
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural triterpenoid-aided identification of the druggable interface of HMGB1 occupied by TLR4†
Pingping
Shen
a,
Xuewa
Jiang
a,
Yi
Kuang
a,
Weiwei
Wang
b,
Richa
Raj
a,
Wei
Wang
c,
Yuyuan
Zhu
d,
Xiaochun
Zhang
e,
Boyang
Yu
f and
Jian
Zhang
 *af
*af
aDepartment of Resources Science of Traditional Chinese Medicines, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 210009, P. R. China. E-mail: 1020071849@cpu.edu.cn; Fax: +86-25-86185158; Tel: +86-25-86185157
bNanjing Hospital of Chinese Medicine Affiliated to Nanjing University of Chinese Medicine, Nanjing 210046, P. R. China
cDepartment of Chemistry, College of Liberal Arts and Sciences, University of Illinois Chicago, Chicago, IL, USA
dThe Center for Chemical Biology, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China
eSchool of Pharmaceutical Sciences, Tsinghua University, Beijing, 100084, P. R. China
fJiangsu Key Laboratory of TCM Evaluation and Translational Research, China Pharmaceutical University, Nanjing 211198, P. R. China
First published on 7th June 2024
Abstract
HMGB1 interacts with TLR4 to activate the inflammatory cascade response, contributing to the pathogenesis of endogenous tissue damage and infection. The immense importance of HMGB1–TLR4 interaction in the immune system has made its binding interface an area of significant interest. To map the binding interface of HMGB1 occupied by TLR4, triterpenoids that disrupt the HMGB1–TLR4 interaction and interfere with HMGB1-induced inflammation were developed. Using the unique triterpenoid PT-22 as a probe along with photoaffinity labeling and site-directed mutagenesis, we found that the binding interface of HMGB1 was responsible for the recognition of TLR4 located on the “L” shaped B-box with K114 as a crucial hot-spot residue. Amazingly, this highly conserved interaction surface overlapped with the antigen-recognition epitope of an anti-HMGB1 antibody. Our findings propose a novel strategy for better understanding the druggable interface of HMGB1 that interacts with TLR4 and provide insights for the rational design of HMGB1–TLR4 PPI inhibitors to fine tune immune responses.
1. Introduction
Protein–protein interactions (PPIs) are critical in most biological processes, including antigen–antibody recognition, cellular signal transduction, and innate and adaptive immune regulation.1 In innate immunity, pattern recognition receptors (PRRs) can recognize the damage-associated molecular patterns (DAMPs) or pathogen associated molecular patterns (PAMPs) to initiate local and systemic inflammation.2 As a prototypical DAMP, high mobility group box 1 (HMGB1) normally resides in the nucleus but it can be actively secreted and passively released outside cells.3 Extracellular HMGB1 functions as a critical immune mediator by interacting with toll-like receptor 4 (TLR4) to activate the inflammatory response, leading to a massive release of cytokines and chemokines.4 The ongoing activation of the HMGB1–TLR4 axis facilitates tissue damage that further upregulates HMGB1 levels and forms a positive feedback loop, causing detrimental outcomes such as inflammatory and autoimmune diseases, hepatic and renal injuries, and cardiovascular diseases.5 Administration of anti-HMGB1 antibodies significantly prevents these pathologically relevant cellular events in wild-type mice, but this effect disappears in TLR4-deficient mice.6With the ever-increasing number of experimentally validated PPIs, structural insight into the PPI interface is essential for a better understanding of the fundamentals of protein–protein association and its potential as a therapeutic target. Despite intensive efforts in X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, cryo-electron microscopy (CryoEM), and computational approaches, success in determining native protein interfaces that are biologically relevant remains limited.7,8 The regulation of HMGB1–TLR4 interaction has drawn intense focus on physiological and pathological research, where the binding interface represents a potent target for therapeutic intervention. Only a few studies have reported the first 20 amino acids (89–108 aa) of the B-box domain in HMGB1 as the minimal sequence necessary to activate the TLR4 signaling pathway.9 Challenges persist in identifying the structural elements within HMGB1 responsible for TLR4 recognition and immune response regulation due to the lack of known complex structures.
Natural products are secondary metabolites biosynthesized through multi-enzyme reactions in living organisms, historically recognized as privileged structures to interact with protein targets.10 Pentacyclic triterpenoids (PTs), a class of natural bioactive substances, are generally considered part of plant defense systems with valuable pharmacological properties.11 Structurally, they typically have a higher molecular mass, a larger number of sp3 carbon atoms but lower calculated lipid/water partition coefficients, and greater molecular rigidity. These properties, especially the higher rigidity and hydrophobicity of PTs, are beneficial to target large and featureless protein surfaces. Cellular and animal-level studies have confirmed that oleanolic acid (OA)12 and glycyrrhizin (GL)13 exhibit significant inhibitory activity against HMGB1 and potential therapeutic efficacy in clinical trials for hepatitis. Additionally, the natural friedelane type PT, celastrol, has been shown to possess neuroprotective and anti-inflammatory effects by directly acting on HMGB1.14 Thus, naturally derived PTs are considered to be promising probes for exploring the HMGB1–TLR4 interaction interface.
To expand the structural diversity of triterpenoids, biotransformation as a green and efficient tool was utilized, where microbes with peculiar enzymes can yield bioactive compounds by metabolic networks in a continuous and self-optimizing manner.15 In our long-term research on the microbial transformation of PTs, nearly 300 natural derivatives with different structural types were prepared.16–18 To develop specific triterpenoid leads targeting the HMGB1–TLR4 interaction, the cell-based large-scale screening method was established on HMGB1- and LPS-induced acute inflammatory cell models. As a result, triterpenoids that selectively interfered with the HMGB1–TLR4 interaction and inhibited the subsequent cytokine secretion were found. Using triterpenoid leads as chemical probes in combination with computational prediction and experimental assessment including molecular dynamics (MD) simulations and site-directed mutation, the druggable interface within HMGB1 that was recognized by TLR4 was elucidated, which would provide a structural basis for the rational design of new therapeutic agents.
2. Results
2.1. HMGB1-specific triterpenoid probes inhibiting TLR4 downstream signaling
Although GL is the first reported and structurally well-characterized natural HMGB1 inhibitor, it exhibits a low affinity and specificity for HMGB1 (KD ∼ 150 μM).19 To improve these limitations, reversible interactions can be strategically introduced, e.g., hydrogen bonds, which have been proven crucial for the high selectivity and lead-likeness of small molecules.20,21 In this context, microbial transformation as a robust tool was applied to add polar groups into triterpenoid skeletons with excellent regioselectivity and stereoselectivity. A natural library consisting of hundreds of chemicals was constructed.22–24 The catalytic sites covered A, B, C, D, and E rings of oleanane, ursolane and lupane-type triterpenoids, greatly enriching the structural diversity of PTs (Fig. S1, ESI†). Taking these triterpenoids as tool molecules is conducive to capturing extensive HMGB1–TLR4 interfaces that lack ligand binding pockets. However, unlike traditional drug target enzymes or receptors, developing a more effective approach for screening potential HMGB1–TLR4 regulators is necessary.In contrast to endogenous HMGB1-initiated sterile inflammation, external LPS plays a crucial role in host infection and pathogenicity. Consistently, they are both recognized by the TLR4 receptor to promote activation of NF-κB and the release of cytokines, leading to an inflammatory cascade.2 To discover specific triterpenoids, cell models of RAW 264.7 macrophage-based nitrite release induced by HMGB1 and LPS were established with GL as a positive control (Fig. 1A). In these two cell models, the inflammatory mediator production was blocked by TAK-242, a specific TLR4 inhibitor25,26 (Fig. S2, ESI†), thus confirming that the effect of LPS and HMGB1 was TLR4-dependent. Furthermore, cell-based phenotypic screening of the in-house PT compound library was carried out (Table S1, ESI†). Structure–activity relationship analysis highlighted that the chemical cluster with an A-ring cleaved fragment on the triterpenoid skeleton from microbial-catalyzed Baeyer–Villiger oxidation by Streptomyces olivaceus CICC 23628 exhibited better inhibition of HMGB1-induced inflammation than LPS (Fig. S3, ESI†).
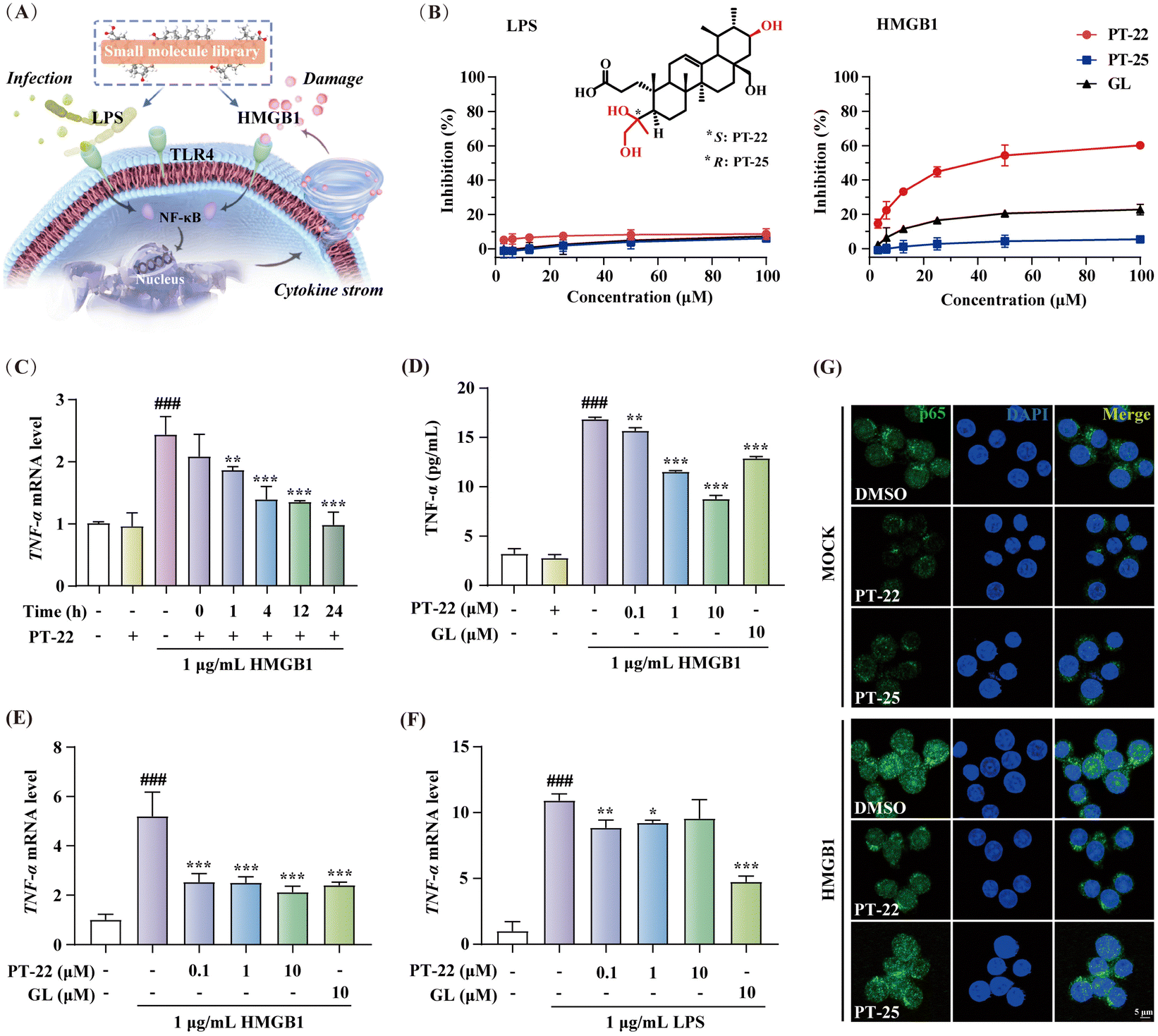 | ||
| Fig. 1 Discovery of triterpenoid probes with HMGB1–TLR4 dependent anti-inflammatory effects. (A) Compound screening strategy on two inflammatory cell models induced by HMGB1 and LPS. (B) The dose-dependent inhibitory curves of PT-22 and PT-25 in LPS or HMGB1-activated nitrite release on RAW 264.7 with GL as the positive control and their chemical structures. (C) The effect of co-incubation time of 1 μM PT-22 with HMGB1 on TNF-α mRNA expression in THP-1 cells. The relative levels of TNF-α mRNA in THP-1 cells were measured by quantitative real-time PCR (qRT-PCR). (D) The dose-dependent relationship of PT-22 on HMGB1-triggered TNF-α release detected by ELISA. (E) The dose-dependent inhibition of PT-22 on HMGB1-induced TNF-α mRNA transcription. (F) The inhibitory effect of PT-22 on LPS-activated inflammation. (G) Effect of 1 μM PT-22 or PT-25 on the nuclear translocation of NF-κB p65 induced by 1 μg mL−1 HMGB1. Data shown are mean ± SD from three independent experiments. ###P < 0.001 compared to the control group. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to the model group, as calculated by Student's t-test. Gene expression was normalized to GAPDH. | ||
Structurally, this type of A-ring cleaved triterpenoid derivative is similar to steroids, a kind of wide-spectrum anti-inflammatory agent, equipped with higher drug-likeness compared to the substrate according to the number of hydrogen bond donors, acceptors and rotatable bonds. Among them, PT-22 (3,4-seco-urs-12-en-4(S), 21β, 24, 28-tetrol-3-oic acid) showed a unique chiral architecture and was found to inhibit HMGB1-induced nitrite elevation, rather than LPS (IC50 > 100 μM) (Fig. 1B). In addition, PT-22 alone did not show such an effect during these periods and was not cytotoxic. However, as a diastereoisomer of PT-22 that differs only in the absolute configuration of vicinal diol at the C-4 position, PT-25 did not inhibit HMGB1-triggered nitrite release. These findings indicated that the stereochemical configuration of triterpenoids is crucial for their inhibition of HMGB1-initiated inflammatory responses.
To confirm the specific inhibitory effect on HMGB1-dependent inflammation, we pre-incubated PT-22 with HMGB1 in different time periods before exposure to human acute monocytic leukemia THP-1 cells. The results showed that the upregulation of TNF-α mRNA levels profoundly decreased in a time-dependent manner (Fig. 1C). Besides, PT-22 at concentrations of 0.1 to 10 μM gradually suppressed the HMGB1-activated mRNA transcription and secretion of TNF-α (Fig. 1D and E). In contrast, there was no dose-dependent inhibitory effect on LPS-induced inflammation (Fig. 1F). In RAW 264.7 cells, pre-incubation of HMGB1 with PT-22 also significantly improved its anti-inflammatory activity, with the most apparent effect observed for 4 h (Fig. S4, ESI†). Additionally, it was found that PT-22 strikingly interfered with HMGB1-initiated nuclear translocation of NF-κB p65, while PT-25 did not (Fig. 1G). Overall, PT-22 displayed a specific inhibition on the HMGB1–TLR4 signal and was considered a potential probe for further investigation.
2.2. Verification of triterpenoid probe interfering with HMGB1–TLR4 interaction
Given the evidence that the inhibition of PT-22 on the transcription and release of pro-inflammatory cytokines, which depends on the HMGB1–TLR4 signal, we hypothesized that it might disturb the interaction between HMGB1 and TLR4. To test the assumption, we employed surface plasmon resonance (SPR), which allows the detection of ligand binding to immobilized proteins (Fig. 2A). As shown in Fig. 2B, the concentration-dependent binding curves of HMGB1 to immobilized TLR4 were monitored. The dissociation constant (KD) value was determined to be 7.41 × 10−8 M, suggesting that both HMGB1 and TLR4 proteins were in their active form. Subsequently, we pre-incubated different concentrations of PT-22 with HMGB1 flowing through the TLR4 sensor. As shown in Fig. 2C, the response signals between HMGB1 and TLR4 was dramatically inhibited in a concentration-dependent manner. Furthermore, the SPR signals of different concentrations of HMGB1 and TLR4 interactions were significantly decreased after adding 1 μM PT-22. As a result, the calculated binding affinity of HMGB1–TLR4 was reduced approximately 10-fold with a KD value of 7.96 × 10−7 M (Fig. 2D).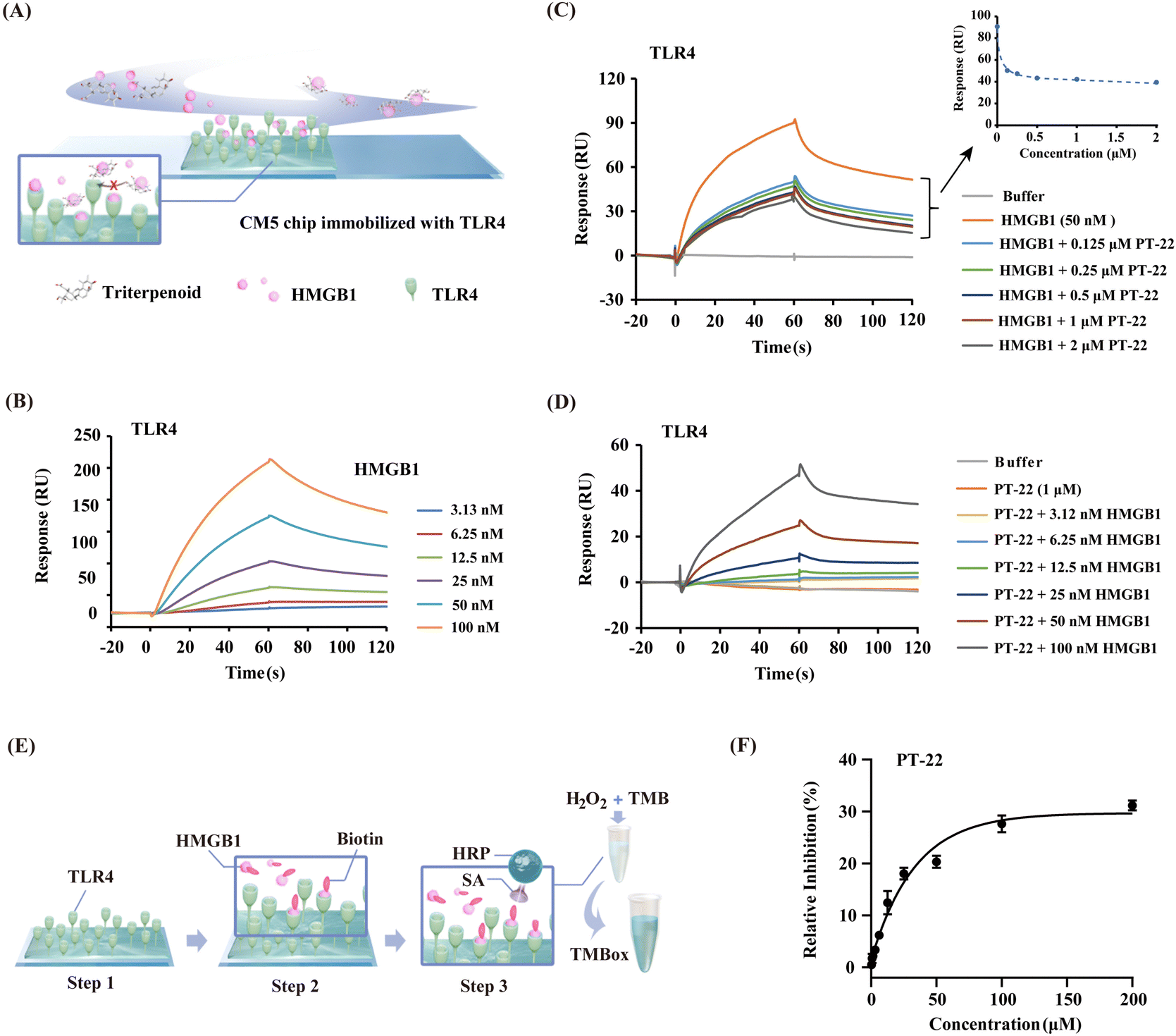 | ||
Fig. 2 Triterpenoid probe PT-22 interferes with the interaction between HMGB1 and TLR4. (A) Scheme of the competition SPR assay. (B) Validation of the HMGB1–TLR4 interaction (the extracellular domain of TLR4 was immobilized on the CM5 sensor chip). The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model was used to assess binding kinetics. (C) SPR sensorgrams representing the inhibition of PT-22 (0, 0.125, 0.25, 0.5, 1, and 2 μM) on the interaction of HMGB1 (50 nM) to immobilized TLR4. (D) The inhibitory effect of 1 μM PT-22 on the binding of HMGB1 at different concentrations (3.12, 6.25, 12.5, 25, 50, and 100 nM) with TLR4. (E) Schematic diagram of the principle of a competitive ELISA assay. (F) The inhibitory curve of different concentrations of PT-22 (0.39, 0.78, 1.5, 3.1, 6.2, 12.5, 25, 50, 100, and 200 μM) on the HMGB1–TLR4 interaction. :1 binding model was used to assess binding kinetics. (C) SPR sensorgrams representing the inhibition of PT-22 (0, 0.125, 0.25, 0.5, 1, and 2 μM) on the interaction of HMGB1 (50 nM) to immobilized TLR4. (D) The inhibitory effect of 1 μM PT-22 on the binding of HMGB1 at different concentrations (3.12, 6.25, 12.5, 25, 50, and 100 nM) with TLR4. (E) Schematic diagram of the principle of a competitive ELISA assay. (F) The inhibitory curve of different concentrations of PT-22 (0.39, 0.78, 1.5, 3.1, 6.2, 12.5, 25, 50, 100, and 200 μM) on the HMGB1–TLR4 interaction. | ||
To further corroborate our findings, a competitive ELISA method was also developed and the principle of this assay is shown in Fig. 2E. The binding of biotin-labeled HMGB1 to TLR4 coated on a 96-well plate could be detected directly by the color change of TMB solution (Fig. S5, ESI†). The optical density (OD) values increased with the increase in the concentration of biotinylated HMGB1, indicating that there is a direct binding between HMGB1 and immobilized TLR4 (Fig. S6A, ESI†). Next, recombinant HMGB1 was used as a positive control to test the reliability of the competition binding assay. In the presence of HMGB1, the interaction between biotin-labeled HMGB1 and TLR4 was significantly disrupted (Fig. S6B, ESI†). Thus, a rapid and sensitive method for evaluating the inhibitory potency of HMGB1–TLR4 interactions was established. Incubating PT-22 with biotin-labeled HMGB1 for 1 h before mixing with loaded TLR4 resulted in a dose-dependent inhibition of the HMGB1–TLR4 interaction (Fig. 2F). These observations indicated the potential of PT-22 as a modulator of HMGB1–TLR4 and thus could be used as a probe to clarify the potential binding interface of PPIs.
2.3. Triterpenoid probe target HMGB1 to disrupt its interaction with TLR4
In light of the intervention of PT-22 on the HMGB1–TLR4 PPI, we presumed that it might bind with HMGB1 or TLR4. In support of this hypothesis, SPR-based binding assays were conducted. As shown in Fig. 3A, the dose-dependent association and disassociation curves of PT-22 with HMGB1 were observed. The KD value was calculated as 8.75 × 10−5 M, lower than the well-known HMGB1 inhibitor GL (Fig. S7A, ESI†). However, there was no observable interaction between HMGB1 and PT-25 in the parallel experiment (Fig. S7B, ESI†). In addition, no specific binding of PT-22 to immobilized TLR4 was detectable up to a concentration of 100 μM (Fig. S8, ESI†), excluding TLR4 as the target of PT-22. Although TLR4 is the dominant receptor sensed by extracellular HMGB1 to mediate inflammation, antagonism of TLR4 renders organisms susceptible to infectious or injurious insults caused by other exogenous ligands e.g., LPS.27 Herein, the triterpenoid probe targeting the central node of this signaling network, HMGB1, is biologically rational to abolish the downstream inflammatory responses.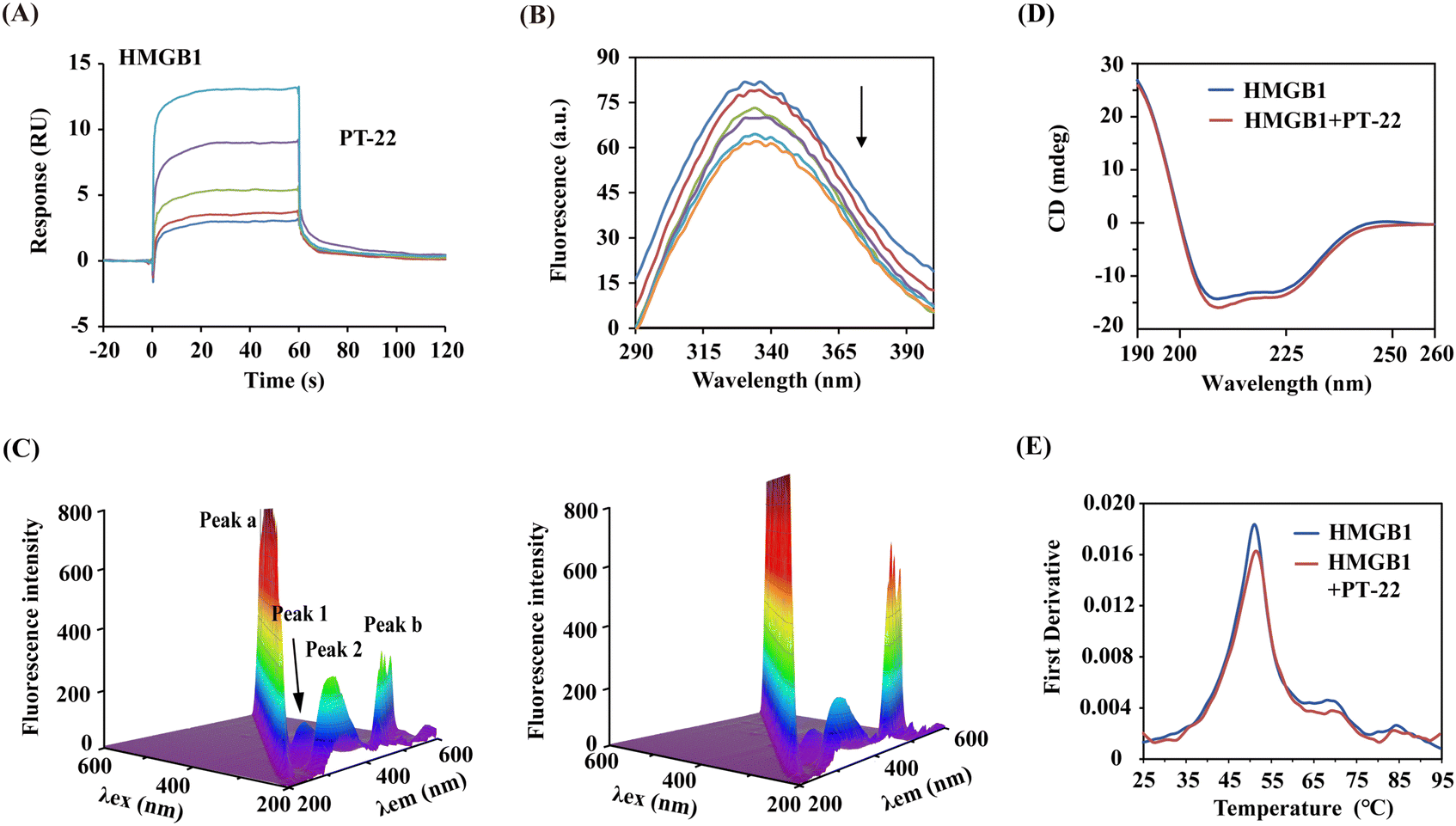 | ||
| Fig. 3 The direct binding of PT-22 induced the local conformation and thermal stability changes of HMGB1. (A) Association and dissociation curves of PT-22 with immobilized HMGB1. (B) Effect of PT-22 (0, 50, 100, 150, 200, and 250 μM) on the fluorescence spectra of 3 μM HMGB1. (C) 3D fluorescence spectrum of HMGB1 at a concentration of 3 μM without (left) or with PT-22 (20 μM) (right). (D) CD spectra of HMGB1 alone or co-incubated with 20 μM PT-22. (E) Thermal stability of HMGB1 (10 μM) in the presence or absence of 100 μM PT-22. | ||
To further decipher the PT-22–HMGB1 interaction, a series of spectra under simulated physiological conditions were monitored. As shown in Fig. 3B, the addition of PT-22 was found to quench the intrinsic fluorescence of HMGB1 in a dose-dependent manner, meaning the micro-environmental variation in the vicinity of aromatic residues. In three-dimensional (3D) fluorescence spectra, the intensity of peak 1 decreased (Fig. 3C), indicating that the surrounding microenvironment of tryptophan and tyrosine in HMGB1 was disturbed by PT-22. Meanwhile, the fluorescence intensity of peak 2 also decreased, which was related to the change in the tertiary structure of HMGB1. Furthermore, the presence of PT-22 increased the fluorescence intensity of peak a, suggesting an increase in the diameter of HMGB1 due to the formation of the ligand–HMGB1 complex, thus enhancing scattering.
The predominant secondary structure of HMGB1 is reported to be an α-helix with characteristic double circular dichroism (CD) signal peaks at 208 and 222 nm.28,29 In Fig. 3D, the addition of excess PT-22 induced a reduction of the peak signal of HMGB1 from 40.06% to 37.89% near 208 nm, indicating a slight decrease in the α-helical content of HMGB1. Thermal shifts of target proteins affected by ligands have been shown to correlate with drug efficacy and are widely used to characterize ligand binding in structural biology.30 Herein, the thermal stability of the HMGB1 protein was also evaluated by nano-differential scanning fluorometry (nanoDSF). It was found that triterpenoid probe PT-22 could improve the Tm (melting temperature) value of HMGB1 from 51.1 °C to 51.5 °C (Fig. 3E). Overall, the binding of PT-22 to HMGB1 was accompanied by local structural changes of HMGB1 without strikingly distorting its secondary structure and thermostability. These data revealed the intrinsic conformational dynamics of HMGB1 that are likely to affect its binding with TLR4.
2.4. Prediction of the HMGB1 interface that interacts with TLR4 by PeSTo and MD
Next, to explore the possible interface of HMGB1–TLR4 interaction in atomistic detail, a protein structure transformer (PeSTo), a newly reported machine learning approach,31 was employed. The results showed that most interfacial residues involved in the interaction of TLR4 are located on the pro-inflammatory B-box domain of HMGB1 (Fig. 4A), suggesting the reliability of the predicted surface. Among them, K90 and K96 are likely to occur on the binding interface, and R97, P98 and P99 were predicted to have a moderate probability as shown in Table S4 (ESI†). In addition, K88, F89, E74, D33 and D158 are also likely to be involved in protein interactions. The obtained HMGB1–TLR4 complex by PeSTo was saved as the starting structure for a subsequent 200-ns MD simulation. The root mean square deviation (RMSD) plot reached equilibrium from 150 ns during the simulation and remained stable thereafter (Fig. S9, ESI†). Upon inspection of the HMGB1–TLR4 structure, it was found that the TLR4 protein showed direct contact with A-box and B-box domains in HMGB1 (Fig. 4B). The surface area buried at the HMGB1–TLR4 interface was calculated as 1227.04 Å2, which is larger than the typical protein–ligand contact area (300–1000 Å2). Based on previous reports32 and the above prediction, we primarily focus on the binding interface on the B-box. Specifically, the outer region on the HMGB1 surface remains partially solvent-accessible upon binding, consisting of polar and charged residues (K114, R110, K96, K165, K87, and E84). The central region of the interface (F103, I122, A126, P92, and V125) is occluded from solvent upon association, which is crucial for the binding affinity. Detailed analysis of the complex interface indicated that the externally charged residues can provide high stability for the HMGB1–TLR4 complex through hydrogen bonding interactions. In contrast, the central hydrophobic residues determine the specificity of the complex through hydrophobic contributions and van der Waals forces.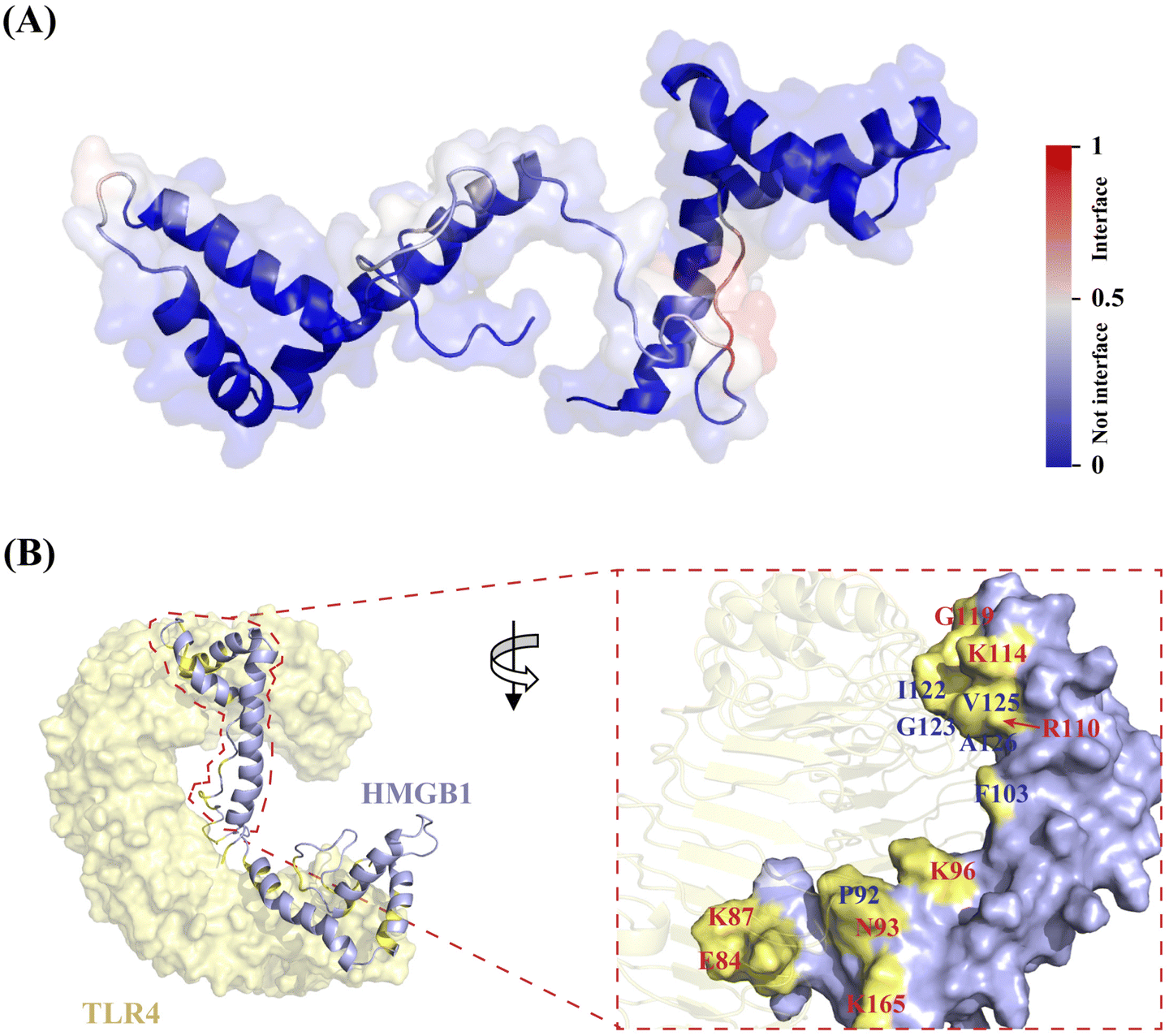 | ||
| Fig. 4 Architecture of HMGB1–TLR4 interaction interface through PeSTo and MD simulation. (A) PeSTo-based prediction of binding interface of HMGB1 (PDB: 2YRQ) with TLR4 (PDB: 3FXI) (the confidence of the predictions is represented with a gradient of color from blue for non-interfaces to red for interfaces). (B) Structural basis of HMGB1 recognition by TLR4 through MD simulation (the interaction interface is shown in yellow, where the central hydrophobic residues are labeled in blue, while the surrounding hydrophilic residues are labeled in red). | ||
To further identify the PT-22–HMGB1-binding region, MD simulation was performed in the presence of different concentrations of ligands. The results showed that the fluctuation degree of the RMSD of the HMGB1 backbone decreased after adding PT-22 as shown in Fig. 5A, indicating an improvement in the stability of the system. Nevertheless, residues 79–89 in the linker region between the A-box and the B-box exhibit large root mean square fluctuation (RMSF) values (Fig. S10, ESI†). These structural fluctuations were associated with the shift of these two boxes with respect to each other, as shown by the dominant conformations in Fig. 5B. In a ratio of HMGB1 to PT-22 of 1:1, the ligand was found to be in close contact with the L-shaped α-helical B-box domain in the equilibrium state. Interestingly, with the ratio increasing from 1:10 to 1:100, most PT-22 molecules preferentially gather around the B-box.
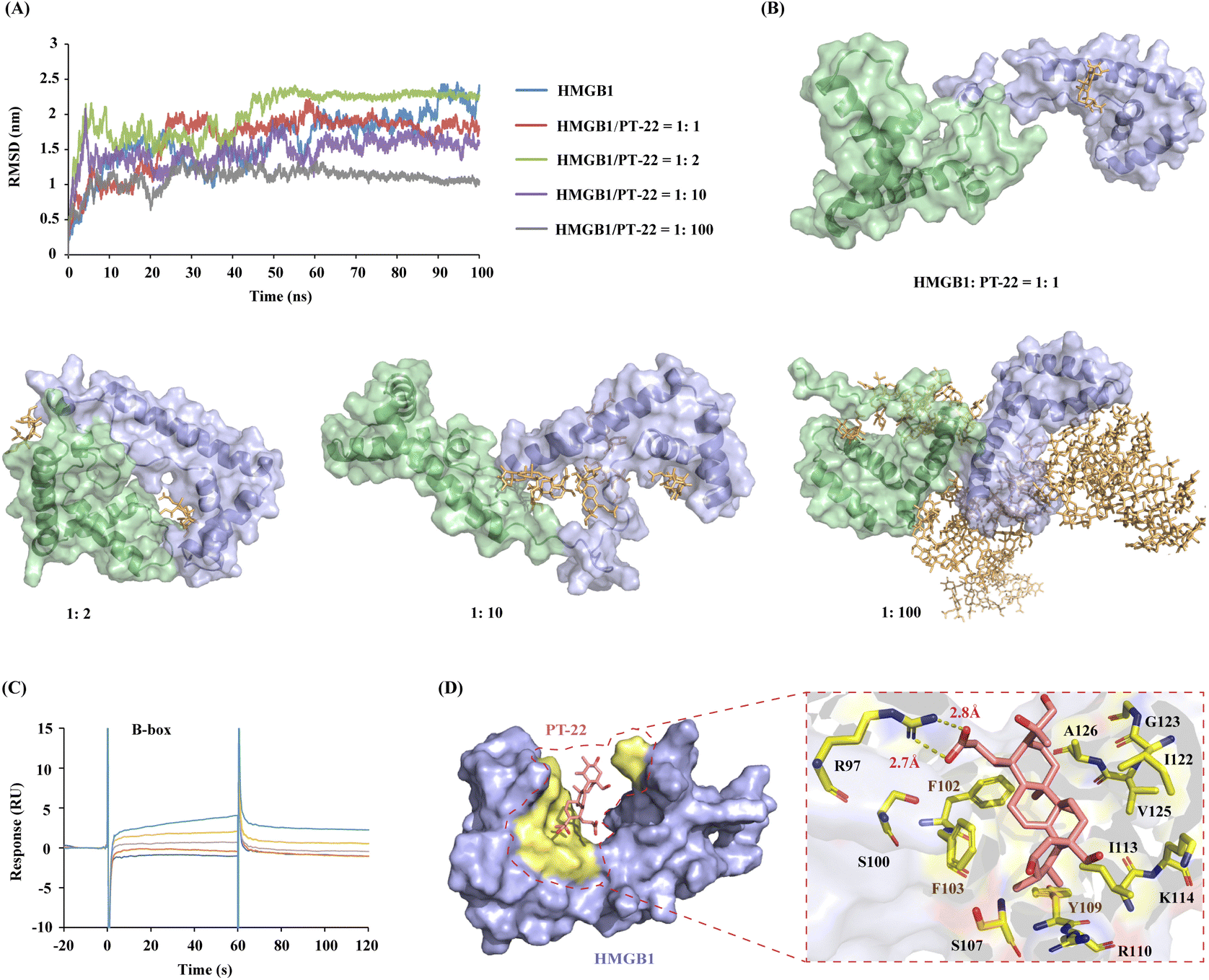 | ||
| Fig. 5 Triterpenoid probe PT-22 specifically binds to the B-box domain of HMGB1. (A) RMSD curves of HMGB1 in the presence of different ratios of PT-22. (B) Binding conformation of HMGB1 (the A-box shown as the surface in cyan and the B-box shown as the surface in blue) with different concentrations of PT-22 (shown as stick in orange). (C) SPR plots of PT-22 (6.25, 12.5, 25, 50, and 100 μM) binding with the B-box. (D) Critical interactions of PT-22 (shown as stick in pink) and HMGB1 (shown as the surface in blue, where interfacial residues are in yellow). | ||
Next, molecular docking and MD simulation of PT-22 with two independent domains, A-box and B-box, were carried out, respectively. The binding free energy (ΔG) was calculated through molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA).33 It can be seen from Fig. S11 (ESI†) that ΔG of PT-22 binding with the A-box was calculated as −0.48 kJ mol−1. However, the interaction of the B-box with PT-22 is tighter with a ΔG value of −35.64 kJ mol−1. SPR further confirmed the result that PT-22 exhibited a strong binding force with the B-box (Fig. 5C), whereas no concentration-dependent response signals were observed with the A-box (Fig. S12, ESI†). Previous studies have proven that the pro-inflammatory function of HMGB1 was held within the B-box domain. Herein, we found that co-incubation of PT-22 attenuated B-box-induced pro-inflammatory cytokine TNF-α mRNA expression and release in human and mouse macrophages (Fig. S13, ESI†).
Having established that the dominant interaction between PT-22 and HMGB1 occurs at the TLR4-binding domain on the B-box, to further obtain the structural information in the PT-22–HMGB1 interface, the snapshot of the lowest energy in the 100-ns MD simulation was analyzed. It can be seen from Fig. 5D thatPT-22-binding HMGB1 surfaces are enriched with aliphatic (I122 and G123) and charged (R97, R110, K114) residues. Besides, the aromatic (F102, F103, and Y109) residues also present a higher preference, which might have participated in the intrinsic fluorescence quenching of HMGB1 induced by triterpenoids. Furthermore, the carboxyl group of C-3 in PT-22 forms two stable hydrogen bonds with R97 of HMGB1 with lengths of 2.7 Å and 2.8 Å, further reinforcing their interactions. These essential residues in the binding regions of triterpenoid-HMGB1 constitute a specific interface where they are highly complementary in shape.
Given the competitive inhibition of triterpenoids on the HMGB1–TLR4 interaction, we next map the PT-22-binding region on HMGB1 to the aforementioned HMGB1–TLR4 binding interface. To our delight, several shared interfacial residues were found, including F103, R110, K114, I122, G123, V125 and A126, which are mainly located at the core of the HMGB1–TLR4 interface. These residues are evolutionarily conserved in the HMGB1 protein, enabling it to recognize TLR4 and initiate innate immune responses. Using the MM/PBSA method,33 we decomposed the binding free energy into the corresponding residues. Among them, the energy contribution of R110, K114, and I122 is the most significant, which are −15.88, −21.86, and −23.27 kJ mol−1, respectively. Further experimental validation of the predicted HMGB1–TLR4 binding interface is expected.
2.5. Validation of the HMGB1 interface that interacts with TLR4 using the triterpenoid probe
Our computational analysis indicated that the triterpenoid probe shared several characteristic residues with TLR4 on the HMGB1 surface. To further prove these findings, photoaffinity labeling (PAL) was applied for target identification as well as elucidating ligand binding sites (Fig. 6A). Firstly, probe PT-22-BP containing a ‘‘minimalist’’ diazirine-alkyne as a photo-activatable crosslinking moiety was strategically synthesized (Fig. 6B).34 The end of alkyne was used as a functional tag for a bio-orthogonal click reaction with azide-linked biotin to enrich the target protein. Before the PAL experiment, the photoreactivity of PT-22-BP in MeOH was examined by high-resolution mass spectrometry (HR-MS). As a consequence of UV light exposure, the diazirine undergoes photolysis to form a highly reactive carbene with the loss of N2 yielding a methanol adduct (Fig. S17, ESI†), confirming the photoprobe's utility. With the synthesized PT-22-BP in hand, the SPR assay was performed to evaluate its binding affinity with HMGB1. To our delight, PT-22-BP exhibited binding potency comparable to that of the original compound (Fig. S19, ESI†).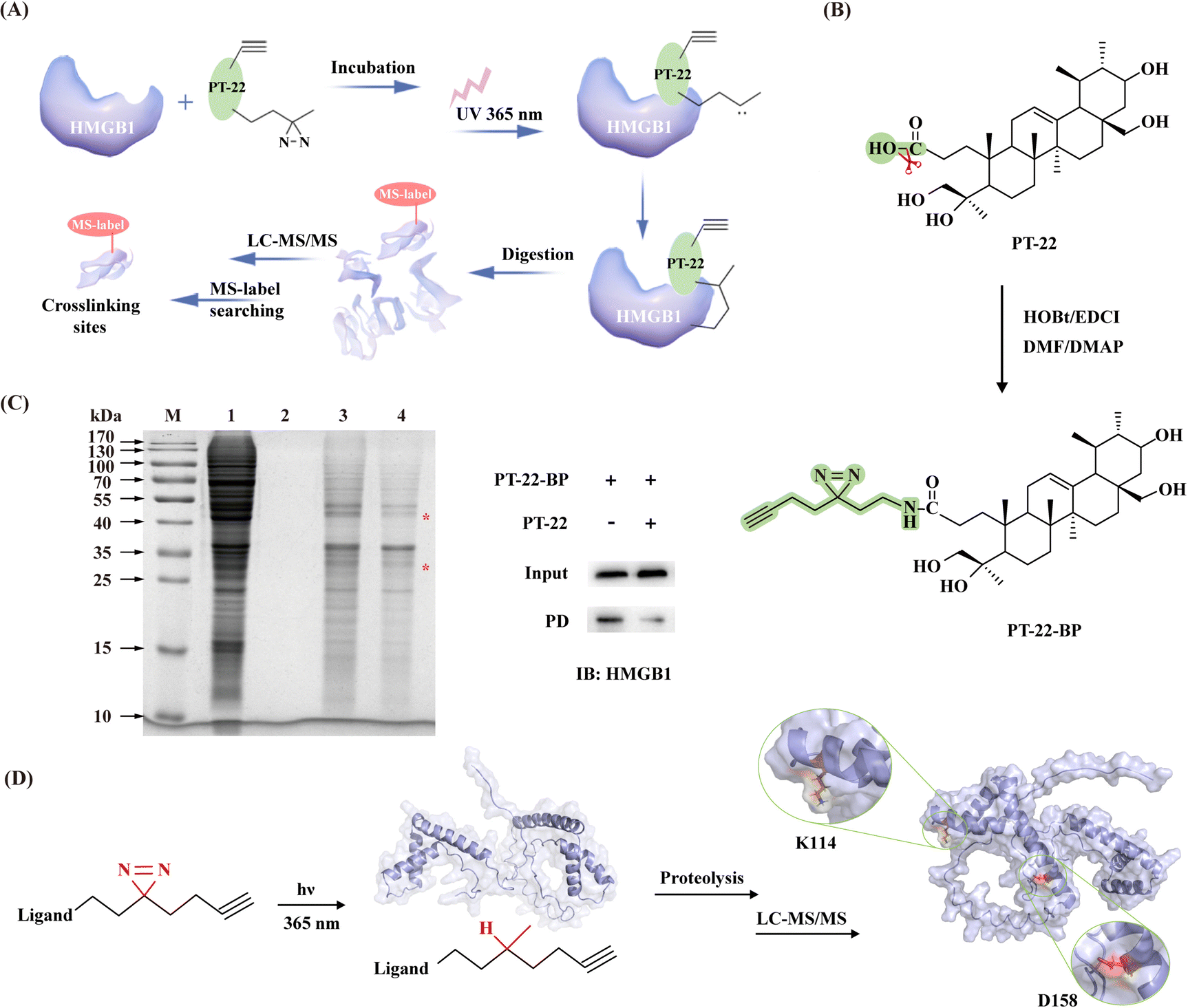 | ||
| Fig. 6 Identification of interfacial residues on HMGB1 with the triterpenoid as the probe. (A) Scheme of the triterpenoid probe covalently attaches the target protein via sequential photocrosslinking capture and click-mediated biotin harvest. (B) Synthesis of PT-22-BP (green represents a photoaffinity group). (C) SDS-PAGE analysis of PT-22-BP-labeled proteomes with or without PT-22 competition (M: marker, 1: cell lysate, 2: control group, 3: experimental group, and 4: competitive binding group). Right: Immunoblotting analysis of the streptavidin-based enrichment of biotin-tagged molecules. (The asterisk denotes specific binding.) (D) Identification of the binding site of PT-22-BP on HMGB1 (shown as the cartoon in blue) using photoaffinity labeling in combination with LC-MS/MS. | ||
Next, we sought to validate the target engagement of PT-22 in cell lysates using competitive labeling, facilitating the effective exclusion of nonspecific binding proteins. PT-22-BP was incubated with the crude protein extract followed by UV activation specifically cross-linked to HMGB1. And SDS-PAGE were analyzed (Fig. S20, ESI†). A direct comparison of the labeling patterns of PT-22-BP with and without PT-22 competition showed two distinct bands around 30 kDa and 50 kDa (Fig. 6C). Among them, the protein in the 30-kDa band was selected to assess the specific binding event of PT-22-BP with HMGB1 by immunoblotting. The results showed that HMGB1 was efficiently bound with PT-22-BP, whereas the excess PT-22 as a soluble competitor prevented the specific cross-linking, confirming the interaction of the triterpenoid probe with HMGB1.
To further identify essential interaction regions and critical residues, the purified recombinant HMGB1 protein was incubated with PT-22-BP. Once activated by UV light (365 nm), the probe was inserted into C–H, O–H or N–H bonds of residues proximal to the binding sites in HMGB1 for covalent labeling. The photo-crosslinked complex was further subjected to LC-MS/MS analysis. A molecular weight increase of 597.4466 Da in D158 was detected in the peptide 158DIAAYR163, indicating that PT-22-BP was labeled on D158 (Fig. 6D). Similarly, K114 was found as another potential photocrosslinking site in the 113IKGEHPGLSIGDVAK127 peptide (Fig. S21, ESI†). It is worth noting that these two residues lie in the B-box domain and are 100% conserved between humans and mice by sequence alignment analysis. These results were highly consistent with the computational prediction that K114 appears at the triterpenoid-binding HMGB1 interface and the HMGB1–TLR4 interface.
Inspired by these findings, we then applied ANCHOR (https://structure.pitt.edu/anchor/), a web-based tool search for anchor residues in protein–protein interfaces suitable for small molecule intervention.35 According to this result, K114 in the box-like domain acted as a binding anchor, preferentially bound to the surface of TLR4 through hydrogen bonding and electrostatic force that stabilized the overall complex structure (Fig. S22, ESI†).
2.6. TLR4-binding HMGB1 interface and its antigen-recognition epitope
Immune recognition of antigens by antibodies represents an essential class of specific PPIs in biological systems. Recently, it has been reported that the anti-HMGB1 IgM autoantibody neutralizes extracellular HMGB1 via specifically binding to a conserved epitope, namely HMW4 (HMGB199–113, PSAFFLFCSEYRPKI).36 The surface area involved in antigen–antibody recognition lies in the B-box composed of three α-helices, including the hydrophobic residues (Phe, Pro, and Ile) and polar and charged residues (Ser, Arg, and Lys) (Fig. S23A, ESI†). TLR4, as a cell surface receptor, is indispensable for HMGB1-induced innate immunity activation and cytokine production. Therefore, extracellular HMGB1 is both a ligand for TLR4 and an antigen for the anti-HMGB1 antibody. In contrast to the antigen–antibody, the HMGB1–TLR4 heterodimer interface tends to be flat and wide and consists of a series of discontinuous residues (Fig. S23B, ESI†). Consistently, some residues like K114 at the HMGB1–TLR4 binding interface were found to be adjacent to antigen-recognition epitopes of the anti-HMGB1 antibody.However, the application of neutralizing antibodies is limited to the extracellular HMGB1 protein due to its large molecular weight. Unlike TLR4 or anti-HMGB1 antibodies, the existing triterpenoids only occupy a relatively smaller binding interface of HMGB1 (Fig. S23C, ESI†), which disturbs the interaction of proteins with TLR4. Using a direct biophysical binding assay, we further found that the anti-HMGB1 antibodies function similarly to PT-22 (Fig. S24, ESI†), that is, once they bind with HMGB1, it leads to the failure of TLR4 to recognize HMGB1 and activate inflammation. In addition, the composition of the large antigen–antibody interfaces is not significantly different from that of the accessible HMGB1 surface of triterpenoids. Among them, Lys makes overall similar contributions to the ligand–receptor and two kinds of protein–protein interfaces. In particular, K114 is not only involved in the interaction of HMGB1 with its ligand triterpenoid and receptor TLR4 but also closely related to the conserved epitope of HMGB1 antibody recognition.
In short, HMGB1 was found to use almost the same area to recognize these two different receptors or ligands, and the structural features of each complex interface are nearly identical. In particular, the HMGB1 interface that interacts with TLR4 highly overlaps with the recognition region of the anti-HMGB1 antibody. The most consistently discernible property is the higher evolutionary conservation of residues at the interface of HMGB1-ligand and HMGB1-receptors, including TLR4 and anti-HMGB1 neutralizing antibodies. The strong conservation of interfacial residues further highlights the importance of the core region of HMGB1 in ligand or receptor recognition. Altogether, these three highly unified interfaces share common characteristics that make them great significance in inflammatory signal transduction and immune regulation (Fig. S23D, ESI†).
2.7. Mutation verification of druggability of the HMGB1 interface occupied by TLR4
To explore the druggability of the identified HMGB1–TLR4 interface, we mutated the key anchor residue K114 at the HMGB1 protein surface. The HMGB1 mutant (K114A) was prepared, and its binding affinity with TLR4 was also detected by the SPR-based assay (Fig. 7A). The results showed that the replacement of residue K114 with Ala created a significant decrease of binding affinity (Fig. S25, ESI†). In addition, compared with wild type (WT), the HMGB1 mutant also exhibited a ten-fold decreased affinity toward PT-22 (Fig. S26, ESI†). Considering the above results, we then evaluated its influence on the pro-inflammatory activity of HMGB1. The results showed that the mutant K114A displayed a dramatically decreased activity in triggering transcription (Fig. 7B) and release (Fig. 7C) of TNF-α in THP-1 compared with WT. In this regard, the pro-inflammatory effect of the HMGB1 mutant on RAW 264.7 was also significantly reduced (P < 0.001, Fig. S27, ESI†), indicating the key role of K114 in the HMGB1–TLR4 interaction and the downstream inflammatory response. On this basis, the inhibition of PT-22 on inflammatory responses induced by the HMGB1 mutant was also assessed. The results showed that PT-22 did not inhibit HMGB1 mutant-induced TNF-α elevation in two macrophages (P > 0.05).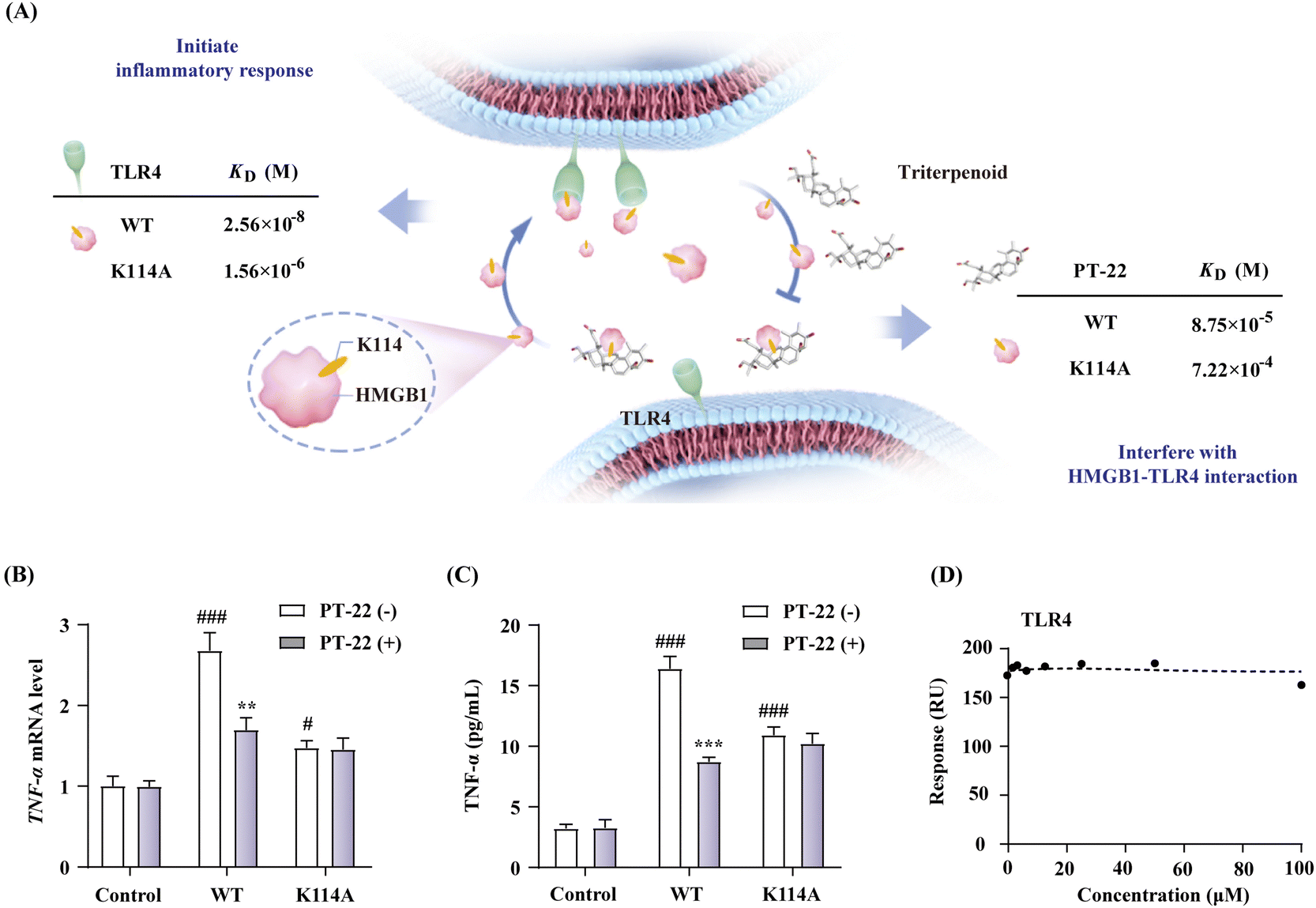 | ||
| Fig. 7 (A) Binding affinity of HMGB1 with receptor TLR4 and ligand PT-22 before and after mutation. (B) Effect of the HMGB1 mutant (1 μg mL−1) on TNF-α mRNA expression in THP-1 cells with or without PT-22 (1 μM). (C) The effect of mutant on the TNF-α release of 1 μg mL−1 HMGB1 and inhibition of PT-22 (1 μM) on RAW 264.7. (D) Competitive inhibition of PT-22 on the interaction of the HMGB1 mutant and TLR4. Data shown are mean ± SD from three independent experiments. #P < 0.05, ###P < 0.001 compared to the control group. **P < 0.01, ***P < 0.001 compared to the model group, as calculated by Student's t-test. Gene expression was normalized to GAPDH. | ||
TLR4, an integral part of innate and adaptive immune systems, can rapidly respond to the invasion of extracellular LPS and intracellular HMGB1. In this work, we aimed to understand the druggable interface of the HMGB1–TLR4 interaction to attenuate HMGB1-mediated sterile inflammation (Fig. 8B). First, two different cell models were successfully established in LPS- and HMGB1-stimulated RAW 264.7 cells. The triterpenoid that selectively interferes with the HMGB1–TLR4 dependent signal other than LPS-TLR4 axis was found. Binding of the triterpenoid to HMGB1 induces a conformational change in HMGB1, which hinders its interaction with TLR4 and further prevents inflammation initiation. With triterpenoid PT-22 as the probe, the hot-spot residues involved in protein surface recognition of HMGB1–TLR4 were mapped, including a single anchoring residue (K114) in the helical peptide of HMGB1. Mutation at the essential residue significantly reduced the HMGB1–TLR4 interaction and related cytokine release, suggesting that their recognition through this specific interface is indispensable for inflammatory diseases. Our study enhances the comprehension of the recognition interface of HMGB1–TLR4 in the immune system, which is crucial for regulating signal transduction networks and rational design of new therapeutic agents.
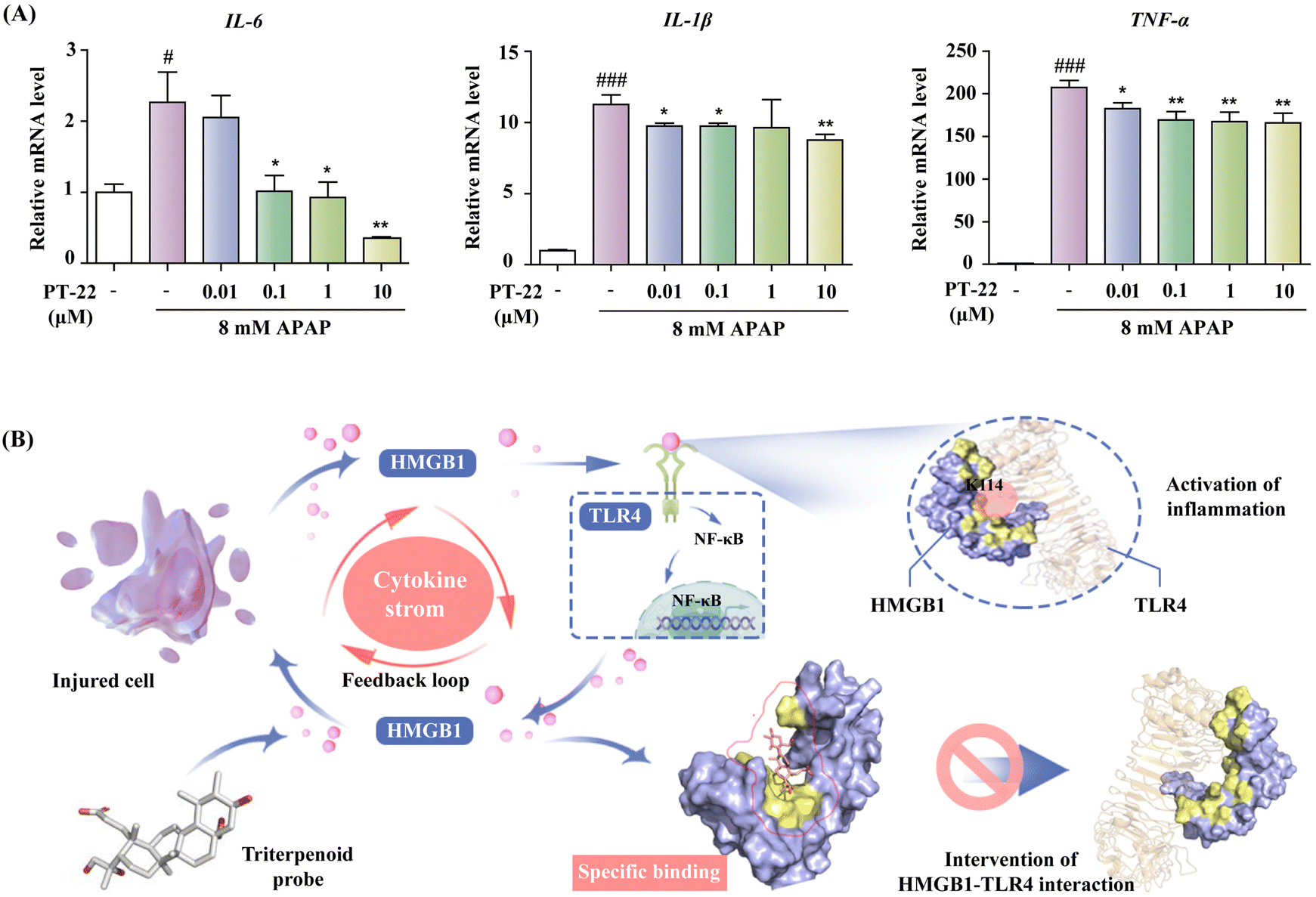 | ||
| Fig. 8 (A) Inhibition of PT-22 on the IL-6, IL-1β and TNF-α mRNA levels of THP-1 cells stimulated by the APAP-damaged hepatocyte supernatant. (B) Schematic model of the process in which PT-22 modulates the HMGB1–TLR4 signaling. PT-22 inhibited the HMGB1–TLR4 PPI through binding to a previously unknown interface on HMGB1 involving K114, a binding determinant of the HMGB1–TLR4 complex, inhibiting the pro-inflammatory cascade to implement the treatment of endogenous injury and exogenous infection. Data shown are mean ± SD from three independent experiments. #P < 0.05, ###P < 0.001 compared to the control group. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the model group, as calculated by Student's t-test. Gene expression was normalized to GAPDH. | ||
Mutation at K114 of HMGB1 interrupted the HMGB1–TLR4 complex and alleviated inflammatory responses, suggesting that the interaction between HMGB1 and TLR4 through this specific interface is indispensable for the immune regulation process. Furthermore, we evaluated the ability of PT-22 to interfere with the interaction of the HMGB1 mutant with TLR4 at the molecular level (Fig. S28, ESI†). It was surprisingly found that the substitution of K114 with Ala completely abolished the intervention of PT-22 to HMGB1–TLR4 PPI (Fig. 7D). The results further confirmed the potential of K114 as the critical interfacial residue of the triterpenoid probe to compete with TLR4 on the druggable surface of HMGB1. Altogether, the essentiality of K114 to maintain the interaction of HMGB1 and TLR4 and its participation in cytokine secretion provides a significant therapeutic opportunity for using HMGB1–TLR4 PPI inhibitors in acute and chronic inflammatory diseases.
Encouraged by above results, we next established a highly HMGB1-dependent cell model of acute liver failure induced by acetaminophen (APAP) to assess the potency of PT-22 in neutralizing released HMGB1.37 The hepatocyte supernatant was collected and added to THP-1 cells (Fig. S29, ESI†), which was activated by releasing the pro-inflammatory cytokines in TLR4 downstream. To confirm that the cytokine secretion of THP-1 was mediated by HMGB1 released from necrotic hepatocytes, we incubated the culture medium exposed to APAP with the A-box, an HMGB1-specific antagonist. The results showed that the production of TNF-α and IL-1β was greatly reduced in the presence of the A-box (P < 0.05) (Fig. S30, ESI†). Besides, PT-22 was found to dose-dependently inhibit the transcription and release of pro-inflammatory mediators (Fig. 8A), which are attributed to the neutralization of HMGB1 in the APAP-induced liver injury. Given that the modulator of HMGB1–TLR4 PPI was effective in treating HMGB1-related diseases, it is reasonable to assume that the PPI interface identified using the triterpene probe is valuable for drug design.
3. Discussion
Investigating HMGB1–TLR4 interactions holds great potential for therapeutic applications, since they mediate intricate cell signaling networks in physiological and pathological states. However, the design and development of drugs targeting HMGB1–TLR4 is hindered by the lack of a functionally relevant protein–protein binding interface. In this study, we found that triterpenoids derived from microbial transformation selectively interfered with NF-κB activation and cytokine production of macrophages induced by HMGB1 instead of LPS, despite both of them being activators of TLR4. Using the specific triterpenoid as a chemical probe, we observed the hot spot K114 in an α-helical peptide protruding from the HMGB1 surface preferentially bound to TLR4. On this basis, we coincidently found that TLR4 shared at least part of its characteristic binding region with the neutralizing antibody of HMGB1, where the interface is amenable to inhibition by triterpenoids. On the whole, our work provides a paradigm for the fast and efficient characterization of the druggable interface of HMGB1–TLR4, which will deepen our understanding of the interaction of endogenous DAMP–PRR axis in the innate immune system.Author contributions
Pingping Shen, Jian Zhang and Xuewa Jiang designed the experiments. Pingping Shen, Yi Kuang, Weiwei Wang and Richa Raj carried out the experiments. Xuewa Jiang and Wei Wang analyzed and interpreted the data. Pingping Shen, Xiaochun Zhang, Boyang Yu, and Richa Raj wrote and revised the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Nature Science Foundation of China (NSFC no. 21302052) and the “Program for New Century Excellent Talents in University” awarded to Prof. Jian Zhang (NECT-11-0739). This study was also supported by the “Jiangsu Funding Program for Excellent Postdoctoral Talent” awarded to Pingping Shen (2022ZB316).References
- S. Celis, F. Hobor, T. James, G. J. Bartlett, A. A. Ibarra, D. K. Shoemark, Z. Hegedüs, K. Hetherington, D. N. Woolfson, R. B. Sessions, T. A. Edwards, D. M. Andrews, A. Nelson and A. J. Wilson, Chem. Sci., 2021, 12, 4753–4762 RSC.
- J. Zindel and P. Kubes, Annu. Rev. Pathol., 2020, 15, 493–518 CrossRef CAS PubMed.
- D. Tang, R. Kang, H. J. Zeh and M. T. Lotze, Nat. Rev. Immunol., 2023, 23, 824–841 CrossRef CAS PubMed.
- S. VanPatten and Y. Al-Abed, J. Med. Chem., 2018, 61, 5093–5107 CrossRef CAS PubMed.
- H. E. Harris, U. Andersson and D. S. Pisetsky, Nat. Rev. Rheumatol., 2012, 8, 195–202 CrossRef CAS PubMed.
- H. Yang, H. S. Hreggvidsdottir, K. Palmblad, H. Wang, M. Ochani, J. Li, B. Lu, S. Chavan, M. Rosas-Ballina, Y. Al-Abed, S. Akira, A. Bierhaus, H. Erlandsson-Harris, U. Andersson and K. J. Tracey, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11942–11947 CrossRef CAS PubMed.
- S. Matsumoto, S. Ishida, M. Araki, T. Kato, K. Terayama and Y. Okuno, Nat. Mach. Intell., 2021, 3, 153–160 CrossRef.
- Y. Hu, K. Cheng, L. He, X. Zhang, B. Jiang, L. Jiang, C. Li, G. Wang, Y. Yang and M. Liu, Anal. Chem., 2021, 93, 1866–1879 CrossRef CAS PubMed.
- J. Li, R. Kokkola, S. Tabibzadeh, R. Yang, M. Ochani, X. Qiang, H. E. Harris, C. J. Czura, H. Wang, L. Ulloa, H. Wang, H. S. Warren, L. L. Moldawer, M. P. Fink, U. Andersson, K. J. Tracey and H. Yang, Mol. Med., 2003, 9, 37–45 CAS.
- A. E. Modell, S. L. Blosser and P. S. Arora, Trends Pharmacol. Sci., 2016, 37, 702–713 CrossRef CAS PubMed.
- L. Si, K. Meng, Z. Tian, J. Sun, H. Li, Z. Zhang, V. Soloveva, H. Li, G. Fu, Q. Xia, S. Xiao, L. Zhang and D. Zhou, Sci. Adv., 2018, 4, eaau8408 CrossRef CAS PubMed.
- Y. Han, Z. Tong, C. Wang, X. Li and G. Liang, Eur. J. Pharmacol., 2021, 893, 173811 CrossRef CAS PubMed.
- L. Mollica, F. De Marchis, A. Spitaleri, C. Dallacosta, D. Pennacchini, M. Zamai, A. Agresti, L. Trisciuoglio, G. Musco and M. E. Bianchi, Chem. Biol., 2007, 14, 431–441 CrossRef CAS PubMed.
- D.-D. Liu, P. Luo, L. Gu, Q. Zhang, P. Gao, Y. Zhu, X. Chen, Q. Guo, J. Zhang, N. Ma and J. Wang, J. Neuroinflammation, 2021, 18, 174 CrossRef CAS PubMed.
- Y. Li, J. Wang, L. Li, W. Song, M. Li, X. Hua, Y. Wang, J. Yuan and Z. Xue, Nat. Prod. Rep., 2023, 40, 1303–1353 RSC.
- W. Wang, W. Wang, H. Ge, G. Li, P. Shen, S. Xu, B. Yu and J. Zhang, Bioorg. Chem., 2020, 99, 103826 CrossRef CAS PubMed.
- W. Wang, W. Wang, H. Ge, G. Li, P. Shen, S. Xu, B. Yu and J. Zhang, Bioorg. Chem., 2020, 99, 103826 CrossRef CAS PubMed.
- Y. Zhu, P. Shen, J. Wang, X. Jiang, W. Wang, R. Raj, H. Ge, W. Wang, B. Yu and J. Zhang, Bioorg. Med. Chem., 2021, 52, 116494 CrossRef CAS PubMed.
- J. Sgrignani, V. Cecchinato, E. M. A. Fassi, G. D'Agostino, M. Garofalo, G. Danelon, M. Pedotti, L. Simonelli, L. Varani, G. Grazioso, M. Uguccioni and A. Cavalli, J. Med. Chem., 2021, 64, 13439–13450 CrossRef CAS PubMed.
- P. Shen, J. Zhou, X. Jiang, H. Ge, W. Wang, B. Yu and J. Zhang, ACS Omega, 2022, 7, 18745–18751 CrossRef CAS PubMed.
- P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC.
- P. Shen, J. Zhang, Y. Zhu, W. Wang, B. Yu and W. Wang, Bioorg. Med. Chem., 2020, 28, 115465 CrossRef CAS PubMed.
- X. Zhou, P. Shen, W. Wang, J. Zhou, R. Raj, Z. Du, S. Xu, W. Wang, B. Yu and J. Zhang, J. Agric. Food Chem., 2021, 69, 6791–6798 CrossRef CAS PubMed.
- P. Shen, W. Wang, S. Xu, Z. Du, W. Wang, B. Yu and J. Zhang, J. Agric. Food Chem., 2020, 68, 5910–5916 CrossRef CAS PubMed.
- A. Shibata, K. Sugiura, Y. Furuta, Y. Mukumoto, O. Kaminuma and M. Akiyama, J. Autoimmun., 2017, 80, 28–38 CrossRef CAS PubMed.
- C. Engelmann, A. Habtesion, M. Hassan, A. J. C. Kerbert, L. Hammerich, S. Novelli, M. Fidaleo, A. Philips, N. Davies, S. Ferreira-Gonzalez, S. J. Forbes, T. Berg, F. Andreola and R. Jalan, J. Hepatol., 2022, 77, 1325–1338 CrossRef CAS PubMed.
- A. Poltorak, X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton and B. Beutler, Science, 1998, 282, 2085–2088 CrossRef CAS PubMed.
- W. L. Anggayasti, K. Ogino, E. Yamamoto, E. Helmerhorst, K. Yasuoka and R. L. Mancera, Comput. Struct. Biotechnol. J., 2020, 18, 1160–1172 CrossRef CAS PubMed.
- P. Shen, Y. Peng, X. Zhou, X. Jiang, R. Raj, H. Ge, W. Wang, B. Yu and J. Zhang, LWT, 2022, 169, 113983 CrossRef CAS.
- D. Martinez Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef PubMed.
- L. F. Krapp, L. A. Abriata, F. Cortés Rodriguez and M. Dal Peraro, Nat. Commun., 2023, 14, 2175 CrossRef CAS PubMed.
- S. Sun, M. He, S. VanPatten and Y. Al-Abed, J. Biomol. Struct. Dyn., 2019, 37, 3721–3730 CrossRef CAS PubMed.
- L. Wang, L. Zhang, L. Li, J. Jiang, Z. Zheng, J. Shang, C. Wang, W. Chen, Q. Bao, X. Xu, Z. Jiang, J. Zhang and Q. You, Sci. Adv., 2019, 5, eaax2277 CrossRef CAS PubMed.
- T. Seifert, M. Malo, J. Lengqvist, C. Sihlbom, E. M. Jarho and K. Luthman, J. Med. Chem., 2016, 59, 10794–10799 CrossRef CAS PubMed.
- L. M. C. Meireles, A. S. Dömling and C. J. Camacho, Nucleic Acids Res., 2010, 38, W407–W411 CrossRef CAS PubMed.
- Y. Geng, G. Munirathinam, S. Palani, J. E. Ross, B. Wang, A. Chen and G. Zheng, J. Immunol., 2020, 205, 407–413 CrossRef CAS PubMed.
- P. Lundbäck, J. D. Lea, A. Sowinska, L. Ottosson, C. M. Fürst, J. Steen, C. Aulin, J. I. Clarke, A. Kipar, L. Klevenvall, H. Yang, K. Palmblad, B. K. Park, K. J. Tracey, A. M. Blom, U. Andersson, D. J. Antoine and H. Erlandsson Harris, Hepatology, 2016, 64, 1699–1710 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00062e |
| This journal is © The Royal Society of Chemistry 2024 |