
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrevention of amyloid β fibril deposition on the synaptic membrane in the precuneus by ganglioside nanocluster-targeting inhibitors†
Erika
Miyamoto
a,
Hideki
Hayashi
 b,
Shigeo
Murayama
cd,
Katsuhiko
Yanagisawa
e,
Toshinori
Sato
*a and
Teruhiko
Matsubara
*a
b,
Shigeo
Murayama
cd,
Katsuhiko
Yanagisawa
e,
Toshinori
Sato
*a and
Teruhiko
Matsubara
*a
aDepartment of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kouhoku-ku, Yokohama 223-8522, Japan. E-mail: matsubara@bio.keio.ac.jp
bDepartment of Applied Biochemistry, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan
cBrain Bank for Aging Research, Tokyo Metropolitan Institute for Geriatrics and Gerontology, 35-2 Sakae-cho, Itabashi-ku, Tokyo 173-0015, Japan
dBrain Bank for Neurodevelopmental, Neurological and Psychiatric Disorders, United Graduate School of Child Development, Osaka University, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan
eResearch and Development Center for Precision Medicine, University of Tsukuba, 1-2 Kasuga, Tsukuba, Ibaraki 305-8550, Japan
First published on 4th April 2024
Abstract
Alzheimer's disease (AD), a progressive neurodegenerative condition, is one of the most common causes of dementia. Senile plaques, a hallmark of AD, are formed by the accumulation of amyloid β protein (Aβ), which starts to aggregate before the onset of the disease. Gangliosides, sialic acid-containing glycosphingolipids, play a key role in the formation of toxic Aβ aggregates. In membrane rafts, ganglioside-bound complexes (GAβ) act as nuclei for Aβ assembly, suggesting that GAβ is a promising target for AD therapy. The formation of GAβ-induced Aβ assemblies has been evaluated using reconstituted planar lipid membranes composed of synaptosomal plasma membrane (SPM) lipids extracted from human and mouse brains. Although the effects of gangliosides on Aβ accumulation in the precuneus have been established, effects on Aβ fibrils have not been determined. In this study, Aβ42 fibrils on reconstituted membranes composed of SPM lipids prepared from the precuneus cortex of human autopsied brains were evaluated by atomic force microscopy. In particular, Aβ42 accumulation, as well as the fibril number and size were higher for membranes with precuneus lipids than for membranes with calcarine cortex lipids. In addition, artificial peptide inhibitors targeting Aβ-sensitive ganglioside nanoclusters cleared Aβ assemblies on synaptic membranes in the brain, providing a novel therapeutic strategy for AD.
Introduction
The world's population has been aging rapidly in recent years. With this aging population comes an increase in age-related diseases, such as dementia. One of the most common causes of dementia, Alzheimer's disease (AD), is a progressive neurodegenerative disease1 characterized by senile plaques, neurofibrillary tangles, and brain atrophy.2 Senile plaques are formed by the deposition of aggregated amyloid β-protein (Aβ), a peptide of 39–43 amino acids, which begins to aggregate before the onset of AD.3,4 The physiological concentrations of Aβ40 and Aβ42 in cerebrospinal fluids of patients with AD are 6–30 nM (Aβ40) and 8–60 pM (Aβ42), respectively, whereas those in healthy individuals are 0.25–8 nM (Aβ40). Aβ aggregates into various forms, such as oligomers and fibrils, not only by self-aggregation but also via lipid membranes and metal ions.5–10 Aβ aggregates are highly toxic and are deeply involved in the onset and progression of AD.11–15Gangliosides, sialic acid-containing glycosphingolipids, are involved in the formation of highly toxic Aβ aggregates.16 On membrane rafts, membrane microdomains composed of sphingomyelin (SM) and cholesterol, gangliosides assemble and form nanoclusters with high density.17–20 We have shown that a ganglioside-bound complex (GAβ) is formed by the interaction of Aβ monomers with nanoclusters composed of gangliosides, such as Galβ1–3GalNAcβ1–4(Neu5Acα2–3)Galβ1–4Glcβ1–1′Cer (GM1),21–24 and the complex acts like a seed for Aβ assembly due to conformational transitions of other Aβ monomers.10,25–27 We have used planar lipid bilayers to elucidate the features of Aβ assemblies by atomic force microscopy (AFM). By use of the planar membranes, constructed by the control of membrane pressure using the facultative lipid composition, we can observe lipid rafts and ganglioside nanoclusters at a nanometer scale from surface topographic images. Typically, we prepare a planar lipid bilayer composed of a ganglioside, SM, and cholesterol to mimic the ganglioside nanocluster that interacts with Aβ, and we can identify Aβ fibrils by AFM observation.23,28
Many Aβ-targeting agents have recently been developed; however, it has not been possible to create effective disease-modifying agents.29–33 It is possible that Aβ aggregation in vitro is different from that in vivo.34 Therefore, we have evaluated Aβ assemblies using reconstituted planar lipid membranes composed of lipids extracted from human and mouse brains.35,36 In the brain of patients with AD, Aβ accumulates in a brain region-specific manner. One of the most vulnerable areas for Aβ deposition is the precuneus (PC), and, conversely, one of the most resistant areas is the calcarine.37–39 Since Aβ aggregates are deposited in the PC during the early phase of AD progression,40 this process can provide insight into the mechanism underlying the onset and progression of the AD. We have previously observed Aβ accumulation on reconstituted membranes with synaptosomal plasma membrane (SPM) lipids extracted from the PC bearing amyloid from the human autopsied brain, with no accumulation using lipids extracted from the amyloid-free cortex of calcarine (CC).36 An imbalance in the fatty-acid chain length of ganglioside GD1b between PC and CC extracts can explain this difference in accumulation. However, the generation of Aβ fibrils after a long incubation period (>12 h) has not been evaluated.
In this study, we investigated Aβ assemblies generated on reconstituted membranes composed of SPM lipids extracted from autopsied brains of elderly patients with AD. Aβ fibrils on the membrane were identified by AFM and the effect of linear and cyclic peptide inhibitors that have affinity for Aβ-sensitive ganglioside nanoclusters was evaluated. Our results revealed that toxic Aβ fibrils were not only reduced but were also released from the synaptosomal membrane in the presence of the inhibitor.
Results and discussion
Surface topography of the reconstituted membrane composed of SPM lipids isolated from the precuneus (PC) and calcarine cortexes (CC)
To investigate region-specific Aβ accumulation in the brain, we first prepared two SPM lipid extracts from the PC and CC of human autopsy brains. Following previously described methods, the reconstituted membrane composed of SPM lipids was prepared and the surface topography of the membrane was observed using AFM (Fig. 1(A) and (B)).36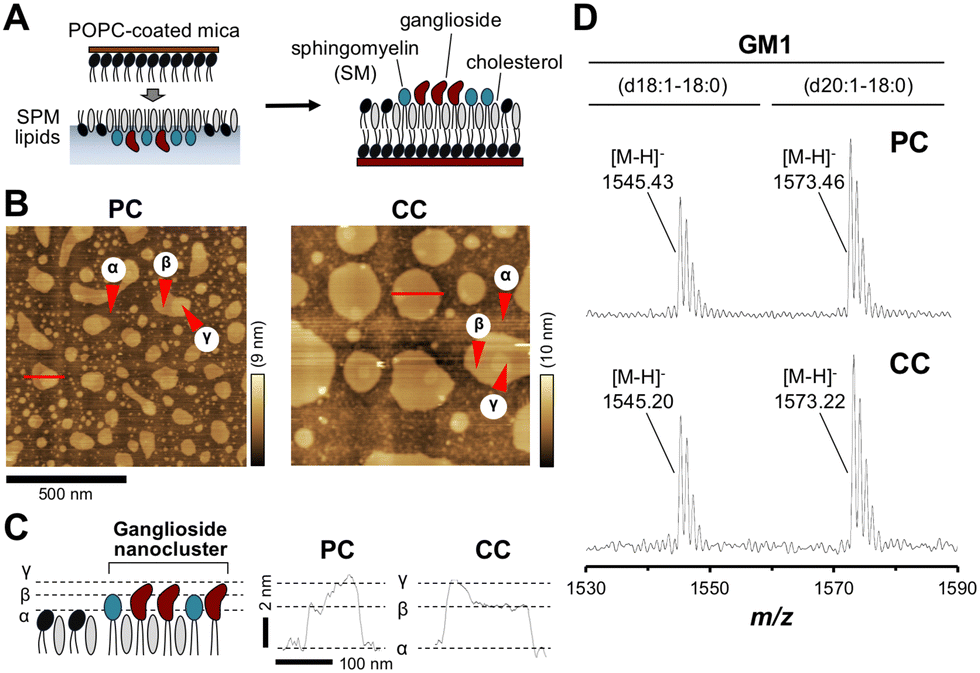 | ||
| Fig. 1 Surface topography of reconstituted membranes prepared with lipids extracted from SPM isolated from autopsied brains. (A) Preparation of the reconstituted lipid bilayer. A monolayer of SPM lipids was deposited on POPC-coated mica to prepare a lipid bilayer. This lipid bilayer was incubated with PBS at 37 °C for 24 h and the surface topography was observed using AFM. (B) Typical AFM images of the reconstituted lipid bilayer with SPM lipids extracted from the precuneus (PC, left) and calcarine cortex (CC, right). (C) Cross-section analysis of the bilayer at the sites marked with red lines in AFM images (panel B). (D) Detection of ganglioside GM1 in SPM lipids by MALDI-TOF/MS. Two molecular species of GM1 with ceramides, d18:1-18:0 and d20:1-18:0, were found in SPM lipids. GM1, Galβ1–3GalNAcβ1–4(Neu5Acα2–3)Galβ1–4Glcβ1–1′Cer. | ||
In the case of the PC, there were two domains β and γ, segregated from the area α of the non-raft region as the liquid-disordered (Ld) phase (Fig. 1(C)). These domains were assigned to the lipid raft with the liquid-ordered (Lo) phase, and the uppermost highest domain γ was further segregated from domain β with a diameter of 10–200 nm. Domain γ with heights of 2 and 4 nm from domain β and area α, respectively, was identified as a ganglioside-enriched domain based on features described in GM1-containing membrane studies.17 In the case of CC, although similar domains were found, the domain γ count was lower than that of PC. The size of domain β in the membrane was larger in CC than in PC. These differences in topography between PC and CC lipids could be explained by the lipid composition. Depending on the membrane environment, such as the type of gangliosides in the membrane, various kinds of Aβ aggregates are formed.16,22,28 It is possible that Aβ aggregation on the CC membrane occurs via a different pathway from that involved in GAβ formation induced by ganglioside nanoclusters on PC membranes.
We have previously determined the ganglioside composition of SPM lipids extracted from the PC or CC of the human brain.36 We confirmed the presence of GM1, the most abundant ganglioside in the brain with a role in GAβ formation, by MALDI-TOF/MS. Two major peaks at m/z 1545.4 and 1573.5 were assigned to the [M − H]− ion of GM1 with ceramide d18:1–18:0 (1544.9, calculated) and d20:1–18:0 (1572.9, calculated), respectively (Fig. 1(D)). These results indicate that the SPM lipids extracted from both regions contain GM1. In addition, two major peaks at m/z 917.1 and 933.1 were assigned to the [M − 2H]2− ion of GD1 with ceramide d18:1–18:0 (917.5, calculated) and d20:0–18:0 (932.5, calculated), respectively (Fig. S1, ESI†). A peak at m/z 1063.9 was assigned to the [M − 2H]2− ion of GT1 with ceramide d18:1–18:0 (1063.0) (Fig. S1, ESI†). These gangliosides were also detected in SPM lipids extracted from PC and CC of human autopsy brains in a previous study.36
Aβ42 binding and accumulation on the reconstituted membrane composed of SPM lipids isolated from PC and CC
To clarify the binding and subsequent accumulation of Aβ42, the reconstituted membrane was incubated with seed-free Aβ42 monomer at 37 °C. In a previous study, Aβ42 accumulation was observed on a reconstituted membrane with amyloid-bearing PC lipids for 12 h; however, no fibrils were generated.36 In this study, after 15 min of incubation, short Aβ42 fibrils (around 50–200 nm long) with a diameter of 10–30 nm were observed on the membrane in an Aβ42 concentration-dependent manner (Fig. 2(A) and Fig. S2, ESI†). The total length and number of fibrils observed on the membrane with PC lipids were 3.2 ± 1.0 μm and 28 ± 4, respectively, at 20 μM Aβ42 (Fig. 2(B) and (C)). The total length and number of fibrils observed on the membrane with CC lipids (1.0 ± 0.3 μm and 7 ± 1, respectively) were lower than those of PC, indicating that Aβ42 assembly is significantly elevated on the membrane with PC lipids. This result is consistent with our previous findings that Aβ42 assembly levels on the membrane with PC lipids are larger than those on the membrane with CC lipids.36 Previously, we determined size and shapes of Aβ fibrils induced by seven kinds of ganglioside nanoclusters (GM1, GM2, GM3, GD1a, GD1b, GT1b, and GQ1b).28 Since SPM lipids contain at least three kinds of gangliosides (GM1, GD1, and GT1) (Fig. 1(D) and Fig. S1, ESI†), Aβ fibrils should have diverse structural features.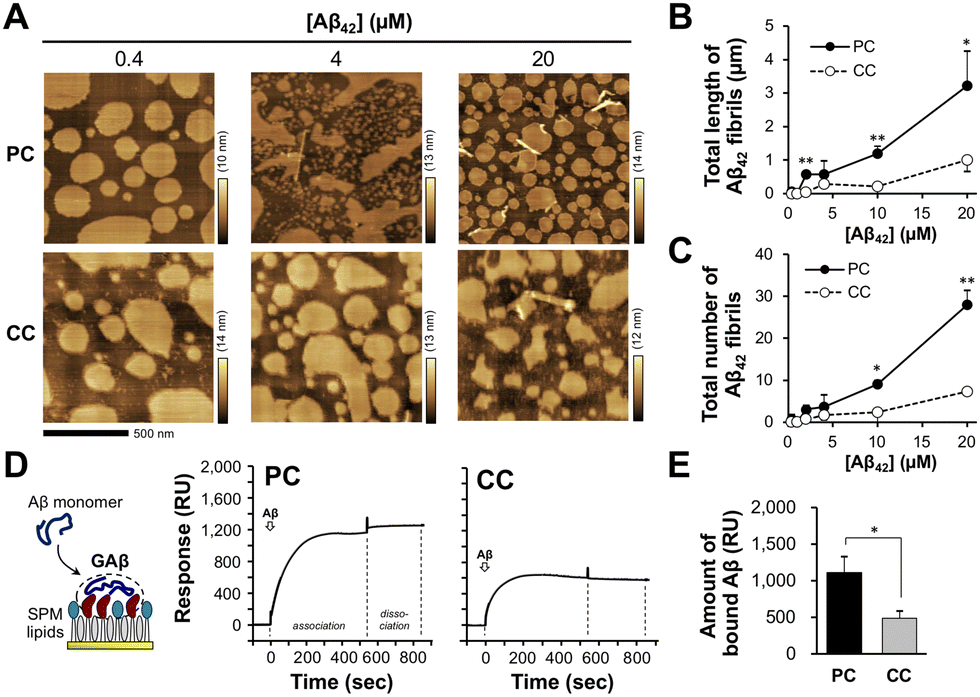 | ||
Fig. 2 Binding and accumulation of Aβ42 on reconstituted SPM lipid membranes. (A) Typical AFM images of the reconstituted lipid bilayer with SPM lipids extracted from the precuneus (PC) and cortex of calcarine (CC) after incubation with Aβ42 (0.4–20 μM) at 37 °C for 15 min. (B) and (C) Total length (B) and number (C) of observed Aβ42 fibrils on the membrane after Aβ42 interaction for 15 min. *p < 0.05, **p < 0.01 compared with CC. (D) Typical SPR sensorgrams of Aβ42 (20 μM) against the reconstituted lipid monolayer. Sensorgrams for an association time of 540![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) s and dissociation time of 300 s are shown. (E) Amount of Aβ42 bound to the lipid monolayer. The response value (RU) at 840 s was used as the amount of bound Aβ42. *p < 0.05. Data are presented as average values ± standard deviation (n = 3). s and dissociation time of 300 s are shown. (E) Amount of Aβ42 bound to the lipid monolayer. The response value (RU) at 840 s was used as the amount of bound Aβ42. *p < 0.05. Data are presented as average values ± standard deviation (n = 3). | ||
We next determined the amounts of Aβ42 bound to the reconstituted lipid membrane by SPR analysis. Reconstituted lipid monolayers composed of the SPM lipids extracted from PC and CC were immobilized on a sensor chip, and SPR signals were then detected in the presence of Aβ42 (20 μM) as the analyte. The binding of Aβ42 to the membranes with PC and CC lipids was observed during the association phase (540 s); however, the release of Aβ42 was not observed during the dissociation phase (300 s) (Fig. 2(D)). The amount of Aβ42 bound to the membrane was higher for PC (1110 ± 220 resonance units (RU)) than for CC (490 ± 95 RU) (Fig. 2(E)). This result indicates that Aβ42 accumulation is promoted on the membrane with PC lipids. The SPR signals for Aβ42 binding to the membrane with PC lipids supports that the formation of GAβ, oligomers, and short fibrils, as shown in Fig. 2(A).
Generation of Aβ42 fibrils on the reconstituted membrane composed of PC lipids
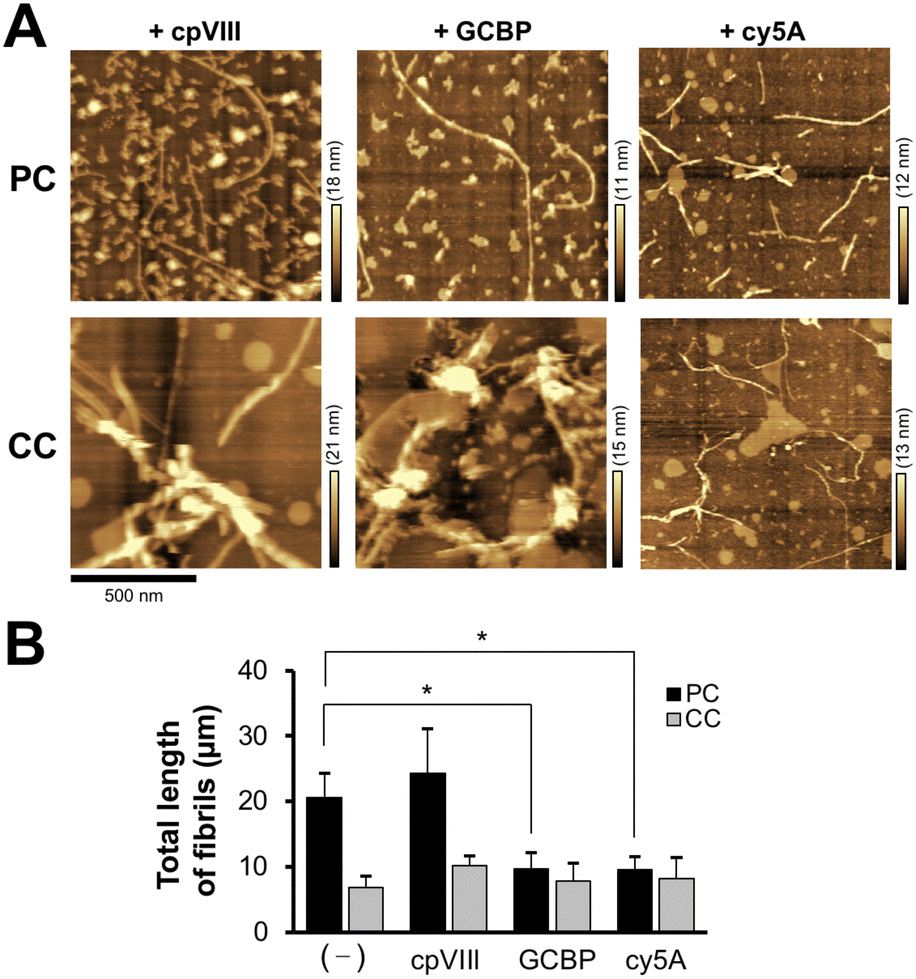
During a short Aβ interaction time (15 min), Aβ42 accumulation was observed; however, long Aβ42 fibrils were not observed on the membranes. When Aβ42 interacted with the membrane at 37 °C for 24 h, long fibrils with diameters of 10–30 nm were generated on the membrane with PC lipids (Fig. 3(A)). At 24 h, there was a six-fold increase in fibril length (20.6 ± 3.8 μm) at 4 μM Aβ42 on the membrane with PC lipids and a 3.5-fold increase in fibril number (98 ± 16) over those at 15 min (Fig. 3(B) and (C)). On the membrane with CC lipids, the fibril length and number were higher at 24 h than at 15 min (6.9 ± 1.8 μm and 45 ± 13, respectively); however, the PC lipid-induced fibrils (50–200 nm range in length) were still larger than those for CC (<50 nm in length) (Fig. S2, ESI†). These results indicated that the formation of Aβ42 into fibrils was promoted in the membrane with PC lipids.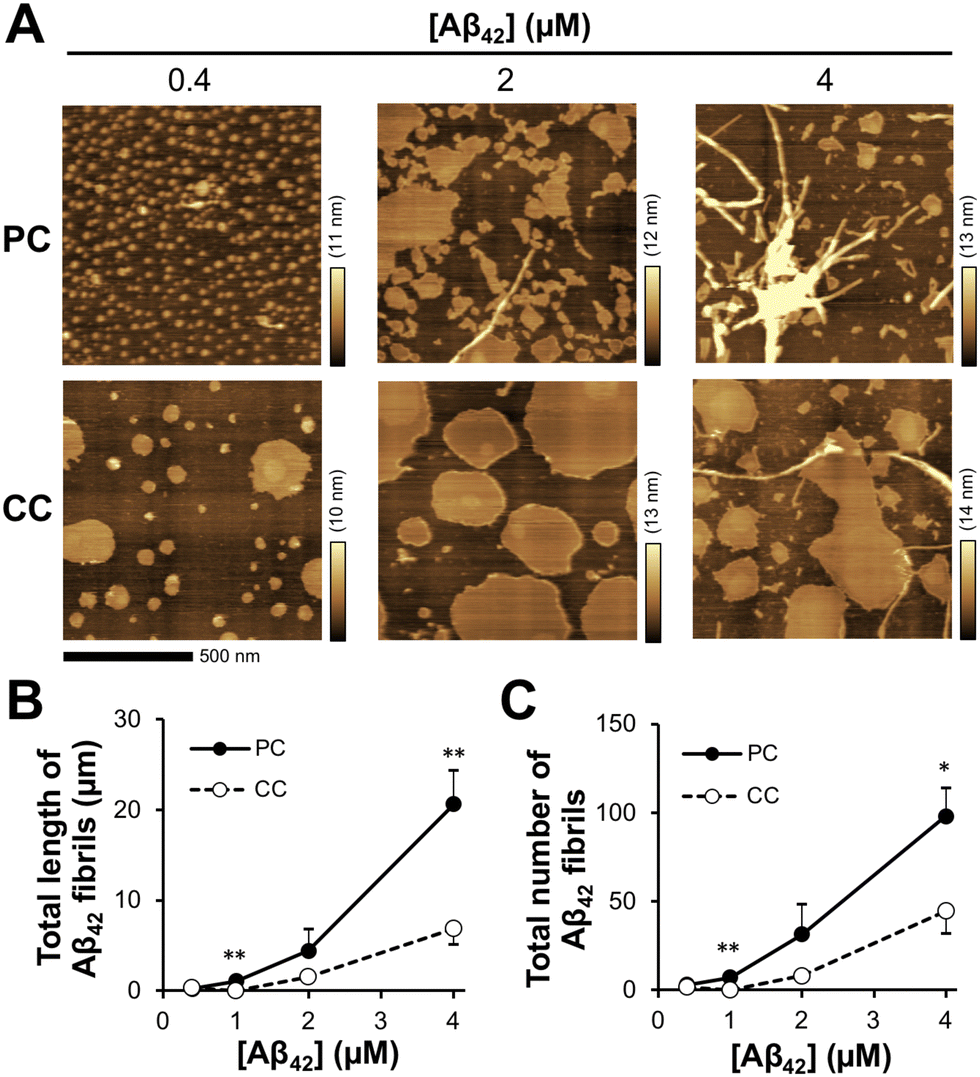 | ||
| Fig. 3 Aβ42 fibril generation on the SPM lipid membrane. (A) Typical AFM images of the reconstituted lipid bilayer with SPM lipids extracted from PC and CC after incubation with Aβ42 (0.4–4 μM) at 37 °C for 24 h. (B) and (C) Total length (B) and number (C) of observed Aβ42 fibrils on the lipid bilayer after Aβ42 interaction for 24 h. *p < 0.05, **p < 0.01 compared with CC. Data are presented as average values ± standard deviation (n = 3). | ||
Toxicity of Aβ assemblies induced on reconstituted membranes
The toxicity of Aβ42 assemblies induced on reconstituted membranes of PC and CC, was evaluated by a calcein-AM assay using SH-SY5Y cells. Cell viability against the membrane with PC lipids incubated with 10 μM Aβ42 for 48 h decreased to 80.2 ± 5.2% (Fig. 4). Toxicity was greater for Aβ42 assemblies induced by the membrane with PC lipids than with CC lipids, consistent with previous results showing that assemblies formed via GAβ complexes on the lipid membrane are highly cytotoxic.26,41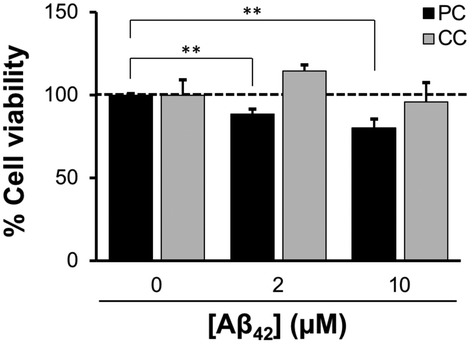 | ||
| Fig. 4 Toxicity of Aβ42 assemblies formed on the reconstituted membranes with SPM lipids. Aβ42 (2 and 10 μM) was incubated on the lipid bilayer on disc at 37 °C for 24 h to generate the Aβ42 assembly. SH-SY5Y cells were then seeded on the Aβ42-deposited lipid bilayer and incubated for 48 h. After incubation, a calcein-AM assay was performed to obtain the cell viability (%). Data are presented as average values ± standard deviation (n = 3). *p < 0.05, **p < 0.01. | ||
Reduction of Aβ assemblies deposited on the membrane with PC lipids in the presence of inhibitors
We hypothesized that GAβ-targeting molecules can inhibit the formation of highly toxic Aβ aggregates or the release of Aβ assemblies from the precuneus, providing therapeutic agents for AD. We have previously demonstrated that GM1 cluster-binding peptide (GCBP), VWRLLAPPFSNRLLP, can inhibit the generation of Aβ assemblies induced on 10 mol% GM1-containing lipid membranes (GM1/SM/cholesterol, 10:45:45) and the release of Aβ fibrils from the membrane.42 GCBP has affinity for a-series gangliosides, such as GM1, GD1a, and GM3.17 Recently, we designed a cysteine-constrained cyclic peptide, cy5A, based on the GCBP sequence; cy5A is an intramolecular cyclic peptide with a disulfide bond between a pair of cysteine residues (Fig. 5(A)).43 To determine the binding of GCBP and cy5A, a monolayer of SPM PC and CC lipids was loaded on plastic discs. The amount of GCBP and cy5A (10 μM) bound to the membrane with PC lipids was 26.3% and 11.6% higher than that for CC lipids, respectively (Fig. 5(B)). This result indicates that Aβ-sensitive ganglioside nanoclusters formed on the membrane with PC lipids.
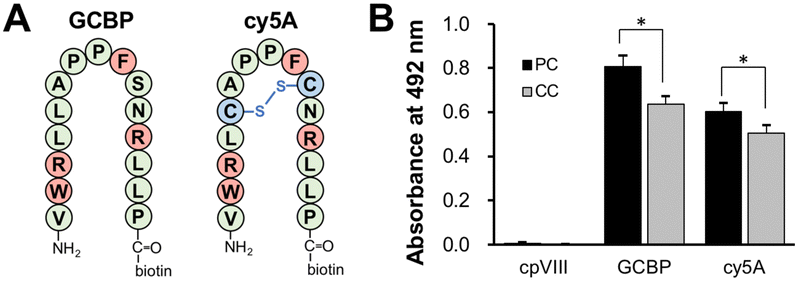 | ||
| Fig. 5 Binding of GCBP and cy5A to ganglioside nanoclusters on the reconstituted lipid monolayer with SPM lipids. (A) Structure of linear (GCBP) and cyclic peptides (cy5A). Leu5 and Ser10 of GCBP were substituted with Cys (blue), and the Cys-substituted peptide was cyclized to give a cyclic peptide cy5A. Four residues, Trp2, Arg3, Phe9, and Arg12, responsible for binding to GM1, are shown in red. (B) The amount of bound peptide was determined using the avidin–biotin complex method. Biotinylated peptides (10 μM) were incubated with the lipid monolayer at 37 °C for 1 h and the amount of each peptide bound to the lipid monolayer was estimated by HRP-conjugated avidin. *p < 0.05, **p < 0.01. Data are presented as average values ± standard deviation (n = 3). | ||
Next, we investigated the behavior of Aβ42 assemblies on the membrane with PC lipids in the presence of GCBP and cy5A. The membranes of PC and CC were incubated with Aβ42 (4 μM) in the presence of peptide (10 μM) at 37 °C for 24 h (Fig. 6(A)). Aβ42 fibril formation induced on the membrane was inhibited by GCBP and cy5A. The total length of Aβ42 fibrils (20.6 ± 3.8 μm) was reduced to 9.7 ± 2.4 μm and 9.6 ± 1.9 μm in the presence of GCBP and cy5A, whereas the control peptide (cpVIII) did not have an inhibitory effect (Fig. 6(B)).
 | ||
| Fig. 6 Inhibition of Aβ fibril formation by ganglioside nanocluster-targeting peptides. (A) Typical AFM images of the reconstituted lipid bilayer with SPM lipids after incubation with Aβ42 (4 μM) in the presence of 10 μM GCBP, cy5A, and cpVIII peptides (control) at 37 °C for 24 h. (B) Total length of Aβ42 fibrils deposited on the lipid bilayer after 24 h of incubation with 4 μM Aβ42 in the presence of the peptide (10 μM) determined from AFM images. *p < 0.05. Data are presented as average values ± standard deviation (n = 3). | ||
Release of Aβ assemblies on the membrane by inhibitors
We further investigated the release of Aβ42 on the membrane with PC lipids using SPR. After the preparation of the Aβ42-accumulated membrane by the interaction of Aβ42, the amount of Aβ42 accumulated on the membrane was 2400 RU (Fig. S3, ESI†). When GCBP and cy5A at 10 μM were allowed to interact with the Aβ42-accumulated membrane, the response values of 550 and 720 RU increased immediately at around 50 s, respectively (Fig. 7(A)). The response was comparable to that of cy5A against a 10% GM1-containing membrane (GM1/SM/cholesterol, 10:45:45) as reported previously (320 RU),43 indicating the binding of peptides to the membrane. The response value then decreased, despite the association phase (200–500 s). This can be explained by the release of Aβ42 accumulated on the membrane by the peptide interaction. The maximum response in the GCBP association phase and the amount of released Aβ42 on the membrane with PC lipids was higher than that on the membrane with CC lipids (Fig. 7(B) and (C)). Similar results were obtained by AFM in the case of the clearance of Aβ fibrils on the 10% GM1-containing membrane.42,43 The control peptide cpVIII did not show any binding or release of Aβ.
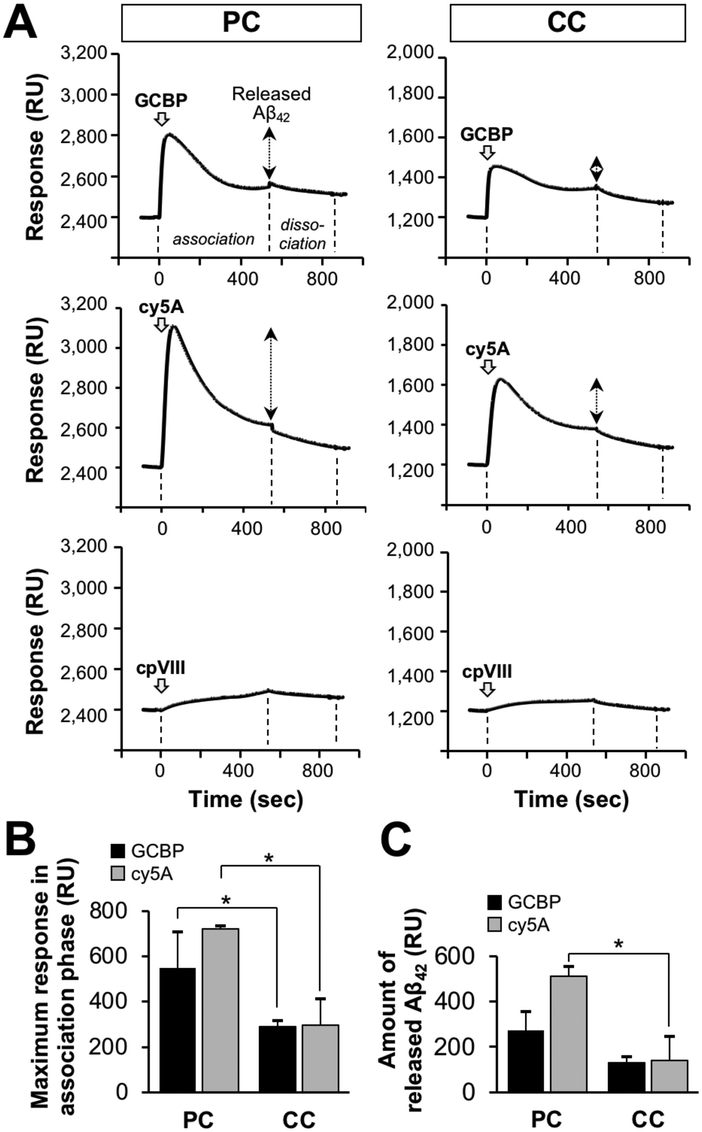 | ||
| Fig. 7 Clearance of Aβ from reconstituted membranes with Aβ-accumulation by ganglioside nanocluster-targeting peptides. (A) SPR sensorgrams indicating the release of fibrils on the reconstituted SPM lipid monolayer after the addition of GCBP, cy5A, and cpVIII. Aβ42-accumulated lipid monolayer was prepared by the interaction of Aβ42 (20 μM) three times (Fig. S2, ESI†), and 10 μM GCBP, cy5A, or cpVIII was then added to the Aβ42-accumulated lipid bilayer for 540s (association phase). After peptide association phase, the dissociation phase of 300 s is indicated. (B) Maximum SPR response during the peptide association phase. This value was obtained from a peak of the sensorgram at around 50 s and assigned to the amount of peptide bound to the Aβ42-accumulated lipid bilayer. (C) Amount of released Aβ42 by the peptide. This value was calculated by subtracting the response at the end of the association phase (540 s) from the maximum response. *p < 0.05. Data are presented as average values ± standard deviation (n = 3). | ||
Proposed model for the clearance of Aβ assembly on synaptic membranes by ganglioside nanocluster-targeting inhibitors
Our results indicated that the reconstituted membrane composed of SPM lipids mimics the synaptic membrane and induces toxic Aβ fibrils through the GAβ complex (Fig. 8). Although SPM lipids contain multiple gangliosides, Aβ assemblies observed in this study were similar to those on GM1- and other ganglioside-containing membranes reported in previous papers.28 These Aβ assemblies have the potential to take β-sheet structures, but this has not been determined in the present stage.23,27 Information on secondary structures of Aβ assemblies induced by ganglioside nanoclusters is limited, and further structural investigations are needed. In addition, the binding of GCBP and cy5A to the membrane with PC lipids specifically inhibited Aβ fibril formation and promoted the release of Aβ42 assemblies. Disruption of the GAβ complex by the binding of peptide inhibitors to ganglioside nanoclusters is a promising mechanism to reduce Aβ deposition, as reported previously.42,43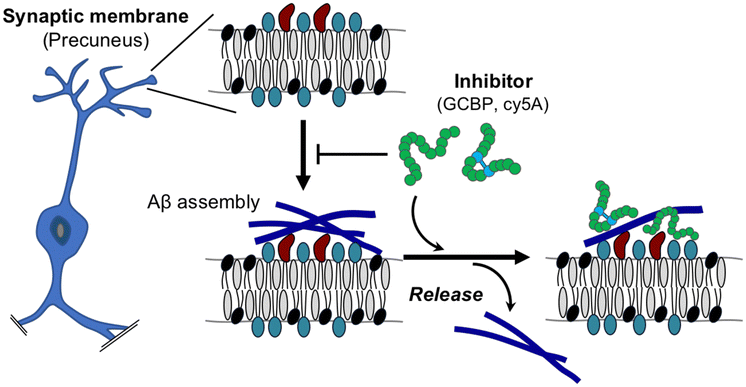 | ||
| Fig. 8 Proposed model of the release of Aβ42 assemblies on the synaptic membrane by ganglioside nanocluster-targeting inhibitors. | ||
Natural products of various polyphenols containing tannic acid, curcumin, and epigallocatechin-3-gallate have inhibitory effects, with IC50 values of 0.012–27 μM for Aβ aggregation in vitro.44 In previous studies, GCBP and cy5A have been shown to inhibit GM1-induced Aβ aggregation with IC50 values of 12 pM and 1.2 fM, respectively.42,43 Alanine scanning has revealed that arginines (Arg3 and Arg12) and hydrophobic amino acids (Trp3 and Phe9) of GCBP and cy5A interact with sialic acid and the galactose ring of gangliosides.17 Although several kinds of gangliosides in SPM lipids likely contribute to Aβ fibril formation, the design of inhibitors, based on our peptides, against ganglioside-induced Aβ assembly, is a promising strategy.
Conclusions
While Aβ aggregation is closely related to the onset and progression of AD, but a detailed and extensive analysis of how Aβ fibrils assemble, as well as the pathway of their subsequent deposition in the brain is currently underway. In addition to the presence of the ε4 allele of the apolipoprotein E gene,45 which is a genetic risk factor for late-onset AD, the roles of microglia and astrocyte cells is important for the clearance of Aβ deposits.46,47 We investigated the properties of Aβ42 fibrils, including their generation, deposition, and release, on reconstituted membranes composed of SPM lipids extracted from PC and CC. Synaptic membranes in PC promoted Aβ aggregation and accumulation via the GAβ-complex. GCBP and cy5A is expected to not only inhibit Aβ accumulation but also prevent repetitive amyloid accumulation on the neuronal membrane in the brain. Our results indicate that the ganglioside nanocluster-targeting inhibitor is a potentially effective therapeutic strategy against AD.Experimental section
Materials
1-Palmitoyl-2-oleoyl-sn-glycelo-3-phosphocholine (POPC; Sigma, St. Louis, MO, USA) was dissolved in chloroform–methanol (4:1, v/v) and stored at −30 °C until use. Synthetic human amyloid-beta protein 1–42 (Aβ42), with a purity of >95%, was purchased from Peptide Institute Co. Ltd (Osaka, Japan).
Tissue source
Specimens of the human precuneus and calcarine cortex were obtained from the Brain Bank for Aging Research at Tokyo Metropolitan Institute of Geriatrics and Gerontology with the approval of the Ethics Committees of the National Center for Geriatrics and Gerontology and Tokyo Metropolitan Institute of Geriatrics and Gerontology. The specimens were neuropathologically characterized by modified methenamine, Gallyas-Braak silver staining, and immunohistochemical staining using anti-Aβ and anti-phosphorylated tau antibodies as reported previously.36Preparation of synaptosomes
Synaptosomes from the autopsied brain were prepared as reported previously with minor modifications.36 Briefly, the gray matter was homogenized in a cold buffer solution (buffer A) composed of 10 mM HEPES, 0.32 M sucrose, and 0.25 mM EDTA (pH 7.4). The post-nuclear supernatant (PNS) was then obtained by centrifuging at 700 × g and 4 °C for 8 min. The PNS was subjected to another centrifugation step at 17400 × g and 4 °C for 20 min. The crude mitochondrial pellet was collected and resuspended in buffer B, consisting of 10 mM HEPES and 0.32 M sucrose (pH 7.4), with manual homogenization. This suspension was then layered over 7.5% and 14% Ficoll solutions in buffer B. Following this, a final centrifugation was conducted at 94000 × g and 4 °C for 30 min. The interface between the 7.5% and 14% Ficoll layers was gathered, suspended in buffer A, and centrifuged at 18400 × g at 4 °C for 15 min. The resulting pellet was identified as synaptosomes.
Preparation of synaptic plasma membrane (SPM) lipids
SPM was prepared as reported previously with minor modifications.36 Briefly, synaptosomes were first suspended in ice-cold 5 mM Tris buffer (pH 8.5) and osmotically shocked by stirring on ice, with vortex mixing. The resulting crude SPM pellet was collected and then re-suspended in buffer B after centrifugation at 42400 × g and 4 °C for 20 min. The resuspended mixture was layered over 25% and 32.5% sucrose in 10 mM HEPES buffer (pH 7.4), and centrifuged at 58800 × g and 4 °C for 30 min. The interface between the 25% and 32.5% sucrose solutions was collected, re-suspended in 10 mM HEPES buffer and centrifuged at 50400 × g at 4 °C for 20 min. The pellet was collected as the SPM. A chloroform/methanol (1:2, v/v) solution was added and extracted lipids from the SPM with chloroform/methanol/water (1:2:0.8, v/v) by vortex mixing for 30 s. The mixture was then centrifuged at 1200 × g at 4 °C for 10 min. The process of extraction and centrifugation was repeated twice. These supernatants were combined and dried up. A solution of chloroform/methanol (4:1, v/v) (400 μL) was further added to the SPM and vortexed for 10 s. The mixture was then centrifuged at 21100 × g and 4 °C for 3 min. The supernatants were collected as SPM lipids and stored at −20 °C until use.
Detection of gangliosides by MALDI-TOF/MS
SPM lipid extracts (10 μL) were added to the bottom of a glass tube and the solvents were dried under a stream of nitrogen to obtain lipid films. Three microliters of chloroform/methanol (1:1, v/v) were added to the lipid films and the lipids were resuspended. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS) analyses were performed in negative-ion and reflectron mode. 2,5-Dihydroxybenzoic acid (Wako Pure Chemical Industries., Osaka, Japan) was used as a matrix as reported previously.48
Peptides
Synthetic linear (GCBP and cpVIII) and cyclic (cy5A) peptide amides (purity >95%) carrying a biotinyl group at the C-terminus with biotinylated lysine are listed in Table S1 (ESI†).42,43 Peptide stock solutions (0.1–1 mM) were prepared in PBS (pH 7.4) and stored at −20 °C until use.Preparation of seed-free Aβ42 solutions
Seed-free Aβ42 solutions were prepared as described previously.42 Briefly, synthetic Aβ42 was dissolved in an ice-cold 0.02% ammonia solution. The solution was then ultracentrifuged at 560000 × g and 4 °C for 3 h to remove undissolved peptide aggregates. One-third of the supernatant was collected as the seed-free fraction (seed-free Aβ42 monomer) and stored in aliquots at −80 °C until use. The concentration of the seed-free Aβ42 solution (20–50 μM) was confirmed using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific Inc., Fitchburg, WI, USA). Prior to the experiments, the aliquots were diluted in PBS (pH 7.4).
Preparation of lipid monolayers and bilayers
The lipid monolayer and bilayer composed of SPM lipids were prepared as described previously.42 A reconstituted lipid monolayer at the air–water interface was prepared on a Langmuir trough (Microtrough G1; Kibron Inc., Finland). As the subphase, MilliQ water at 25 °C was used. To monitor the surface pressure, a Dyne Probe (a metal alloy, 0.5 mm in diameter) from Kibron Inc. was used.For the avidin-biotin complex (ABC) method, surface plasmon resonance (SPR), and cell viability experiments, a monolayer composed of SPM lipids was horizontally loaded onto a plastic disc or a sensor chip, while maintaining a surface pressure of 30 mN m−1. To prepare the lipid bilayer for AFM experiments, the reconstituted lipid monolayer was transferred horizontally at a surface pressure of 30 mN m−1 onto mica coated with POPC (Fig. 1(A)).
Observation of surface topography by AFM
The lipid bilayer on mica was prepared as described previously42 and incubated with seed-free Aβ42 in PBS at 37 °C for 15 min or 24 h. After washing the bilayer with MilliQ water twice, the bilayer was placed in MilliQ water at 25 °C and observed using the SPM-9600 atomic force microscope (Shimadzu Corp., Kyoto, Japan) as described previously.42 Briefly, AFM images (2 × 2 μm, n ≥ 3) of the surface topography of the lipid bilayer were obtained in dynamic mode in water. A silicon nitride cantilever with a silicon tetrahedral tip (Olympus Corporation, Tokyo, Japan) was used for the observation. Three representative images were obtained for further analyses. The number and length of Aβ42 fibrils were measured using ImageJ (ver. 1.52a) (National Institution of Health, Bethesda, MD, USA). In this analysis, Aβ42 assemblies were identified as fibrils when their long-to-short axis aspect ratio exceeded 3.SPR analysis
The SPR analysis was performed with the reconstituted lipid monolayer as reported previously.49 Briefly, the lipid monolayer with a surface pressure of 30 mN m−1 was immobilized on the bare gold surface of the sensor chip (1 × 1 cm) (SIA Kit Au; Cytiva, Tokyo, Japan). After assembly with a sensor chip support, the sensor chip was immediately docked into the Biacore X100 instrument (Cytiva). All measurements were carried out at 25 °C. PBS was passed through a 0.22 μm pore size filter and degassed as the running buffer. After docking, the running buffer was injected over the chip at a flow rate of 10 μL min−1 for over 30 min to stabilize the membrane. Seed-free Aβ42 as the analyte was diluted in running buffer before injection. For the association (540 s) and dissociation phases (300 s) of Aβ42 (20 μM), a flow rate of 10 μL min−1 was used. Each experiment was performed three times.For the inhibition experiment, the Aβ42-accumulated lipid monolayer was prepared by the incubation of Aβ42 (20 μM) three times with the reconstituted membrane by the set of the association (540 s) and dissociation phases (300 s) at a flow rate of 10 μL min−1, (Fig. S3, ESI†). Biotinylated peptides, as inhibitors, were injected over the Aβ42-accumulated lipid monolayer. A pentadeca peptide of the phage body (cpVIII, AETVESCLAKPHTEN(bK)-NH2) was used as a control.50,51
Cell culture and viability assay (calcein-AM assay)
Human neuroblastoma SH-SY5Y cells were cultured with Dulbecco's modified Eagle's medium (DMEM) low glucose (Nacalai Tesque Inc., Kyoto, Japan) containing 10% fetal bovine serum (FBS), 10000 U per mL penicillin, and 10 mg per mL streptomycin at 37 °C under 5% CO2. The calcein-AM assay was performed as described previously.43
To assess the cytotoxicity of Aβ42 assemblies generated on the reconstituted lipid monolayer, the lipid monolayer was prepared on a plastic disc (13 mm in diameter). To induce the aggregation of Aβ42 on the lipid bilayer, the Aβ42 solution (2 and 10 μM) was incubated with the lipid monolayer on the disc in a 24-well plate at 37 °C for 24 h. Then, SH-SY5Y cells (8.0 × 104 cells per well) were seeded onto the disc and incubated at 37 °C for 48 h. Before the calcein-AM assay, the discs on which the cells were incubated on top were washed twice with PBS and cells were transferred to a 24-well clear-bottom black plate. Then, 3′,6′-di(O-acetyl)-4′,5′-bis[N,N-bis(carboxymethyl)aminomethyl]fluorescein (calcein-AM) (Dojindo Laboratories Co., Ltd, Kumamoto, Japan) was dissolved in dimethyl sulfoxide and stored at −30 °C until use. The calcein-AM solution in PBS (250 μM) and the same volume of DMEM Ham's F-12 without phenol red (Nacalai Tesque Inc.) were added each well, and the mixed solution was incubated at 37 °C for 1 h. Fluorescence emission at 520 nm upon excitation at 485 nm was measured using a plate reader.
Statistical analysis
Student's t-tests (unpaired, two-tailed) in Microsoft Excel were used for comparisons between treatment groups in all experiments. Statistical significance was set at p < 0.05 and p < 0.01.Ethical guideline
All experiments were performed in accordance with the Ethical Guidelines for Brain Bank by the Japanese Society of Neuropathology and the Japanese Society of Biological Psychiatry, and approved by the ethics committee at Keio University, Tokyo University of Pharmacy and Life Sciences, Tokyo Metropolitan Institute of Geratrics and Gerontology, and National Center for Geriatrics and Gerontology. Informed consent was obtained from all patients.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant numbers JP22J20821 (E. M.), 22KJ2711 (E. M.), JP16H06277 (CoBiA) (S. M.), 23H04562 (T. M.), and AMED grant number JP19dm0107106 (S. M.).References
- A. Abbott, Nature, 2011, 475, S2–S4 CrossRef CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- C. R. Jack, D. S. Knopman, W. J. Jagust, L. M. Shaw, P. S. Aisen, M. W. Weiner, R. C. Petersen and J. Q. Trojanowski, Lancet Neurol., 2010, 9, 1–20 CrossRef PubMed.
- Y. M. Kuo, M. R. Emmerling, C. Vigo-Pelfrey, T. C. Kasunic, J. B. Kirkpatrick, G. H. Murdoch, M. J. Ball and A. E. Roher, J. Biol. Chem., 1996, 271, 4077–4081 CrossRef CAS PubMed.
- Y. Okada, K. Okubo, K. Ikeda, Y. Yano, M. Hoshino, Y. Hayashi, Y. Kiso, H. Itoh-Watanabe, A. Naito and K. Matsuzaki, ACS Chem. Neurosci., 2019, 10, 563–572 CrossRef CAS PubMed.
- M. Sakono and T. Zako, FEBS J., 2010, 277, 1348–1358 CrossRef CAS PubMed.
- G. Di Natale, G. Sabatino, M. F. M. Sciacca, R. Tosto, D. Milardi and G. Pappalardo, Molecules, 2022, 27, 5066 CrossRef CAS.
- P. Faller, C. Hureau and O. Berthoumieu, Inorg. Chem., 2013, 52, 12193–12206 CrossRef CAS PubMed.
- K. Yanagisawa, J. Neurochem., 2011, 116, 806–812 CrossRef CAS.
- M. E. King, H. M. Kan, P. W. Baas, A. Erisir, C. G. Glabe and G. S. Bloom, J. Cell Biol., 2006, 175, 541–546 CrossRef CAS.
- A. E. Roher, M. O. Chaney, Y. M. Kuo, S. D. Webster, W. B. Stine, L. J. Haverkamp, A. S. Woods, R. J. Cotter, J. M. Tuohy, G. A. Krafft, B. S. Bonnell and M. R. Emmerling, J. Biol. Chem., 1996, 271, 20631–20635 CrossRef CAS.
- N. Yamamoto, E. Matsubara, S. Maeda, H. Minagawa, A. Takashima, W. Maruyama, M. Michikawa and K. Yanagisawa, J. Biol. Chem., 2007, 282, 2646–2655 CrossRef CAS.
- M. Jucker and L. C. Walker, Nature, 2013, 501, 45–51 CrossRef CAS.
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183233 CrossRef CAS PubMed.
- A. K. Srivastava, J. M. Pittman, J. Zerweck, B. S. Venkata, P. C. Moore, J. R. Sachleben and S. C. Meredith, Protein Sci., 2019, 28, 1567–1581 CrossRef CAS.
- T. Matsubara, K. Iijima, T. Kojima, M. Hirai, E. Miyamoto and T. Sato, Langmuir, 2021, 37, 646–654 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- K. Simons and W. L. C. Vaz, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 269–295 CrossRef CAS PubMed.
- S. Sonnino, A. Prinetti, L. Mauri, V. Chigorno and G. Tettamanti, Chem. Rev., 2006, 106, 2111–2125 CrossRef CAS PubMed.
- K. Yanagisawa, A. Odaka, N. Suzuki and Y. Ihara, Nat. Med., 1995, 1, 1062–1066 CrossRef CAS.
- A. Kakio, S. i Nishimoto, K. Yanagisawa, Y. Kozutsumi and K. Matsuzaki, Biochemistry, 2002, 41, 7385–7390 CrossRef CAS.
- T. Matsubara, H. Yasumori, K. Ito, T. Shimoaka, T. Hasegawa and T. Sato, J. Biol. Chem., 2018, 293, 14146–14154 CrossRef CAS PubMed.
- L. P. i Choo-Smith, W. Garzon-Rodriguez, C. G. Glabe and W. K. Surewicz, J. Biol. Chem., 1997, 272, 22987–22990 CrossRef CAS PubMed.
- M. Yagi-Utsumi and K. Kato, Glycoconjugate J., 2015, 32, 105–112 CrossRef CAS PubMed.
- H. Hayashi, N. Kimura, H. Yamaguchi, K. Hasegawa, T. Yokoseki, M. Shibata, N. Yamamoto, M. Michikawa, Y. Yoshikawa, K. Terao, K. Matsuzaki, C. A. Lemere, D. J. Selkoe, H. Naiki and K. Yanagisawa, J. Neurosci., 2004, 24, 4894–4902 CrossRef CAS.
- M. Yagi-Utsumi, S. G. Itoh, H. Okumura, K. Yanagisawa, K. Kato and K. Nishimura, ACS Chem. Neurosci., 2023, 14, 2648–2657 CrossRef CAS PubMed.
- T. Matsubara, M. Nishihara, H. Yasumori, M. Nakai, K. Yanagisawa and T. Sato, Langmuir, 2017, 33, 13874–13881 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- F. Panza, M. Lozupone, G. Logroscino and B. P. Imbimbo, Nat. Rev. Neurol., 2019, 15, 73–88 CrossRef.
- K. Rajasekhar, M. Chakrabarti and T. Govindaraju, Chem. Commun., 2015, 51, 13434–13450 RSC.
- C. H. van Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer and T. Iwatsubo, N. Engl. J. Med., 2023, 388, 9–21 CrossRef CAS.
- T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309–1318 CrossRef CAS PubMed.
- S. H. W. Scheres, B. Ryskeldi-Falcon and M. Goedert, Nature, 2023, 621, 701–710 CrossRef CAS PubMed.
- T. Matsubara, K. Iijima, N. Yamamoto, K. Yanagisawa and T. Sato, Langmuir, 2013, 29, 2258–2264 CrossRef CAS.
- N. Oikawa, T. Matsubara, R. Fukuda, H. Yasumori, H. Hatsuta, S. Murayama, T. Sato, A. Suzuki and K. Yanagisawa, PLoS One, 2015, 10, 1–19 Search PubMed.
- H. Braak and E. Braak, Acta Neuropathol., 1991, 82, 239–259 CrossRef CAS PubMed.
- M. D. Ikonomovic, W. E. Klunk, E. E. Abrahamson, J. Wuu, C. A. Mathis, S. W. Scheff, E. J. Mufson and S. T. DeKosky, Neurology, 2011, 77, 39–47 CrossRef CAS PubMed.
- R. A. Sperling, B. C. Dickerson, M. Pihlajamaki, P. Vannini, P. S. LaViolette, O. V. Vitolo, T. Hedden, J. A. Becker, D. M. Rentz, D. J. Selkoe and K. A. Johnson, NeuroMol. Med., 2010, 12, 27–43 CrossRef CAS.
- S. Palmqvist, M. Schöll, O. Strandberg, N. Mattsson, E. Stomrud, H. Zetterberg, K. Blennow, S. Landau, W. Jagust and O. Hansson, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- E. Takada, K. Okubo, Y. Yano, K. Iida, M. Someda, A. Hirasawa, S. Yonehara and K. Matsuzaki, ACS Chem. Neurosci., 2020, 11, 796–805 CrossRef CAS PubMed.
- T. Matsubara, M. Nakai, M. Nishihara, E. Miyamoto and T. Sato, ACS Chem. Neurosci., 2022, 13, 1868–1876 CrossRef CAS PubMed.
- E. Miyamoto, T. Sato and T. Matsubara, ACS Chem. Neurosci., 2023, 14, 4199–4207 CrossRef CAS PubMed.
- Y. Porat, A. Abramowitz and E. Gazit, Chem. Biol. Drug Des., 2006, 67, 27–37 CrossRef CAS PubMed.
- G. Bu, Nat. Rev. Neurosci., 2009, 10, 333–344 CrossRef CAS PubMed.
- M. R. Brown, S. E. Radford and E. W. Hewitt, Front. Mol. Neurosci., 2020, 13, 609073 CrossRef CAS PubMed.
- R. G. Nagele, J. Wegiel, V. Venkataraman, H. Imaki, K. C. Wang and J. Wegiel, Neurobiol. Aging, 2004, 25, 663–674 CrossRef CAS.
- T. Sato, M. Takashiba, R. Hayashi, X. Zhu and T. Yamagata, Carbohydr. Res., 2008, 343, 831–838 CrossRef CAS PubMed.
- T. Matsubara, A. Onishi and T. Sato, Bioorg. Med. Chem., 2012, 20, 6452–6458 CrossRef CAS PubMed.
- T. Matsubara, M. Sumi, H. Kubota, T. Taki, Y. Okahata and T. Sato, J. Med. Chem., 2009, 52, 4247–4256 CrossRef CAS.
- T. Matsubara, R. Otani, M. Yamashita, H. Maeno, H. Nodono and T. Sato, Biomacromolecules, 2017, 18, 355–362 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00038b |
| This journal is © The Royal Society of Chemistry 2024 |