossRef CAS PubMed.
- W. S. Chan, Anticancer Res., 1996, 1887–1892 CAS.
- T. Patrice, Photodynamic Therapy, The Royal Society of Chemistry, 1st edn, 2003 Search PubMed.
- B. Elias, C. Creely, G. W. Doorley, M. M. Feeney, C. Moucheron, A. Kirsch-DeMesmaeker, J. Dyer, D. C. Grills, M. W. George, P. Matousek, A. W. Parker, M. Towrie and J. M. Kelly, Chem. – Eur. J., 2008, 14, 369–375 CrossRef CAS PubMed.
- B. Elias and A. Kirsch-De Mesmaeker, Coord. Chem. Rev., 2006, 250, 1627–1641 CrossRef CAS.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet, A. Decottignies, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2018, 24, 19216–19227 CrossRef CAS PubMed.
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. MacNeil and J. A. Thomas, Chem. Sci., 2019, 10, 3502–3513 RSC.
- C. Reichardt, S. Monro, F. H. Sobotta, K. L. Colón, T. Sainuddin, M. Stephenson, E. Sampson, J. Roque, H. Yin, J. C. Brendel, C. G. Cameron, S. McFarland and B. Dietzek, Inorg. Chem., 2019, 58, 3156–3166 CrossRef CAS PubMed.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- P. M. Keane and J. M. Kelly, Coord. Chem. Rev., 2018, 364, 137–154 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- A. Rilak Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- P. Kaspler, S. Lazic, S. Forward, Y. Arenas, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2016, 15, 481–495 CrossRef CAS PubMed.
- F. Li, M. Feterl, J. M. Warner, A. I. Day, F. R. Keene and J. G. Collins, Dalton Trans., 2013, 42, 8868–8877 RSC.
- S. H. Lai, W. Li, J. H. Yao, B. J. Han, G. B. Jiang, C. Zhang, C. C. Zeng and Y. J. Liu, J. Photochem. Photobiol., B, 2016, 158, 39–48 CrossRef CAS PubMed.
- E. Dobrinskikh, K. Okamura, J. B. Kopp, R. B. Doctor and J. Blaine, Am. J. Physiol. Renal. Physiol., 2014, 306, F941–951 CrossRef CAS PubMed.
- T. Moriyama, T. Takei, M. Itabashi, K. Uchida, K. Tsuchiya and K. Nitta, J. Cell. Biochem., 2015, 116, 1060–1069 CrossRef CAS PubMed.
- S. M. Cloonan, R. B. Elmes, M. Erby, S. A. Bright, F. E. Poynton, D. E. Nolan, S. J. Quinn, T. Gunnlaugsson and D. C. Williams, J. Med. Chem., 2015, 58, 4494–4505 CrossRef CAS PubMed.
- P. J. Peters, A. J. Mironov, D. Peretz, E. van Donselaar, E. Leclerc, S. Erpel, S. J. DeArmond, D. R. Burton, R. A. Williamson, M. Vey and S. B. Prusiner, J. Cell Biol., 2003, 162, 703–717 CrossRef CAS PubMed.
- L. Cao, R. Zhang, W. Zhang, Z. Du, C. Liu, Z. Ye, B. Song and J. Yuan, Biomaterials, 2015, 68, 21–31 CrossRef CAS PubMed.
- J. Q. Wang, P. Y. Zhang, C. Qian, X. J. Hou, L. N. Ji and H. Chao, J. Biol. Inorg. Chem., 2014, 19, 335–348 CrossRef CAS PubMed.
- Q. Yu, Y. Liu, L. Xu, C. Zheng, F. Le, X. Qin, Y. Liu and J. Liu, Eur. J. Med. Chem., 2014, 82, 82–95 CrossRef CAS PubMed.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- Z. Zhao, Z. Luo, Q. Wu, W. Zheng, Y. Feng and T. Chen, Dalton Trans., 2014, 43, 17017–17028 RSC.
- C. T. Poon, P. S. Chan, C. Man, F. L. Jiang, R. N. Wong, N. K. Mak, D. W. Kwong, S. W. Tsao and W. K. Wong, J. Inorg. Biochem., 2010, 104, 62–70 CrossRef CAS PubMed.
- J. Zhang, K. L. Wong, W. K. Wong, N. K. Mak, D. W. Kwong and H. L. Tam, Org. Biomol. Chem., 2011, 9, 6004–6010 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gilles, Chem. – Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- Y. Ding, Q. Wu, K. Zheng, L. An, X. Hua and W. Mei, RSC Adv., 2015, 5, 63330–63337 RSC.
- Z. P. Zeng, Q. Wu, F. Y. Sun, K. D. Zheng and W. J. Mei, Inorg. Chem., 2016, 55, 5710–5718 CrossRef CAS PubMed.
- Z. Wang, C. Tiruppathi, J. Cho, R. D. Minshall and A. B. Malik, IUBMB Life, 2012, 63, 659–667 CrossRef PubMed.
- X. Hao, J. Wu, Y. Shan, M. Cai, X. Shang, J. Jiang and H. Wang, J. Phys.: Condens. Matter, 2012, 24, 164207 CrossRef PubMed.
- M. J. Reilly, J. D. Larsen and M. O. Sullivan, Mol. Pharmaceutics, 2012, 9, 1280–1290 CrossRef CAS PubMed.
- A. W. Wong, S. J. Scales and D. E. Reilly, J. Biol. Chem., 2007, 282, 22953–22963 CrossRef CAS PubMed.
- F. R. Svensson, J. Andersson, H. L. Åmand and P. Lincoln, J. Biol. Inorg. Chem., 2012, 17, 565–571 CrossRef CAS PubMed.
- R. R. Ye, Z. F. Ke, C. P. Tan, L. He, L. N. Ji and Z. W. Mao, Chem. Eur. J., 2013, 19, 10160–10169 CrossRef CAS PubMed.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- Y. Liu, R. Hammitt, D. A. Lutterman, L. E. Joyce, R. P. Thummel and C. Turro, Inorg. Chem., 2009, 48, 375–385 CrossRef CAS PubMed.
- Photodynamic therapy, ed. T. Patrice, 2003 Search PubMed.
- E. Baggaley, M. R. Gill, N. H. Green, D. Turton, I. V. Sazanovich, S. W. Botchway, C. Smythe, J. W. Haycock, J. A. Weinstein and J. A. Thomas, Angew. Chem., Int. Ed., 2014, 53, 3367–3371 CrossRef CAS PubMed.
- P. Zhang, H. Huang, Y. Chen, J. Wang, L. Ji and H. Chao, Biomaterials, 2015, 53, 522–531 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278–23290 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
- S. H. Lai, G. B. Jiang, J. H. Yao, W. Li, B. J. Han, C. Zhang, C. C. Zeng and Y. J. Liu, J. Inorg. Biochem., 2015, 152, 1–9 CrossRef CAS PubMed.
- K. J. Kilpin, C. M. Clavel, F. Edafe and P. J. Dyson, Organometallics, 2012, 31, 7031–7039 CrossRef CAS.
- S. Monro, J. Scott, A. Chouai, R. Lincoln, R. Zong, R. P. Thummel and S. A. McFarland, Inorg. Chem., 2010, 49, 2889–2900 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, M. Susan and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS PubMed.
- A. Juarranz, J. Espada, J. C. Stockert, A. Villanueva, S. Polo, V. Dominguez and M. Canete, Photochem. Photobiol., 2001, 73, 283–289 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00206c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |