
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetabolically activated proteostasis regulators that protect against erastin-induced ferroptosis†
Gabriel M.
Kline‡
a,
Nicole
Madrazo‡
 b,
Christian M.
Cole
a,
Meera
Pannikkat
b,
Michael J.
Bollong
a,
Jessica D.
Rosarda
bc,
Jeffery W.
Kelly
*ad and
R. Luke
Wiseman
*b
b,
Christian M.
Cole
a,
Meera
Pannikkat
b,
Michael J.
Bollong
a,
Jessica D.
Rosarda
bc,
Jeffery W.
Kelly
*ad and
R. Luke
Wiseman
*b
aDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, USA. E-mail: jkelly@scripps.edu
bDepartment of Molecular and Cellular Biology, The Scripps Research Institute, La Jolla, CA 92037, USA. E-mail: wiseman@scripps.edu
cDepartment of Anatomy, Physiology, and Genetics, Uniformed Services University of the Health Sciences, Bethesda, MD 20814, USA
dSkaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037, USA
First published on 17th July 2024
Abstract
We previously showed that the proteostasis regulator compound AA147 (N-(2-hydroxy-5-methylphenyl)benzenepropanamide) potently protects against neurotoxic insults, such as glutamate-induced oxytosis. Though AA147 is a selective activator of the ATF6 arm of the unfolded protein response in non-neuronal cells, AA147-dependent protection against glutamate toxicity in cells of neuronal origin is primarily mediated through activation of the NRF2 oxidative stress response. AA147 activates NRF2 through a mechanism involving metabolic activation of AA147 by endoplasmic reticulum (ER) oxidases, affording an AA147-based quinone methide that covalently targets the NRF2 repressor protein KEAP1. Previous results show that the 2-amino-p-cresol A-ring of AA147 is required for NRF2 activation, while the phenyl B-ring of AA147 is amenable to modification. Here we explore whether the protease-sensitive amide linker between the A- and B-rings of this molecule can be modified to retain NRF2 activation. We show that replacement of the amide linker of AA147 with a carbamate linker retains NRF2 activation in neuronal cells and improves protection against neurotoxic insults, including glutamate-induced oxytosis and erastin-induced ferroptosis. Moreover, we demonstrate that inclusion of this carbamate linker facilitates identification of next-generation AA147 analogs with improved cellular tolerance and activity in disease-relevant assays.
Introduction
Imbalances in neuronal redox homeostasis are implicated in the onset and pathogenesis of etiologically diverse neurodegenerative diseases, including ischemic stroke and Alzheimer's disease.1–4 In these disorders, loss of redox homeostasis leads to neuronal death through multiple regulated cell death pathways, such as oxytosis and ferroptosis.5,6 These closely related cell death pathways proceed through similar mechanisms involving glutathione depletion, the accumulation of reactive oxygen species (ROS), increased lipid peroxidation, and cytotoxic calcium influx.7,8 Since the ability to counteract ROS-induced damage in the brain declines with age, oxytosis and ferroptosis are prevalent in age-related neurodegenerative disorders.6,9 This has led to considerable interest in developing pharmacologic approaches to mitigate oxytosis/ferroptosis-associated neurotoxicity in many age-related neurodegenerative disorders.10We previously identified the proteostasis regulator small molecule AA147 (N-(2-hydroxy-5-methylphenyl)benzenepropanamide) as a compound that protects neuronal cells against glutamate-induced oxytosis.11 AA147 was originally identified as a highly selective activator of the ATF6 signaling arm of the unfolded protein response (UPR) in cells of non-neuronal origin (e.g. HEK293T, HUH7, and HepG2). However, subsequent studies showed that AA147 possesses the ability to activate the oxidative stress-responsive transcription factor NRF2 with selectivity over ATF6 signaling, but only in neuron-derived cells at later time points following compound treatment.11–13 NRF2 is a cap-n-collar basic leucine zipper transcription factor that controls expression of genes comprising the oxidative stress response.14 Under basal conditions, association with the cullin ring E3 ligase adaptor protein Kelch-like ECH-associated protein 1 (KEAP1) with NRF2 leads to NRF2 ubiquitination and subsequent proteasomal degradation, suppressing transcription factor activity.15 However, covalent modification of sensor cysteines on KEAP1 with either reactive oxygen species (ROS) induced by oxidative stress or by electrophilic small molecules (e.g. diroximel fumarate) decreases NRF2 ubiquitination and proteasomal degradation, allowing nuclear translocation of NRF2 and activation of the NRF2-regulated transcriptional program.14–18
We previously showed that AA147 activates NRF2 through a mechanism involving the metabolic activation of AA147 by ER oxidases to generate a quinone methide in the A-ring of AA147 that covalently modified KEAP1.11 Further, we demonstrated that the AA147-dependent protection against glutamate-induced toxicity in neuronal cell culture models could be primarily attributed to activation of NRF2. Additionally, AA147 mitigates neuronal damage in mouse models of ischemic stroke – a condition associated with high levels of oxidative stress – through a mechanism involving NRF2 activation.19,20 This indicates that AA147-dependent NRF2 activation represents a potential strategy to broadly mitigate ROS-associated damage implicated in numerous neurodegenerative diseases.
Previous structure–activity relationship (SAR) studies on the AA147 scaffold (Fig. S1A, ESI†) have primarily focused on identifying compounds with improved activation of protective ATF6 signaling. These studies have shown that the 2-amino-p-cresol A-ring of AA147 is converted to a quinone methide by an ER oxidase enzyme(s), leading to covalent modification of ER protein disulfide isomerases, required for ATF6 activation. The phenyl B-ring of AA147 is more tolerant to chemical modification regarding AA147 analog-dependent ATF6 activation. However, little effort has been directed towards modifying the amide linker between the A- and B-rings of AA147 to improve compound-dependent ATF6 or NRF2 activity or selectivity. Furthermore, previous SAR work has not focused on optimizing the NRF2 activation selectively observed in neuron-derived models.
Here, we generated a series of AA147 analogs by chemical synthesis containing alterations to the linker region. We then screened these compounds to identify those with improved potential for neuroprotection through NRF2 activation. Through these efforts, we identified AA28, an AA147 analog where the amide linker is replaced with a carbamate, as a compound that robustly activates NRF2 in neuronal models.21 Further, we demonstrate that AA28-dependent NRF2 activation shows improved protection against neurodegenerative insults including glutamate-induced oxytosis and erastin-induced ferroptosis in HT22 cells. These results identify carbamate-containing AA28 as an improved scaffold that can be used to further optimize neuroprotection afforded by metabolically activatable proteostasis regulators. To demonstrate this, we generated a series of AA28 analogs with changes to the B-ring to identify compounds with improved protection and transcriptional selectivity towards mitigating neurotoxic insults.
Results
Replacement of the AA147 amide linker with a carbamate linker does not disrupt compound activity in neuron-derived cell lines
The requirement for metabolic activation of AA147 presents an opportunity to tune the reactivity of AA147 and render analogs more potent while simultaneously addressing potential metabolic liabilities of the compound.22,23 To address this, we performed SAR studies on the AA147 scaffold aimed at improving ATF6 and NRF2 activation. We synthesized a library of AA147 analogs with alterations to the AA147 linker region (Fig. 1(A)). These analogs were initially screened at a concentration of 10 μM to determine their ability to activate the ATF6-selective ERSE-firefly luciferase (FLuc) reporter in HEK293T cells. Intriguingly, we found that replacing the amide linkage with the isostructural carbamate, affording AA28, showed similar ERSE-FLuc reporter activation to that observed upon AA147 treatment (Fig. 1(B)). Notably, the urea isostere, AA27, showed no significant reporter activation. AA28 activated the ATF6-selective ERSE-FLuc reporter with an EC50 of 2.9 μM (Fig. 1(C)), which is similar to that observed for AA147 (1.1 μM).13 Further, AA28 induced expression of ATF6 target genes (e.g., BiP) in HEK293T cells to the same levels observed upon AA147 treatment, as measured by quantitative PCR (qPCR; Fig. S1B, ESI†). These results indicate that AA28 and AA147 show similar activity in HEK293T cells.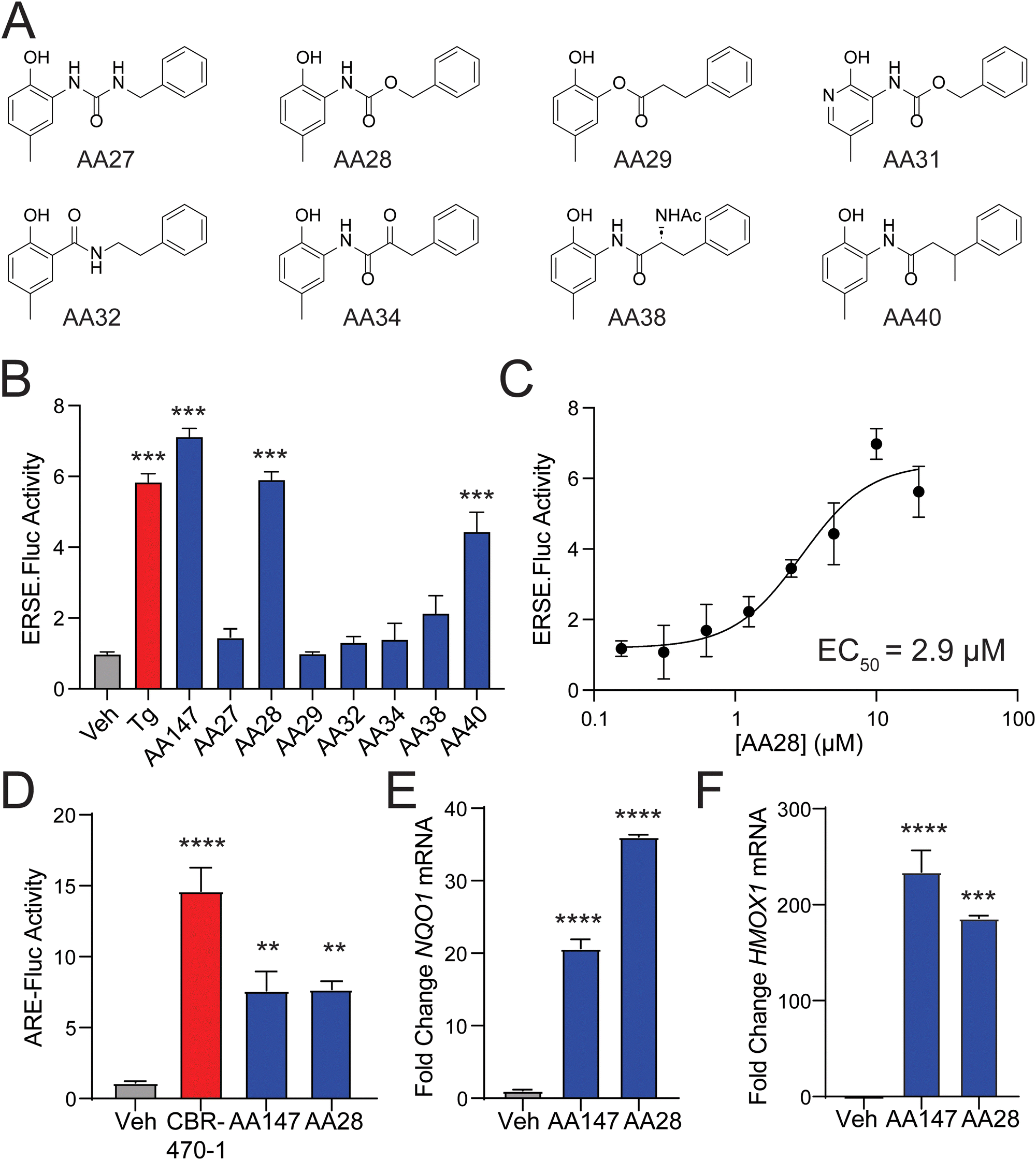 | ||
| Fig. 1 An amide to carbamate substitution within the AA147 linker does not impair compound activity. (A) Chemical structures of AA147 analogs containing changes to the linker region. (B) Luciferase activity in HEK293T cells stably expressing the ATF6-seletive ERSE-FLuc reporter treated for 18 h with the indicated compound (10 μM). The global ER stressor thapsigargin (Tg; 0.5 μM) is shown as a control. Error bars show SEM for n = 6 biological replicates. ***p < 0.001 from one-way ANOVA. (C) Luciferase activity in HEK293T cells stably expressing the ATF6-seletive ERSE-FLuc reporter treated for 18 h with the indicated dose of AA28. Error bars show SEM for n = 3 replicates. The EC50 is shown. (D) Luciferase activity in IMR32 cells transiently expressing the NRF2-selective ARE-FLuc reporter treated for 16 h with the indicated compound (5 μM). The NRF2-activating compound CBR-470-1 (5 μM) is shown as a control. Error bars show SEM for n = 3 replicates. ****p < 0.001, **p < 0.01 from one-way ANOVA. (E) and (F) Expression, measured by qPCR of the NRF2 target genes NQO1 and HMOX1 in IMR32 cells treated for 16 h with the indicated compound (2.5 μM). ****p < 0.0001 from one-way ANOVA. | ||
We previously showed that AA147 activates the protective NRF2 oxidative stress response (OSR) in neuron-derived cells.11 Thus, we sought to determine whether the carbamate-containing AA28 possessed a similar ability to activate the NRF2-regulated OSR in neuron-derived cell lines. We initially confirmed that both AA147 and AA28 activated the ATF6-selective ERSE-FLuc reporter in human neuroblast-derived IMR32 cells (Fig. S1C, ESI†). Similar results were observed by qPCR, monitoring expression of the ATF6 target gene BiP (Fig. S1D, ESI†). Next, we tested the activation of the NRF2-responsive ARE-FLuc reporter in IMR32 cells treated with 10 μM AA147 or AA28. Both compounds robustly activated this NRF2 transcriptional program reporter (Fig. 1(D)). The potent NRF2-activating compound CBR-470-1 was used as a positive control.24 Both AA147 and AA28 also induced expression of the NRF2 target genes NQO1 and HMOX1 in IMR32 cells, as measured by qPCR (Fig. 1(E) and (F)). Similar results were observed in mouse hippocampus-derived HT22 cells (Fig. S1E and F, ESI†). Collectively, these results show that replacement of the AA147 amide linker with an isostructural carbamate linker retains the transcriptional selectivity of AA147 to activate the NRF2 and ATF6 stress-responsive signaling pathways in neuronal cell culture models.
AA28 activates NRF2 through a mechanism involving metabolic activation and covalent targeting of KEAP1
We previously showed that AA147 activates NRF2 transcriptional signaling in neuron-derived cells through a mechanism involving metabolic activation of the 2-amino-p-cresol A-ring by ER oxidases and subsequent covalent targeting of the NRF2 suppressor protein KEAP1 (Fig. 2(A)).11 Thus, we sought to determine if AA28 activated NRF2 transcriptional signaling through a similar mechanism. Initially, we used the selective NRF2 inhibitor ML38525 to confirm that AA28 increases ARE-FLuc activity through an NRF2-dependent mechanism. Cotreatment with ML385 blocked ARE-FLuc activation in IMR32 cells treated with both AA147 and AA28 (Fig. 2(B)). Similar results were observed in HT22 cells (Fig. S2A, ESI†). These results confirm that AA28, like AA147, activates this reporter through an NRF2-dependent mechanism in cells of neuronal origin.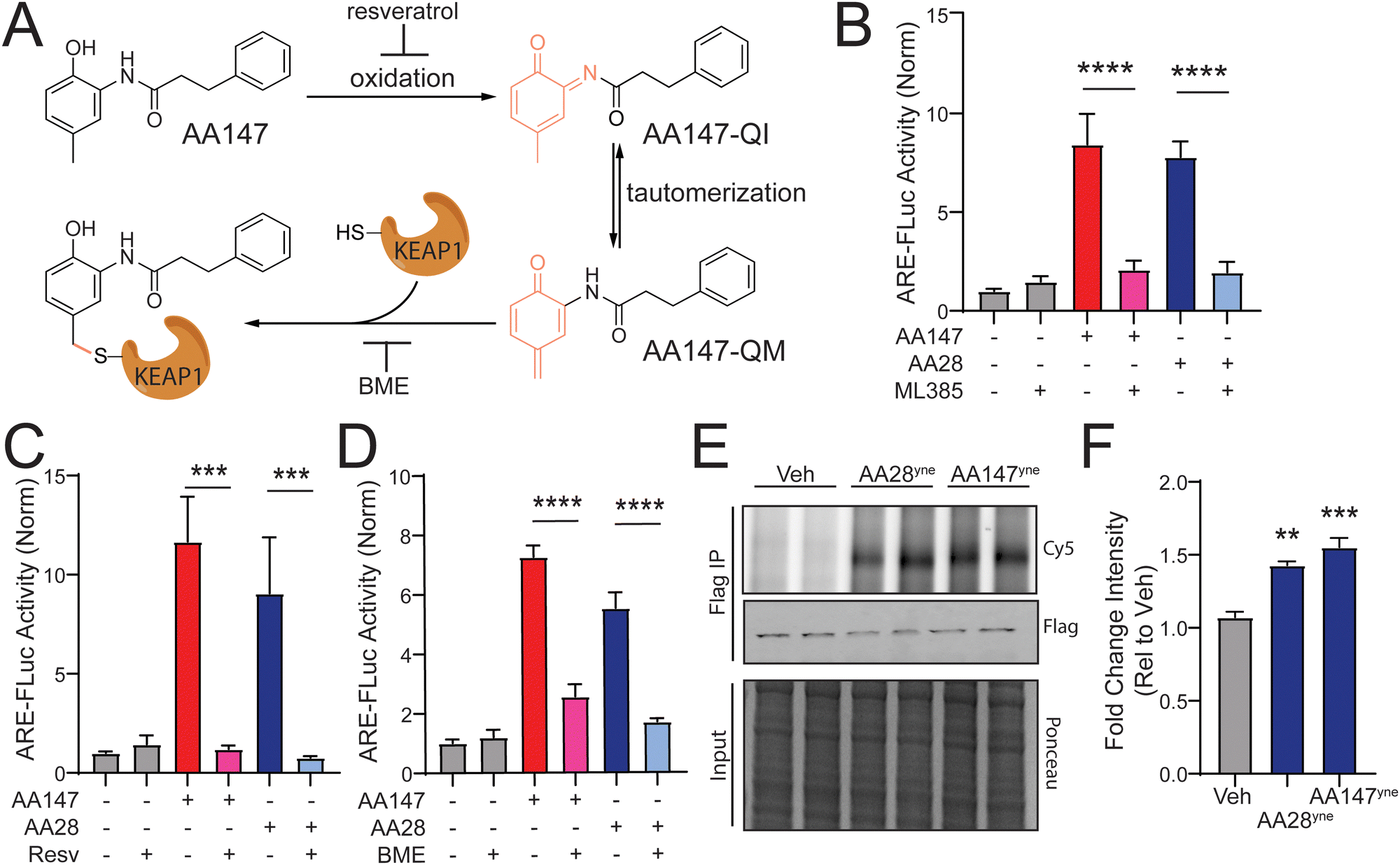 | ||
| Fig. 2 AA28 activates NRF2 through a mechanism involving metabolic activation and covalent modification of KEAP1. (A) AA147 becomes metabolically activated and can modify cysteine 151 on KEAP1. (B)–(D) Luciferase activity in IMR32 cells transiently transfected with ARE-FLuc reporter and pretreated for 16 h with 5 μM AA147 or AA28 in the presence or absence of NRF2-inhibitor ML385 (5 μM), P450 inhibitor resveratrol (10 μM), or β-mercaptoethanol (BME; 55 μM). Error bars show SEM for n = 6 biological replicates. ***p < 0.001, ****p < 0.0001 from one-way ANOVA. (E) Cy5 fluorescence image (top) and FLAG immunoblot (middle) of FLAG immunopurifications prepared from HT22 cells transiently overexpressing wild-type (WT) KEAPFT and treated with vehicle, AA28yne, or AA147yne (10 μM; 6 h). Two independent biological replicates are shown. Small-molecule-modified proteins were conjugated to Cy-5 azide (Cy5-N3) via click chemistry. KEAP1FT levels were confirmed by immunoblotting with FLAG antibody (middle). Total protein from input shown by ponceau stain (bottom). (F) Quantification of Fig. 2(E). Error bars show SEM for n = 3 biological replicates. **p < 0.01 for one-way ANOVA. | ||
Next, we sought to determine the reliance of AA28-dependent ARE-FLuc activation on compound metabolic activation and covalent targeting. Previous results showed that metabolic activation of AA147 and its subsequent covalent targeting of KEAP1 could be blocked by cotreating cells with resveratrol or β-mercaptoethanol (BME), respectively (Fig. 2(A)).11 Co-treatment with either resveratrol or BME blocked AA28-dependent ARE-FLuc activation in IMR32 cells (Fig. 2(C) and (D)). Similar results were observed in HT22 cells (Fig. S2B and C, ESI†). These results support a model whereby AA28, like AA147, activates NRF2 transcriptional signaling through a mechanism involving AA28 metabolic activation by ER oxidases followed by covalent targeting of KEAP1 (Fig. 2(A)).
Finally, we wanted to confirm that AA28 covalently modifies the NRF2 repressor KEAP1, as previously shown for AA147. We synthesized an AA28 analog containing an alkyne moiety on the B-ring (AA28yne; Fig. S2D, ESI†). We confirmed that this compound activated the ARE-FLuc reporter and induced expression of the NRF2 target gene NQO1 in IMR32 cells (Fig. S2E and F, ESI†). Using this compound and the analogous AA147 alkyne-containing analog (AA147yne; Fig. S2D, ESI†), we monitored covalent targeting of FLAG-tagged KEAP1 immunopurified from IMR32 cells by appending a fluorophore to the alkyne moiety on these compounds using a copper-mediated azide–alkyne cycloaddition “click” reaction and visualizing modification by fluorescence.26,27 This showed that AA28ynecovalently modifies FLAG-KEAP1 in HT22 cells at similar levels to AA147yne (Fig. 2(E) and (F)). Importantly, AA28yne-dependent labeling of KEAP1 was inhibited by co-treatment with excess AA28 (Fig. S2E, ESI†). AA28 also effectively competed for proteome labeling by AA147yne, further confirming that AA28 and AA147 target the same protein populations, including KEAP1 (Fig. S2H, ESI†). These results support a model whereby the carbamate-containing AA28 activates NRF2 signaling through a mechanism similar to that reported for AA147 involving metabolic activation and covalent targeting of KEAP1 (Fig. 2(A)).
AA28 is a platform for continued development of metabolically activated proteostasis regulators
We next sought to identify AA28 analogs that show improved NRF2 activation in neuron-derived cells. Unlike the 2-amino-p-methyl cresol A-ring, previous results show that the B-ring of AA147 is amenable to modification for NRF2 activation.11,12 Thus, we predicted that the B-ring of AA28 would behave similarly and synthesized a library of AA28 analogs with B-ring modifications (Fig. 3(A)). We then screened these compounds in dose-responsive format to identify those that show improved activation of the NRF2-responsive ARE-FLuc reporter in IMR32 cells. While modifications changing the lipophilicity of the B-ring were largely disfavored, we found that most analogs achieved some degree of ARE-FLuc activation (Fig. 3(B)). Many of these compounds, including AA58, AA63, AA64, AA65, AA74, and AA78, showed increased ARE-FLuc luciferase reporter amplitude relative to AA28 at different concentrations, warranting further investigation. Intriguingly, 67% (4/6) of these analogs possessed a five-membered B-ring heterocycle.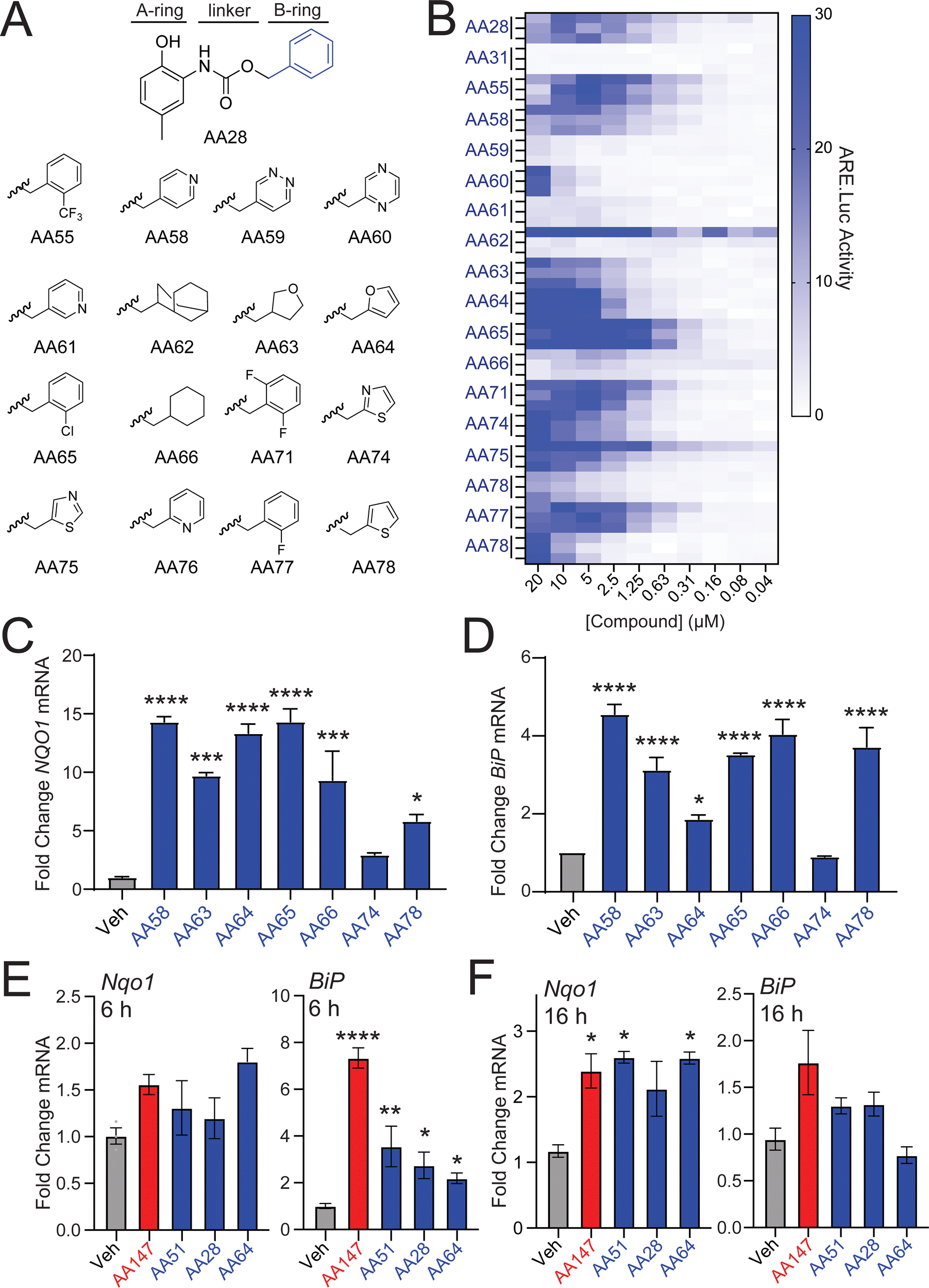 | ||
| Fig. 3 AA28 analogs with B-ring modifications improve NRF2 activation. (A) AA28 analogs containing the indicated B-ring modifications. (B) Heat map showing the activation of the ARE-FLuc NRF2 reporter transiently expressed in IMR32 cells treated for 16 h with the indicated concentration of AA28 analog. Three independent biological replicates are shown in the heat map for all AA28 analogs. (C) and (D) Expression, measured by qPCR, of the NRF2-target gene NQO1 and the ATF6 target gene BiP in IMR32 cells treated for 16 h with the indicated AA28 analog (10 μM). Error bars show SEM for n = 3 biological replicates. *p < 0.05, ***p < 0.001, ****p < 0.0001 for one-way ANOVA. (E) and (F) Expression, measured by qPCR of the NRF2-target gene Nqo1 and the ATF6 target gene Bip, in HT22 cells treated for with the indicated AA28 analog (10 μM) for 6 h (E) or 16 h (F). Error bars show SEM for n = 3 replicates. *p < 0.05, **p < 0.01, ****p < 0.0001 for one-way ANOVA. | ||
We next monitored expression of the NRF2 target gene NQO1 in IMR32 cells treated with these prioritized analogs. AA58, AA63, AA64, AA65, and AA78 all induced expression of NQO1, but AA74 did not, so it was eliminated from further consideration (Fig. 3(C)). Similar results were observed for expression of the ATF6 target gene BiP; however, the furan-containing AA64 showed lower levels of BiP induction (Fig. 3(D)), suggesting that further elaboration of this compound could afford selective NRF2 activation over ATF6 activation. This hypothesis is further supported by the observation that AA64 selectively induced expression of NRF2 target genes (e.g., Nqo1), relative to ATF6 target genes (e.g., BiP) in HT22 cells treated with compound for 6 or 16 h (Fig. 3(E) and (F)). Similar results were observed when monitoring dose-dependent activation of the NRF2-selective ARE-FLuc reporter and the ATF6-selective ERSE-FLuc reporter in HT22 cells (Fig. S3A and B, ESI†). We confirmed that AA64-dependent activation of ARE-FLuc was blocked by cotreatment with resveratrol and BME in both IMR32 and HT22 cells (Fig. S3C–F, ESI†). Furthermore, we confirmed that AA64 competed with AA147yne for proteome-wide labeling in HT22 cells (Fig. S3G, ESI†). This suggests that AA64 shows increased selectivity for NRF2 activation, relative to ATF6 activation, in cells of neuronal origin.
To further define the importance of the furan B-ring in AA64, we prepared an AA147 analog containing a furan B-ring (AA51; Fig. S3H, ESI†) for comparison. We confirmed that AA51 robustly and dose-dependently activated the ARE-FLuc reporter (Fig. S3H, ESI†). Further, AA51 shows similar upregulation of NRF2 target genes (e.g., NQO1) as AA64 in IMR32 and HT22 cells (Fig. 3(E), (F) and Fig. S3I, ESI†). Notably, we do observe lower levels of BiP induction in HT22 cells treated with AA51, as compared to AA147 (Fig. 3(E) and (F)) after 6 and 16 h of treatment, suggesting that this compound, like AA64, shows preferential activation of NRF2 signaling over ATF6 signaling. Collectively, these results indicate that the presence of the furan B-ring altered the selectivity of our compounds towards NRF2 activation relative to ATF6 activation in neuron-derived cell culture models.
Carbamate-containing AA147 analogs protect neuron-derived cells against erastin-induced ferroptosis
AA147-dependent NRF2 activation protects neuron-derived cells such as hippocampal HT22 cells against glutamate-induced oxytosis.11 Thus, we evaluated the potential for our carbamate-containing AA147 analogs AA28, AA64, and AA65 to protect HT22 cells against glutamate through a similar mechanism. Initially, we evaluated the impact of AA147, AA28, AA64, and AA65 on the viability of HT22 cells in the absence of stress. As reported previously,11 treatment of HT22 cells with AA147 (10 μM) for 40 h in the absence of stress reduced viability by ∼25% (Fig. S4A, ESI†). In contrast, treatment with AA28, AA64, or AA65 did not reduce HT22 viability under these conditions. Further, all three of these compounds improved the viability of HT22 cells treated with glutamate to levels similar to or greater than that observed for AA147 (Fig. S4B–D, ESI†). Collectively, these results show that these compounds, like AA147,11 both confer protection against glutamate-induced oxytosis and demonstrate improved cellular tolerance.We next sought to explore the potential of these compounds to protect HT22 cells against other disease-related neurotoxic insults such as erastin – a potent inducer of ferroptotic cell death.28–30 AA28, AA64, and AA65 all protected HT22 cells against erastin-induced toxicity with greater potency than that observed for AA147 (Fig. 4(A)–(D)). Similar results were observed when cell death was monitored by annexin-V (AV) and propidium iodide (PI) staining, with the carbamate-containing AA28 and AA64 analogs demonstrating the best protection in this assay (Fig. S4E, ESI†). Further, we confirmed that AA147, AA28, AA64, and AA65 pretreatment reduced erastin-induced lipid peroxidation, a marker of ferroptotic cell death, to similar levels as ferrostatin-1, a known ferroptosis inhibitor (Fig. 4(E), (F) and Fig. S4F, ESI†).31,32 These results demonstrate that carbamate-containing AA147 analogs robustly protect HT22 cells against erastin-induced ferroptosis.
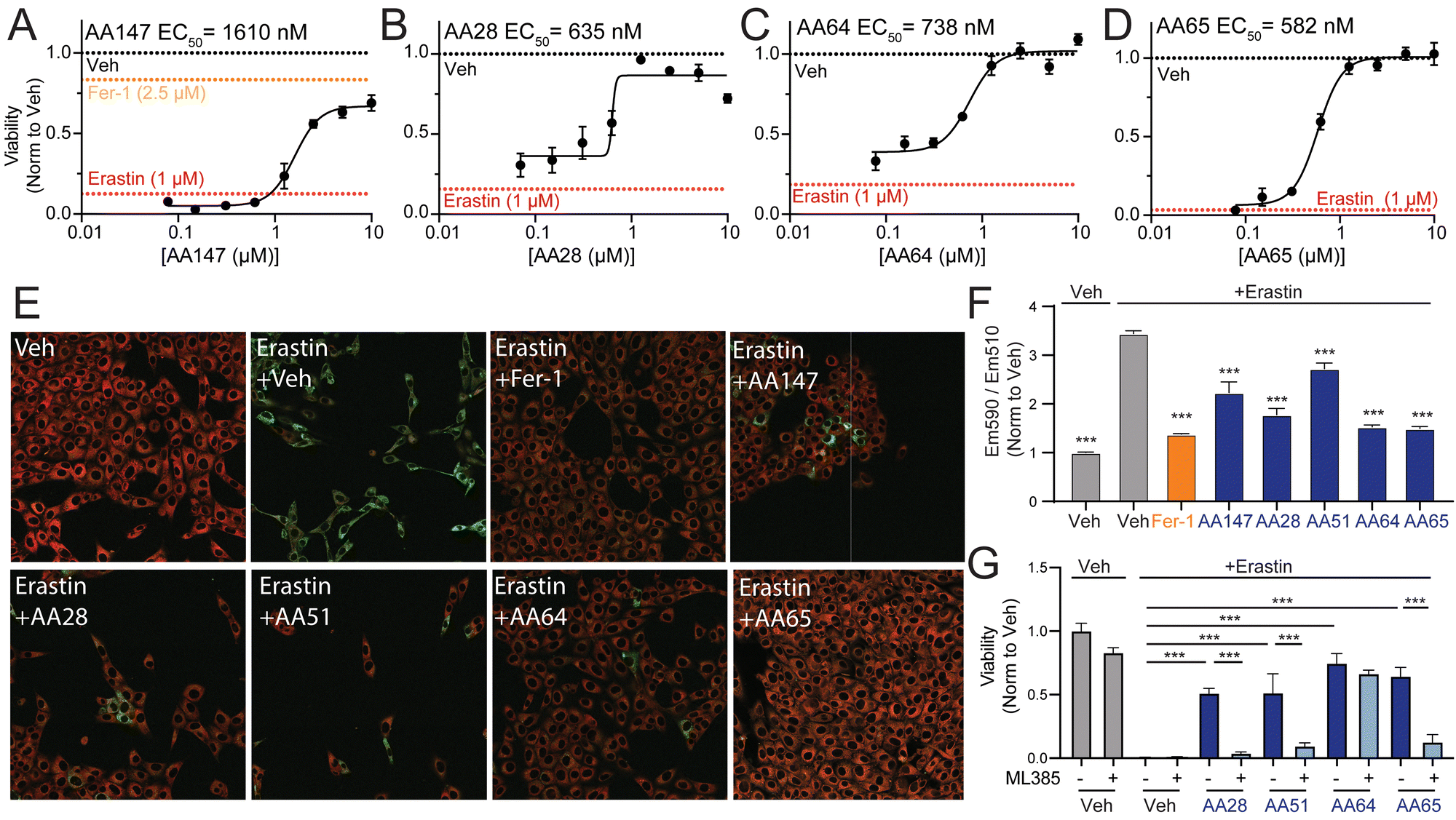 | ||
| Fig. 4 AA147 analogs protect HT22 cells against erastin-induced ferroptosis. (A)–(D) Normalized viability, measured by cell-titer glo (CTG), of HT22 cells pretreated for 16 h at the indicated concentration with AA147 (A), AA28 (B), AA64 (C), or AA65 (D) and then challenged with erastin (1 μM) for 24 h. Basal erastin toxicity for each individual experiment is shown by red dotted line. Protection afforded by the ferroptosis inhibitor ferrostatin-1 (Fer-1; 2.5 μM) is shown by the orange dotted line in (A). Error bars show SEM for n = 3 replicates. **p < 0.01, ***p < 0.005 for one-way ANOVA. EC50 of protection is shown. (E) Representative images showing lipid peroxidation, measured by confocal microscopy and C11-BODIPY staining, of HT22 cells pretreated with 10 μM AA147, AA28, AA51, AA64, or AA65 for 16 h and then challenged with 1 μM erastin for 4 h. Cells treated with the potent ferroptosis inhibitor ferrostatin are shown as a control (Fer-1; 2.5 μM). Images show merged C11-BODIPY (red) and C11-BODIPY oxidized (green). Images showing the individual channels are shown in Fig. S4F (ESI†). (F) Quantification of lipid peroxidation (ratio C11-BODIPYgreen/C11-BODIPYred) from images shown in Fig. 4(E). Error bars show SEM for n = 3 replicates. ***p < 0.005 for one-way ANOVA compared to erastin-treated cells. (G) Viability, as measured by CTG, of HT22 cells pretreated for 16 h with DMSO, AA28, AA51, or AA64 with or without NRF2 inhibitor ML385 (5 μM) and then challenged with 1 μM erastin for 24 h. Error bars show SEM for n = 3 replicates. **p < 0.01, ***p < 0.005 for one-way ANOVA. | ||
We next probed the dependence of this compound-dependent protection against erastin-induced toxicity on the activation of NRF2. Cotreatment with the NRF2 inhibitor ML385 blocked AA147-dependent protection against erastin-induced reductions in HT22 cell viability (Fig. 4(G)).11 Similar results were observed for AA28 and AA65. However, protection afforded by AA64 was insensitive to cotreatment with ML385. Similar results were observed when cell death was monitored by AV/PI staining (Fig. S4E, ESI†). This effect appears to be dependent on the carbamate linker, as the AA147 analog AA51 (Fig. S3H, ESI†) shows protection against erastin-induced toxicity through a mechanism sensitive to co-treatment with the NRF2 inhibitor ML385 (Fig. 4(G) and Fig. S4E, G, ESI†). Further, the NRF2-independent protection afforded by AA64 appears to require the 2-amino-p-cresol A-ring of this compound, as disruption of this structural moiety abrogates protection against erastin-induced toxicity (Fig. S4H, ESI†). This suggests that the protection against erastin-induced ferroptosis afforded by AA64 still involves metabolic activation of the A-ring and covalent protein targeting. These results identify the carbamate-containing AA28 and its analogs as metabolically activated proteostasis regulators that protect against erastin-induced ferroptosis in HT22 cells through both NRF2-dependent and NRF2-independent mechanisms.
Prioritized AA147 analogs are broadly protective against diverse types of ferroptosis-inducing insults
Ferroptosis can be pharmacologically induced through multiple different modes of action. These include perturbation of GSH homeostasis (e.g., erastin), inactivation of glutathione peroxidase 4 (GPX4; e.g., RSL-3), or direct oxidation of the labile iron pool (e.g., FINO2).33–36 We showed that prioritized AA147 analogs, such as AA64 and AA65, protect neuron-derived HT22 cells against erastin-induced ferroptosis (Fig. 4, and Fig. S4, ESI†). Here, we sought to define the potential for these two potent carbamate-containing analogs to protect against other ferroptosis-inducing insults across multiple different cell types. We found that AA64 protects HT22 cells against ferroptosis induced by RSL-3 or FINO2 at levels similar to those observed for the potent ferroptosis inhibitor ferrostatin-1 (Fig. 5(A) and (B)). Similar results were observed for AA65 (Fig. S5A and B, ESI†). This indicates that these compounds broadly protect HT22 against diverse types of ferroptosis-inducing insults. Further, AA64 and AA65 showed protection against ferroptotic insults in multiple other cell types, including the neuroblastoma IMR32 cells, liver-derived Huh7 cells, and fibrosarcoma HT1080 cells (Fig. 5(C)–(F) and Fig. S5C, D, ESI†). Together, these results demonstrate that metabolically activatable proteostasis regulators offer broad protection against a portfolio of ferroptotic insults across multiple different cell types.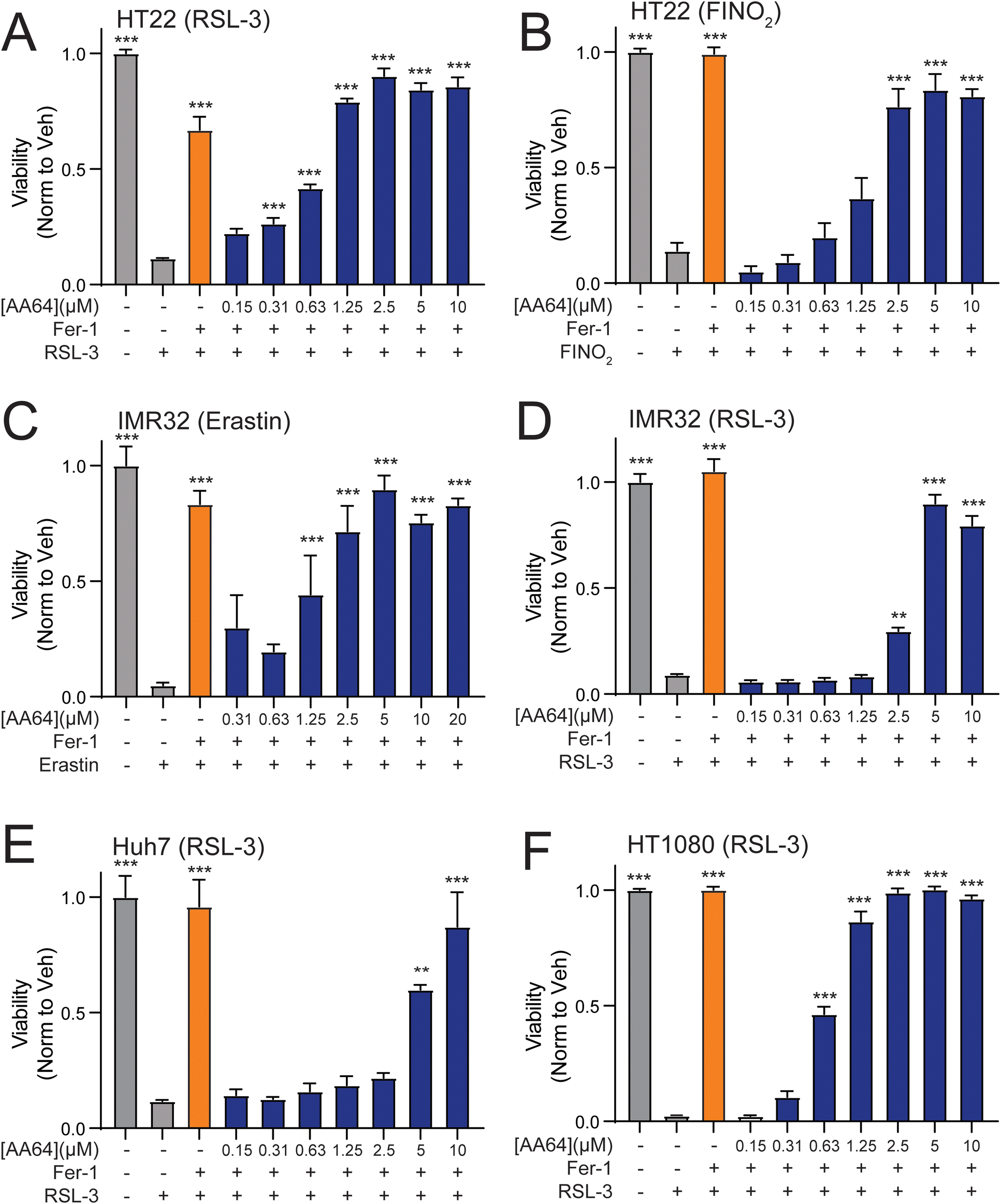 | ||
| Fig. 5 AA28 analogs protect against diverse types of ferroptosis inducers across cell types. (A) and (B) Viability, measured by Cell Titer Glo (CTG), of HT22 cells pretreated for 16 h with AA64 (blue) at the indicated concentration and then challenged with RSL-3 (1.25 μM) (A) or FINO2 (5 μM) (B) for 24 h. Cells pretreated with ferrostatin (Fer-1; 2.5 μM) are shown as a control. Error bars show SEM for n = 3 replicates. **p < 0.01, ***p < 0.001 for one-way ANOVA compared to cells treated with ferroptosis activator alone. (C) and (D) Viability, measured by Cell Titer Glo (CTG), of IMR32 cells pretreated for 16 h with AA64 (blue) at the indicated concentration for 16 h and then challenged with erastin (2.5 μM; C) or RSL-3 (1.25 μM; D) for 24 h. Cells pretreated with ferrostatin (Fer-1; 2.5 μM) are shown as a control. Error bars show SEM for n = 3 replicates. **p < 0.01, ***p < 0.005 for one-way ANOVA compared to cells treated with ferroptosis activator alone. (E) Viability, measured by Cell Titer Glo (CTG), of Huh7 cells pretreated for 16 h with AA64 (blue) at the indicated concentration for 16 h and then challenged with RSL-3 (1.25 μM) for 24 h. Cells pretreated with ferrostatin (Fer-1; 2.5 μM) are shown as a control. Error bars show SEM for n = 3 replicates. **p < 0.01, ***p < 0.01 for one-way ANOVA compared to cells treated with RSL-3 alone. (F) Viability of HT1080 cells, measured by CTG, pretreated for 16 h with AA64 (blue) at the indicated concentration and then challenged with RSL-3 (1.25 μM) for 24 h. Cells treated with ferrostatin (Fer-1; 2.5 μM) are shown as a control. Error bars show SEM for n = 3 replicates. ***p < 0.01 for one-way ANOVA compared to cells treated with RSL-3 alone. | ||
Discussion
Herein, we used medicinal chemistry to establish AA147 analogs with altered linker moieties. Through these efforts, we identified AA28, which contains a carbamate linker, as a compound that shows robust activation of the NRF2 transcriptional response in neuron-derived cell culture models. Furthermore, we showed that AA28 demonstrates improved protection of neuronal cells against glutamate-induced oxytosis and erastin-induced ferroptosis, as compared to AA147. These results establish AA28 as a new scaffold for the development of enhanced proteostasis regulators to mitigate pathologic neurotoxicity.Amide isosteres are commonly used in medicinal chemistry to improve bioactive compounds.37,38 However, our results demonstrate that several of these commonly employed substitutions, (i.e., retro-amide, ester, urea) are not tolerated on the AA147 scaffold. Instead, the carbamate modification proved to be the only amide isostere that retained the unique stress-responsive signaling activity afforded by the parent compound AA147. Interestingly, the closely related urea isostere seemed to abolish compound activity. This suggests that extra hydrogen bond donors, delocalized electron density adjacent to the carbonyl functionality, or potentially conformational restrictions may play an underappreciated role in the efficacy of metabolically activatable proteostasis regulators of this class.39,40 Indeed, there is a significant thermodynamic difference (3–4 kcal mol−1) in the amide resonance of carbamates versus amides.41 As carbamates are included in numerous FDA-approved drugs for imbuing improved physicochemical properties, these compounds may show even greater activity than AA147 when used in vivo.21
Here, we demonstrate that metabolically activated proteostasis regulators containing the 2-amino-p-cresol substructure protect against both glutamate-induced oxytosis and erastin-induced ferroptosis in neuron-derived cell models. Intriguingly, AA28 showed increased activity in these assays as compared to AA147. Activation of the NRF2 oxidative stress response mediates this protection, as evidenced by the blockage of this protection upon cotreatment with the selective NRF2 inhibitor ML385. NRF2 regulates the expression of genes involved in multiple redox-modulating pathways, including iron metabolism, GSH synthesis, and NADPH production, all of which have been shown to be protective against ferroptosis.42–45 Thus, our results indicate that pharmacologic activation of this protective pathway comprising multiple anti-oxidant factors using these AA147 analogs protects neurons against ferroptotic cell death. These results extend the potential application of this class of compounds to other types of neurodegenerative diseases where ferroptosis plays an important role (e.g., Alzheimer's disease).6,46
We demonstrate the potential for using AA28 as a new scaffold to probe the potential benefits for modification of the phenyl B-ring of this compound in neuroprotection. By screening different B-ring analogs of AA28, we identified compound AA64 that replaces the phenyl B-ring with a furan. AA64 showed increased selectivity for NRF2 activation over ATF6 activation, relative to AA147 or AA28, and robust protection against glutamate- and erastin-induced toxicity. Further analysis elucidated that this robust protection extends towards numerous pharmacological activators of ferroptosis, as well as across several distinct cell lines of interest. Surprisingly, the protection afforded by AA64 could not be attributed to NRF2 activation, as cotreatment with the potent NRF2 inhibitor ML385 did not prevent AA64-dependent suppression of erastin-induced neurotoxicity. Several potential hypotheses can explain this discrepancy. Furan derivatives can form electrophilic epoxide- or enedial-based metabolites, which may modify alternate cellular protein targets to confer protection against neurotoxic insults.47 While such electrophiles typically exhibit indiscriminate reactivity towards proteins, we and others have shown pro-electrophilic warheads can show unique and tempered proteome reactivity in certain cellular contexts.12,48,49 This effect could be further enhanced by unique physicochemical properties of the AA28 scaffold, which direct this compound to the ER membrane for metabolic activation. In addition, furan derivatives have been shown to possess a degree of intrinsic antioxidant capacity, suggesting that AA64 could directly suppress ROS accumulation induced by glutamate and erastin treatment.50–52 However, the lack of protection afforded by our AA64 analog missing the 2-amino-p-cresol moiety against erastin-induced ferroptosis suggests covalent protein modification, like AA147, is requisite for protection against ferroptosis. This suggests that the presence of the furan, specifically in the context of the carbamate linker, affords emergent properties to the compound that could contribute to neuroprotection. We are continuing to pursue the mechanistic basis for the NRF2-independent activities of AA64 in subsequent studies.
In conclusion, we leveraged medicinal chemistry to identify AA147 analogs that show enhanced protection against neurotoxic insults such as erastin-induced ferroptosis. Specifically, we identified AA28 as a carbamate-containing scaffold that can be used to develop next generation proteostasis regulators of this class. In addition, our results reveal further insights into how the pro-electrophilic nature of metabolically activatable proteostasis regulators allows targeted stress-responsive signaling.53,54 This contrasts with more reactive covalent NRF2 activators that may have idiosyncratic toxicities mediated through excessive covalent conjugation to proteogenic thiols. Ultimately, our continued development of metabolically activated proteostasis regulators such as AA28 provides new opportunities to therapeutically target NRF2 activity for neuroprotection.
Data availability
ESI† and chemical compound information are available in the online version of the paper.Conflicts of interest
JWK and RLW are scientific advisory board members and shareholders in Protego Biopharma, which has licensed metabolically proteostasis regulators including AA147 for translational development.Acknowledgements
We thank Evan T. Powers for critical reading of this manuscript and Emily P. Bentley for expert editorial feedback. We thank the TSRI Flow Cytometry core for support in the work described in this manuscript. The opinions and assertions expressed herein are those of the author(s) and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences or the Department of Defense. Funding for this work was provided by the National Institutes of Health (AG046495 to JWK and RLW). This is manuscript # 30257 from The Scripps Research Institute.References
- I. Liguori, G. Russo, F. Curcio, G. Bulli, L. Aran, D. Della-Morte, G. Gargiulo, G. Testa, F. Cacciatore, D. Bonaduce and P. Abete, Clin. Interventions Aging, 2018, 13, 757–772 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- A. Ionescu-Tucker and C. W. Cotman, Neurobiol. Aging, 2021, 107, 86–95 CrossRef CAS PubMed.
- H. Sies, C. Berndt and D. P. Jones, in Annual Review of Biochemistry, ed. R. D. Kornberg, 2017, vol. 86, pp.715–748 Search PubMed.
- J. Lewerenz, G. Ates, A. Methner, M. Conrad and P. Maher, Front. Neurosci., 2018, 12, 214 CrossRef PubMed.
- P. Maher, A. Currais and D. Schubert, Cell Chem. Biol., 2020, 27, 1456–1471 CrossRef CAS PubMed.
- X. Jiang, B. R. Stockwell and M. Conrad, Nat. Rev. Mol. Cell Biol., 2021, 22, 266–282 CrossRef PubMed.
- B. R. Stockwell, Cell, 2022, 185, 2401–2421 CrossRef CAS PubMed.
- S. Masaldan, A. I. Bush, D. Devos, A. S. Rolland and C. Moreau, Free Radicals Biol. Med., 2019, 133, 221–233 CrossRef CAS PubMed.
- S. K. Ryan, C. L. Ugalde, A.-S. Rolland, J. Skidmore, D. Devos and T. R. Hammond, Trends Pharmacol. Sci., 2023, 44, 674–688 CrossRef CAS PubMed.
- J. D. Rosarda, K. R. Baron, K. Nutsch, G. M. Kline, C. Stanton, J. W. Kelly, M. J. Bollong and R. L. Wiseman, ACS Chem. Biol., 2021, 16, 2852–2863 CrossRef CAS PubMed.
- R. Paxman, L. Plate, E. A. Blackwood, C. Glembotski, E. T. Powers, R. L. Wiseman and J. W. Kelly, eLife, 2018, 7, e37168 CrossRef PubMed.
- L. Plate, C. B. Cooley, J. J. Chen, R. J. Paxman, C. M. Gallagher, F. Madoux, J. C. Genereux, W. Dobbs, D. Garza, T. P. Spicer, L. Scampavia, S. J. Brown, H. Rosen, E. T. Powers, P. Walter, P. Hodder, R. L. Wiseman and J. W. Kelly, eLife, 2016, 5, e15550 CrossRef PubMed.
- L. Ibrahim, J. Mesgarzadeh, I. Xu, E. T. Powers, R. L. Wiseman and M. J. Bollong, Antioxidants, 2020, 9, 1025 CrossRef CAS PubMed.
- T. Nguyen, P. Nioi and C. B. Pickett, J. Biol. Chem., 2009, 284, 13291–13295 CrossRef CAS PubMed.
- Q. Ma, in Annual Review of Pharmacology and Toxicology, ed. P. A. Insel, 2013, vol. 53, pp.401–+ Search PubMed.
- S. K. Niture, R. Khatri and A. K. Jaiswal, Free Radicals Biol. Med., 2014, 66, 36–44 CrossRef CAS PubMed.
- A. Kobayashi, M. I. Kang, Y. Watai, K. I. Tong, T. Shibata, K. Uchida and M. Yamamoto, Mol. Cell. Biol., 2006, 26, 221–229 CrossRef CAS PubMed.
- C. C. Glembotski, J. D. Rosarda and R. L. Wiseman, Trends Mol. Med., 2019, 25, 538–550 CrossRef CAS PubMed.
- Z. Yuan, L. P. Lu, Y. T. Lian, Y. R. Zhao, T. T. Tang, S. Xu, Z. Yao and Z. Yu, Front. Pharmacol., 2022, 13 DOI:10.3389/fphar.2022.1028002.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- M. J. Bollong, G. Lee, J. S. Coukos, H. Yun, C. Zambaldo, J. W. Chang, E. N. Chin, I. Ahmad, A. K. Chatterjee, L. L. Lairson, P. G. Schultz and R. E. Moellering, Nature, 2018, 562, 600–604 CrossRef CAS PubMed.
- A. Singh, S. Venkannagari, K. H. Oh, Y. Q. Zhang, J. M. Rohde, L. Liu, S. Nimmagadda, K. Sudini, K. R. Brimacombe, S. Gajghate, J. Ma, A. Wang, X. Xu, S. A. Shahane, M. Xia, J. Woo, G. A. Mensah, Z. Wang, M. Ferrer, E. Gabrielson, Z. Li, F. Rastinejad, M. Shen, M. B. Boxer and S. Biswal, ACS Chem. Biol., 2016, 11, 3214–3225 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Chem. Biol., 2008, 15, 234–245 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- S. J. Dixon, D. Patel, M. Welsch, R. Skouta, E. Lee, M. Hayano, A. G. Thomas, C. Gleason, N. Tatonetti, B. S. Slusher and B. R. Stockwell, eLife, 2014, 3, e02523 CrossRef PubMed.
- A. N. von Krusenstiern, R. N. Robson, N. Qian, B. Qiu, F. Hu, E. Reznik, N. Smith, F. Zandkarimi, V. M. Estes, M. Dupont, T. Hirschhorn, M. S. Shchepinov, W. Min, K. A. Woerpel and B. R. Stockwell, Nat. Chem. Biol., 2023, 19, 719–730 CrossRef CAS PubMed.
- E. H. W. Pap, G. P. C. Drummen, V. J. Winter, T. W. A. Kooij, P. Rijken, K. W. A. Wirtz, J. A. F. Op den Kamp, W. J. Hage and J. A. Post, FEBS Lett., 1999, 453, 278–282 CrossRef CAS PubMed.
- W. S. Yang and B. R. Stockwell, Chem. Biol., 2008, 15, 234–245 CrossRef CAS PubMed.
- M. Conrad and D. A. Pratt, Nat. Chem. Biol., 2019, 15, 1137–1147 CrossRef CAS PubMed.
- W. S. Yang, R. SriRamaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell, Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- M. M. Gaschler, A. A. Andia, H. Liu, J. M. Csuka, B. Hurlocker, C. A. Vaiana, D. W. Heindel, D. S. Zuckerman, P. H. Bos, E. Reznik, L. F. Ye, Y. Y. Tyurina, A. J. Lin, M. S. Shchepinov, A. Y. Chan, E. Peguero-Pereira, M. A. Fomich, J. D. Daniels, A. V. Bekish, V. V. Shmanai, V. E. Kagan, L. K. Mahal, K. A. Woerpel and B. R. Stockwell, Nat. Chem. Biol., 2018, 14, 507–515 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- M. J. Deetz, C. C. Forbes, M. Jonas, J. P. Malerich, B. D. Smith and O. Wiest, J. Org. Chem., 2002, 67, 3949–3952 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2020, 63, 2751–2788 CrossRef CAS PubMed.
- D. Kaur, P. Sharma and P. V. Bharatam, J. Mol. Struct., 2005, 757, 149–153 CrossRef CAS.
- A. Anandhan, M. Dodson, A. Shakya, J. Chen, P. Liu, Y. Wei, H. Tan, Q. Wang, Z. Jiang, K. Yang, J. G. N. Garcia, S. K. Chambers, E. Chapman, A. Ooi, Y. Yang-Hartwich, B. R. Stockwell and D. D. Zhang, Sci. Adv., 2023, 9, eade9585 CrossRef CAS PubMed.
- M. Dodson, R. Castro-Portuguez and D. D. Zhang, Redox Biol., 2019, 23, 101107 CrossRef CAS PubMed.
- Z. Fan, A. K. Wirth, D. Chen, C. J. Wruck, M. Rauh, M. Buchfelder and N. Savaskan, Oncogenesis, 2017, 6, e371 CrossRef CAS PubMed.
- X. Sun, Z. Ou, R. Chen, X. Niu, D. Chen, R. Kang and D. Tang, Hepatology, 2016, 63, 173–184 CrossRef CAS PubMed.
- A. Anandhan, M. Dodson, C. J. Schmidlin, P. Liu and D. D. Zhang, Cell Chem. Biol., 2020, 27, 436–447 CrossRef CAS PubMed.
- L. A. Peterson, Chem. Res. Toxicol., 2013, 26, 6–25 Search PubMed.
- H. Cho, Q. Shen, L. H. Zhang, M. Okumura, A. Kawakami, J. Ambrose, F. Sigoillot, H. R. Miller, S. Gleim, A. Cobos-Correa, Y. Wang, P. Piechon, G. Roma, F. Eggimann, C. Moore, P. Aspesi, Jr., F. A. Mapa, H. Burks, N. T. Ross, P. Krastel, M. Hild, T. J. Maimone, D. E. Fisher, D. K. Nomura, J. A. Tallarico, S. M. Canham, J. L. Jenkins and W. C. Forrester, Cell Chem. Biol., 2021, 28, 1407–1419 Search PubMed.
- J. K. Eaton, R. A. Ruberto, A. Kramm, V. S. Viswanathan and S. L. Schreiber, J. Am. Chem. Soc., 2019, 141, 20407–20415 CrossRef CAS PubMed.
- D. M. Goldberg, B. Hoffman, J. Yang and G. J. Soleas, J. Agric. Food Chem., 1999, 47, 3978–3985 CrossRef CAS PubMed.
- R. A. S. Lemke, A. C. Peterson, E. C. Ziegelhoffer, M. S. Westphall, H. Tjellstroem, J. J. Coon and T. J. Donohue, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3450–E3457 CAS.
- Y. Okada and H. Okajima, J. Pharm. Soc. Jpn., 1998, 118, 226–230 CrossRef CAS PubMed.
- S. A. Lipton, T. Rezaie, A. Nutter, K. M. Lopez, J. Parker, K. Kosaka, T. Satoh, S. R. McKercher, E. Masliah and N. Nakanishi, Cell Death Dis., 2016, 7, e2499 CrossRef CAS PubMed.
- R. H. Scannevin, S. Chollate, M. Y. Jung, M. Shackett, H. Patel, P. Bista, W. K. Zeng, S. Ryan, M. Yamamoto, M. Lukashev and K. J. Rhodes, J. Pharmacol. Exp. Ther., 2012, 341, 274–284 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00027g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |