
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to capped RNAs by chemical ligation†
Karolina
Bartosik
and
Ronald
Micura
 *
*
Institute of Organic Chemistry, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: ronald.micura@uibk.ac.at
First published on 13th September 2024
Abstract
A distinctive feature of eukaryotic mRNAs is the presence of a cap structure at the 5′ end. The typical cap consists of 7-methylguanosine linked to the first transcribed nucleotide through a 5′,5′-triphosphate bridge. It plays a key role in many processes in eukaryotic cells, including splicing, intracellular transport, initiation of translation and turnover. Synthetic capped oligonucleotides have served as useful tools for elucidating these physiological processes. In addition, cap mimics with artificial modifications are of interest for the design of mRNA-based therapeutics and vaccines. While the short cap mimics can be obtained by chemical synthesis, the preparation of capped analogs of mRNA length is still challenging and requires templated enzymatic ligation of synthetic RNA fragments. To increase the availability of capped mRNA analogs, we present here a practical and non-templated approach based on the use of click ligation resulting in RNAs bearing a single triazole linkage within the oligo-phosphate backbone. Capped RNA fragments with up to 81 nucleotides in length have thus been obtained in nanomolar yields and are in demand for biochemical, spectroscopic or structural studies.
Introduction
Messenger RNAs (mRNAs) of eukaryotes contain a characteristic 7-methylguanosine (m7G)-5′-ppp moiety at their 5′-end, the so-called “cap”.1,2 In addition, the terminus of mRNA is often heavily methylated at the first four nucleosides, such as in trypanosomatids.3,4 Other aspects of modification include artificially altered mRNA caps and cap mimics, such as those tailored for mRNA vaccines.5 All of these modifications can critically affect translation efficiency, nuclear stability, and binding affinity to the many enzymes that interact with the cap in the cell.1 While the synthesis of modified di- and trinucleotide caps is a domain of synthetic organic chemistry,6–9 the length of mRNAs is typically well beyond the scope of chemical synthesis. Here, we demonstrate that current size limitations of synthetic capped 5′-mRNA can be overcome by chemical ligation (applying click chemistry)10 to generate RNA with single units of triazole backbone linkages (Fig. 1). This approach provides an alternative to synthetic mRNA obtained by enzymatic ligation of chemically synthesized RNA fragments, using T4 DNA ligases and splint oligos.11 Capped RNA fragments from 6 to 81 nucleotides in length, accessible by this approach in nanomolar yields, are in demand for biochemical, spectroscopic or structural studies.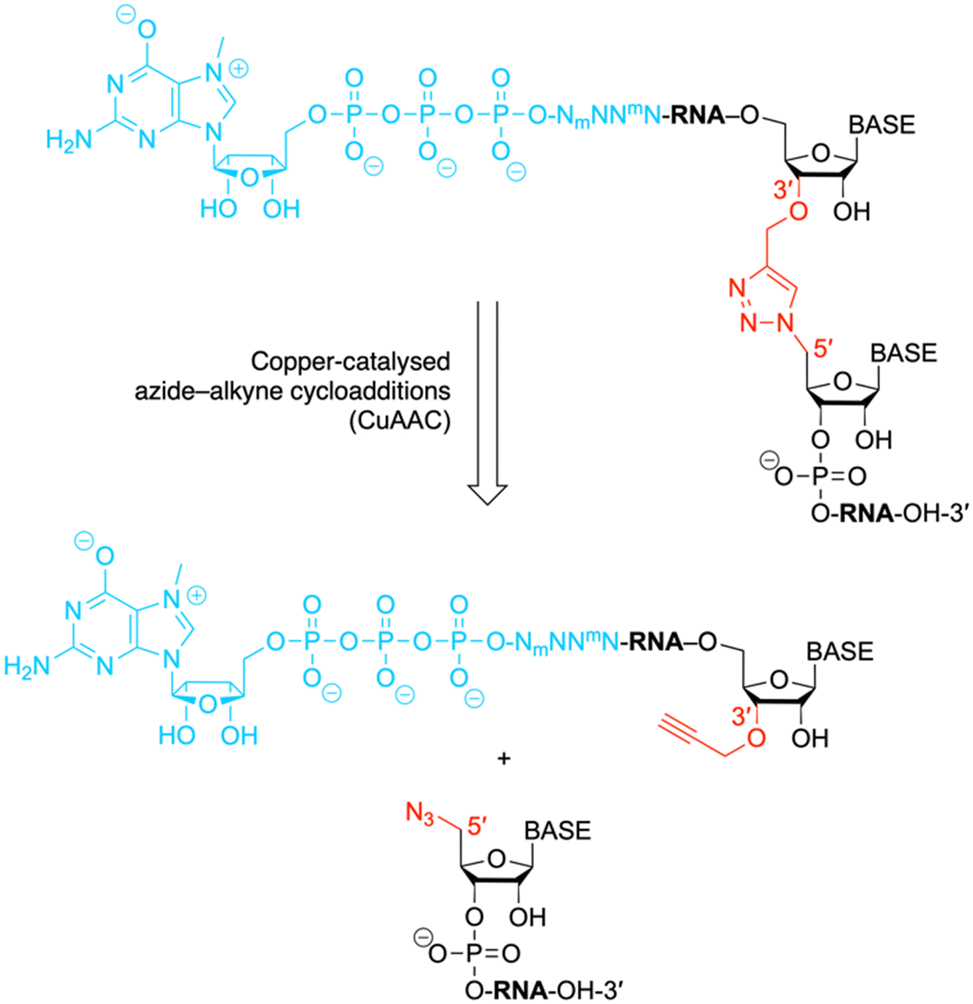 | ||
| Fig. 1 The chemical structure shows an exemplary mRNA with a biocompatible triazole linkage (TrzB)10 as phosphate backbone replacement. The present work describes a chemical ligation approach to generate such target compounds. N, nucleotide; Nm and mN, modified nucleotides. | ||
Moreover, the approach presented here is an important step towards efficient all-chemical synthesis of mRNAs without the use of in vitro transcription (IVT) for RNA synthesis, which usually results in a significant fraction of uncapped transcripts and additionally in double-stranded RNA by-products that cause an undesired immunogenic response.2 mRNA generated by IVT requires intensive purification,2,12 and therefore alternatives—such as the one presented here—for all-chemical mRNA synthesis are of current interest.13,14 It is worth mentioning that first concepts combining chemical cap synthesis, chemical RNA synthesis and click bioconjugation to obtain short mRNA fragments were described by the Jemielity group and date back to 2017.15
Results and discussion
Established approaches to synthesize short RNAs with caps
The most common strategy to synthesize RNA caps in solution is based on the reaction between an activated derivative of 7-methylguanosine 5′-diphosphate (m7GDP) with a 5′-phosphorylated oligoribonucleotide in the presence of metal ions. Several phosphate activating groups (phenylthiol,16 methoxyphenylthio,17,18 5-chloro-8-quinolyl,19 and imidazolide,20–24 in combination with diverse divalent metal catalysts (MgCl2, MnCl2, CaCl2, CdCl2, CuCl2 and ZnCl2),20 have been described. The most practical and widely applied conditions employ the imidazolide group and zinc chloride under anhydrous conditions, providing capped oligoribonucleotides in good to sufficient yields. One drawback is, however, the limitation to rather short (up to 4 nt) RNAs.21,24 Recently, Abe and coworkers developed a chemical method allowing the synthesis of longer sequences bearing cap modification.14 In this method, the fully deprotected 5′-phosphorylated RNAs were reacted with Im-m7GDP using 1-methylimidazole as an activator and DMSO as a solvent. It enabled quantitative preparation of 107 nt capped RNAs within 3 h; a huge excess of capping reagent was required and the RNAs were obtained in picomol amounts only.The instability of m7G under acidic (depurination) and basic conditions (opening of the m7G imidazole ring) makes the chemical solid-phase synthesis of cap RNAs challenging.25 For this reason, some of the methods utilize weak acid-labile26 or disulfide linkers27 between the RNA chain and solid support instead of standard base-labile linker. These methods include the coupling of imidazolide derivative of m7G mono- or di-phosphate with the support-bound 5′-di- or 5′-mono-phosphorylated RNA, respectively. After release from the support, the yield of capped RNAs was rather low. To increase the efficiency of capped RNA synthesis, Debart and coworkers developed a method that combined solid-phase RNA synthesis and enzymatic N7 methylation of the guanine moiety.28 In more detail, the short RNAs (from 4 to 18 nt), prepared on solid support, were phosphorylated at the terminal 5′-OH, activated by imidazole, and then reacted with GDP. The Gppp-RNAs were purified on HPLC before being enzymatically methylated using human (guanine-N7)-methyltransferase. Recently, the same group reported an alternative approach exploiting m7GDP attachment to fully protected resin-bound RNAs.13 The previously employed PivOM-protected monomers28 were replaced by more labile PrOM-protected building blocks, allowing the capped RNAs to be deprotected under mild conditions that prevented m7G degradation. The authors synthesized capped oligoribonucleotides of different sequences and lengths (up to 28 nt).
Intended concept to overcome size limitations in the synthesis of capped RNA by chemical ligation
The ligation of short chemically synthesized RNA strands by copper-catalyzed azide–alkyne cycloadditions (CuAAC coupling) was previously shown to offer a valid and efficient strategy for the generation of large, chemically modified RNA constructs (e.g. ribozymes).10,29 In particular, the TrzB linkage (Fig. 1) – originally introduced by Brown and coworkers – turned out to be useful because it displays high biocompatibility.10,29 This linkage was recently also used for the generation of pools of single guide RNAs (sgRNAs) in the emerging field of CRISPR-Cas (clustered regularly interspaced palindromic repeats) gene editing.30Encouraged by these early studies, we intended to explore the TrzB linkage in the context of mRNA synthesis (Fig. 1). We exemplify our ligation endeavours for cap-4 spliced leader (SL) RNA of Trypanosomatid parasites.3,4,11 In short, a hexanucleotide m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) offering an alkyne group at the 3′ end, is generated and effectively ligated to a chemically synthesized RNA bearing an azido group at the 5′ end, following untemplated cooper-catalyzed azide–alkyne cycloaddition (CuAAC) chemistry (Fig. 1). The resultant cap4 SL RNAs of about 19 to 81 nts in length, containing an artificial triazole backbone at the site of ligation, provide valuable probes for ongoing and future biochemical and structural studies (cryo-EM and X-ray crystallography) of these RNAs in complex with enzymes recognizing mRNAs with the short hypermethylated cap4 modality.
Synthesis of 3′-alkyne m7G-5′ppp-5′-oligonucleotides
To prepare short m7G-ppp-oligoribonucleotides bearing alkyne functionality at the 3′ end, two alternative routes were devised (Fig. 2). The first one followed the procedure that we previously applied to obtain a 39 nt cap-4 SL RNA fragment of Trypanosoma cruzi.11 The novel aspect here was the application of a (commercially available) CPG solid support that provided the terminal 3′-O-propargyl nucleoside of interest. The four methylated nucleosides m26Am, Am, Cm, and m3Um were assembled into the sequence of 5′-DMT-O-(m26Am)(Am)(Cm)-(m3Um)(3′-O-propargyl-A) by standard phosphoramidite chemistry (Fig. 2 and 3 and Table 1)). After 5′-O detritylation, the 5′ hydroxyl group of the CPG-bound protected RNA (RNA 1) was converted into the H-phosphonate RNA derivative by treatment with diphenyl phosphite and subsequent hydrolysis with triethylammonium bicarbonate buffer (RNA 2) (Fig. 2, 3 and Table 1). Then, oxidation and activation resulted in the corresponding 5′-phosphoroimidazolide RNA, that was reacted with the tri-butylammonium salt of GDP in DMF in the presence of ZnCl2 (RNA 3) (Fig. 2, 3 and Table 1). All these steps were performed while the protected RNA was bound to the support, thus allowing the removal of excess reagents by simple washing which makes the synthesis convenient. The final release from the support and deprotection yielded the crude G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A), which was purified by anion exchange (AE) HPLC (RNA 3) (Fig. 2, 3 and Table 1 and Fig. S1, ESI†). Methylation of guanine at position N7 was performed enzymatically using Ecm1 methyltransferase in the presence of AdoMet as the methyl donor (Fig. 2 and 3). The conversion of G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 3) into m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 4) proceeded very clean and quantitative in less than 1 h of incubation at 37 °C (Fig. 2 and 3 and Table 1 and Fig. S1, ESI†).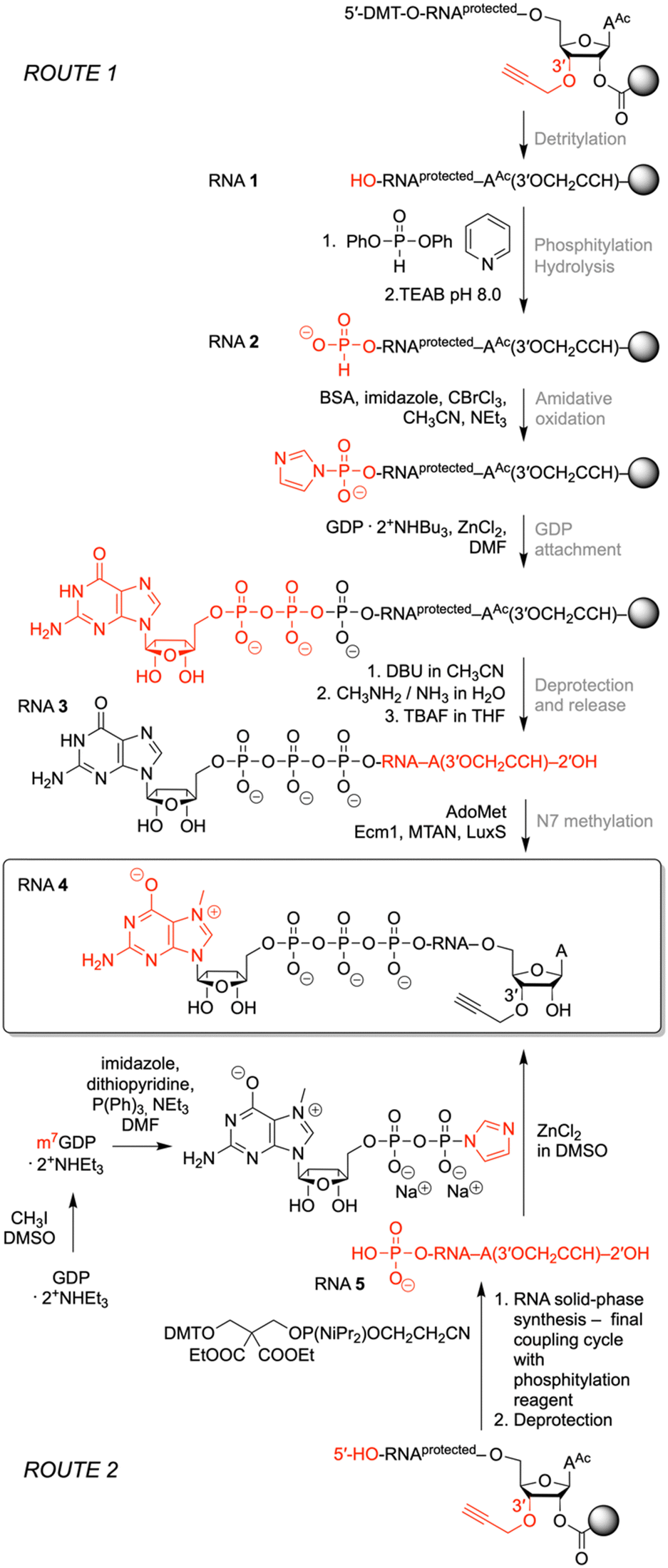 | ||
| Fig. 2 Two routes for the preparation of m7Gppp-RNA 3′-alkyne: solid-phase synthesis of Gppp-RNA 3′-alkyne followed by enzymatic N7 methylation (top), and alternatively, the synthesis of 5′-p-RNA 3′-alkyne followed by reaction with Im-m7GDP (bottom). Reagents and conditions: ROUTE 1: detritylation: 3% dichloroacetic acid in Cl2HCCHCl2; phosphitylation: 0.1 M diphenyl phosphite in py, rt, 10 min; hydrolysis: 0.1 M TEAB in H2O/ACN (6/4 v/v), rt, 20 min; amidative oxidation: imidazole, N,O-bis(trimethylsilyl)acetamide, CBrCl3, TEA, ACN, rt, 1 h; GDP attachment: 0.28 M GDP· 2Bu3NH+ and 0.5 M ZnCl2 in DMF, rt, 16 h; deprotection and release: (i) 1.0 M DBU/ACN, rt, 5 min, (ii) 40% aq MeNH2/30% aq NH3 (1/1 v/v), 40 °C, 4 h, (iii) 1 M TBAF/THF, 37 °C, 16 h; N7 methylation: phosphate buffer (1.5 M NaCl, 200 mM Na2HPO4, pH 7.4), AdoMet, Ecm1 methyltransferase, MTAN nucleosidase, LuxS lyase, 37 °C, 1 h. ROUTE 2: GDP methylation: CH3I/DMSO, rt, 3 h; activation: imidazole, 2,2′-dithiopyridine, Ph3P, TEA, DMF, rt, 24 h; cap installation: Im-m7GDP/DMSO, ZnCl2, rt, 24 h or 55 °C, 3 h. | ||
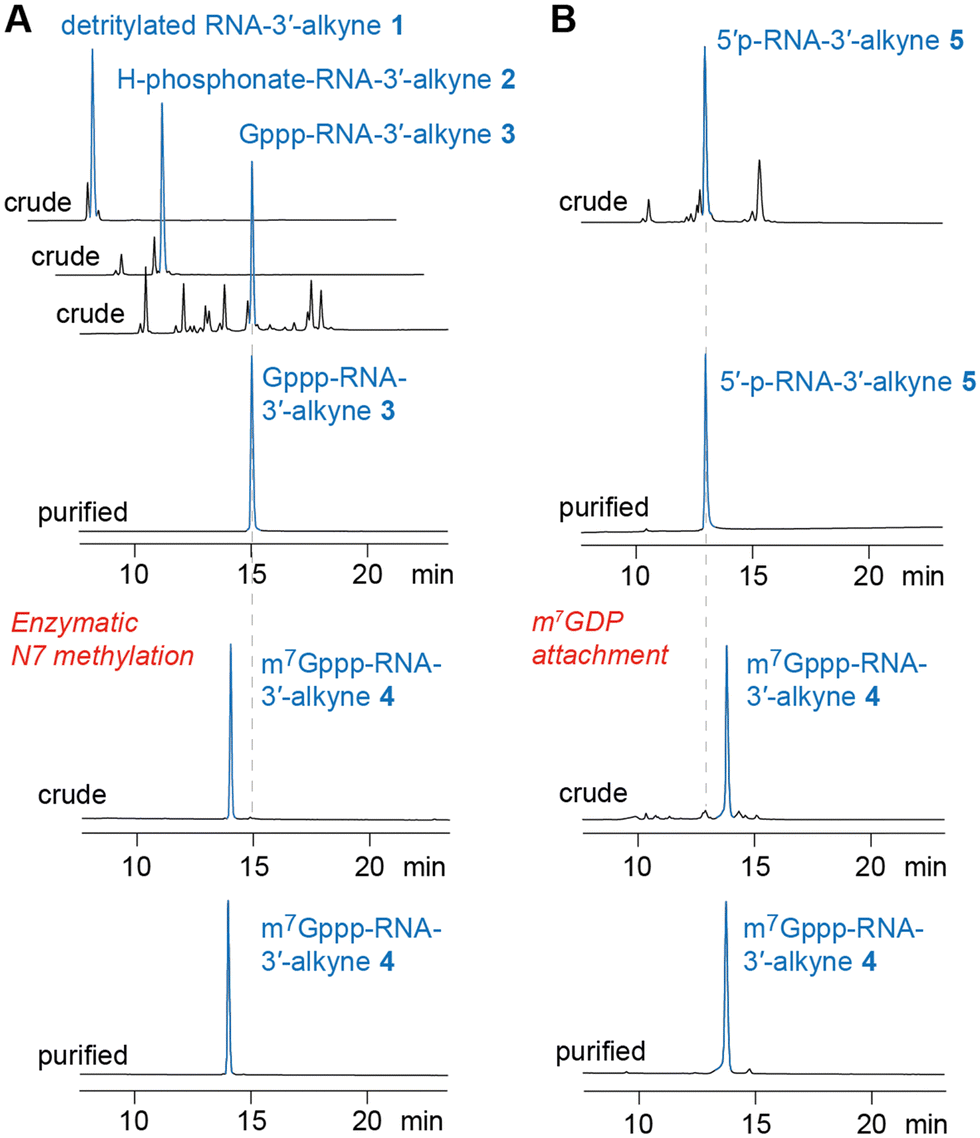 | ||
| Fig. 3 HPLC analysis of m7Gppp-RNA 3′-alkyne preparations (exemplified for m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A, RNA 4). (A) Path for chemoenzymatic synthesis. (B) Path for all-chemical synthesis (for details see Fig. 2 and Table 1 and the ESI†). | ||
| RNA No. | Sequence | nt | Scale | Isolated amounta nmol | m.w. (calc.) [amu] | m.w. (found) [amu] |
|---|---|---|---|---|---|---|
| a Isolated amount refers to the amount of product that is produced from a single batch at the annotated scale and obtained after AE HPLC purification. b For analytical purposes (HPLC), RNA from a small portion of the solid-support was deprotected and released to judge the quality of the individual reaction steps (see Fig. 3). c Hp relates to H-phosphonate group. | ||||||
| Short capped RNAs (Route 1) | ||||||
| 1 | 5′-HO-(m26Am)(Am)(Cm)(m3Um)A-3′-alkyneb | 5 | — | — | 1673.3 | 1672.7 |
| 2 | 5′-Hp-(m26Am)(Am)(Cm)(m3Um)A-3’-alkynebc | 5 | — | — | 1737.2 | 1736.6 |
| 3 | Gppp-(m26Am)(Am)(Cm)(m3Um)A-3′-alkyne | 6 | 1.0 μmol | 75 | 2178.4 | 2178.2 |
| 4 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-3′-alkyne | 6 | 20 nmol | 18 | 2192.4 | 2191.9 |
| Short capped RNAs (Route 2) | ||||||
| 5 | 5′-p-(m26Am)(Am)(Cm)(m3Um)A-3′-alkyne | 5 | 1.0 μmol | 140 | 1753.2 | 1752.6 |
| 4 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-3′-alkyne | 6 | 20 nmol | 12 | 2192.4 | 2191.7 |
| 5′-azide RNAs | ||||||
| 6 | 5′-N3-ACGCUAUUAUUGA-OH-3′ | 13 | 1.0 μmol | 114 | 4111.5 | 4111.3 |
| 7 | 5′-N3-ACGCUAUUAUUGAUACAGUUUCUGU-ACUAUAUUG-OH-3′ | 34 | 1.0 μmol | 80 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770.4 770.4 |
10770.4 |
| Triazole-linked (TrzB) RNAs | ||||||
| 8 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-TrzB-ACG-CUAUUAUUGA-OH-3′ | 19 | 5 nmol | 3.0 | 6303.9 | 6304.1 |
| 9 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-TrzB-ACG-CUAUUAUUGAUACAGUUUCUGUACUAUA-UUG-OH-3′ | 40 | 5 nmol | 2.5 | 12962.9 |
12963.1 |
| 12 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-TrzB-ACGCUAUUAUUAGAACAGUUUCUGUACUAUAUUGGUAUGAGAAGCUCCCAGUAGC-OH-3′ | 61 | 5 nmol | 1.5 | 19783.0 |
19782.5 |
| 13 | m7Gppp-(m26Am)(Am)(Cm)(m3Um)A-TrzB-ACGCUAUUAUUAGAACAGUUUCUGUACUAUAUUGGUAUGAGAAGCUCCCAGUAGCAGCUGGGCCAACACACGCAU-OH-3′ | 81 | 5 nmol | 1.0 | 26232.9 |
26232.2 |
For some laboratories, the use of enzymes might affect the attractiveness of the above approach. Therefore, we focused on an exclusively chemical access to short m7Gppp-RNA-3′-alkynes as alternative. We considered the reaction of 5′-phosphorylated RNA-3′-alkynes with the chemical capping agent 7-methylguanosine 5′-diphosphate imidazolide (Im-m7GDP)21,24 appropriate to generate the target m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 4) (Fig. 2 and 3 and Table 1). Indeed, the 5′-phosphorylated pentamer 5′-p-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 5) (Fig. 2 and 3 and Table 1) was readily obtained by RNA solid-phase synthesis on the same solid support as used above and a typical building block for 5′-O-phosphorylation (3-DMTrO-2,2-bis(ethoxy-carbonyl)propyl 2-cyanoethyl N,N-diisopropylphosphor-amidite).31,32 Subsequently, the RNA was deprotected under standard conditions (AMA, 40 °C, 4h; 1 M TBAF, THF) and 5′-p-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 5) isolated by AE HPLC.
The synthesis of the Im-m7GDP was performed as described earlier20,33 whereby the triethylammonium salt of GDP was methylated using iodomethane, and the resulting N7-methylated GDP was precipitated before having been purified by RP-HPLC (Fig. 2). Then, m7GDP was converted to the imidazolide of the diphosphate using imidazole in the presence of an activation system including 2,2′dithiopyridine and triphenylphosphine. Im-m7GDP was isolated as the sodium salt.
For the capping reaction, we were inspired by the approach published by Jemielity and coworkers for the synthesis of cap2 trinucleotide.23 We performed the reaction between 5′-p-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) (RNA 5) and Im-m7GDP at room temperature utilizing DMSO as a solvent and ZnCl2 as a catalyst. We increased the excess of reagents (to 50 equiv. of Im-m7GDP and to 25 equiv. of ZnCl2), which resulted in a shortening of the reaction time from 48 h to 24 h, obtaining the same yields (Fig. 2). Even more conveniently, the reaction time was further decreased (to 3 hours) at elevated temperature (55 °C). After AE HPLC purification, the integrity of m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)-(3′-O-propargyl-A) (RNA 4) was confirmed by ESI MS analysis (Table 1 and Fig. S1, ESI†).
Synthesis of 5′-azido-modified oligoribonucleotides
To prepare oligonucleotides bearing an azide moiety at the 5′ end, we oriented ourselves on earlier reported method, however, instead of relying on RNA solid-phase synthesis with 2′-O-silyl/N-tac-protected29 or 2′-O-PivOM/N-Pac-protected15 monomers, we implemented the standard 2′-O-silyl- and N-acetyl phosphoramidite building blocks. The azido group was incorporated on solid support by treating the 5′-OH deprotected RNA with methyltriphenoxyphosphonium iodide (MTPI), followed by incubation in a saturated solution of sodium azide in DMF (Fig. 4). After the azidination step, the RNAs were deprotected and cleaved from the support by using aqueous ammonia/methylamine solutions, and subsequently, tetrabutylammonium fluoride in THF. The crude RNAs were purified by ion-exchange chromatography (Table 1 and Fig. S1, ESI†), affording pure 5′-azido-functionalized oligoribo-nucleotides (RNA 6 and 7) in good overall yields (Table 1).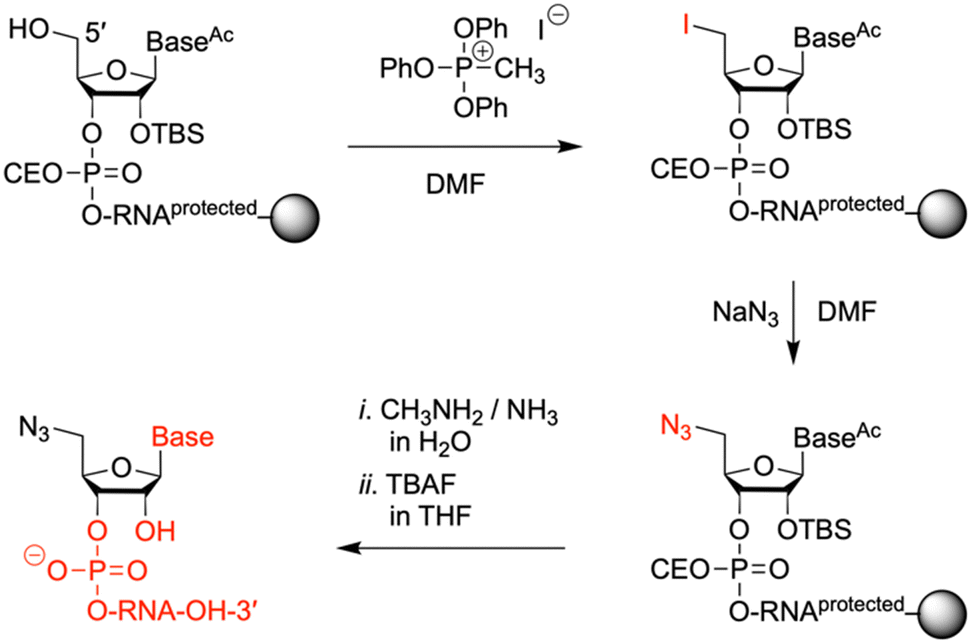 | ||
| Fig. 4 Solid-phase synthesis of 5′-azido modified oligoribonucleotides. Reagents and conditions: (1) 0.5 M MTPI/DMF, rt, 1 h; (2) sat. NaN3 solution in DMF, 55 °C, 5 h; (3) (i) 40% aq MeNH2/30% aq NH3 (1/1 v/v), 40 °C, 4 h, (ii) 1 M TBAF/THF, 37 °C, 16 h. | ||
Non-templated click ligation
Having the capped RNA-3′-alkynes and 5′-azide-RNAs in our hands, we set out to ligate these fragments according to the original plan. Fig. 5A illustrates a typical CuAAC reaction setup with CuSO4, ascorbic acid, and water-soluble tris(3-hydroxypropyltriazolylmethyl)-amine (THPTA) as CuI stabilizing ligand to react the 3′-alkyne-functionalized m7Gppp-hexanucleotide (RNA 4) with the 13 nt RNA 6 providing the 5′ azido group. The use of denaturation conditions (50% dimethyl sulfoxide in H2O) removes the need for a ligation template and simplifies the system.30 The click ligation proceeded in 1 h at room temperature with almost complete conversion of substrates to the triazole-linked cap4 mimic as reflected in the corresponding AE-HPLC chromatogram of the reaction mixture (Fig. 5B). After HPLC purification, the integrity of the triazole-linked 19 nt long cap4-RNA analog (RNA 8) was confirmed by LC-ESI mass spectrometry (Table 1). Also for longer cap4-RNA target sequences the CuAAC RNA ligation work satisfyingly. After 1 h of incuabtion of RNA 4 and RNA 7 about 80% product yield was cleanly formed (Fig. 5C). The resulting 40 nt RNA (RNA 9) with triazole backbone at the site of ligation was isolated by AE HPLC and the expected molecular weight confirmed by ESI mass spectrometry (Table 1).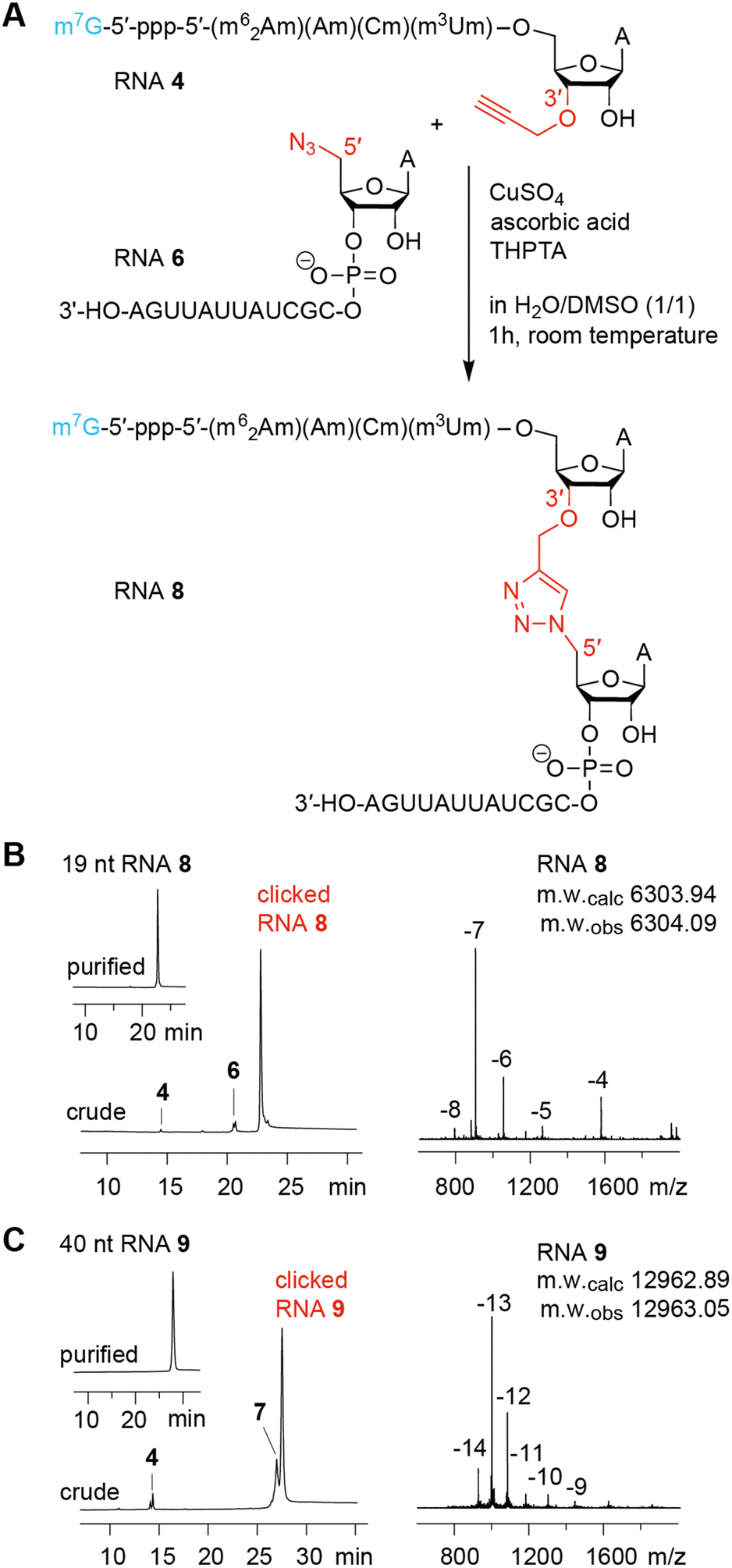 | ||
| Fig. 5 Chemical ligation using CuAAC reaction of 3′-alkyne modified cap-RNA fragments and 5′-azide modified RNA fragments. (A) Exemplary reaction scheme for a 19 nt cap4 RNA. (B) AE HPLC traces for the click ligation of the 19 nt cap4 RNA (left). The crude trace shows the reaction mixture after 1 h at room temperature; the upper left inset displays the purified RNA product; ESI mass spectrum of the product RNA (right). (C) Same as B but for a 40 nt cap4 RNA. | ||
To push the limits of the approach we approached 61 and 81 nt cap4 RNA targets, again based on the ligation of the short 6 nt 5′-fragment of m7G-5′-ppp-5′-(m26Am)(Am)(Cm)(m3Um)(3′-O-propargyl-A) and synthetic 55 and 75 nt 3′-azide RNA fragments, respectively (Fig. S3, ESI†). Also in this case we obtained significant amounts of ligation products, however, AE HPLC encountered limitation with respect to separation of unreacted RNA from the corresponding cap4-RNA (Fig. S3, ESI†). Nevertheless, the correct molecular weight of the product could be detected unequivocally (Fig. S3, ESI†).
Conclusions
The azide–alkyne cycloaddition is an immensely powerful technique for labeling and bioconjugation of nucleic acids due to the facile formation, high chemical stability, and compatibility of triazole linkages in cellular systems.8,10 More specifically, several examples of artificial triazole backbones, that can mimic natural phosphodiester linkages have been reported so far.8,10,29 For our studies, we chose a triazole linkage presented in Fig. 1 and 5A, due to its compatibility with DNA and RNA polymerases.29,34–36 Brown et al. reported that such a triazole backbone can be also tolerated in functionally critical regions of the single guide (sg) RNA and enable effective Cas9-mediated DNA cleavage in vitro and in cells with no unexpected off-target effects.30 Moreover, this linkage has the synthetic advantage of being efficiently formed from 5′-azido- and 3′-alkyne-modified oligonucleotides that are readily accessible by the standard RNA synthesis using phosphoramidite chemistry.The strategy outlined herein represents a significant advancement towards achieving efficient all-chemical synthesis of messenger RNAs (mRNAs) without resorting to in vitro transcription (IVT) methods for RNA production or enzymatic ligation of RNA fragments. IVT-based RNA synthesis typically yields a substantial fraction of uncapped transcripts and unwanted double-stranded RNA byproducts, triggering undesired immunogenic responses.2 This also necessitates rigorous purification procedures.2,12
Further, we point out that our strategy retains the native structure of the cap including the first four frequently modified nucleosides; the artificial linkage is located just downstream from that sequence motif that appears critical for recognition of the native capping enzyme machinery and hence the functionality of the cap. This aspect differs to other studies that contain the triazole linkage within the first 5 nucleosides of capped RNA.8
Finally, it should be noted that the mRNA targets synthesised here were selected with structural biology applications in mind;37 however, we believe that these triazole-linked RNAs could be active in translation. This is suggested by the findings of Hiroyuki Isobe and coworkers on a closely related type of triazole-linked RNA.38 Nevertheless, more extensive studies are needed to unlock the full potential of triazole-linked mRNAs for applications in cellular translation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Andrea Rentmeister (LMU) for a generous gift of Emc1, MTAN, LuxS enzymes. This work was funded in part by the Austrian Science Fund FWF [P31691, 10.55776/P31691; F8011-B, 10.55776/F80], the Austrian Research Promotion Agency FFG [West AustrianBio NMR 858017], and the Tyrolean Science Fund TWF [F.33309/2021 to K. Ba.]. For open access purposes, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission.Notes and references
- A. Ramanathan, G. B. Robb and S.-H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 Search PubMed.
- M. Warminski, A. Mamot, A. Depaix, J. Kowalska and J. Jemielity, Acc. Chem. Res., 2023, 56, 2814–2826 Search PubMed.
- A. Bochler, J. B. Querido, T. Prilepskaja, H. Soufari, A. Simonetti, M. L. D. Cistia, L. Kuhn, A. R. Ribeiro, L. S. Valášek and Y. Hashem, Cell Rep., 2020, 33, 108534 Search PubMed.
- S. Meleppattu, H. Arthanari, A. Zinoviev, A. Boeszoermenyi, G. Wagner, M. Shapira and M. Léger-Abraham, Nucleic Acids Res., 2018, 46, 3791–3801 Search PubMed.
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 Search PubMed.
- M. Shanmugasundaram, A. Senthilvelan and A. R. Kore, Chem. Rec., 2022, 22, e202200005 Search PubMed.
- A. Wojtczak, P. J. Sikorski, K. Fac-Dabrowska, A. Nowicka, M. Warminski, D. Kubacka, E. Nowak, M. Nowotny, J. Kowalska and J. Jemielity, J. Am. Chem. Soc., 2018, 140, 5987–5999 Search PubMed.
- M. Kozarski, K. Drazkowska, M. Bednarczyk, M. Warminski, J. Jemielity and J. Kowalska, RSC Adv., 2023, 13, 12809–12824 Search PubMed.
- F. Muttach, N. Muthmann and A. Rentmeister, Beilstein J. Org. Chem., 2017, 13, 2819–2832 Search PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 Search PubMed.
- J. Leiter, D. Reichert, A. Rentmeister and R. Micura, ChemBioChem, 2020, 21, 265–271 Search PubMed.
- M. Inagaki, N. Abe, Z. Li, Y. Nakashima, S. Acharyya, K. Ogawa, D. Kawaguchi, H. Hiraoka, A. Banno, Z. Meng, M. Tada, T. Ishida, P. Lyu, K. Kokubo, H. Murase, F. Hashiya, Y. Kimura, S. Uchida and H. Abe, Nat. Commun., 2023, 14, 2657 Search PubMed.
- M. Noël, T. Guez, Y. Thillier, J. Vasseur and F. Debart, ChemBioChem, 2023, 24, e202300544 Search PubMed.
- N. Abe, A. Imaeda, M. Inagaki, Z. Li, D. Kawaguchi, K. Onda, Y. Nakashima, S. Uchida, F. Hashiya, Y. Kimura and H. Abe, ACS Chem. Biol., 2022, 17, 1308–1314 Search PubMed.
- M. Warminski, J. Kowalska and J. Jemielity, Org. Lett., 2017, 19, 3624 Search PubMed.
- I. Nakagawa, S. Konya, S. Ohtani and T. A. Hata, Synthesis, 1980, 556–557 Search PubMed.
- T. Kamimura, Y. Osaki, M. Sekine and T. Hata, Tetrahedron Lett., 1984, 25, 2683–2686 Search PubMed.
- M. Sekine, S. Nishiyama, T. Kamimura, Y. Osaki and T. Hata, Bull. Chem. Soc. Jpn., 1985, 58, 850–860 Search PubMed.
- I. I. Koukhareva and A. V. Lebedev, Nucleosides, Nucleotides & Nucleic Acids, 2004, 23, 1667–1680 Search PubMed.
- H. Sawai, H. Wakai and A. Nakamura-Ozaki, J. Org. Chem., 1999, 64, 5836–5840 Search PubMed.
- M. Lewdorowicz, Y. Yoffe, J. Zuberek, J. Jemielity, J. Stepinski, R. Kierzek, R. Stolarski, M. Shapira and E. Darzynkiewicz, RNA, 2004, 10, 1469–1478 Search PubMed.
- K. Piecyk, R. E. Davis and M. Jankowska-Anyszka, Tetrahedron Lett., 2012, 53, 4843–4847 Search PubMed.
- E. Veliath, B. L. Gaffney and R. A. Jones, Nucleosides, Nucleotides Nucleic Acids, 2014, 33, 40–52 Search PubMed.
- W. Ziemkiewicz, M. Warminski, R. Wojcik, J. Kowalska and J. Jemielity, J. Org. Chem., 2022, 87, 10333–10348 Search PubMed.
- S. Lee, B. R. Bowman, Y. Ueno, S. Wang and G. L. Verdine, J. Am. Chem. Soc., 2008, 130, 11570–11571 Search PubMed.
- M. Kadokura, T. Wada, K. Seio, T. Moriguchi, J. Huber, R. Lührmann and M. Sekine, Tetrahedron Lett., 2001, 42, 8853–8856 Search PubMed.
- J. Jemielity, P. Heinonen, H. Lönnberg and E. Darzynkiewicz, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 601–605 Search PubMed.
- Y. Thillier, E. Decroly, F. Morvan, B. Canard, J.-J. Vasseur and F. Debart, RNA, 2012, 18, 856–868 Search PubMed.
- A. H. El-Sagheer and T. Brown, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15329–15334 Search PubMed.
- L. Taemaitree, A. Shivalingam, A. H. El-Sagheer and T. Brown, Nat. Commun., 2019, 10, 1610 Search PubMed.
- A. Guzaev, H. Salo, A. Azhayev and H. Lönnberg, Tetrahedron, 1995, 51, 9375–9384 Search PubMed.
- U. Pradere, F. Halloy and J. Hall, Chem. – Eur. J., 2017, 23, 5210–5213 Search PubMed.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119–1131 Search PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Commun., 2011, 47, 12057–12058 Search PubMed.
- C. N. Birts, A. P. Sanzone, A. H. El-Sagheer, J. P. Blaydes, T. Brown and A. Tavassoli, Angew. Chem., Int. Ed., 2014, 53, 2362–2365 Search PubMed.
- X. Chen, A. H. El-Sagheer and T. Brown, Chem. Commun., 2014, 50, 7597–7600 Search PubMed.
- H. Bernhard, H. Petržílková, B. Popelářová, K. Ziemkiewicz, K. Bartosik, M. Warmiński, L. Tengo, H. Gröger, L. G. Dolce, R. Micura, J. Jemielity and E. Kowalinski, 2024, bioRxiv, preprint, bioRxiv2024.05.04.591051, DOI:10.1101/2024.05.04.591051.
- T. Fujino, T. Suzuki, K. Okada, K. Kogashi, K. Yasumoto, K. Sogawa and H. Isobe, J. Org. Chem., 2016, 81, 8967–8976 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for RNA solid-phase synthesis, purification, and characterization; procedures for RNA ligation, HPLC traces; ESI mass spectra. See DOI: https://doi.org/10.1039/d4cb00165f |
| This journal is © The Royal Society of Chemistry 2024 |