
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA covalent compound selectively inhibits RNA demethylase ALKBH5 rather than FTO†
Gan-Qiang
Lai‡
ac,
Yali
Li‡
a,
Heping
Zhu‡
b,
Tao
Zhang
 a,
Jing
Gao
a,
Hu
Zhou
abc and
Cai-Guang
Yang
*abcd
a,
Jing
Gao
a,
Hu
Zhou
abc and
Cai-Guang
Yang
*abcd
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: yangcg@simm.ac.cn
bSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dShandong Laboratory of Yantai Drug Discovery, Bohai Rim Advanced Research Institute for Drug Discovery, Yantai 264117, China
First published on 19th February 2024
Abstract
N 6-Methyladenosine (m6A) is the most prevalent mRNA modification and is required for gene regulation in eukaryotes. ALKBH5, an m6A demethylase, is a promising target, particularly for anticancer drug discovery. However, the development of selective and potent inhibitors of ALKBH5 rather than FTO remains challenging. Herein, we used a targeted covalent inhibition strategy and identified a covalent inhibitor, TD19, which selectively inhibits ALKBH5 compared with FTO demethylase in protein-based and tumor cell-based assays. TD19 irreversibly modifies the residues C100 and C267, preventing ALKBH5 from binding to m6A-containing RNA. Moreover, TD19 displays good anticancer efficacy in acute myeloid leukemia and glioblastoma multiforme cell lines. Thus, the ALKBH5 inhibitor developed in this study, which selectively targets ALKBH5 compared with FTO, can potentially be used as a probe for investigating the biological functions of RNA demethylase and as a lead compound in anticancer research.
Introduction
N 6-Methyladenosine (m6A) is the most abundant methylation on eukaryotic mRNA;1 it is dynamically and reversibly regulated by the m6A methyltransferase complex (METTL3-METTL14/WTAP),2 m6A demethylases (fat mass and obesity-associated protein (FTO) and alkylation protein AlkB homolog 5 (ALKBH5)),3 and m6A binding proteins (such as YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3).4 The m6A modification has emerged as a key player in diverse diseases such as cancer,5 neurodegenerative disorders,6 and metabolic diseases;7 it affects gene expression by regulating mRNA splicing, localization, transport, translation, and degradation.8 ALKBH5 plays crucial roles in physiological processes in eukaryotes, including hypoxic stress and spermatogenesis.9 Dysregulated ALKBH5 expression is associated with tumorigenesis promotion in glioblastoma stem-like cells,10 and ALKBH5 overexpression in patients with glioblastoma multiforme (GBM) or acute myeloid leukemia (AML) is associated with poor outcomes.11 Therefore, ALKBH5 is a potential anticancer drug target and the development of ALKBH5 inhibitors has drawn much attention in recent years.12Both RNA demethylases, FTO and ALKBH5, belong to the AlkB family of nonheme Fe(II)- and 2-oxoglutarate (2OG)-dependent dioxygenases.13 In addition, both FTO and ALKBH5 have been established as oncogenic factors that elevated the demethylation of varying methylated mRNA targets in AML and GBM tumor cells. So, either the selective inhibitors for the individual RNA demethylase or the dual inhibitors for FTO/ALKBH5 are of significant importance since they could be used as chemical probes for each RNA demethylase and drug candidates for anticancer therapies. It remains unknown whether the dual inhibitors for FTO/ALKBH5 exhibit more potent antitumor activities than the selective inhibitors. Although 2OG analogs such as IOX314 and citrate15 and other inhibitors such as MV1035,16ALK-04,17cmp-3, cmp-6,1820m,19 and Ena15,20 generally exhibit good inhibitory activity on ALKBH5 demethylation in vitro, there still lacks highly selective and potent ALKBH5 inhibitors in vivo.21 Moreover, the inhibitory activities of these inhibitors on tumor cells are far from satisfactory. The tumor cells treated with ALKBH5 inhibitors did not exhibit the same phenotype as ALKBH5-knockdown cells. Although the recently reported DDO-2728 exhibited significantly improved selectivity on ALKBH5 over FTO in vitro and antitumor potency in vivo,22 the current challenges highlight the importance of exploring alternative compounds to selectively inhibit ALKBH5.
Targeted covalent inhibition (TCI) is an effective strategy for achieving selective enzyme modulation.23 In this study, we identified a covalent inhibitor, TD19, that targets specific cysteine residues in ALKBH5 and inhibits ALKBH5 without affecting FTO. TD19 inhibited the proliferation of both AML and GBM cell lines, underscoring its potential as a chemical tool for investigating the biological functions of ALKBH5 and as a candidate for anticancer drug in future.
Results and discussion
Tideglusib analog TD19 inhibits ALKBH5 rather than FTO
Five cysteine residues (C100, C200, C227, C230, and C267), surround the catalytic center in ALKBH5 (Fig. 1a). They are conserved across different species, such as mouse and zebrafish. However, these cysteines are not conserved in other AlkB homologs, including FTO, ALKBH2, and ALKBH3 (Fig. S1a, ESI†).24 Interestingly, the capability for a disulfide bond formation between C230 and C267 endows ALKBH5 with the ability to distinguish single-stranded and double-stranded dm6A-modified nucleic acid substrates.15 We thus wondered whether chemically modifying these cysteines would selectively inhibit ALKBH5 without displaying off-target effects on other homologous demethylases.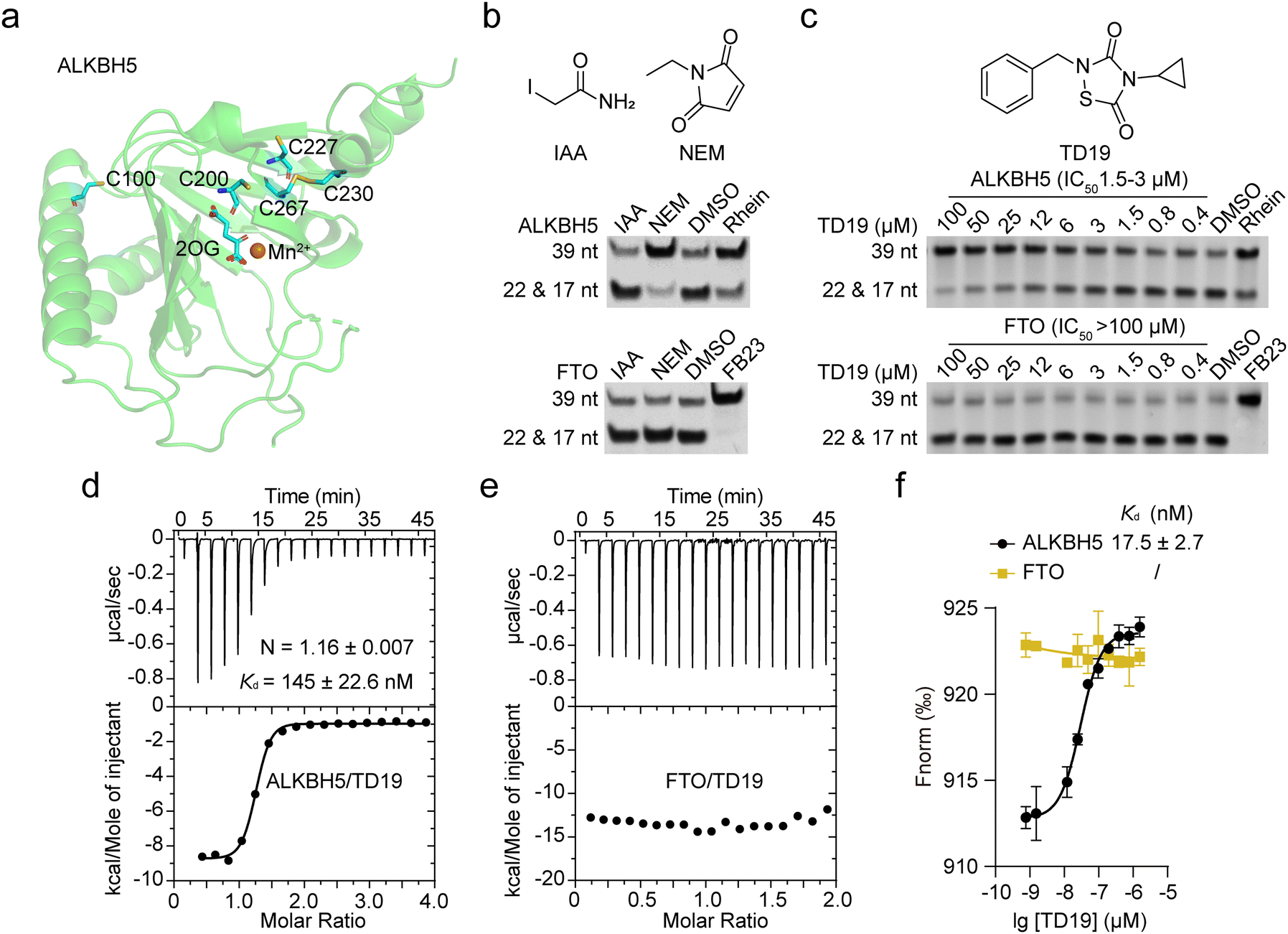 | ||
| Fig. 1 TD19 selectively inhibits ALKBH5 over FTO. (a) ALKBH5 structure (PDB ID: 4NRO) showing the five cysteines. The protein appears as green cartoon, 2OG and cysteines appear as sticks, and Mn2+ is the orange sphere. (b) Inhibitory effect of IAA and NEM (50 μM) on the demethylation of dm6A-modified nucleic acid substrates by ALKBH5 and FTO, assessed in a PAGE-based assay. (c) Quantification of TD19 inhibition on ALKBH5 and FTO in the PAGE-based assay. Rhein and FB23 were assayed as controls. (d) and (e) Binding of TD19 to ALKBH5 and FTO assessed by ITC titration. The dissociation constant (Kd) and stoichiometry factor (N) are indicated. (f) Binding affinity curves between TD19 and ALKBH5 or FTO in MST assay. | ||
We initially evaluated the inhibitory effects of iodoacetamide (IAA) and N-ethylmaleimide (NEM), two widely used cysteine alkylating agents, on ALKBH5 and FTO demethylation using a PAGE-based assay.25 The nonselective RNA demethylase inhibitor rhein and the specific FTO inhibitor FB23 were assayed as controls.26 Interestingly, NEM (50 μM) inhibited the demethylation of dm6A-modified nucleic acid substrates by ALKBH5 but not FTO, while IAA did not inhibit RNA demethylases (Fig. 1b). These data suggest that small-molecule modulators bearing proper electrophilic groups can selectively inhibit ALKBH5 through the chemical alkylation of cysteines. We then screened our in-house collection of covalent warhead-containing compounds on the inhibition of ALKBH5 and FTO. We found that TD19, an analog of tideglusib, whose analogs were previously identified as inhibitors of Staphylococcus aureus sortase A,27 selectively inhibited ALKBH5 over FTO. TD19 inhibited ALKBH5-induced demethylation with an IC50 of 1.5–3 μM while minimally inhibiting FTO and ALKBH3, even at 100 μM, as determined in the PAGE-based assay (Fig. 1c and Fig. S1b, ESI†).
To investigate the structure–activity relationship of the thiadiazolidinone scaffold of TD19 on the inhibitory activity toward ALKBH5, we prepared several TD19 analogs. As shown in Table S1 (ESI†), we kept the benzyl group of TD19 and changed the cyclopropyl to methyl, propyl, 2-chloroethyl, butyl, phenethyl, and benzyl moieties (compounds S1–S6, obtained in good yields). Like TD19, most of these analogs weakly inhibited FTO demethylation, with IC50 values above 50 μM. However, none of them inhibited ALKBH5 demethylation more potently than TD19. In addition, S7 and S8 analogs displayed a lower ALKBH5 demethylation inhibitory activity than TD19 (Table S1, ESI†). Thus, we chose TD19 to study the mode of action of this type of selective ALKBH5 inhibitor using protein binding and activity assays and tumor cell-based assays.
Next, we assessed the interaction between TD19 and RNA demethylases. The estimated dissociation constant (Kd) of TD19 binding to ALKBH5 was quantitatively estimated to be 145 ± 22.6 nM in an isothermal titration calorimetry (ITC) assay (Fig. 1d). Similarly, TD19 displayed a Kd of 17.5 ± 2.7 nM for binding to ALKBH5 in a microscale thermophoresis (MST) assay (Fig. 1f). However, neither in the ITC nor MST analyses did TD19 exhibit detectable binding to the FTO demethylase (Fig. 1e and f). These data indicate that TD19 selectively binds to ALKBH5 compared with FTO in vitro.
TD19 irreversibly inhibits ALKBH5
Next, we investigated the mode of action of the selective ALKBH5 inhibitor TD19. We preincubated human and zebrafish ALKBH5 (referred to as fALKBH5) in the presence of TD19 at different concentrations for various durations (0, 15, 30, and 60 min) and subsequently measured the IC50 values of TD19 in a modified FRET-based m6A detection assay (Fig. S2a, ESI†).28TD19 time-dependently inhibited ALKBH5. Its IC50 values (after 60 min of preincubation) were 1.1 ± 0.3 μM for ALKBH5 and 0.9 ± 0.4 μM for fALKBH5 (Fig. 2a and Fig. S2b, ESI†). Next, we found that the TD19-induced inhibition of ALKBH5 and fALKBH5 (after 60 min of preincubation) persisted after replacing the buffer to eliminate the unbound compounds (Fig. 2b and Fig. S2c, ESI†).29 Meanwhile, the rhein-induced inhibition of ALKBH5 demethylation is always time-independent and reversible (Fig. S2d, ESI†). Moreover, we observed that TD19 reacts with endogenous thiols such as GSH in a time- and concentration-dependent manner in vitro (data not shown), which indicated that the cellular effects of the endogenous thiols on TD19 should be carefully considered in future study.30 Based on these results, we suggested that TD19 inhibits ALKBH5 demethylation through sulfhydryl modification, similar to the RGS4 (regulator of G-protein signaling 4) inhibition mechanism by the tideglusib analog CCG-50014 (Scheme 1).31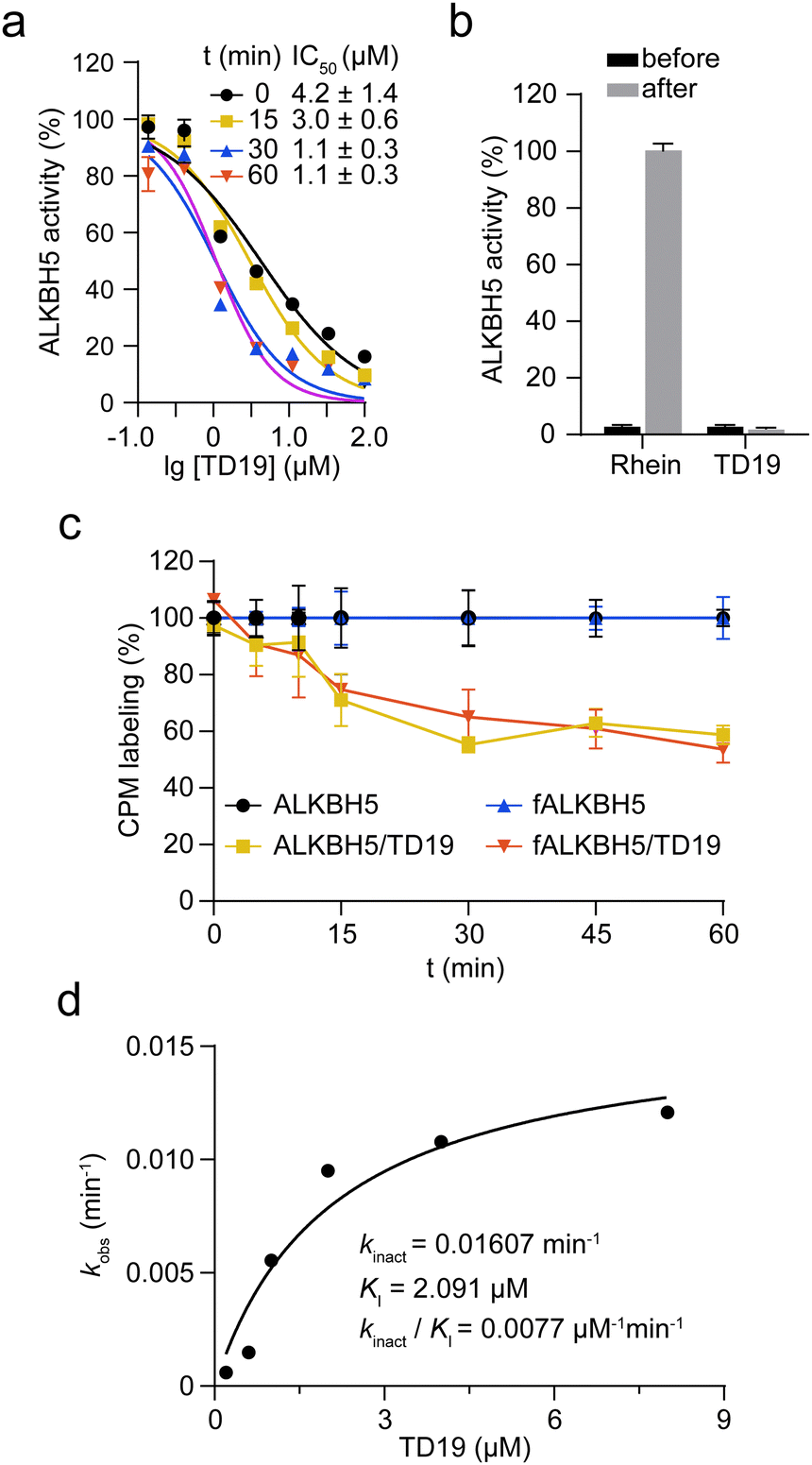 | ||
| Fig. 2 TD19 irreversibly inhibits ALKBH5. (a) Time-dependent irreversible inhibition of ALKBH5 by TD19, with IC50 values. (b) Effect of 5 μM TD19 and 100 μM rhein on the demethylation by ALKBH5 in a buffer replacement assay. The control samples (treated with DMSO) were used as the 100% activity reference. (c) Quantification of the CPM labeling of cysteines in ALKBH5 and fALKBH5 in the presence and absence of TD19. (d) Kinetic analysis of the TD19-induced ALKBH5 inhibition in a FRET-based m6A assay. | ||
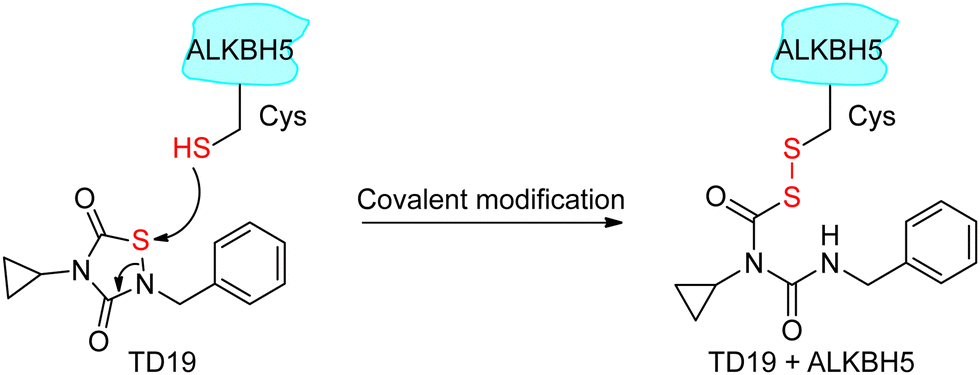 | ||
| Scheme 1 Proposed reaction of TD19 with a thiol in ALKBH5. | ||
To assess the importance of the sulfur–nitrogen (S–N) bond in TD19 for covalently binding to the cysteines in ALKBH5, we replaced it with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (compound S9). Interestingly, S9 completely lost its inhibitory activity toward ALKBH5, with an IC50 value of >50 μM (Table S2, ESI†). In addition, we prepared the S–S bond-containing compound S10, designed based on the ring-opening mechanism of the parental scaffold of TD19; this compound also showed a decreased inhibitory activity on ALKBH5 demethylation (Table S2, ESI†). These results demonstrated that the S–N bond in TD19 is required for the covalent inhibition of ALKBH5.
C bond (compound S9). Interestingly, S9 completely lost its inhibitory activity toward ALKBH5, with an IC50 value of >50 μM (Table S2, ESI†). In addition, we prepared the S–S bond-containing compound S10, designed based on the ring-opening mechanism of the parental scaffold of TD19; this compound also showed a decreased inhibitory activity on ALKBH5 demethylation (Table S2, ESI†). These results demonstrated that the S–N bond in TD19 is required for the covalent inhibition of ALKBH5.
To investigate the amount of ALKBH5 cysteines affected by TD19, we performed a labeling assay using 7-diethylamino-3-(4-maleimidophenyl)-4-methylcoumarin (CPM) as a cysteine-reactive probe.32 The covalent addition of TD19 to cysteine residues renders the thiol group unavailable to react with CPM, reducing the fluorescent signal. The fluorescence intensity of ALKBH5 pre-treated with TD19 was around 60% of that of DMSO-treated ALKBH5, suggesting that TD19 modified approximately two of the five cysteines around the ALKBH5 catalytic center (Fig. 2c). We also conducted a FRET-based assay to determine the inactivation constant (KI) and maximum inactivation rate constant (kinact) of TD19.33 The KI and kinact values of TD19 were 2.091 μM and 0.01607 min−1, respectively, giving a kinact/KI value of 0.0077 μM−1 min−1 (Fig. 2d). These data suggest that TD19 covalently modifies ALKBH5 in a two-step manner, with reversible binding as the first step and covalent bond formation as the second step. To evaluate the selectivity of TD19 toward other cysteine proteases, we then evaluated the inhibitory effect of TD19 on papain, another well-known cysteine protease.31 We used the broad-spectrum cysteine protease inhibitor L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E64), and the cysteine alkylator IAA as positive controls. TD19 (10 μM) did not inhibit papain, whereas E64 and IAA exhibited significant inhibition (Fig. S2e, ESI†), suggesting that TD19 selectively inhibits ALKBH5 over other cysteine proteases, such as papain.
TD19 modifies C100 and C267 in ALKBH5
To scrutinize the formation of the covalent bond between TD19 and ALKBH5, we performed mass spectrometry (MS) analysis. After treating ALKBH5 with TD19, we observed a higher mass peak with a mass difference of +248.53 Da, which matched the molecular weight of TD19 (248.30 Da), indicating the covalent addition of TD19 on ALKBH5 (Fig. 3a and b). To precisely identify the cysteine residue binding to TD19, we conducted HPLC-MS/MS analysis. The results revealed that TD19 bound to either the C100 or C267 residue (Fig. 3c and d). Additionally, we analyzed the effect of TD10 (a negative inhibitor of either ALKBH5 or FTO) on ALKBH5 using MS (Fig. S3a and b, ESI†). MS scanning revealed no additional peak in ALKBH5 treated with TD10 (Fig. S3c, ESI†).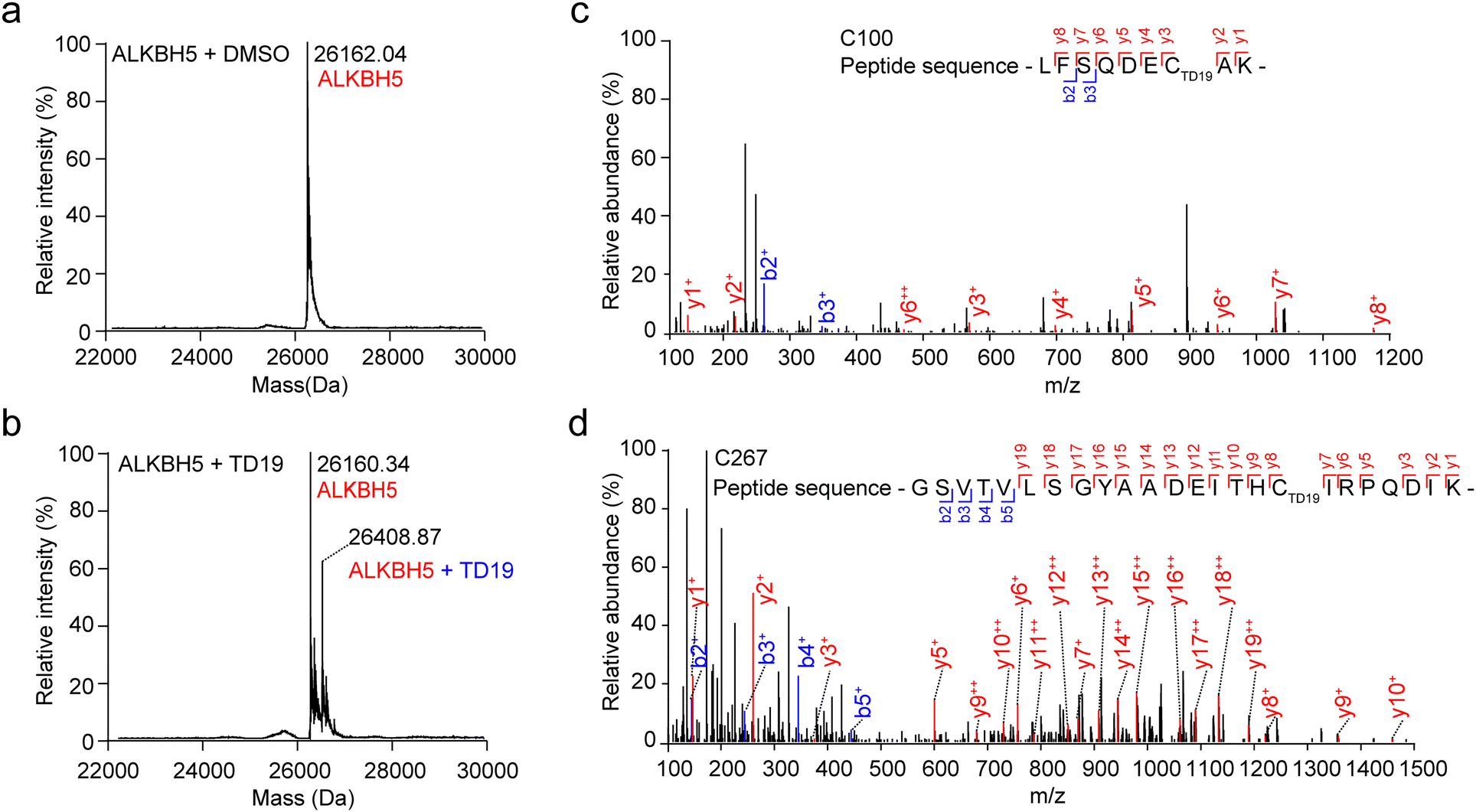 | ||
| Fig. 3 Identification of the modification sites of TD19 in ALKBH5. (a) and (b) Mass spectrometry spectra of ALKBH5 incubated with DMSO or TD19. (c) and (d) HPLC-MS/MS results indicating that TD19 modified C100 and C267 in ALKBH5. | ||
TD19 modification on C267 mainly contributed to the ALKBH5 inhibition
We then investigated the impact of C100 and C267 modification by TD19 on the enzymatic inhibition of ALKBH5. An inhibition assay revealed that, after a 60-min incubation period, the IC50 values of TD19 for the ALKBH5 mutants C100S, C267S, and C100SC267S were 3.2 ± 0.6 μM, 7.6 ± 3.4 μM, and 10.1 ± 3.2 μM, respectively (Fig. 4a–c). This result suggests that TD19 retains its binding affinity with cysteine mutants and demonstrates inhibitory activity. Furthermore, the modification of TD19 on C267 contributes more to the inhibitory effect than that on C100. Similarly, TD19 displayed reduced inhibitory effects on the corresponding mutants of fALKBH5 (where C68 and C235 in fALKBH5 correspond to C100 and C267 in ALKBH5, respectively), further underscoring the mechanism of TD19 modification on C100 and C267 residues in ALKBH5 (Fig. S4a, ESI†).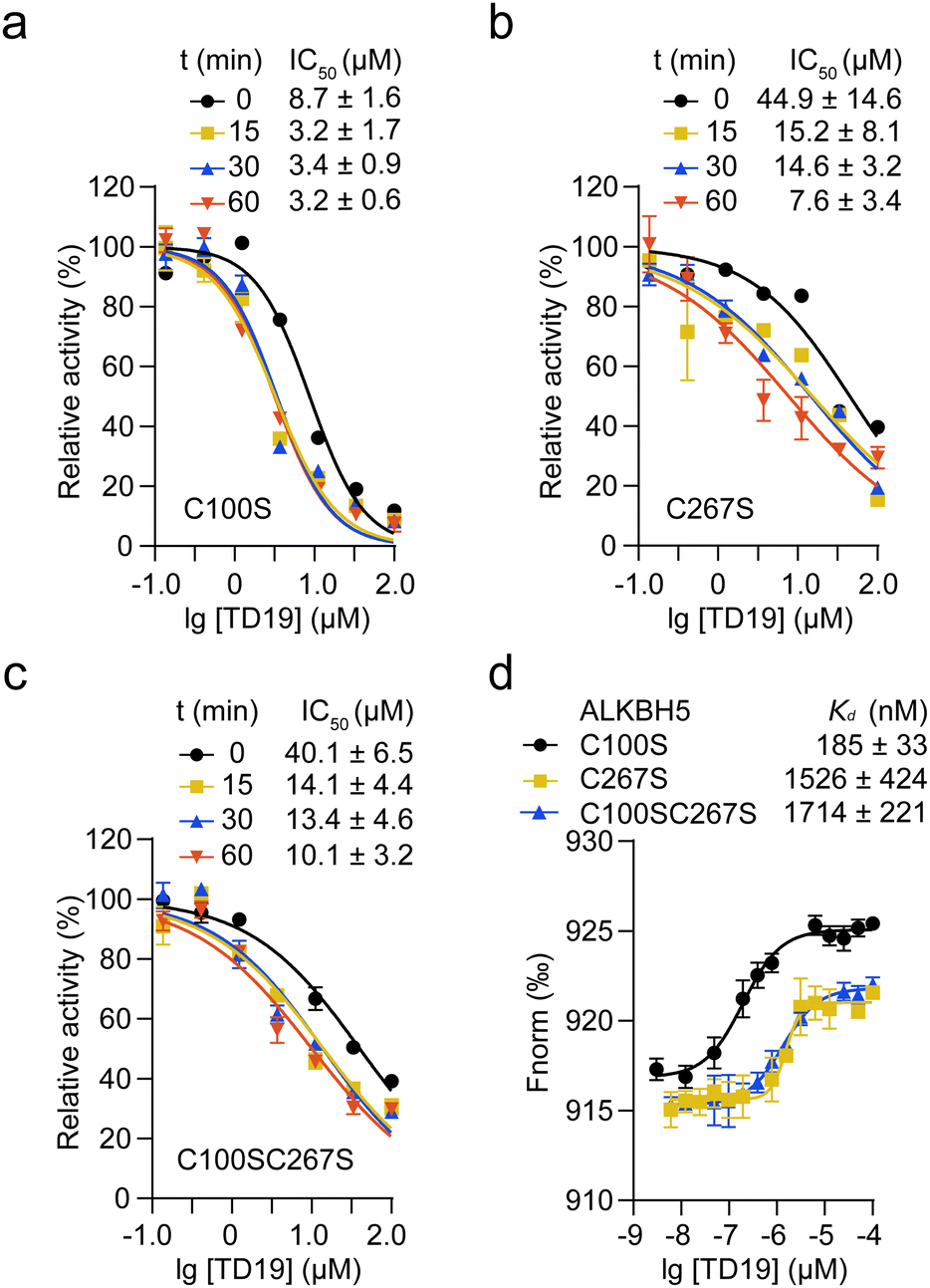 | ||
| Fig. 4 Effect of cysteine mutations on the inhibition of ALKBH5 by TD19. (a)–(c) Inhibition of TD19 on the ALKBH5 mutants C100S, C267S, and C100SC267S after different incubation times. (d) Binding affinity between TD19 and the ALKBH5 mutants by MST assay. | ||
Next, we performed an MST assay to determine the binding stoichiometry of TD19 and ALKBH5 mutants. The Kd values between TD19 and the ALKBH5 mutants C100S, C267S, and C100SC267S were 185 ± 33, 1526 ± 424, and 1714 ± 221 nM, respectively (Fig. 4d). In addition, we quantified the Kd values for the binding of TD19 to the ALKBH5 mutants C100S, C267S, and C100SC267S as 335 ± 68, 825 ± 209, and 1110 ± 260 nM, respectively, via an ITC assay. (Fig. S4b–d, ESI†). These results suggest that the modification of TD19 on C267 contributes more to the binding capacity of TD19 toward ALKBH5 than that on C100. Additionally, we estimated the stoichiometry factor (N) of TD19 titration to the C100SC267S mutant to be 0.74 ± 0.02 by ITC assay, indicating that TD19 still bound to ALKBH5 in the absence of the two cysteine residues. This observation suggests that TD19 covalently modifies ALKBH5 in a two-step manner, consistent with previous findings (Fig. 2d).
TD19 inhibits ALKBH5 binding to m6A-RNA substrate
To investigate the mechanism of ALKBH5 inhibition by TD19, we determined the Kd values for ALKBH5 and the m6A-containing RNA substrate in the presence and absence of TD19 in an ITC assay.34 Without TD19, ALKBH5 bound to m6A-RNA with a Kd of 23.1 ± 8.8 μM (Fig. 5a). However, no binding was detected when ALKBH5 was preincubated with TD19 (Fig. 5b). In addition, we conducted a pull-down assay to evaluate the effect of TD19 on the interaction between ALKBH5 and a dm6A-containing DNA substrate as previously described.35 ALKBH5 is known to weakly bind to DNA substrates in the absence of 2OG. Therefore, we assayed the sample without 2OG as the control. TD19 inhibited the binding between ALKBH5 and the DNA substrate in a concentration-dependent manner, but minimally disrupted the C100SC267S mutant binding to the DNA substrate (Fig. 5c). These data show that the modification of ALKBH5 by TD19 inhibits RNA demethylase binding to the methylated RNA/DNA substrate.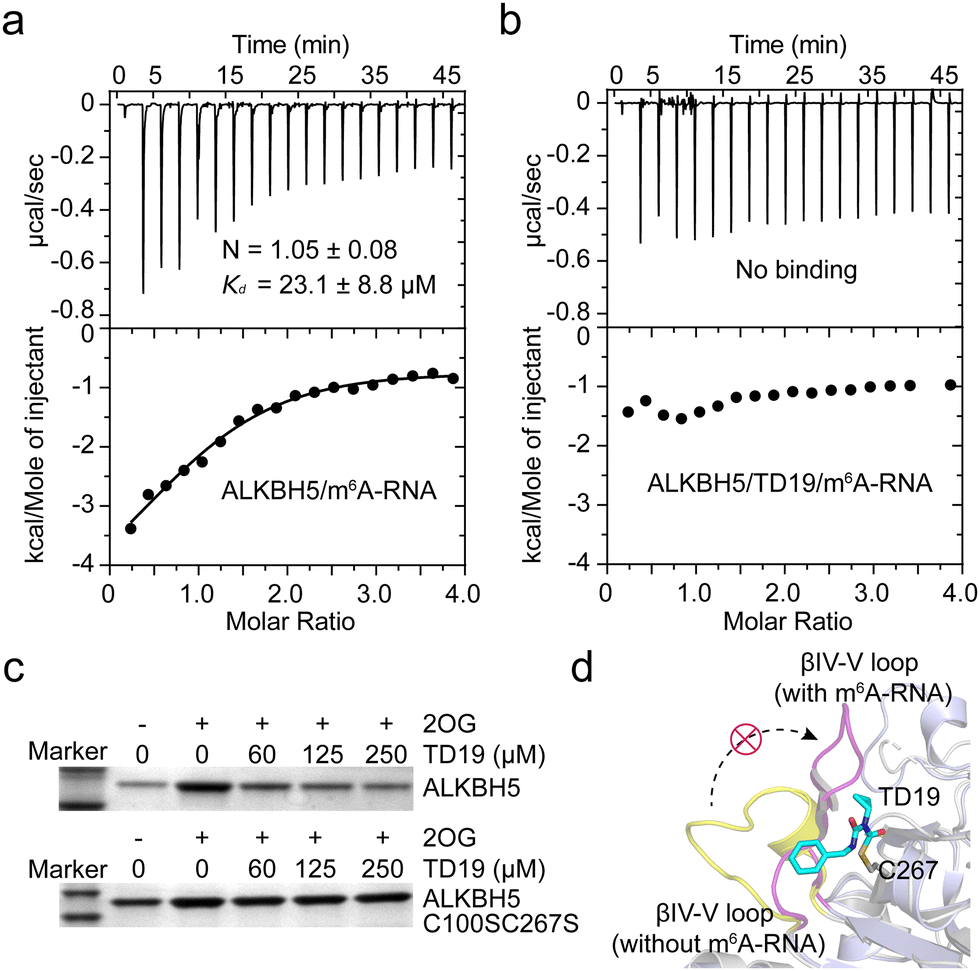 | ||
| Fig. 5 Effect of TD19 on the interaction between ALKBH5 and substrate. (a) and (b) Effect of TD19 on the binding of ALKBH5 with an m6A-RNA substrate in ITC titration. (c) Effect of TD19 on the interaction between dm6A-containing DNA oligonucleotide and ALKBH5 WT and the C100SC267S mutant in a pull-down assay. (d) Structural superimposition of the ALKBH5/m6A-RNA complex structure (light blue cartoon, PDB ID: 7WKV) and the apo structure of ALKBH5 (white cartoon, PDB ID: 4NRO). TD19 (colored in cyan) was docked with C267 of the apo ALKBH5 structure. The βIV-V loops with and without the m6A-RNA substrate were colored in purple and yellow, respectively. | ||
To investigate the potential interaction mode between TD19 and ALKBH5, we conducted a docking study using the reported ALKBH5 structure as a model.15 In the covalent docking model of TD19 and C100 in ALKBH5, two hydrogen bonds were observed (Fig. S5a and b, ESI†). One is formed between the carbonyl group in TD19 and the –NH of Arg93, with a distance of 3.2 Å, the other is formed between the –NH in TD19 and the carbonyl group of Arg250, with a distance of 3.4 Å. In the covalent docking model of TD19 and C267 in ALKBH5, a hydrogen bond was observed between the carbonyl group in TD19 and the –NH in the Gln233 residue, with a distance of 2.6 Å (Fig. S5c and d, ESI†). Additionally, the phenyl ring of TD19 forms a π–π stacking interaction with Phe232 (Fig. S5c, ESI†). Next, we performed a superimposition of the ALKBH5/m6A-RNA complex and the apo ALKBH5 structure and found that the βIV-V loop (residues 229–242) underwent a large conformational change in the ALKBH5/m6A-RNA complex compared to the apo ALKBH5 structure. The docking model suggested that TD19 modification on C267 hindered the conformational change in the βIV-V loop, potentially impeding ALKBH5 binding to the methylated RNA substrate (Fig. 5d). Nevertheless, resolving the structure of the TD19/ALKBH5 complex would give more precise insights on mode of action of TD19.
TD19 targets ALKBH5 and regulates the cellular m6A for anticancer
ALKBH5 is required for tumor growth in AML11 and GBM.10 To assess the anticancer potential of TD19, we evaluated its effect on the viability of two AML (NB4 and MOLM13) and two GBM cell lines (U87 and A172), using FB23-2 as a control TD19 exhibited good antiproliferative effects on all four tumor cell lines in a dose-dependent manner. The IC50 values were 15.1 ± 1.4, 9.5 ± 3.0, 7.2 ± 0.4, and 22.3 ± 1.7 μM in the NB4, MOLM13, U87, and A172 cell lines, respectively (Fig. 6a). The FTO inhibitor FB23-2 displayed better activities than TD19 (Fig. S6a, ESI†). To corroborate the direct interaction between TD19 and ALKBH5 in cells, we conducted a drug affinity responsive target stability (DARTS) assay. As expected, the presence of TD19 in NB4 and U87 cells rendered ALKBH5, but not FTO, resistant to protease hydrolysis (Fig. 6b), suggesting that TD19 directly and selectively targeted ALKBH5 in cell lysates. We also evaluated the effect of TD19 on the m6A modification in tumor cell lines. The treatment with TD19 increased m6A levels in NB4, MOLM13, U87, and A172 cells, as detected through an m6A dot blot assay (Fig. 6c, and Fig. S6b, ESI†). Conversely, treating the cells with the negative control compound TD10 minimally altered m6A levels (Fig. 6d and Fig. S6c, ESI†). The abundance of the m1A modification in NB4 and U87 cells remained unchanged upon treatment with TD19, showing that TD19 did not inhibit ALKBH3-mediated m1A demethylation in tumor cells (Fig. 6e).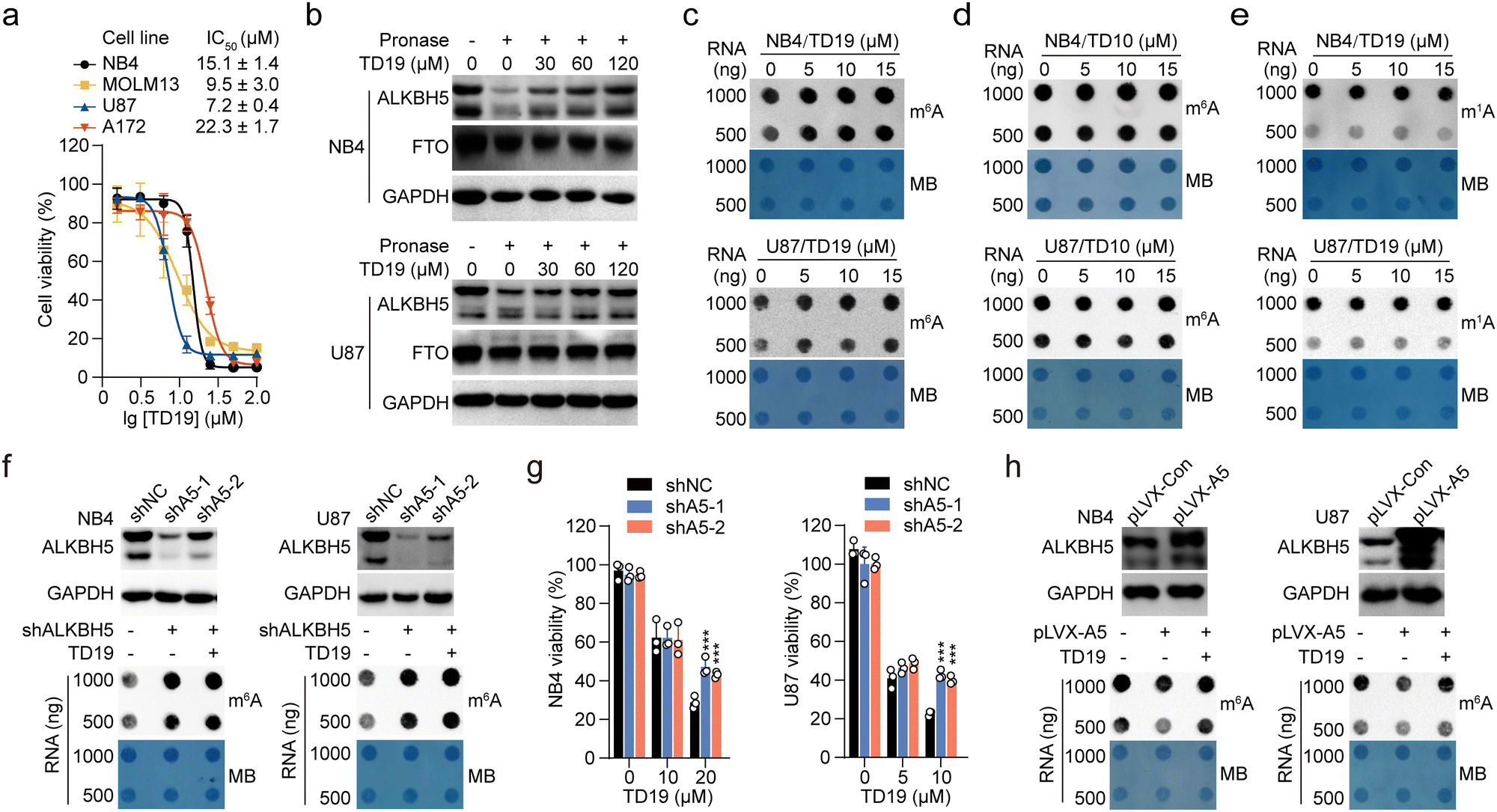 | ||
| Fig. 6 TD19 regulates m6A modification and shows anticancer activity in tumor cells. (a) Effect of TD19 on the viability of AML and GBM cells. The cells were treated with TD19 for 72 h at the indicated concentrations before MTT/CCK8 measurement. (b) Representative gel images showing the effect of TD19 on the hydrolysis of ALKBH5 and FTO by pronase in the DARTS assay. The ALKBH5 and FTO levels in NB4 and U87 cells were quantified by western blot. (c) and (d) Effect of TD19 (c) and TD10 (d) on m6A abundance in NB4 and U87 cells. The cells were treated with the compounds for 72 h before quantification of m6A in total RNA through a dot blot assay. Methylene blue was used as a loading control for RNA samples. (e) Effect of TD19 on m1A abundance in NB4 and U87 cells, assessed by dot blot assay. (f) Effect of TD19 on m6A abundance in ALKBH5 knockdown NB4 and U87 cells, assessed through an m6A dot blot assay. (g) Effect of TD19 on the viability of ALKBH5 knockdown NB4 and U87 cells. The cells were treated with TD19 for 72 h. (h) Effect of TD19 on m6A abundance in NB4 and U87 cells overexpressing ALKBH5, assessed through an m6A dot blot assay. Statistical significance is denoted as ***p < 0.001 using the unpaired Student's t-test. The error bars represent the mean ± SD, n = 3. | ||
We also investigated the target engagement of TD19 in tumor cells. We constructed stable cell lines with decreased ALKBH5 mRNA expression by utilizing two short hairpin RNAs (shA5-1 and shA5-2, collectively referred to as shALKBH5) (Fig. 6f). Compared with the control shRNA (referred to as shNC), shALKBH5 significantly increased the m6A abundance detected by dot blot assay in NB4 and U87 cells (Fig. 6f). Interestingly, TD19 treatment minimally affected m6A levels in shALKBH5-treated cells, implying that TD19 regulates cellular m6A levels in an ALKBH5-dependent manner (Fig. 6f and Fig. S6d, ESI†). Moreover, TD19 at 10–20 μM exhibited a significantly lower inhibitory effect on the viability of shALKBH5-treated AML and GBM cells than on shNC-treated ones, implying that the TD19 inhibition of cell proliferation is contingent on ALKBH5 protein levels (Fig. 6g). To further evaluate the on-target effects of TD19 on the regulation of m6A abundance in tumor cells, we transfected NB4 and U87 cells using lentiviruses containing a wild-type ALKBH5 plasmid (referred to as pLVX-A5). This transfection significantly reduced the m6A level in the tumor cells. However, TD19 treatment counteracted the decrease of the m6A abundance resulting from ALKBH5 overexpression (Fig. 6h). Collectively, these results indicate that the regulation of m6A by TD19 is dependent on ALKBH5 in tumor cells.
Lastly, we investigated whether TD19 treatment regulated the expression of downstream target genes of ALKBH5 using real-time quantitative polymerase chain reaction (RT-qPCR). Two direct target genes, AXL (AXL receptor tyrosine kinase) in AML and FOXM1 (Forkhead box M1) in GBM, which promote tumorigenesis, were selected for validation. In line with the positive regulation of AXL and FOXM1 expression by ALKBH5 in AML and GBM cells, treatment with TD19 or shALKBH5 significantly reduced the mRNA abundance of the two genes (Fig. S6e and f5, ESI†). These findings show that TD19 targets ALKBH5 and regulates the mRNA expression of downstream genes.
Conclusions
ALKBH5 and FTO are the two known m6A demethylases in RNA epigenetics. They are usually highly expressed in AML and GBM tumor cells. Overexpression of m6A demethylases is associated with poor outcomes in patients with these diseases. These results highlight the therapeutic significance for the development of small-molecule inhibitors that selectively targets certain m6A demethylase because FTO and ALKBH5 are homologous demethylases.36 The development of selective FTO inhibitors by us and others has greatly progressed,37 while the development of selective and highly potent inhibitors for ALKBH5 remains challenging.Our identification of TD19 as a covalent and selective inhibitor for ALKBH5 rather than FTO has revealed a general path to the selective inhibition of ALKBH5 by covalent modification on cysteines using an established TCI strategy. Although several crystal structures of the apo ALKBH5 protein and the ALKBH5/substrate complex have been resolved, the structure-guided development of selective ALKBH5 inhibitors has made little progress. The highly conserved ligand binding pockets shared by the ALKBH5 and FTO demethylases easily explain the slow development of highly selective ALKBH5 inhibitors. Inspired by the unexpected observation of the covalent modification of IOX3 on C200 in the crystal structure of ALKBH5, we wondered whether the cysteine residues near the substrate binding site could serve as covalent modification sites for the selective inhibition of ALKBH5 over other nonheme Fe(II)/2OG-dependent demethylases. Interestingly, the commercially available alkylating agent NEM selectively inhibited the demethylation of ALKBH5 but not FTO, supporting the idea of covalent modification on cysteines for the selective inhibition of ALKBH5. We then showed that TD19 covalently bound to the C100 and C267 ALKBH5 residues, disrupting the binding between ALKBH5 and m6A-RNA substrates, thus selectively inhibiting ALKBH5 demethylation over FTO, and modulating m6A levels and downstream genes expression in AML and GBM tumor cell lines.
We are aware that demonstrating the chemical traceability of the active cysteines in ALKBH5 in the cellular environment is warranted. It is required to develop highly selective covalent inhibitors of ALKBH5 with minimal off-target effects, which might be caused by the nonselective modification on undesired cysteines. The substantiation of this validation necessitates the systematic profiling of cysteine activity and accessibility within ALKBH5, a task well-suited to the application of chemical proteomics methodologies, notably activity-based protein profiling.38 In addition, a large-scale high-throughput screening of the covalent compound library would provide more hit scaffolds for synthesis optimization. Furthermore, we acknowledge the necessity for additional investigation to intricately dissect the selectivity of TD19 against a comprehensive array of pharmacologically relevant targets, encompassing GSK3β, the target of the related inhibitor tideglusib.39
In summary, our findings establish TD19 as a selective and potent covalent inhibitor of ALKBH5 that effectively interferes with m6A regulation in tumor cells. TD19 is a promising chemical tool to explore ALKBH5 functions and a starting point for developing anticancer drug candidates selectively targeting ALKBH5 rather than FTO.
Author contributions
C.-G.Y. conceptualization, project administration, writing – original draft, writing – review & editing, funding acquisition. G.-Q.L., H.Z. data curation, validation, methodology, writing – original draft, writing – review & editing. Y.L. data curation, validation, methodology, writing – original draft, writing – review & editing, funding acquisition. T.Z., J.G., H.Z. data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the staff of the Large-scale Protein Preparation System at the National Facility for Protein Science (NFPS) in Shanghai, Shanghai Advanced Research Institute, Chinese Academy of Sciences, China for providing technical support and assistance in data collection and analysis of ITC and MST assays. This work was supported by the National Natural Science Foundation of China (22007093 to Y. L. and 92153303 to C.-G. Y.), the National Key Research and Development Program of China (2022YFC2705005 to C.-G. Y.).Notes and references
- C. He, Nat. Chem. Biol., 2010, 6, 863–865 CrossRef CAS PubMed; I. A. Roundtree, M. E. Evans, T. Pan and C. He, Cell, 2017, 169, 1187–1200 CrossRef PubMed.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef CAS PubMed; L. Lan, Y. J. Sun, X. Y. Jin, L. J. Xie, L. Liu and L. Cheng, Angew. Chem., Int. Ed., 2021, 60, 18116–18121 CrossRef PubMed.
- G. F. Jia, Y. Fu, X. Zhao, Q. Dai, G. Q. Zheng, Y. Yang, C. Q. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed; G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C. M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W. L. Wang, S. H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y. J. Hao, X. Yang, W. M. Zhao, W. M. Tong, X. J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y. G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef PubMed.
- Z. Zou, C. Sepich-Poore, X. Zhou, J. Wei and C. He, Genome Biol., 2023, 24, 17 CrossRef CAS PubMed.
- Y. Huang, R. Su, Y. Sheng, L. Dong, Z. Dong, H. Xu, T. Ni, Z. S. Zhang, T. Zhang, C. Li, L. Han, Z. Zhu, F. Lian, J. Wei, Q. Deng, Y. Wang, M. Wunderlich, Z. Gao, G. Pan, D. Zhong, H. Zhou, N. Zhang, J. Gan, H. Jiang, J. C. Mulloy, Z. Qian, J. Chen and C.-G. Yang, Cancer Cell, 2019, 35, 677–691 CrossRef CAS PubMed.
- R. Castro-Hernández, T. Berulava, M. Metelova, R. Epple, T. Peña Centeno, J. Richter, L. Kaurani, R. Pradhan, M. S. Sakib, S. Burkhardt, M. Ninov, K. E. Bohnsack, M. T. Bohnsack, I. Delalle and A. Fischer, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2204933120 CrossRef PubMed.
- B. Zhang, H. Jiang, Z. Dong, A. Sun and J. Ge, Genes Dis., 2021, 8, 746–758 CrossRef CAS PubMed.
- H. Shi, J. Wei and C. He, Mol. Cell, 2019, 74, 640–650 CrossRef CAS PubMed.
- A. Thalhammer, Z. Bencokova, R. Poole, C. Loenarz, J. Adam, L. O'Flaherty, J. Schödel, D. Mole, K. Giaslakiotis, C. J. Schofield, E. M. Hammond, P. J. Ratcliffe and P. J. Pollard, PLoS One, 2011, 6, e16210 CrossRef CAS PubMed.
- S. Zhang, B. S. Zhao, A. Zhou, K. Lin, S. Zheng, Z. Lu, Y. Chen, E. P. Sulman, K. Xie, O. Bögler, S. Majumder, C. He and S. Huang, Cancer Cell, 2017, 31, 591–606 CrossRef CAS PubMed.
- J. Wang, Y. Li, P. Wang, G. Han, T. Zhang, J. Chang, R. Yin, Y. Shan, J. Wen, X. Xie, M. Feng, Q. Wang, J. Hu, Y. Cheng, T. Zhang, Y. Li, Z. Gao, C. Guo, J. Wang, J. Liang, M. Cui, K. Gao, J. Chai, W. Liu, H. Cheng, L. Li, F. Zhou, L. Liu, Y. Luo, S. Li and H. Zhang, Cell Stem Cell, 2020, 27, 81–97 CrossRef CAS PubMed; C. Shen, Y. Sheng, A. C. Zhu, S. Robinson, X. Jiang, L. Dong, H. Chen, R. Su, Z. Yin, W. Li, X. Deng, Y. Chen, Y. C. Hu, H. Weng, H. Huang, E. Prince, C. R. Cogle, M. Sun, B. Zhang, C. W. Chen, G. Marcucci, C. He, Z. Qian and J. Chen, Cell Stem Cell, 2020, 27, 64–80 CrossRef PubMed.
- D. Shen, B. Wang, Y. Gao, L. Zhao, Y. Bi, J. Zhang, N. Wang, H. Kang, J. Pang, Y. Liu, L. Pang, Z. S. Chen, Y. C. Zheng and H. M. Liu, Acta Pharm. Sin. B, 2022, 12, 2193–2205 CrossRef CAS PubMed.
- M. A. Kurowski, A. S. Bhagwat, G. Papaj and J. M. Bujnicki, BMC Genomics, 2003, 4, 48 CrossRef PubMed.
- W. Aik, J. S. Scotti, H. Choi, L. Gong, M. Demetriades, C. J. Schofield and M. A. McDonough, Nucleic Acids Res., 2014, 42, 4741–4754 CrossRef CAS PubMed.
- C. Feng, Y. Liu, G. Wang, Z. Deng, Q. Zhang, W. Wu, Y. Tong, C. Cheng and Z. Chen, J. Biol. Chem., 2014, 289, 11571–11583 CrossRef CAS PubMed.
- A. Malacrida, M. Rivara, A. Di Domizio, G. Cislaghi, M. Miloso, V. Zuliani and G. Nicolini, Bioorg. Med. Chem., 2020, 28, 115300 CrossRef CAS PubMed.
- N. Li, Y. Kang, L. Wang, S. Huff, R. Tang, H. Hui, K. Agrawal, G. M. Gonzalez, Y. Wang, S. P. Patel and T. M. Rana, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20159–20170 CrossRef CAS PubMed.
- S. Selberg, N. Seli, E. Kankuri and M. Karelson, ACS Omega, 2021, 6, 13310–13320 CrossRef CAS PubMed.
- Z. Fang, B. Mu, Y. Liu, N. Guo, L. Xiong, Y. Guo, A. Xia, R. Zhang, H. Zhang, R. Yao, Y. Fan, L. Li, S. Yang and R. Xiang, Eur. J. Med. Chem., 2022, 255, 115368 CrossRef PubMed.
- H. Takahashi, H. Hase, T. Yoshida, J. Tashiro, Y. Hirade, K. Kitae and K. Tsujikawa, Chem. Biol. Drug Des., 2022, 100, 1–12 CrossRef CAS PubMed.
- Y. Huang, W. Xia, Z. Dong and C.-G. Yang, Acc. Chem. Res., 2023, 56, 3010–3022 CrossRef CAS PubMed.
- Y.-Z. Wang, H.-Y. Li, Y. Zhang, R.-X. Jiang, J. Xu, J. Gu, Z. Jiang, Z.-Y. Jiang, Q.-D. You and X.-K. Guo, J. Med. Chem., 2023, 66, 15944–15959 CrossRef CAS PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 CrossRef CAS PubMed.
- W. Chen, L. Zhang, G. Zheng, Y. Fu, Q. Ji, F. Liu, H. Chen and C. He, FEBS Lett., 2014, 588, 892–898 CrossRef CAS PubMed.
- B. Chen, F. Ye, L. Yu, G. Jia, X. Huang, X. Zhang, S. Peng, K. Chen, M. Wang, S. Gong, R. Zhang, J. Yin, H. Li, Y. Yang, H. Liu, J. Zhang, H. Zhang, A. Zhang, H. Jiang, C. Luo and C.-G. Yang, J. Am. Chem. Soc., 2012, 134, 17963–17971 CrossRef CAS PubMed.
- L. Liu, Y. Kong, L. He, X. Wang, M.-M. Wang, H. Xu, C.-G. Yang, Z. Su, J. Zhao, Z.-W. Mao, Y. Huang and H.-K. Liu, Chin. J. Chem., 2022, 40, 1156–1164 CrossRef CAS; Q. Li, Y. Huang, X. Liu, J. Gan, H. Chen and C.-G. Yang, J. Biol. Chem., 2016, 291, 11083–11093 CrossRef PubMed.
- T. Yang, T. Zhang, X. N. Guan, Z. Dong, L. Lan, S. Yang and C.-G. Yang, J. Med. Chem., 2020, 63, 8442–8457 CrossRef CAS PubMed.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- R. A. Copeland, A. Basavapathruni, M. Moyer and M. P. Scott, Anal. Biochem., 2011, 416, 206–210 CrossRef CAS PubMed.
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072–10079 CrossRef CAS PubMed.
- L. L. Blazer, H. Zhang, E. M. Casey, S. M. Husbands and R. R. Neubig, Biochemistry, 2011, 50, 3181–3192 CrossRef CAS PubMed.
- J. C. Hunter, D. Gurbani, S. B. Ficarro, M. A. Carrasco, S. M. Lim, H. G. Choi, T. Xie, J. A. Marto, Z. Chen, N. S. Gray and K. D. Westover, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8895–8900 CrossRef CAS PubMed.
- J. M. Strelow, SLAS Discovery, 2017, 22, 3–20 CrossRef CAS PubMed.
- S. Kaur, N. Y. Tam, M. A. McDonough, C. J. Schofield and W. S. Aik, Nucleic Acids Res., 2022, 50, 4148–4160 CrossRef CAS PubMed.
- J. A. Purslow, T. T. Nguyen, B. Khatiwada, A. Singh and V. Venditti, Sci. Adv., 2021, 7, eabi8215 CrossRef CAS PubMed.
- L. L. Zhou, H. Xu, Y. Huang and C.-G. Yang, RSC Chem. Biol., 2021, 2, 1352–1369 RSC; Y. Li, R. Su, X. Deng, Y. Chen and J. Chen, Trends Cancer, 2022, 8, 598–614 CrossRef CAS PubMed; G.-Q. Lai, L.-L. Zhou and C.-G. Yang, Chin. J. Chem., 2020, 38, 420–421 CrossRef.
- Y. Liu, G. Liang, H. Xu, W. Dong, Z. Dong, Z. Qiu, Z. Zhang, F. Li, Y. Huang, Y. Li, J. Wu, S. Yin, Y. Zhang, P. Guo, J. Liu, J. J. Xi, P. Jiang, D. Han, C.-G. Yang and M. M. Xu, Cell Metab., 2021, 33, 1221–1233 CrossRef CAS PubMed; Z. Liu, Z. Duan, D. Zhang, P. Xiao, T. Zhang, H. Xu, C. H. Wang, G. W. Rao, J. Gan, Y. Huang, C.-G. Yang and Z. Dong, J. Med. Chem., 2022, 65, 10638–10654 CrossRef PubMed; P. Xiao, Z. Duan, Z. Liu, L. Chen, D. Zhang, L. Liu, C. Zhou, J. Gan, Z. Dong and C.-G. Yang, J. Med. Chem., 2023, 66, 9731–9752 CrossRef PubMed.
- R. J. Grams and K.-L. Hsu, Trends Pharmacol. Sci., 2022, 43, 249–262 CrossRef CAS PubMed.
- J. M. Dominguez, A. Fuertes, L. Orozco, M. del Monte-Millan, E. Delgado and M. Medina, J. Biol. Chem., 2012, 287, 893–904 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Tables S1, S2 and all the experimental methods. See DOI: https://doi.org/10.1039/d3cb00230f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |