
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile bioluminescent probe with tunable color†
Zachary R.
Torrey
 a,
Lila P.
Halbers
b,
Lorenzo
Scipioni
c,
Giulia
Tedeschi
c,
Michelle A.
Digman
*c and
Jennifer A.
Prescher
*abd
a,
Lila P.
Halbers
b,
Lorenzo
Scipioni
c,
Giulia
Tedeschi
c,
Michelle A.
Digman
*c and
Jennifer A.
Prescher
*abd
aDepartment of Chemistry, University of California Irvine, Irvine, CA 92697, USA. E-mail: jpresche@uci.edu
bDepartment of Pharmaceutical Sciences, University of California Irvine, Irvine, CA 92697, USA
cDepartment of Biomedical Engineering, University of California Irvine, Irvine, CA 92697, USA. E-mail: mdigman@uci.edu
dDepartment of Molecular Biology & Biochemistry, University of California Irvine, Irvine, CA 92697, USA
First published on 20th September 2024
Abstract
Bioluminescence is a powerful method for imaging in vivo, but applications at the microscale are far from routine. This is due, in part, to a lack of versatile tools for visualizing dynamic events. To address this void, we developed a new platform—Bioluminescence Resonance Energy mAKe over with a Fluorescence-Activating absorption-Shifting Tag (BREAKFAST). BREAKFAST features a bright luciferase combined with a chemogenetic tag (pFAST) for rapid color switching. In the presence of luciferin and a discrete fluorogenic ligand, signal is observed via resonance energy transfer. We evaluated spectral outputs with various fluorogens and established the utility of BREAKFAST for combined fluorescence and bioluminescence imaging. Dynamic, four-color visualization was achieved with sequential ligand addition and spectral phasor analysis. We further showed selective signal quenching with a dark fluorogen. Collectively, this work establishes a new method for bioluminescence imaging at the cellular scale and sets the stage for continued probe development.
Introduction
Optical imaging probes enable real-time readouts of cellular and molecular processes. Among the most well-known examples are the small molecule fluorophores and fluorescent proteins.1–3 These probes emit light upon external illumination and are widely used for imaging biological targets. The now-expansive palette of fluorescent reporters can be readily multiplexed for intricate looks at signaling networks and other multicomponent behaviors.4,5 Some limitations in scope remain, though, due to the requirement for external light. Prolonged exposure can also result in photobleaching and reduced cell viability.6Many obstacles to long-term imaging can be circumvented with bioluminescence. This process involves the enzymatic oxidation of a small molecule luciferin by a luciferase enzyme.7–10 No excitation light is required, alleviating issues associated with phototoxicity, bleaching, and autofluorescence. For these reasons, bioluminescence is a go-to method for serial imaging at the macroscale—in tissues and whole organisms.10,11 Imaging at the microscale has long been desired, but historically challenging. Classic luciferases exhibit slow turnover rates, resulting in dim emission and “hazy” images.7,8
Some barriers to bioluminescence microscopy have been overcome with the advent of higher-turnover luciferases (e.g., NanoLuc).12 The improved photon outputs enable faster imaging speeds and resolution on par with conventional fluorescence methods. Additionally, an expanded palette of bioluminescent reporters has been developed.13 The probes primarily comprise fluorescent protein fusions (e.g., enhanced Nano-lanterns) that span the visible spectrum. The lanterns provide altered emission wavelengths via bioluminescence resonance energy transfer (BRET).12–14 Such BRET probes have been used to track sub-cellular features and biomolecules over various length and time scales.7–9,13–16 Additional multiplexing is possible thanks to advances in imaging workflows.10,17–20 Further gains in multi-component detection have come with improved hardware and associated algorithms for visualizing dim emitters.14,18
Despite these achievements, current bioluminescent reporters are far fewer in number than their fluorescent counterparts. Bioluminescent probes also suffer from broad, often unresolvable emission spectra, complicating the number that can be visualized at once. This same issue has plagued the seamless integration of bioluminescence and fluorescence for imaging across multiple length scales. Current reporters are also mostly limited to a single color.21 New spectral outputs typically require a different genetic construct for each desired wavelength, or extensive protein engineering. Some probe sets have overcome this re-engineering requirement. A signature example is H-Luc, a luciferase-HaloTag fusion capable of BRET.22 Different spectral outputs are achieved via covalent attachment of fluorophore ligands. H-Luc is largely limited to single-color readouts during a given imaging session, though, due to the covalent nature of the label.
We aimed to develop a potentially more versatile imaging strategy using chemogenetic tags. We envisioned fusing NanoLuc to a small protein capable of transient interactions with fluorogenic ligands. In this scenario, ligand binding would result in signal turn-on, with the wavelength dictated by the unique small molecule. A spectrum of colors could thus be obtained from one genetically encodable construct. The reversible nature of fluorogen binding would also mitigate issues with photobleaching of the acceptor.
We were drawn to a popular chemogenetic tag—fluorescence-activating and absorption-shifting tag (FAST). FAST enables dynamic color modulation via reversible fluorogen binding.23 The fluorogens comprise small molecule acceptors that mimic fluorescent protein chromophores, making them good energy acceptors. Since the molecules are “off” when unbound, lengthy washouts are not required and greater signal-to-noise ratios can be achieved. FAST acceptors that act as quenchers are also available and have enabled super resolution studies and other applications.1,24–30
Here we describe the new bioluminescent platform, Bioluminescence Resonance Energy mAKe over with a Fluorescence-Activating absorption-Shifting Tag (BREAKFAST). BREAKFAST features a NanoLuc donor fused to a promiscuous FAST variant (pFAST), which serves as the acceptor.30 The resulting probe combines bioluminescence with the color-shifting ability of a chemogenetic tag. Thus, from a single construct, different spectral outputs can be readily and rapidly achieved. We developed and examined various NanoLuc-pFAST fusions and measured their optical properties with multiple fluorogens. Each ligand provided a unique spectral output that could be distinguished using both fluorescence and bioluminescence microscopy in combination with spectral phasor analysis. Furthermore, we were able to perform dynamic cell imaging, changing emission color in real time upon the addition of new fluorogens. Bioluminescent signal was also “erased” by the addition of a dark fluorogen, providing a novel mechanism to turn probes on and off in cells. Overall, this work showcases the versatility of BREAKFAST for optical imaging in cells.
Results and discussion
To craft a user-friendly and versatile imaging platform, we were drawn to NanoLuc, an engineered marine luciferase, and a promiscuous FAST variant (pFAST). NanoLuc emits bright blue light upon incubation with furimazine (Fz, Fig. 1a). pFAST is a well-established chemogenetic tag with a repertoire of unique fluorogenic 4-hydroxybenzylidene rhodanine (HBR) ligands (Fig. 1b). Non-covalent binding of the analogs results in signal turn-on, spanning a spectrum of outputs. We hypothesized that the HBR derivatives would be suitable BRET acceptors for NanoLuc. The emission spectrum of BREAKFAST would thus vary depending on the ligand present (Fig. 1c). Importantly, many HBR analogs are also readily accessible via commercial sources or straightforward syntheses.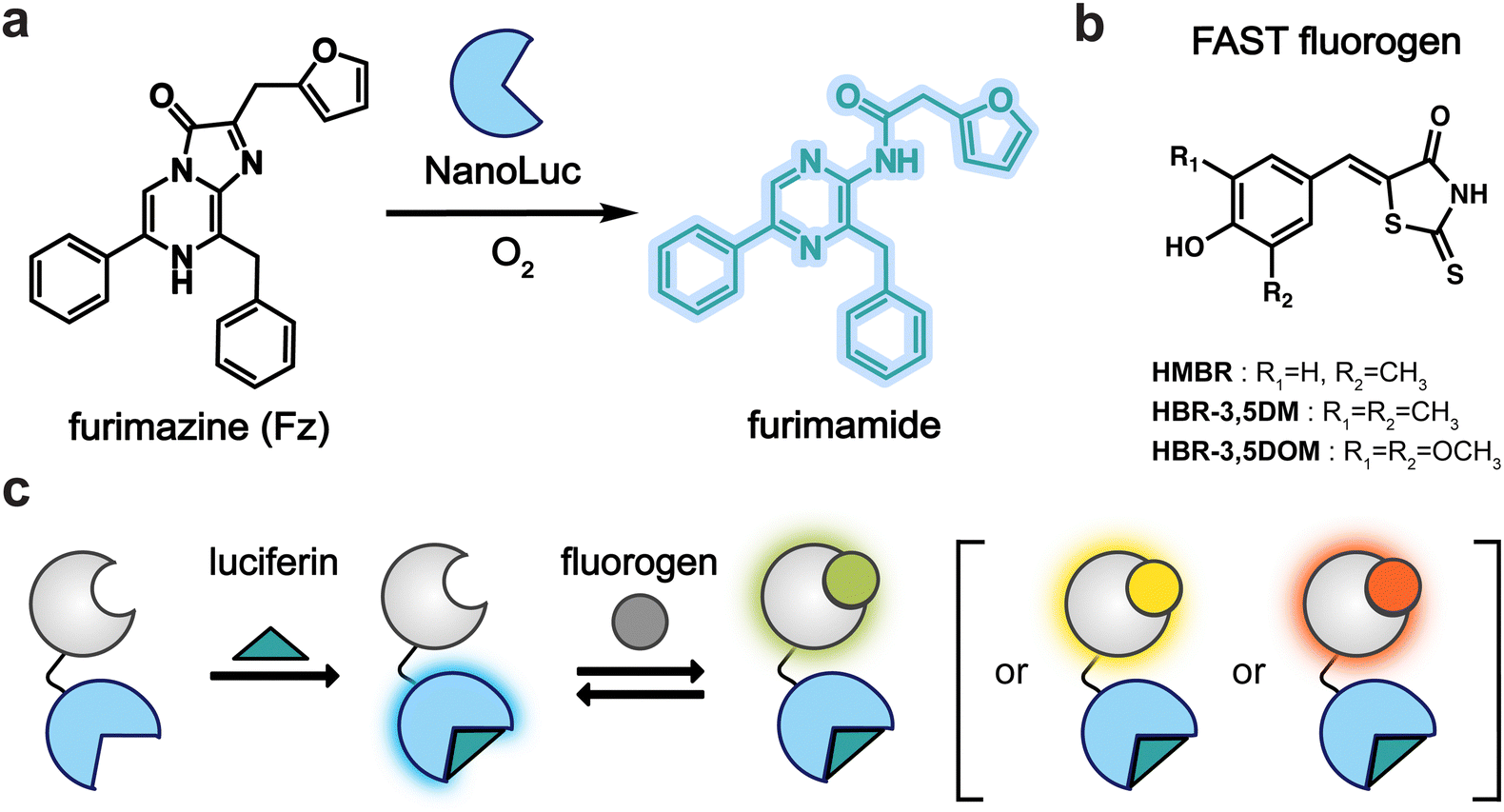 | ||
| Fig. 1 BREAKFAST components and overall design. (a) NanoLuc luciferase catalyzes the oxidation of furimazine (Fz) to produce furimamide and light. (b) Fluorogenic ligands that reversibly bind to pFAST. (c) Tuning BREAKFAST emission via fluorogen addition. | ||
We prepared an initial pFAST-NanoLuc fusion (BREAKFAST) and evaluated its optical properties with a small panel of fluorogens. While multiple HBR ligands are readily accessible, three were used in this study: HMBR, HBR-3,5DM, and HBR-3,5DOM (Fig. S1, ESI† and Fig. 2a). These molecules span a desirable range of colors and binding affinities for cellular application. When BREAKFAST was incubated with Fz alone, emission from NanoLuc was observed (max emission at ∼460 nm, Fig. 2b). When fluorogens were added, BRET was observed with new peaks present in the emission spectra (540–604 nm) (Fig. 2c–e). Fusion of NanoLuc to pFAST did not drastically alter the emission wavelength of the donor or acceptor on its own, suggesting that the two components can also be used independently of one another. BRET efficiency declined with decreasing ligand concentration, suggesting a dependence on fluorogen binding (Fig. S2, ESI†). The affinities of the fluorogenic ligands for BREAKFAST were also on par with those measured previously for pFAST itself (Fig. S3, ESI†). We capped the ligand concentrations at 50 μM, based on prior reports that higher doses of unbound probe could perturb cell function.30
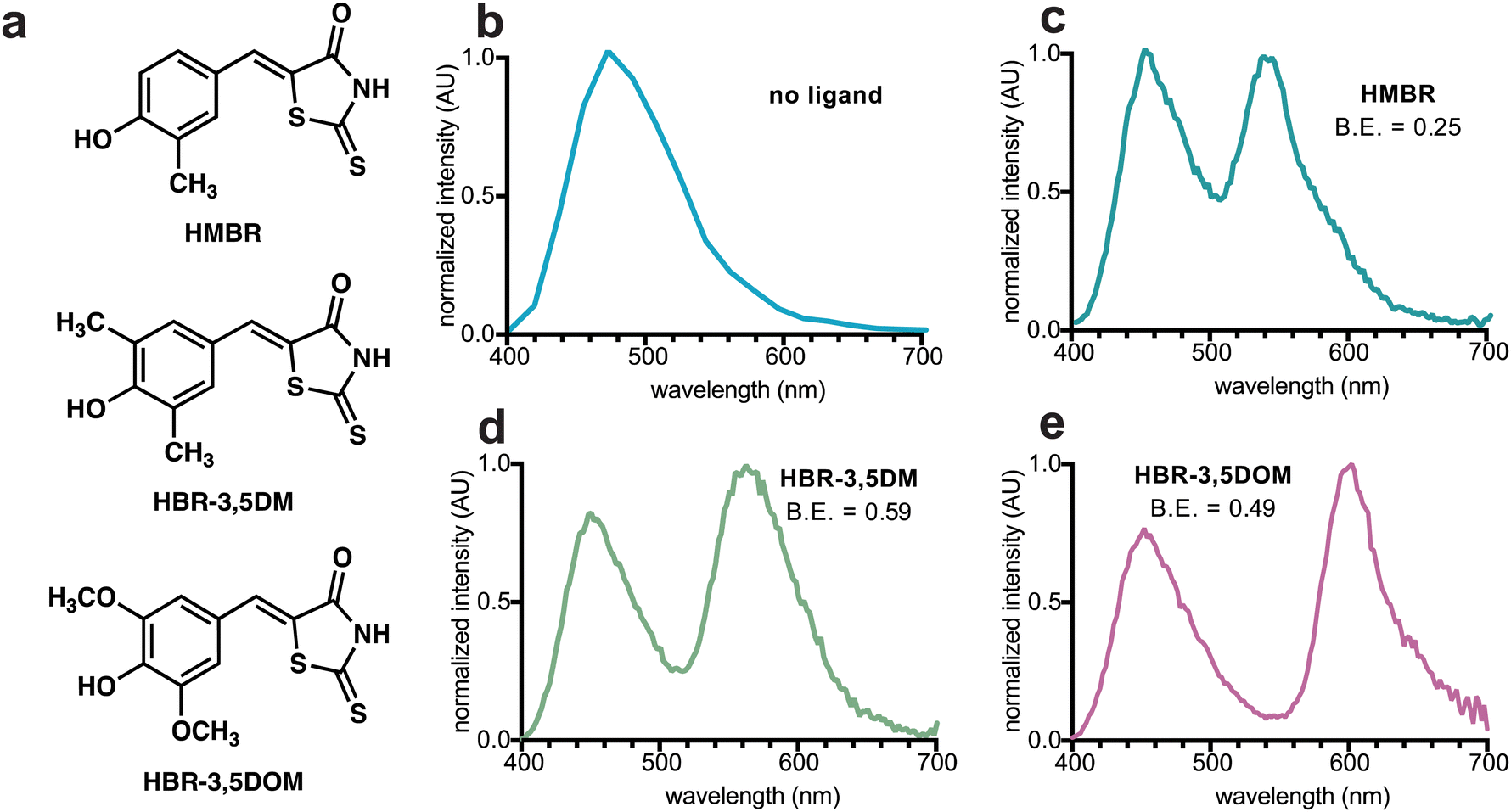 | ||
Fig. 2 Bioluminescence emission spectra with BREAKFAST probes. (a) Structures of candidate fluorogens. Spectra were acquired with BREAKFAST in the presence of (b) Fz only, (c) Fz and HMBR, (d) Fz and HBR-3,5DM or (e) Fz and HBR-3,5DOM. For all measurements, Fz and fluorogen were used at equimolar ratios (50 μM or 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 dilution of commercial stocks). B.E. = BRET efficiency. :50 dilution of commercial stocks). B.E. = BRET efficiency. | ||
We further examined the impact of the NanoLuc-pFAST linkage on the bioluminescent output. Nagai previously showed that trimming the N-terminus of NanoLuc could enhance energy transfer to some fluorescent protein acceptors.13 We thus sequentially removed five N-terminal amino acids from the NanoLuc portion of the pFAST fusion. The resulting constructs exhibited altered emission spectra, albeit with lower desired BRET efficiency (Fig. S4, ESI†). We further examined a fusion comprising a more flexible Gly-Ser linker. This construct also resulted in reduced BRET efficiency (Fig. S4, ESI†). Based on these results, we moved forward with the initial BREAKFAST design.
BREAKFAST is suitable for both bioluminescence and fluorescence detection. As noted earlier, though, multi-color imaging with luciferase probes is complicated by overlapping emission spectra. This issue is exacerbated with BREAKFAST, as sub-saturating ligand concentrations result in incomplete BRET and thus further complex emission patterns. We previously implemented a method – spectral phasor analysis – to rapidly unmix overlapping emitters using unique spectral shapes. Phasor analysis considers the entire spectral emission of a probe across the visible range (400–700 nm) to provide a unique spectral signature that can be represented in the so-called “phasor space”, aiding more facile probe assignment.
To apply the multiplexing technique, HEK293 cells were first transfected with BREAKFAST and then subjected, sequentially, to the pFAST ligands. Upon fluorescence excitation, all three ligands were readily observed and resolved using spectral phasor analysis (Fig. 3a). Each image represents the same field of view, with a media exchange in between. BREAKFAST-expressing cells were further examined using bioluminescence. The cells were incubated with Fz and one of the three fluorogens. Signal was then collected directly (with no excitation) upon ligand addition. As shown in Fig. 3b, the unique spectral outputs from the cells registered as unique phasor locations. It is worth noting that while HMBR and HBR-3,5DM have similar (and overlapping) emission profiles (Fig. 2), they can be readily discerned on the phasor plot. The amount of energy transferred varies depending on the fluorogen, with the phasor location appearing closer (higher BRET) or farther away (lower BRET) from the acceptor location identified in the fluorescence experiments. HBR-3,5DM exhibited the highest level of BRET efficiency, likely due to its favorable spectral overlap with NanoLuc and quantum yield.30
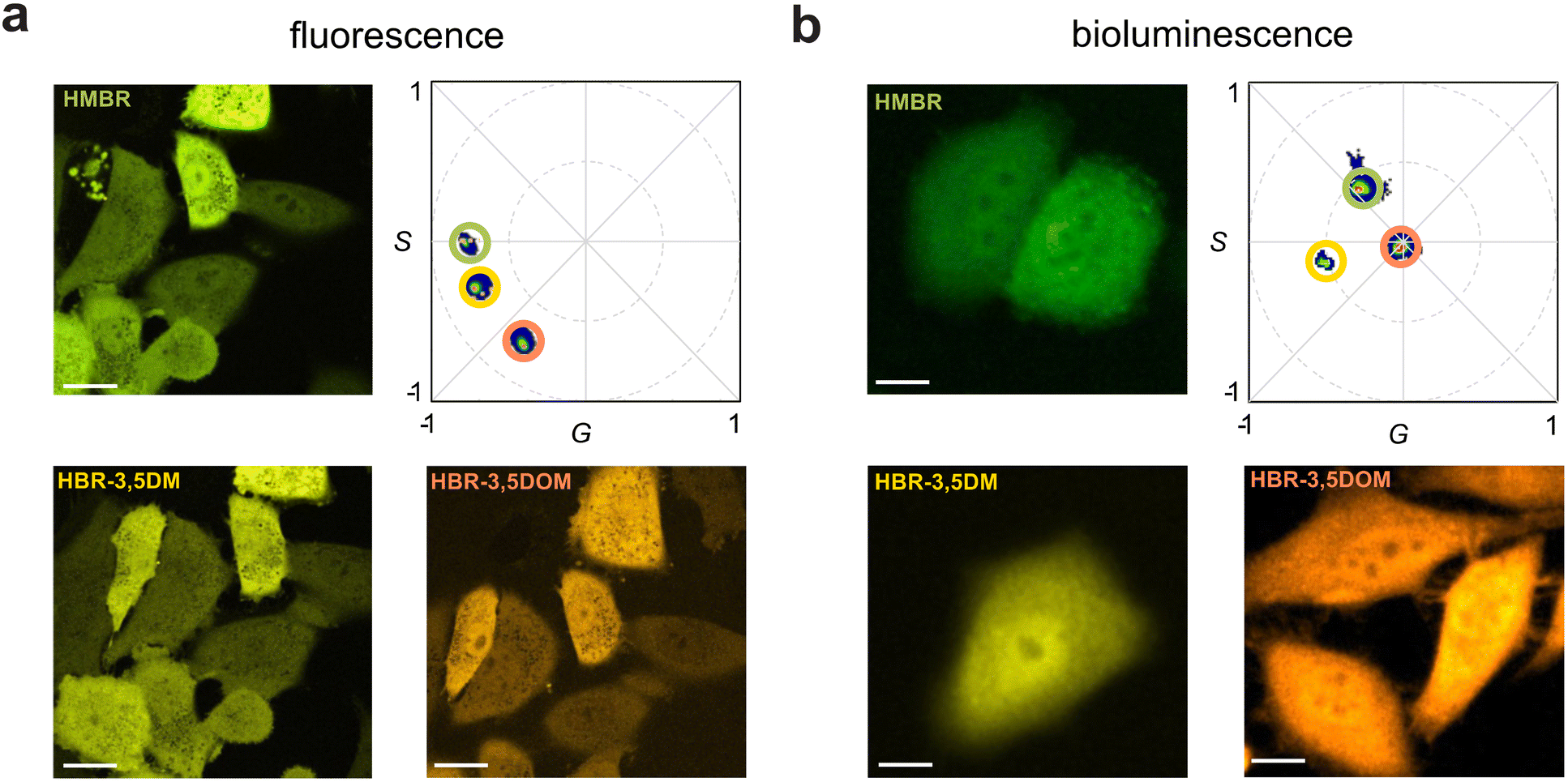 | ||
| Fig. 3 BREAKFAST can be resolved with both fluorescence and bioluminescence imaging using phasor analysis. HEK293 cells expressing BREAKFAST were incubated with fluorogens. (a) Fluorescence images of cells treated with HMBR, HBR-3,5DM, or HBR-3,5DOM and the associated phasor plots. (b) Bioluminescence images of cells treated with Fz and either HMBR, HBR-3,5DM, or HBR-3,5DOM. The associated phasor plots are also shown. For (a) and (b), scale bars = 10 μm. | ||
Our data suggested that BREAKFAST was well suited for dynamic color modulation, using the fluorogenic ligands to dial in desired wavelengths over time. As an initial demonstration, we seeded BREAKFAST-expressing A549 cells in a flow chamber. The cells were then imaged over a 14-min period while flowing in different fluorogenic ligands (at a rate of 100 μL per minute, Fig. 4). Distinct phasor signals were observed upon each ligand addition. The phasor locations were slightly shifted from those examined in isolation (Fig. 3). This was likely due to incomplete exchange under flow or potential mixing of ligands in the flow chamber tubing. Similar observations were made in replicate experiments and when the fluorogens were added in a different order (Fig. S5 and S6, ESI†). Collectively, these results demonstrate that BREAKFAST can enable dynamic color switching and rapid microscale imaging.
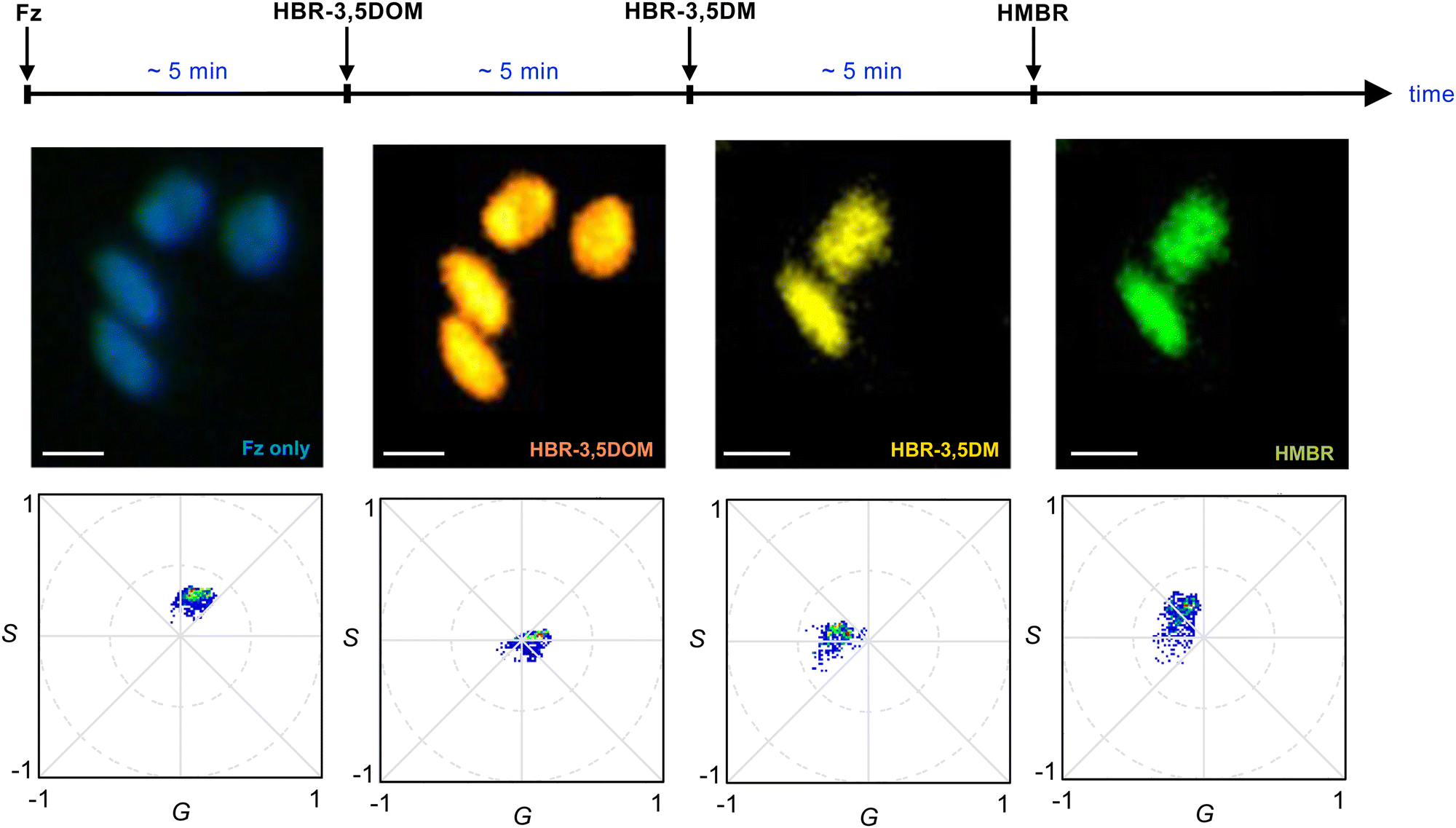 | ||
| Fig. 4 Color tuning with BREAKFAST probes. A549 cells were engineered to express NanoLuc-pFAST in the nucleus. Luciferin (Fz) and fluorogens were then flowed in as depicted, and images were acquired between each addition. Distinct spectral outputs were observed depending on the ligand present. Some cells detached in the imaging chamber under flow. For all images, scale bars = 10 μm. | ||
The ease of spectral tuning suggested that BREAKFAST could also enable bioluminescence quenching. Turning “off” distinct bioluminescent signals is challenging, as most methods inhibit the luciferase active site (common to all reporters). We hypothesized that more precise modulation could be achieved using BREAKFAST in combination with dark fluorogens. Such molecules absorb energy but dissipate it in the form of heat (instead of photons). Binding of dark FAST ligands would thus quench bioluminescence emission, and potentially be useful for detecting low-abundant reporters in the presence of other, brighter signals. To test this possibility, we used HBIR-3M as a quenching ligand. This molecule was previously validated as a dark acceptor in fluorescence lifetime assays.30HBIR-3M was also capable of quenching BREAKFAST signal in vitro (Fig. S7, ESI†).
We moved to examine whether bioluminescence signal could be “erased and replaced” in cells (Fig. 5a). HeLa cells were engineered to express both a nuclear-localized variant of BREAKFAST, along with an ER-localized yellow enhanced nano-Lantern (YeNL). We treated the cells with either Fz only or Fz plus the quencher. Clear signal reduction was observed for the BREAKFAST-localized probe (Fig. 5b), although the quenching was incomplete. We attribute this outcome to both suboptimal quencher accumulation in the nucleus, along with interference from out-of-focus luminescence collected in the pixels. It should also be noted that higher concentrations of quencher in the media result in non-specific photon absorption, lowering overall intensity.30 These results suggest that optimization of ligand concentrations and imaging workflows is required for a given experiment. Excitingly, though, quenched bioluminescent signal could be recovered upon the addition of a competent fluorogen. As shown in Fig. 5 and Fig. S7 (ESI†), the BREAKFAST color signature changed in response to HMBR. Spectral phasor analysis further confirmed the color assignments (Fig. S8, ESI†). The quench and rescue experiment was further reproduced across multiple biological replicates (Fig. S9, ESI†).
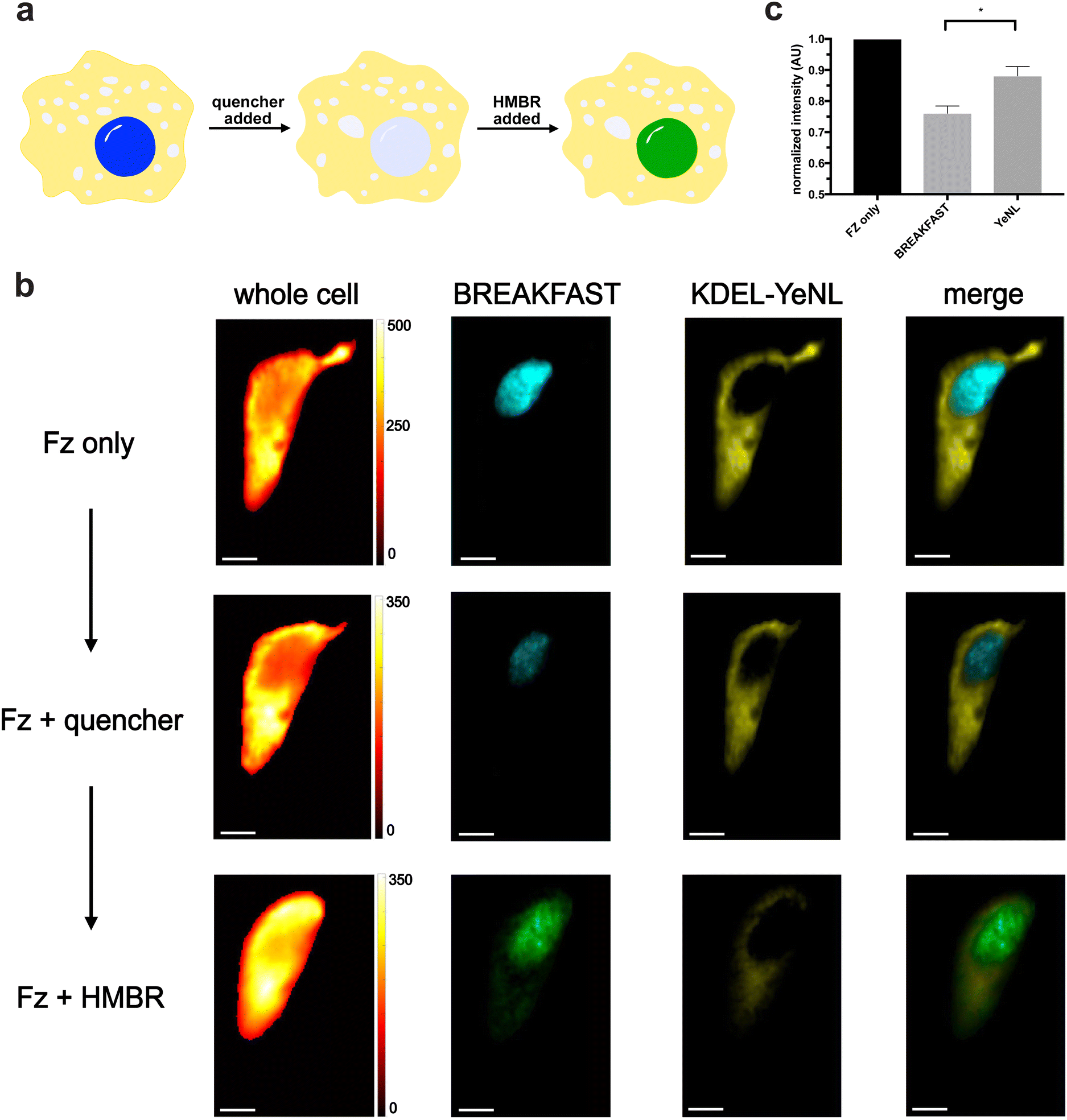 | ||
| Fig. 5 Reversible bioluminescence quenching. (a) Cartoon representation of the quench and rescue experiment. (b) HeLa cells were transiently co-transfected with ER-localized YeNL (KDEL-YeNL) and nuclear localized BREAKFAST reporters. Cells were treated with Fz only, followed by HBIR-3M (quencher), and finally HMBR (fluorogen). Vertical scale bars for the whole cell images represent pixel intensity (in a.u.). Images are representative of n = 3 independent experiments. Scale bars = 10 μm. (c) Quantification of bioluminescence signal quenching for BREAKFAST and YeNL in (b). | ||
Conclusions
In this work, we developed a customizable imaging tool—BREAKFAST—that is suitable for bioluminescence and fluorescence microscopy. BREAKFAST comprises an established and modular chemogenetic tag (pFAST) fused to an ultrabright luciferase (NanoLuc). The reporter was evaluated in the presence of luciferin and a panel of fluorogens. Different ligand combinations produced distinct spectral outputs that were readily distinguished via phasor analysis. Thus, from a single genetic reporter, multiple colors are possible. Dynamic color tuning was achieved by flowing in small molecule fluorogens over time. Additionally, it was possible to control illumination levels with non-emissive quenchers. BREAKFAST can thus serve as an on-demand bioluminescence “eraser”. The customizability of the platform will enable rapid access to different spectral outputs without the need for additional protein engineering.Data availability
The data underlying this article are available in the article and in the ESI.† The luciferase-pFAST plasmid has also been deposited with Addgene.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was supported by an Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Family Foundation (to J. A. P. and M. A. D.). We would like to thank members of the Digman and Prescher laboratories for valuable discussions and guidance.Notes and references
- M. Venkatachalapathy, V. M. Belapurkar, M. Jose, A. Gautier and D. Nair, Nanoscale, 2019, 11, 3626–3632 RSC.
- J. B. Grimm and L. D. Lavis, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- E. A. Rodriguez, R. E. Campbell, J. Y. Lin, M. Z. Lin, A. Miyawaki, A. E. Palmer, X. Shu, J. Zhang and R. Y. Tsien, Trends Biochem. Sci., 2017, 42, 111–129 CrossRef CAS PubMed.
- Y. Qian, O. T. Celiker, Z. Wang, B. Guner-Ataman and E. S. Boyden, Cell, 2023, 186, 5656–5672.e21 CrossRef CAS PubMed.
- Y. Zhang, M. Rózsa, Y. Liang, D. Bushey, Z. Wei, J. Zheng, D. Reep, G. J. Broussard, A. Tsang, G. Tsegaye, S. Narayan, C. J. Obara, J. X. Lim, R. Patel, R. Zhang, M. B. Ahrens, G. C. Turner, S. S.-H. Wang, W. L. Korff, E. R. Schreiter, K. Svoboda, J. P. Hasseman, I. Kolb and L. L. Looger, Nature, 2023, 615, 884–891 CrossRef CAS PubMed.
- H. Schneckenburger, P. Weber, M. Wagner, S. Schickinger, V. Richter, T. Bruns, W. S. L. Strauss and R. Wittig, J. Microsc., 2012, 245, 311–318 CrossRef CAS PubMed.
- A. C. Love and J. A. Prescher, Cell Chem. Biol., 2020, 27, 904–920 CrossRef CAS PubMed.
- H.-W. Yeh and H.-W. Ai, Annu. Rev. Anal. Chem., 2019, 12, 129–150 CrossRef CAS PubMed.
- C. M. Rathbun and J. A. Prescher, Biochemistry, 2017, 56, 5178–5184 CrossRef CAS PubMed.
- S. J. Williams and J. A. Prescher, Acc. Chem. Res., 2019, 52, 3039–3050 CrossRef CAS PubMed.
- Z. Yao, B. S. Zhang and J. A. Prescher, Curr. Opin. Chem. Biol., 2018, 45, 148–156 CrossRef CAS PubMed.
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. Machleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, ACS Chem. Biol., 2012, 7, 1848–1857 CrossRef CAS PubMed.
- K. Suzuki, T. Kimura, H. Shinoda, G. Bai, M. J. Daniels, Y. Arai, M. Nakano and T. Nagai, Nat. Commun., 2016, 7, 13718 CrossRef CAS PubMed.
- Z. Yao, C. K. Brennan, L. Scipioni, H. Chen, K. K. Ng, G. Tedeschi, K. Parag-Sharma, A. L. Amelio, E. Gratton, M. A. Digman and J. A. Prescher, Nat. Methods, 2022, 19, 893–898 CrossRef CAS PubMed.
- S. Inagaki, H. Tsutsui, K. Suzuki, M. Agetsuma, Y. Arai, Y. Jinno, G. Bai, M. J. Daniels, Y. Okamura, T. Matsuda and T. Nagai, Sci. Rep., 2017, 7, 42398 CrossRef CAS PubMed.
- J. Yang, D. Cumberbatch, S. Centanni, S. Shi, D. Winder, D. Webb and C. H. Johnson, Nat. Commun., 2016, 7, 13268 CrossRef PubMed.
- Y. Su, J. R. Walker, Y. Park, T. P. Smith, L. X. Liu, M. P. Hall, L. Labanieh, R. Hurst, D. C. Wang, L. P. Encell, N. Kim, F. Zhang, M. A. Kay, K. M. Casey, R. G. Majzner, J. R. Cochran, C. L. Mackall, T. A. Kirkland and M. Z. Lin, Nat. Methods, 2020, 17, 852–860 CrossRef CAS PubMed.
- C. M. Rathbun, A. A. Ionkina, Z. Yao, K. A. Jones, W. B. Porterfield and J. A. Prescher, ACS Chem. Biol., 2021, 16, 682–690 CrossRef CAS PubMed.
- L. Malacrida, S. Ranjit, D. M. Jameson and E. Gratton, Annu. Rev. Biophys., 2021, 50, 575–593 CrossRef CAS PubMed.
- F. Fereidouni, A. N. Bader and H. C. Gerritsen, Opt. Express, 2012, 20, 12729–12741 CrossRef CAS PubMed.
- E. C. Greenwald, S. Mehta and J. Zhang, Chem. Rev., 2018, 118, 11707–11794 CrossRef CAS PubMed.
- J. Hiblot, Q. Yu, M. D. B. Sabbadini, L. Reymond, L. Xue, A. Schena, O. Sallin, N. Hill, R. Griss and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 14556–14560 CrossRef CAS PubMed.
- M.-A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Quérard, B. Pan, J. Rossignol, K. Moncoq, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. Le Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497–502 CrossRef CAS PubMed.
- C. Li, M.-A. Plamont, H. L. Sladitschek, V. Rodrigues, I. Aujard, P. Neveu, T. Le Saux, L. Jullien and A. Gautier, Chem. Sci., 2017, 8, 5598–5605 RSC.
- C. Li, A. G. Tebo, M. Thauvin, M.-A. Plamont, M. Volovitch, X. Morin, S. Vriz and A. Gautier, Angew. Chem., Int. Ed., 2020, 59, 17917–17923 CrossRef CAS PubMed.
- C. Li, A. Mourton, M.-A. Plamont, V. Rodrigues, I. Aujard, M. Volovitch, T. Le Saux, F. Perez, S. Vriz, L. Jullien, A. Joliot and A. Gautier, Bioconjugate Chem., 2018, 29, 1823–1828 CrossRef CAS PubMed.
- A. Gautier, Acc. Chem. Res., 2022, 55, 3125–3135 CrossRef CAS PubMed.
- A. G. Tebo, B. Moeyaert, M. Thauvin, I. Carlon-Andres, D. Böken, M. Volovitch, S. Padilla-Parra, P. Dedecker, S. Vriz and A. Gautier, Nat. Chem. Biol., 2021, 17, 30–38 CrossRef CAS PubMed.
- A. Gautier, L. Jullien, C. Li, M.-A. Plamont, A. G. Tebo, M. Thauvin, M. Volovitch and S. Vriz, In Multiplexed Imaging: Methods and Protocols, ed. E. Zamir, Springer US, New York, NY, 2021, pp. 253–265 Search PubMed.
- H. Benaissa, K. Ounoughi, I. Aujard, E. Fischer, R. Goïame, J. Nguyen, A. G. Tebo, C. Li, T. Le Saux, G. Bertolin, M. Tramier, L. Danglot, N. Pietrancosta, X. Morin, L. Jullien and A. Gautier, Nat. Commun., 2021, 12, 6989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00101j |
| This journal is © The Royal Society of Chemistry 2024 |