
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific RNA modification via initiation of in vitro transcription reactions with m6A and isomorphic emissive adenosine analogs†
Deyuan
Cong
,
Kfir B.
Steinbuch
 ,
Ryosuke
Koyama
,
Tyler V.
Lam
,
Jamie Y.
Lam
and
Yitzhak
Tor
*
,
Ryosuke
Koyama
,
Tyler V.
Lam
,
Jamie Y.
Lam
and
Yitzhak
Tor
*
Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0358, USA. E-mail: ytor@ucsd.edu
First published on 27th March 2024
Abstract
The templated enzymatic incorporation of adenosine and its analogs, including m6A, thA and tzA into RNA transcripts, has been explored. Enforced transcription initiation with excess free nucleosides and the native triphosphates generates 5′-end modified transcripts, which can be 5′-phosphorylated and ligated to provide full length, singly modified RNA oligomers. To explore structural integrity, functionality and utility of the resulting non-canonical purine-containing RNA constructs, a MazF RNA hairpin substrate has been synthesized and analyzed for its susceptibility to this endonuclease. Additionally, RNA substrates, containing a singly incorporated isomorphic emissive nucleoside, can be used to monitor the enzymatic reactions in real-time by steady state fluorescence spectroscopy.
Introduction
Transcription initiation typically relies on specific promotor sequences.1,2 Although it has been thought to have limited tolerance for modifications, a rather broad repertoire of non-canonical 5′-capped structures appears to be accommodated.3 Additionally, phage-based polymerases (e.g., T7 RNA pol) have been shown to enable the incorporation of modified or non-canonical nucleosides/tides via in vitro transcription, resulting in 5′-modified oligomers.4–6 These initiators frequently lead, however, to RNA constructs that cannot be 5′-ligated to provide internal alterations.7–10In vitro transcription reactions, which site-specifically incorporate alternate nucleosides with a free 5′-hydroxy group, are therefore particularly attractive for the constructions of internally site-specifically modified oligomers as they can be ligated to other RNAs of any length. Herein, we examine transcription initiation with m6A together with thA and tzA, isomorphic emissive adenosine surrogates, which are based on the thieno[3,4-d]pyrimidine11 and isothiazolo[4,3-d]pyrimidine cores,12 respectively, and extension of such 5′-modified RNA fragments via enzymatic ligation reactions.We have previously reported that thGTP acts as a GTP surrogate and can successfully initiate and sustain in vitro T7 RNA polymerase transcription reactions.13,14 This resulted in the replacement of all guanosine residues with thG. To site-specifically incorporate thG15 and tzG,16 large excess of thG or tzG can be used to initiate such transcription reactions. The resulting 5′-modified RNA constructs can then be phosphorylated and ligated to provide site-specifically modified oligomers.15,16 The ability of such enzymes to initiate transcription with non-canonical or unnatural adenosine analogs remains, however, rather underexplored.17,18 Herein, enzymatic pathways for the incorporation of adenosine, its N6-methylated derivative (m6A), and two unnatural fluorescent adenosine surrogates (thA and tzA) into RNA are investigated and optimized (Fig. 1a and Fig. S1–S3, ESI†). To evaluate the structural integrity and functionality of the resulting modified RNA oligomers, the thA/tzA/m6A-containing RNA constructs were subjected to MazF endoribonuclease-mediated cleavage reaction (Fig. 1b). Additionally, we exploit the inherent responsiveness of the emissive adenosine surrogates to facilitate real-time monitoring of the site-specific enzymatic cleavage of the fluorescent oligomers.
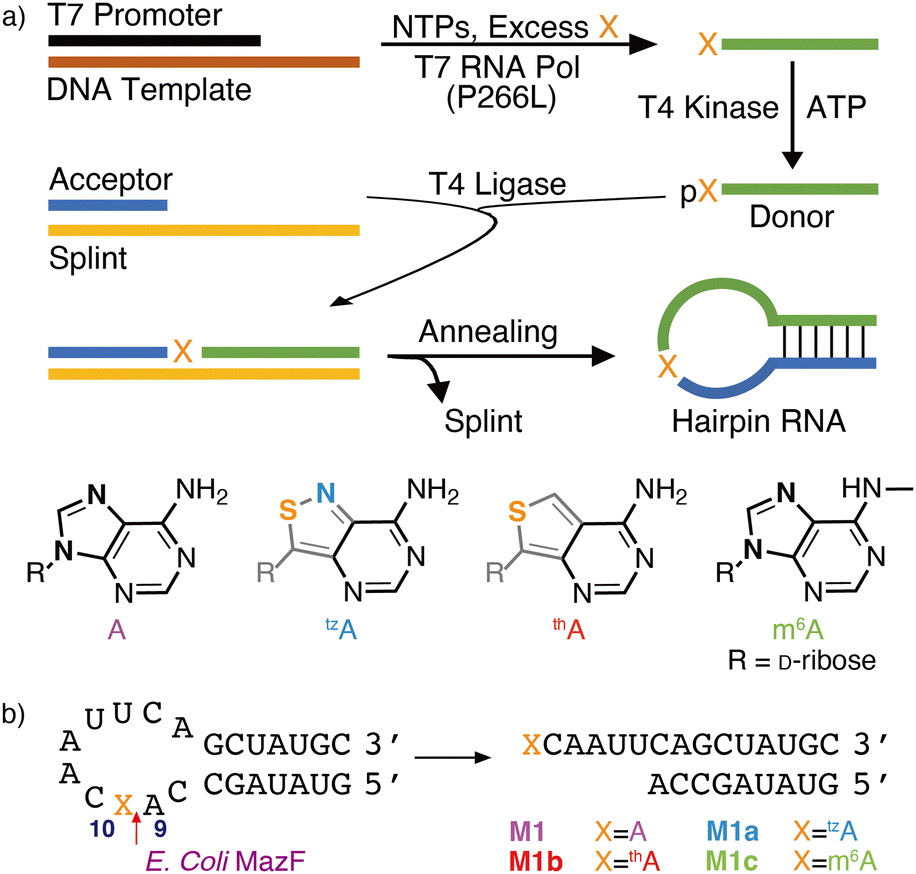 | ||
| Fig. 1 (a) Enzymatic pathway to site-specifically modified RNAs, replacing an adenosine residue with m6A, tzA, thA. (b) A stem-loop substrate shown to be targeted by E. coli's MazF. M1, M1a, M1b, M1c are the native, tzA, thA and m6A modified stem-loop substrate, respectively. | ||
Results and discussion
Synthesis of tzA nucleoside analog
Improvements were made to the reported synthesis of tzA to better converge it with the synthesis of tzG, the guanine surrogate, to lower the number of steps, and to increase the overall yield (Scheme 1).12 By introducing a single step to the protected inosine analog 5, from 4a, the key intermediate of the tzG pathway, the syntheses of tzA and tzG are diverged in a later step, eliminating four steps from the previous syntheses. Previously, the primary thiol 1 was reacted with one of the two reagents, 2a or 2b, both of which had to be synthesized. The products were then reduced to set the stereocenter at the anomeric carbon and to yield key intermediates 4a and 4b for the syntheses of tzG and tzA, respectively (Scheme 1).12 Here, by using formamidine acetate and triethylamine in ethanol, compound 5 was constructed in 70% yield from the key intermediate 4a, eliminating the need for 2b. The last three steps of the synthesis include the thianation of 4a to yield the thioamide 6, an amination reaction to substitute the thiocarbonyl with an amine group, and finally, a benzyl deprotection step to afford the final product tzA (Scheme 1). Alterations made to the thianation reaction conditions and to the workup of the benzyl deprotection (ESI,† Section S1.7), resulted in an increased overall yield for these last three steps (72% vs. 32%).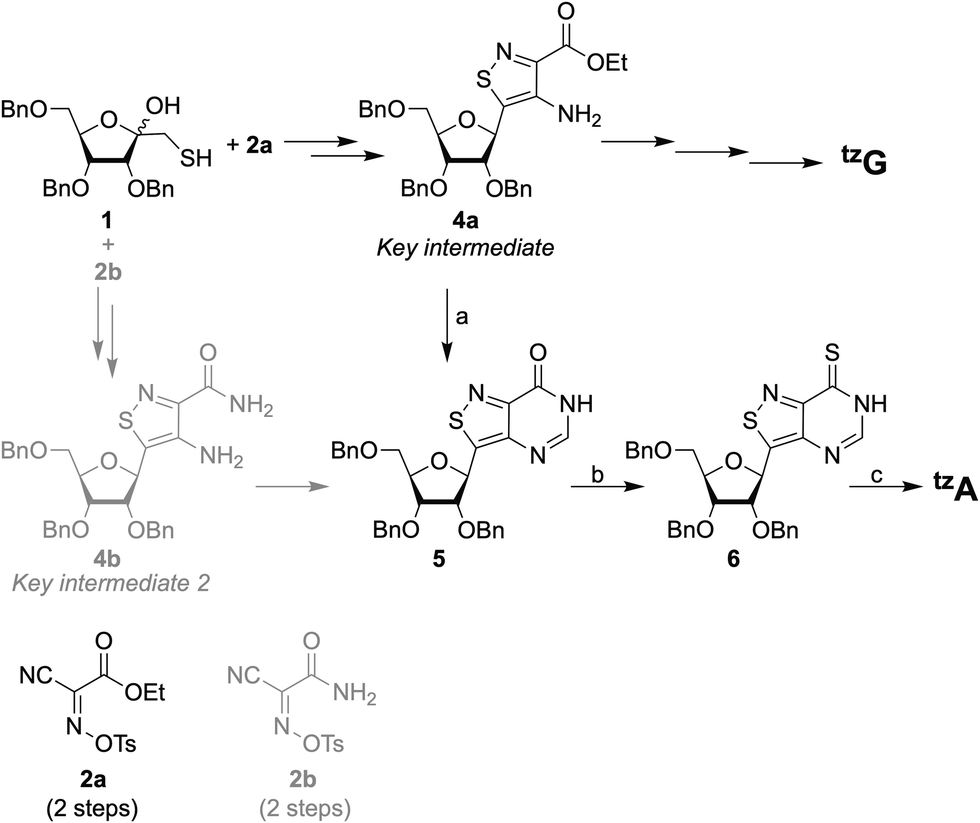 | ||
| Scheme 1 Previous (gray) and current (black) synthetic pathways to tzG and tzA, reagents and conditions: (a) formamidine acetate, Et3N, EtOH, reflux, 70%; (b) P2S5, pyridine, 90 °C, 2 h; (c) (i) NH3, MeOH, 70 °C, 16 h; (ii) HSCH2CH2SH, BF3·OEt2, DCM, rt, 72 h, 72% (for steps b and c). | ||
Enzymatic synthesis of singly modified RNA constructs
To assemble singly modified RNA constructs, where a specific A residue is strategically substituted by thA, tzA, or m6A, a large excess of the corresponding nucleosides (i.e., thA, tzA or m6A, respectively) is utilized over native ATP during transcription. It enforces initiation with A analogs and the resulting 5′-end altered transcripts can be monophosphorylated and then ligated to produce longer site-specifically modified RNA constructs (Fig. 1).Transcription initiation reactions with excess free nucleosides X (where X = A, thA, tzA, or m6A) were performed using template 1, all native NTPs, and mutant T7 RNA polymerase P266L (Fig. 2a and Fig. S1–S3, ESI†), and were separated by denaturing 20% urea-PAGE (Fig. 2b and Fig. S1–S3, ESI†). UV shadowing was used to visualize the full-length product and truncated transcripts (Fig. 2b). Following extraction, all purified 15-mer transcripts T1, T2, T3, T4, and T5 initiated by ATP, A, thA, tzA, and m6A respectively, were quantified via UV absorption at 260 nm and analyzed by MALDI-TOF mass spectrometry (Fig. 2c and Fig. S4–S6, ESI†). The amounts of purified transcripts T2, T3, T4, and T5 obtained from 100 μL transcription reaction were determined to be 3.1, 1.4, 1.4 and 1.3 nmol, respectively. The relative yield was calculated by dividing the concentration of the X-initiated transcript by the concentration of transcript T1, and was determined to be 0.90, 0.40, 0.40 and 0.39, for transcripts T2, T3, T4, and T5, respectively.
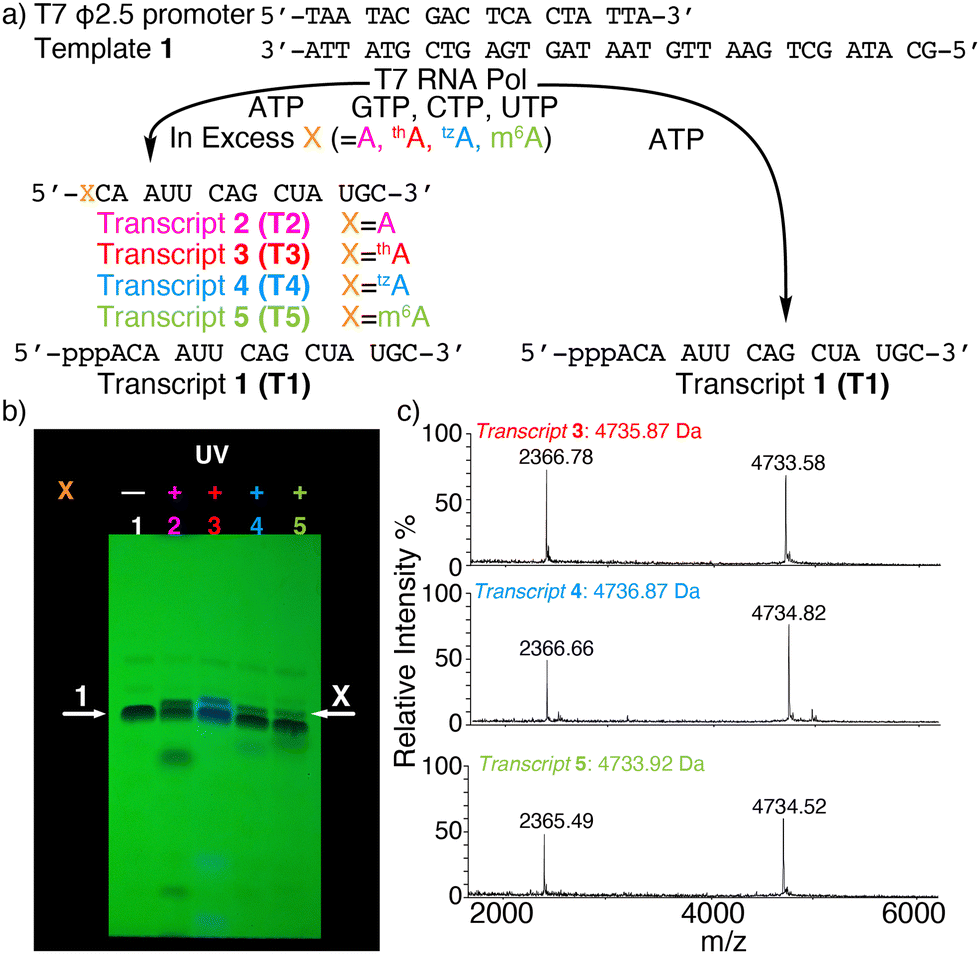 | ||
| Fig. 2 (a) Transcription reactions in the presence of natural NTPs using the ϕ2.5 T7 promotor and template shown, initiated with large excess (see ESI†) or without X (X = A, thA, tzA, or m6A). (b) PAGE of transcription reactions using 2.5 mM NTPs with or without modified nucleosides X. Lane 1: native NTPs only. Lane 2: transcription initiation with A (18.75 mM). Lane 3: transcription initiation with thA (18.75 mM). Lane 4: transcription initiation with tzA (15 mM). Lane 5: transcription initiation with m6A (32 mM). White arrows indicate the expected product transcript 1 (arrow 1) and modified transcripts T2, T3, T4, and T5 (arrow X). UV shadowing is done upon illumination at 254 nm; (c) MALDI-TOF MS of transcript T3, T4, T5. | ||
The purified 5′-end modified transcripts T3, T4, and T5 were then phosphorylated by T4 kinase and splint ligated by T4 DNA ligase to produce the desired site-specifically labelled modified RNA constructs (Fig. 1 and Fig. S7–S9, ESI†). Following purification by denaturing PAGE, the isolated 24-mer oligomers M1a, M1b and M1c, containing tzA, thA, and m6A respectively, were characterized by ESI-TOF mass spectrometry (Fig. S10 and S11, ESI†), then digested by S1 nuclease and dephosphorylated using alkaline phosphatase. The nucleoside mixture was subsequently analyzed by HPLC. Comparison of the chromatogram obtained for the standard (containing C, U, A, G, and X) to that of the enzymatically synthesized 24-mer oligomer digest confirmed the presence and stoichiometry of the incorporated modified nucleosides (Fig. S12, ESI†).
MazF-mediated cleavage reactions
MazF, an Escherichia coli toxin, is capable of cleaving the 5′ or 3′ phosphodiester linkage of the initial A residue in an ACA sequence.19,20 To independently assess the viability of the modified RNAs as MazF substrates, the native RNA (M1) and A10-modified RNA substrates M1a, M1b or M1c, containing tzA, thA or m6A at this position, respectively, were subjected to the enzyme (Fig. 1b). The hairpin RNA substrates were first thermally denatured and refolded in a 40 mM sodium phosphate buffer and the MazF-mediated reactions were carried out at 37 °C. After 1 h, the reaction mixtures were quenched by adding gel loading buffer and were resolved via a 20% PAGE (Fig. 3a). Several shorter RNA oligonucleotides, reflecting known fragments of the native substrates, served as markers and assisted in determining the cleavage site (Fig. 3: “Ref” lane, ESI† Section S1.6.2).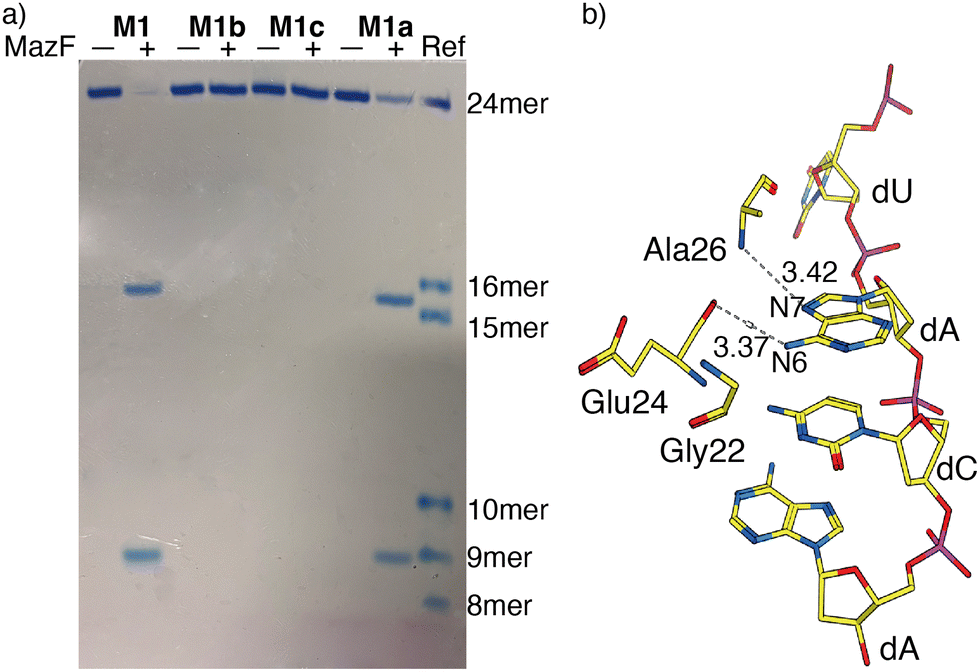 | ||
| Fig. 3 (a) MazF-mediated cleavage of native (M1) and tzA, thA or m6A modified RNA substrates (M1a, M1b or M1c, respectively). The gel was stained by stain-all overnight and de-stained in MilliQ water. The reference sequences, of known length, are the following: 16-mer oligomer: 5′-A ACA AUU CAG CUA UGC-3′, 15-mer oligomer: 5′-ACA AUU CAG CUA UGC-3′, 10-mer oligomer: 5′-GUA UAG CCA A-3′, 9-mer oligomer: 5′-GUA UAG CCA-3′, 8-mer oligomer: 5′-GUA UAG CC-3′. (b) X-Ray co-crystal structure of MazF and d(ACAU), a DNA substrate analogue, illustrating the contacts between the nucleobases and the protein backbone. (PDB ID code: 5CR2) | ||
As seen in Fig. 3a, MazF cleaves the phosphodiester bond between residues A9 and A10 in the native and tzA modified substrates (M1 and M1a, respectively). This suggests tzA facilitates similar folding for M1a compared to M1 and does not extensively interfere with the enzyme's activity, although somewhat longer reactions times were needed for the former. In contrast, the thA-containing and m6A-containing substrates (M1b and M1c, respectively) didn’t show any cleavage when incubated with MazF at 37 °C for 1 h (Fig. 3a).
The observations reported above suggest thA is either not properly accommodated or is significantly perturbing the folding of modified strand M1b, preventing MazF cleavage, as observed for M1c, an m6A modified sequence, which has been established to be resistant to MazF.21 Compared to tzA, thA lacks the basic nitrogen atom corresponding to the purines’ N7 position, which can impact both the RNA substrate's folding and recognition. As seen in the crystal structure of E. coli's MazF in complex with an uncleavable DNA substrate mimic (Fig. 3b),22 the backbone amide residues of Ala26 and carbonyl of Glu24 form H-bonds through the Hoogsteen edge of the first adenine base's N7 and N6 positions, respectively. This is the residue that is replaced by either thA or m6A at the A10 position of the modified hairpin. It is thus reasonably to assume that the missing “N7” H bonding acceptor in the thA-containing hairpin RNA (M1b), which is replaced by a C–H linkage, compromises substrate recognition and cleavage of the thACA sequence by MazF.
To examine the relative cleavage kinetics of M1 and M1a by MazF, reaction aliquots were collected at designated time points and immediately quenched by adding a gel loading buffer, followed by rapid thermal denaturation (90 °C) and cooling to room temperature. Reaction mixtures were then resolved via a 20% PAGE and the gel was stained with 1 × SYBR gold solution for 30 min (Fig. 4a and b). Bands were visualized using Typhoon 5 and their integrated intensities were measured using ImageJ. The M1 substrate was fully consumed within 50 min. Only 84% of the M1a substrate were, however, consumed within 120 min, where the reaction had plateaued (Fig. 4c). The apparent rate constant, obtained by fitting pseudo first order curves to the integrated PAGE bands plotted against time, were 7.3 × 10−2 min−1 and 4.3 × 10−2 min−1 for substrate M1 and M1a, respectively (Fig. 4c). The reaction's half time were 10 min and 16 min for substrate M1 and M1a, respectively (Table 1). The cleavage reaction rate was thus not dramatically impacted by replacing a native A10 with tzA, suggesting that strand M1a could potentially serve as fluorescent probe for monitoring MazF-mediated cleavage reaction by steady state fluorescence spectroscopy.
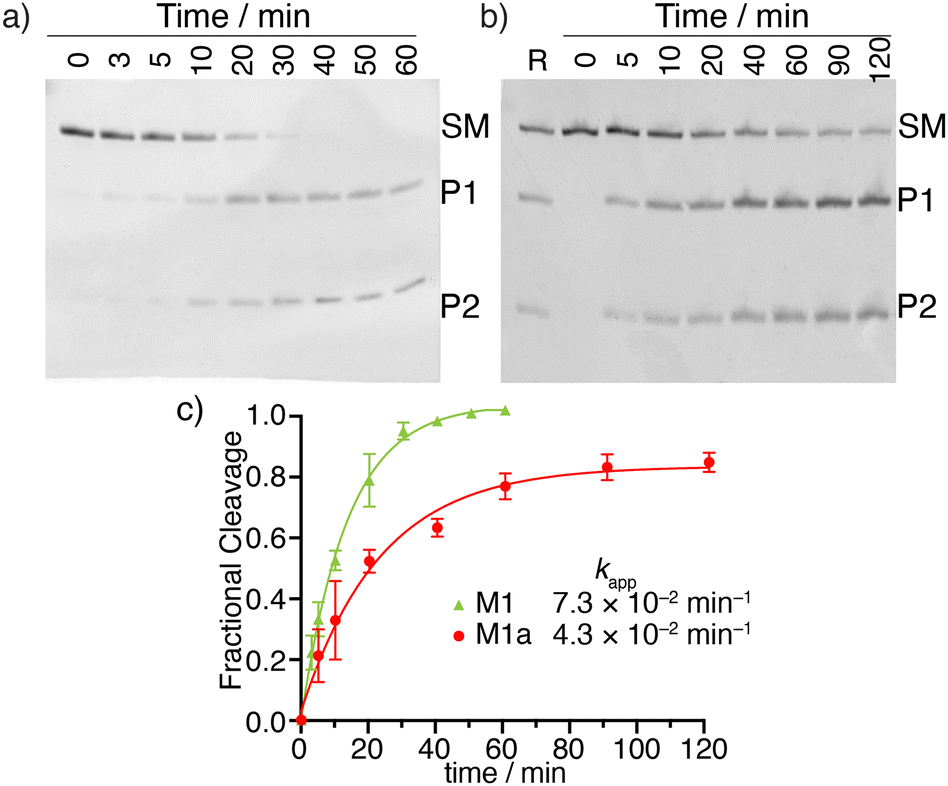 | ||
| Fig. 4 Kinetics of MazF-mediated cleavage of native and tzA modified RNA substrates (M1 and M1a, respectively). (a) MazF-mediated cleavage of M1. (b) MazF-mediated cleavage reaction of M1a. (c) Kinetic profiles of MazF-mediated cleavage reactions of M1 (green), M1a (red). Gels were stained by 1 × SYBR gold solution for 30 min. Lanes R correspond to native reaction indicating the cleavage site of M1a strand. Reactions were done in triplicates and a representative gel is shown per experiment. Error bars indicate SD. | ||
| SYBR golda | Fluorescenceb | ||
|---|---|---|---|
| M1 | M1a | M1a | |
| a Reaction rate constants were obtained by SYBR gold staining protocol. b Reaction rate constants were obtained by measuring fluorescence change. c k app is the pseudo-first-order rate constant (×10−2 min−1). d t 1/2 is reaction's half-life (min). Data are presented as mean ± SD. | |||
| k app | 7.3 ± 0.5 | 4.3 ± 0.4 | 3.5 ± 0.5 |
| t 1/2 | 10 ± 1 | 16 ± 2 | 20 ± 3 |
| R 2 | 0.995 | 0.989 | 0.986 |
Fluorescence-monitored MazF-mediated cleavage reactions
Having demonstrated the feasibility of M1a as a MazF substrate, its enzymatic cleavage was monitored by steady-state fluorescence spectroscopy, under identical experimental conditions to the ones used above. The emission spectrum of each sample was recorded at given time intervals and deconvoluted using origin (Fig. 5a). A significant fluorescence enhancement is seen as the cleavage reaction progresses. Unstacking and solvent exposure of the tzA residue at the product's 5′-terminus,12 as the M1a substrate loop is nicked, is likely the reason for the enhanced emission seen (Fig. 5a).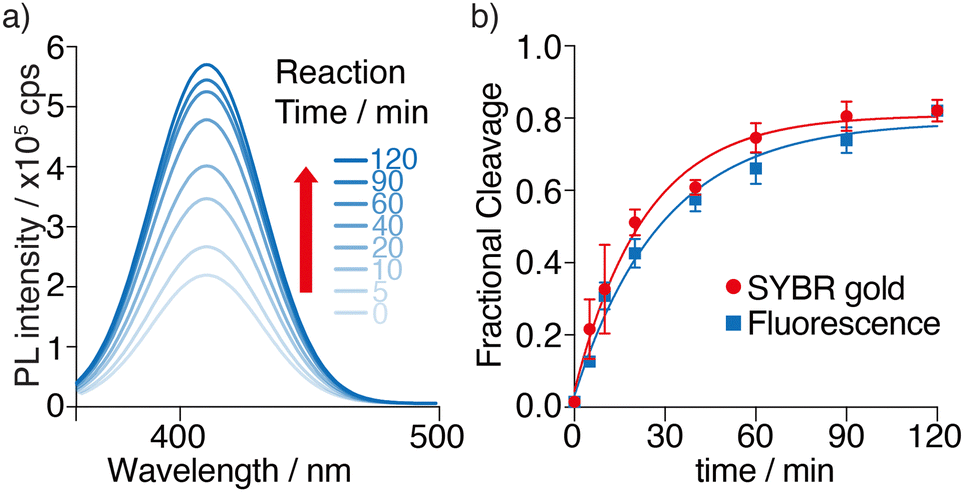 | ||
| Fig. 5 MazF-mediated cleavage reaction of substrate M1a monitored by fluorescence as function of time. (a) Deconvolution of emission spectra of cleavage reaction of tzA substrate M1a. Excitation wavelength was 341 nm. The area of each spectrum was integrated and plotted vs. time depicting the rate of the enzymatic reaction. (b) Cleavage of M1a by MazF at various time points monitored by fluorescence (blue) and SYBR gold (red). Reactions were done in triplicates and a representative deconvoluted emission spectrum is shown. Error bars indicate SD. | ||
To confirm that the fluorescence-monitored data align with the trend and rates determined and visualized through PAGE and SYBR Gold staining, the data sets were normalized and plotted on the same graph (Fig. 5b). The reaction rates, determined by fitting pseudo-first order curves to the integrated area of each deconvoluted emission spectrum plotted against time, yielded an apparent kinetics rate constant kapp value of 3.5 × 10−2 min−1, and a reaction's half time t1/2 value of 20 min for substrate M1a (Fig. 5b and Table 1). Notably, a good agreement between the PAGE and fluorescence-monitored quantification was seen (Fig. 5b), thus supporting that fluorescence changes reflect the same molecular event and can potentially facilitate real-time monitoring of MazF cleavage reactions.
We note that a fluorophore/quencher-based substrate has previously been constructed to monitor the MazF-mediated endonuclease activity23 and was used to demonstrate the resistance of the m6ACA sequence.21 This method, however, is not easily exploited for shedding light on the impact of individual residues on the enzymatic cleavage. A doubly, terminally labelled RNA substrate could limit, in certain circumstances, the landscape of possible explorations, when compared to an internally labelled RNA substrate containing a faithful adenosine surrogate.
Conclusions
RNAs with site-specific modifications, such as epigenetic modifications or fluorescent labels, have proved to be powerful tools for probing structure, function, and mechanisms.24–29 These sequence-specifically modified RNAs are typically fabricated by solid-state synthesis. We submit, though, that when accommodated, site specific incorporation by enforced in vitro transcription initiation reactions can reach a broader community, not necessarily adapted to organic synthesis and solid-phase based assembly methodologies.Certain nucleotides, such as m6Ap18 and others4,5,30 have been shown to initiate transcription. To the best of our knowledge, enzymatic incorporation pathways for free adenosine analogs have not yet been reported. Our studies show that thA, tzA, two isomorphic fluorescent nucleoside analogs, and m6A, as free nucleosides, are acceptable transcription initiators for T7 RNA polymerase, resulting in the generation of 5′-end modified RNA constructs. The resulting modified strands can be phosphorylated by T4 kinase and ligated by T4 ligase, enabling a simple fabrication of internally, singly substituted modified RNAs.
The site-specifically modified RNAs that contain the emissive and responsive tzA can be used to monitor enzymatic reactions by steady-state fluorescence, yielding significantly enhanced emission upon cleavage, and can potentially provide insight into RNA folding and enzyme-substrate recognition features, as demonstrated here for MazF-mediated reactions. The approach reported here could also be extended to the detection of other enzymatic reactions and could potentially provide a platform for inhibitors discovery.31
Author contributions
Deyuan Cong: data curation, formal analysis, investigation, methodology, software, validation, writing of original draft; Kfir B. Steinbuch: data curation, methodology, formal analysis, investigation, writing of original draft; Ryosuke Koyama: data curation, methodology, formal analysis; Tyler V. Lam: methodology, formal analysis. Jamie Y. Lam: methodology; Yitzhak Tor: conceptualization, funding acquisition, project administration, supervision, writing for reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institutes of Health for generous support (through grant R35 GM139407) and the UCSD Chemistry and Biochemistry MS Facility. We are grateful to Venkat Gopalan (OSU) for plasmids and inspiring conversations.Notes and references
- R. G. Roeder, Trends Biochem. Sci., 1996, 21, 327–335 CrossRef CAS PubMed.
- J. T. Kadonaga, K. A. Jones and R. Tjian, Trends Biochem. Sci., 1986, 11, 20–23 CrossRef CAS.
- J. G. Bird, Y. Zhang, Y. Tian, N. Panova, I. Barvík, L. Greene, M. Liu, B. Buckley, L. Krásný, J. K. Lee, C. D. Kaplan, R. H. Ebright and B. E. Nickels, Nature, 2016, 535, 444–447 CrossRef CAS PubMed.
- D. Williamson, M. J. Cann and D. R. W. Hodgson, Chem. Commun., 2007, 5096–5098, 10.1039/B712066D.
- F. Huang, J. He, Y. Zhang and Y. Guo, Nat. Protoc., 2008, 3, 1848–1861 CrossRef CAS PubMed.
- E. Paredes and S. R. Das, ChemBioChem, 2011, 12, 125–131 CrossRef CAS PubMed.
- S. Fusz, S. G. Srivatsan, D. Ackermann and M. Famulok, J. Org. Chem., 2008, 73, 5069–5077 CrossRef CAS PubMed.
- B. Seelig and A. Jäschke, Tetrahedron Lett., 1997, 38, 7729–7732 CrossRef CAS.
- R. Fiammengo, K. Musílek and A. Jäschke, J. Am. Chem. Soc., 2005, 127, 9271–9276 CrossRef CAS PubMed.
- I.-H. Kim, S. Shin, Y.-J. Jeong and S. S. Hah, Tetrahedron Lett., 2010, 51, 3446–3448 CrossRef CAS.
- D. Shin, R. W. Sinkeldam and Y. Tor, J. Am. Chem. Soc., 2011, 133, 14912–14915 CrossRef CAS PubMed.
- A. R. Rovira, A. Fin and Y. Tor, J. Am. Chem. Soc., 2015, 137, 14602–14605 CrossRef CAS PubMed.
- L. S. McCoy, D. Shin and Y. Tor, J. Am. Chem. Soc., 2014, 136, 15176–15184 CrossRef CAS PubMed.
- K. B. Steinbuch and Y. Tor, in Handbook of Chemical Biology of Nucleic Acids, ed. N. Sugimoto, Springer Nature Singapore, Singapore, 2022, pp. 1–24 DOI:10.1007/978-981-16-1313-5_17-1.
- Y. Li, A. Fin, L. McCoy and Y. Tor, Angew. Chem., Int. Ed., 2017, 56, 1303–1307 CrossRef CAS PubMed.
- D. Cong, Y. Li, P. T. Ludford III and Y. Tor, Chem. – Eur. J., 2022, 28, e202200994 CrossRef CAS PubMed.
- J. Hertler, K. Slama, B. Schober, Z. Özrendeci, V. Marchand, Y. Motorin and M. Helm, Nucleic Acids Res., 2022, 50, e115 CrossRef CAS PubMed.
- K. D. Meyer, D. P. Patil, J. Zhou, A. Zinoviev, M. A. Skabkin, O. Elemento, T. V. Pestova, S. B. Qian and S. R. Jaffrey, Cell, 2015, 163, 999–1010 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, H. Hara, I. Kato and M. Inouye, J. Biol. Chem., 2005, 280, 3143–3150 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, K. P. Hoeflich, M. Ikura, G. Qing and M. Inouye, Mol. Cell, 2003, 12, 913–923 CrossRef CAS PubMed.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- V. Zorzini, A. Mernik, J. Lah, Y. G. Sterckx, N. De Jonge, A. Garcia-Pino, H. De Greve, W. Versées and R. Loris, J. Biol. Chem., 2016, 291, 10950–10960 CrossRef CAS PubMed.
- N. R. Wang and P. J. Hergenrother, Anal. Biochem., 2007, 371, 173–183 CrossRef CAS PubMed.
- C. R. Allerson, S. L. Chen and G. L. Verdine, J. Am. Chem. Soc., 1997, 119, 7423–7433 CrossRef CAS.
- S. A. Strobel, Curr. Opin. Struct. Biol., 1999, 9, 346–352 CrossRef CAS PubMed.
- C. S. Chow, S. K. Mahto and T. N. Lamichhane, ACS Chem. Biol., 2008, 3, 30–37 CrossRef CAS PubMed.
- K. Onizuka, Y. Taniguchi, T. Nishioka and S. Sasaki, Nucleic Acids Symp. Ser., 2009, 53, 67–68 CrossRef CAS PubMed.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874–7878 CrossRef CAS PubMed.
- S. C. Alexander and N. K. Devaraj, Biochemistry, 2017, 56, 5185–5193 CrossRef CAS PubMed.
- G.-H. Lee, H. K. Lim, W. Jung and S. S. Hah, Bull. Korean Chem. Soc., 2012, 33, 3861–3863 CrossRef CAS.
- R. A. Mizrahi, D. Shin, R. W. Sinkeldam, K. J. Phelps, A. Fin, D. J. Tantillo, Y. Tor and P. A. Beal, Angew. Chem., Int. Ed., 2015, 54, 8713–8716 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00045e |
| This journal is © The Royal Society of Chemistry 2024 |