
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBispecific FpFs: a versatile tool for preclinical antibody development†
Matthew
Collins‡
a,
Nkiru
Ibeanu‡
bc,
Wiktoria Roksana
Grabowska
d,
Sahar
Awwad
bc,
Peng T.
Khaw
c,
Steve
Brocchini
 b and
Hanieh
Khalili
*bd
b and
Hanieh
Khalili
*bd
aSchool of Health, Sport and Bioscience, University of East London, London, UK
bSchool of Pharmacy, University College London, London, UK. E-mail: hanieh.khalili@uwl.ac.uk
cNational Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, London EC1V 9EL, UK
dSchool of Biomedical Science, University of West London, London, W5 5RF, UK
First published on 27th September 2024
Abstract
We previously described FpFs ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) (Fab–PEG–Fab) as binding mimetics of IgGs. FpFs are prepared with di(bis-sulfone) conjugation reagents
(Fab–PEG–Fab) as binding mimetics of IgGs. FpFs are prepared with di(bis-sulfone) conjugation reagents ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) that undergo disulfide rebridging conjugation with the accessible disulfide of each Fab (Scheme 1). We have now prepared bispecific FpFs
that undergo disulfide rebridging conjugation with the accessible disulfide of each Fab (Scheme 1). We have now prepared bispecific FpFs ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) (bsFpF and Fab1–PEG–Fab2) as potential bispecific antibody mimetics with the intent that bsFpFs could be used in preclinical antibody development since sourcing bispecific antibodies may be challenging during preclinical research. The di(bis-sulfone) reagent was first used to prepare a bsFpF by the sequential conjugation of a first Fab and then a second Fab to another target (Scheme 2). Seeking to improve bsFpF synthesis, the asymmetric conjugation reagent, bis-sulfone bis-sulfide
(bsFpF and Fab1–PEG–Fab2) as potential bispecific antibody mimetics with the intent that bsFpFs could be used in preclinical antibody development since sourcing bispecific antibodies may be challenging during preclinical research. The di(bis-sulfone) reagent was first used to prepare a bsFpF by the sequential conjugation of a first Fab and then a second Fab to another target (Scheme 2). Seeking to improve bsFpF synthesis, the asymmetric conjugation reagent, bis-sulfone bis-sulfide ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) , with different thiol conjugation reactivities at each terminus (Scheme 4) was examined and the bsFpFs appeared to be formed at similar conversion to the di(bis-sulfone) reagent . To explore the advantages of using common intermediates in the preparation of bsFpF families, we investigated bsFpF synthesis with a protein conjugation–ligation approach (Scheme 5). Reagents with a bis-sulfone moiety for conjugation on one PEG terminus and a ligation moiety on the other terminus were examined. Bis-sulfone PEG trans-cyclooctene (TCO)
, with different thiol conjugation reactivities at each terminus (Scheme 4) was examined and the bsFpFs appeared to be formed at similar conversion to the di(bis-sulfone) reagent . To explore the advantages of using common intermediates in the preparation of bsFpF families, we investigated bsFpF synthesis with a protein conjugation–ligation approach (Scheme 5). Reagents with a bis-sulfone moiety for conjugation on one PEG terminus and a ligation moiety on the other terminus were examined. Bis-sulfone PEG trans-cyclooctene (TCO) ![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) and bis-sulfone PEG tetrazine (Tz)
and bis-sulfone PEG tetrazine (Tz) ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) were used to prepare several bsFpFs targeting various therapeutic targets (TNF-α, IL6R, IL17, and VEGF) and tissue affinity targets (hyaluronic acid and collagen II). Surface plasmon resonance (SPR) binding studies indicated that there was little difference between the dissociation rate constant (kd) for the unmodified Fab, mono-conjugated PEG–Fab and the corresponding Fab in a bsFpF. The Fab association rate (ka) in the bsFpF was slower than for PEG–Fab, which may be because of mass differences that influence SPR results. These observations suggest that each Fab will bind to its target independently of the other Fab and that bsFpF binding profiles can be estimated using the corresponding PEG–Fab conjugates.
were used to prepare several bsFpFs targeting various therapeutic targets (TNF-α, IL6R, IL17, and VEGF) and tissue affinity targets (hyaluronic acid and collagen II). Surface plasmon resonance (SPR) binding studies indicated that there was little difference between the dissociation rate constant (kd) for the unmodified Fab, mono-conjugated PEG–Fab and the corresponding Fab in a bsFpF. The Fab association rate (ka) in the bsFpF was slower than for PEG–Fab, which may be because of mass differences that influence SPR results. These observations suggest that each Fab will bind to its target independently of the other Fab and that bsFpF binding profiles can be estimated using the corresponding PEG–Fab conjugates.
Introduction
We previously described an antibody mimetic that is called FpF (Fab–PEG–Fab), where a poly(ethylene glycol) (PEG) linker mimics the IgG hinge.1,2 The IgG hinge enables flexibility of the two antigen-binding fragments (Fabs) to provide enhanced binding that can be derived from bivalency to facilitate rebinding to slow dissociation rates.3,4 The hinge region comprises single polypeptide chains that can be susceptible to degradation.5 FpFs (structures 1) share comparable solution sizes, binding characteristics,1 functional activity2 and enhanced stability6 with IgG antibodies using PEG linkers with molecular weights in the range of 5–20 kDa. FpFs were designed to maintain the Fab topology and flexibility that has evolved in IgG antibodies with increased stability by substituting the hinge region with a PEG linker that has been stably covalently conjugated to each Fab. With these characteristics of FpFs in mind, we decided to examine the synthesis and binding properties of bispecific antibody mimetics called bsFpFs (Fab1–PEG–Fab2).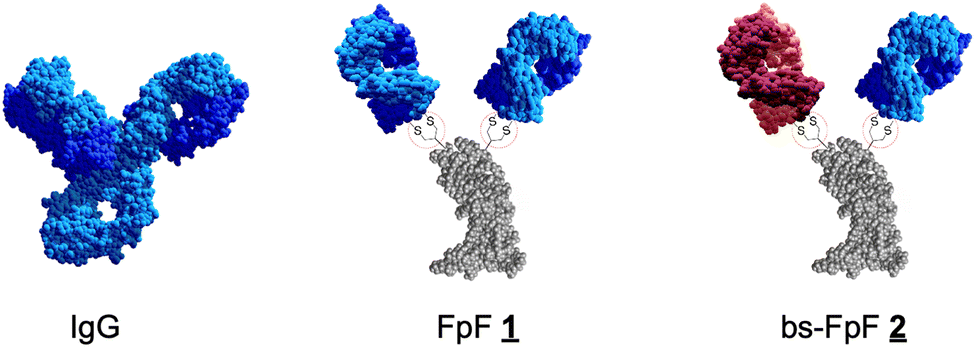
FpFs are prepared using the di(bis-sulfone) reagent to conjugate 2 Fabs that can be obtained by the proteolytic digestion of IgGs (Scheme 1).1 Each bis-sulfone conjugation moiety in the di(bis-sulfone) reagent undergoes site specific bis-alkylation conjugation with the two thiols from the accessible Fab disulfide furthest from the target binding region by a sequence of addition–elimination reactions to give thiol ethers that are more stable than the initial disulfide (Scheme S1, ESI†).7,8 Di(bis-sulfone) has also been used to dimerise other proteins by bis-alkylation of cysteine thiols9,10 including an analogous Fc-fusion mimetic10 which involved conjugating the extracellular receptor binding domains of an Fc-fusion protein. The Fc-fusion mimetic is called a RpR for receptor–PEG–receptor and displayed better binding characteristics compared to the corresponding parent Fc-fusion, aflibercept.
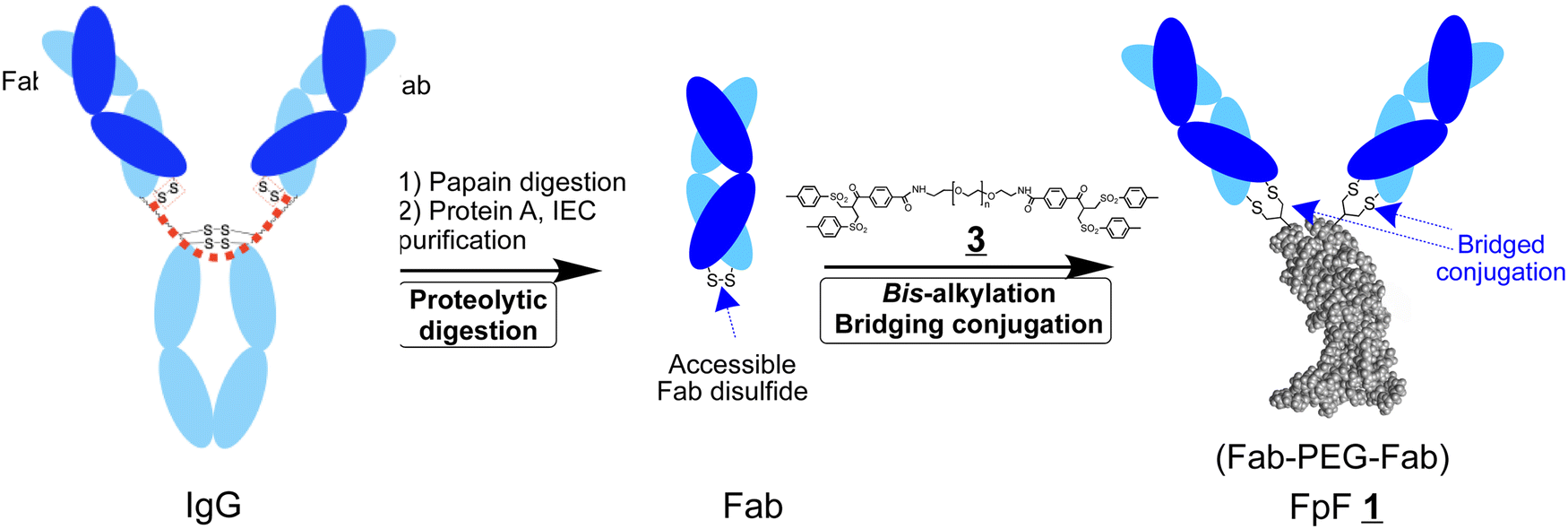 | ||
| Scheme 1 IgG representation showing how the two Fabs topologically are shown to be at the two ends of a flexible linear molecule (note the red dotted line). Proteolytic digestion of the IgG provides the corresponding Fab after purification which can be conjugated to each end of the di(bis-sulfone) reagent to give FpF . | ||
Bispecific antibody-based medicines are clinically proven modalities used to treat cancer, heamophilia and neovascular retinal diseases11–16 with many clinical candidates undergoing development with encouragement from the regulatory authorities. Faricimab is a bispecific antibody that binds to 2 ligands in the vitreous cavity to treat retinal diseases. Intravitreal injections are administered in a small volume (50 μL) and are difficult for patients to endure. Increasing residence time in the vitreous is broadly proportional to the concentration of an antibody-based medicine in the formulation.17 Utilising a high concentration of a single (bispecific) antibody in an intravitreal injection provides better clinical benefits than multiple intravitreal injections of a combination of antibodies.
Bispecific antibodies can also bring two targets together to cause an enhanced biological function not possible by using a combination of 2 separate antibodies. Such spatial–temporal properties have been shown to bring (i) two cells together (e.g. blinatumomab and epcoritamab)13,14 to enhance immune recognition and (ii) two proteins together that are necessary to maintain the coagulation cascade (e.g. emicizumab).16 BsFpFs utilise PEG linkers of a sufficient molecular weight designed to optimise spatial–temporal relationships.
IgGs are multifunctional molecules that can also exert immune-related effector functions through Fc. Many clinically used mono-specific antibodies exert an effector function which is important for their clinical efficacy. Some Fc effector functions can cause inadvertent immune-driven agonism,18,19 but IgG4 subtypes are sometimes used to abrogate unwanted Fc-induced effector functionality.18 Many applications where the spatial–temporal properties of a bispecific antibody can be exploited do not require Fc effector functions.
The concept of bispecific antibodies has been considered for decades20 as well as their preparation by chemical conjugation.21–23 Purely recombinant strategies are used for the preparation of clinically used bispecific antibodies,24 but challenges remain to optimise the structure and format of bispecific antibodies for researchers in early preclinical research.24–26 The use of chemical conjugation and modification strategies to aid in the development of therapeutic proteins including antibody-based molecules is widespread, clinically proven and is being driven by much creative research studies.27–34 There is also intense interest in developing protein–protein and protein–drug conjugates35–38 as the combined use of recombinant and conjugation technologies may yield complex molecules for study and potential development.
Our goal is to develop a practical method to prepare bsFpFs, making these valuable tools readily accessible to researchers interested in a wide range of preclinical research including drug target development, drug delivery, tissue engineering and immunocytochemistry. We first explored the di(bis-sulfone) reagent to prepare bispecific FpFs (Scheme 2) in a conjugation-only approach. Since the preparation of bsFpFs with the di(bis-sulfone) reagent requires the sequential addition of 2 different Fabs, there can be limitations due to the formation of small amounts of homodimeric FpF during the addition of the first Fab, so we also examined a conjugation–ligation strategy for the preparation of bispecific FpFs (Scheme 6). The conjugation–ligation strategy enabled the synthesis of an extensive panel of seventeen different bsFpFs, targeting a diverse range of therapeutic molecules implicated in ocular inflammation (TNF-α, IL6R, IL17, and VEGF) and ocular drug delivery (hyaluronic acid and collagen II). To the best of our knowledge, this represents the first synthesis of such a broad spectrum of bsFpFs which would be more costly and difficult to achieve by recombinant means alone.
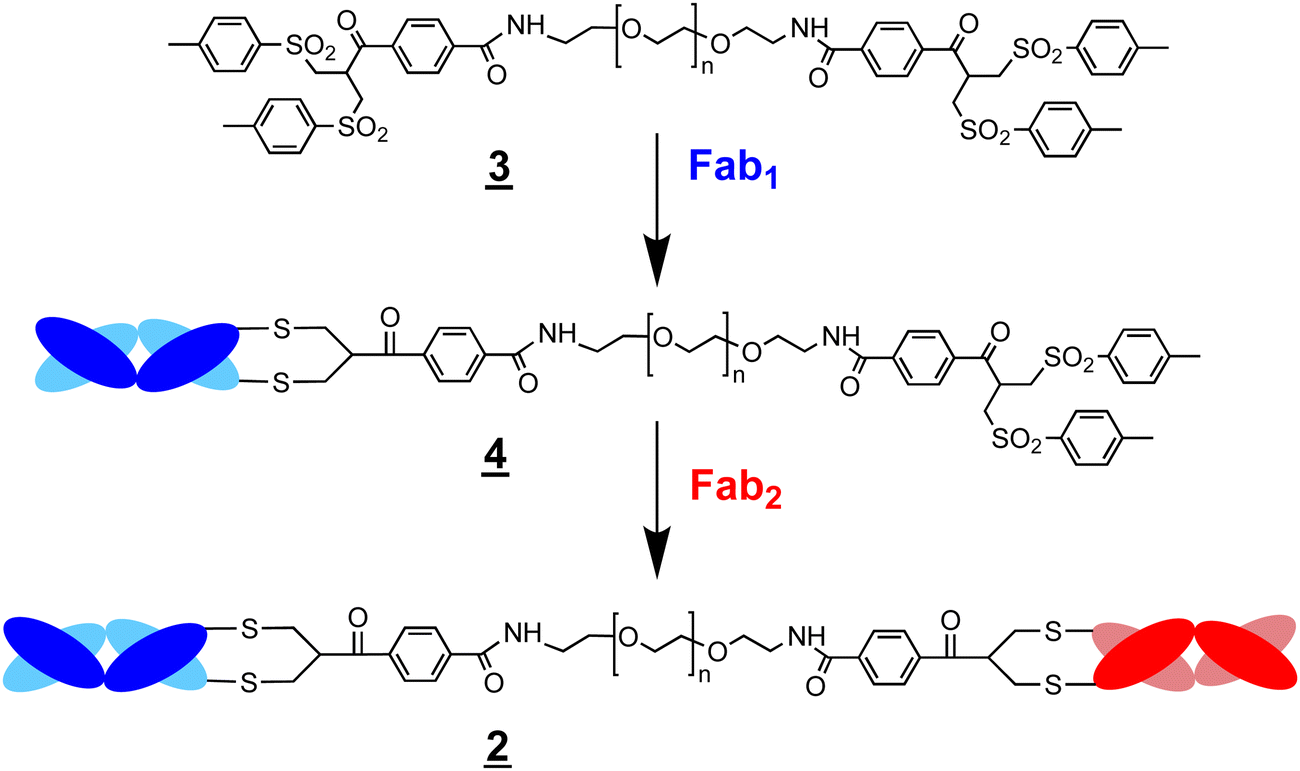 | ||
Scheme 2 Preparation of a bsFpF by the conjugation of Fab1 with an excess di(bis-sulfone) reagent . The intermediate conjugate ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) can be purified by IEX and in some cases by spin filtration to remove the excess starting reagent and unreacted Fab1. Conjugation of Fab2 to the intermediate conjugate gives the bsFpF . can be purified by IEX and in some cases by spin filtration to remove the excess starting reagent and unreacted Fab1. Conjugation of Fab2 to the intermediate conjugate gives the bsFpF . | ||
Experimental section
Materials
Bevacizumab (Avastin®, 25 mg mL−1, anti-VEGF IgG), infliximab (Remicade®, 10 mg mL−1, anti-TNFα IgG), tocilizumab (Avtemra®, 20 mg mL−1, anti-IL6R IgG) and ranibizumab (Lucentis®, 10 mg mL−1, anti-VEGF Fab) were obtained from the pooled remaining contents of vials that had been used clinically. Secukinumab (Cosentyx®, 150 mg) was purchased commercially. Phosphate buffered saline (PBS; 0.16 M NaCl, 0.003 M KCl, 0.008 M Na2HPO4 and 0.001 M KH2PO4) was prepared with tablets purchased from Oxoid. Acetate buffer A (100 mM sodium acetate, pH 4.0) and acetate buffer B (100 mM sodium acetate, 1 M NaCl, pH 4.0) were prepared for ion-exchange chromatography. Novex bis–tris 4–12% gels, sharp blue standard protein markers, NuPAGE MOPS running buffer, NuPAGE LDS sample buffer and SilverXpress silver staining kit were purchased from Invitrogen. InstantBlue was purchased from Expedeon Ltd. Perchloric acid (0.1 M) and barium chloride (5.0%) solutions for barium iodide staining were prepared in the lab. A PD-10 column, cation exchange columns (HiTrap SP HP 1.0 mL) and a Superdex 200 prep grade size exclusion column (34.0 μm particle size) along with Biacore consumables were all purchased from GE Healthcare. Anti-human IgG (Fab specific)-peroxidase, 3,3′,5,5′-tetramethylbenzidine (TMB) and human vascular endothelial growth factor (hVEGF165) were purchased from Sigma Aldrich.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 papain:IgG ratio. The digestion mixture was placed in an incubator at 37 °C for 30 minutes. The crude digestion mixture was then purified using protein L chromatography (Hitrap Protein L) using buffer A (100 mM sodium phosphate, 150 mM sodium chloride, 500 mL, pH 7.2) and buffer B (100 mM glycine, 500 mL, pH 2.5) and an elution gradient.
:20 papain:IgG ratio. The digestion mixture was placed in an incubator at 37 °C for 30 minutes. The crude digestion mixture was then purified using protein L chromatography (Hitrap Protein L) using buffer A (100 mM sodium phosphate, 150 mM sodium chloride, 500 mL, pH 7.2) and buffer B (100 mM glycine, 500 mL, pH 2.5) and an elution gradient.
Eluted fractions were monitored using SDS–PAGE and further purified using an SEC (Superdex 200 Increase 10/300 GL, flow rate of 0.5 mL min−1 and PBS as the mobile phase). Eluted fractions were collected and monitored using SDS–PAGE.
![[3 with combining low line]](https://www.rsc.org/images/entities/b_char_0033_0332.gif) .
FabVEGF (1.0 mg in 1.0 mL of conjugation buffer; 20 mM sodium phosphate, 10 mM EDTA, pH 7.6) was first incubated with DTT (1.0 mg) at ambient temperature without shaking for 30 min. DTT was removed by elution over a PD-10 column. Into 1.0 equivalent of reduced-FabVEGF was added the di(bis-sulfone) PEG10 reagent 1 (5 equivalents, 1 mg) and the solution was incubated for 1 h at ambient temperature. The PEGylation reaction mixture was then purified using a Macrocap SP cation exchange column (Macrocap SP, 5 mL). The IEX-purified bis sulfone–PEG10–FabVEGF was then incubated with the pre-DTT treated FabHER2 (1.0 mg in 3.3 mL after the PD-10 column). The formation of bispecific FabVEGF–PEG10–FabHER2 was then monitored for 2, 15 and 48 hours at ambient temperature using SDS–PAGE. The FabVEGF–PEG10–FabHER2 was purified using single-step size-exclusion chromatography (SEC), and SEC fractions were analysed by SDS–PAGE.
(0.1 mL, 4.0 mg mL−1 in distilled water, 1.0 eq.) was then added to the reduced FabVEGF solution (1.0 mg in 3.3 mL). The PEGylation solution was incubated at ambient temperature for approximately 6 h without shaking. In a separate vial, FabIL6R (1.0 mg in 1.0 mL of conjugation buffer; 20 mM sodium phosphate, 10 mM EDTA, pH 7.6) was incubated with DTT (1.0 mg) and the DTT was removed by elution over a PD-10 column. Bis-sulfone–PEG–Tz (0.1 mL, 2.0 mg mL−1 in distilled water) was then added to the reduced FabIL6R solution (1.0 mg in 3.3 mL). The PEGylation solution was incubated at ambient temperature for approximately 6 h without shaking. Both PEG–Fab conjugates (FabVEGF–PEG TCO
.
FabVEGF (1.0 mg in 1.0 mL of conjugation buffer; 20 mM sodium phosphate, 10 mM EDTA, pH 7.6) was first incubated with DTT (1.0 mg) at ambient temperature without shaking for 30 min. DTT was removed by elution over a PD-10 column. Into 1.0 equivalent of reduced-FabVEGF was added the di(bis-sulfone) PEG10 reagent 1 (5 equivalents, 1 mg) and the solution was incubated for 1 h at ambient temperature. The PEGylation reaction mixture was then purified using a Macrocap SP cation exchange column (Macrocap SP, 5 mL). The IEX-purified bis sulfone–PEG10–FabVEGF was then incubated with the pre-DTT treated FabHER2 (1.0 mg in 3.3 mL after the PD-10 column). The formation of bispecific FabVEGF–PEG10–FabHER2 was then monitored for 2, 15 and 48 hours at ambient temperature using SDS–PAGE. The FabVEGF–PEG10–FabHER2 was purified using single-step size-exclusion chromatography (SEC), and SEC fractions were analysed by SDS–PAGE.
(0.1 mL, 4.0 mg mL−1 in distilled water, 1.0 eq.) was then added to the reduced FabVEGF solution (1.0 mg in 3.3 mL). The PEGylation solution was incubated at ambient temperature for approximately 6 h without shaking. In a separate vial, FabIL6R (1.0 mg in 1.0 mL of conjugation buffer; 20 mM sodium phosphate, 10 mM EDTA, pH 7.6) was incubated with DTT (1.0 mg) and the DTT was removed by elution over a PD-10 column. Bis-sulfone–PEG–Tz (0.1 mL, 2.0 mg mL−1 in distilled water) was then added to the reduced FabIL6R solution (1.0 mg in 3.3 mL). The PEGylation solution was incubated at ambient temperature for approximately 6 h without shaking. Both PEG–Fab conjugates (FabVEGF–PEG TCO ![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) and FabIL6R–PEG Tz ) were purified using a Macrocap SP cation exchange column (Macrocap SP, 5 mL). Fractions (1.0 mL) were analysed by SDS–PAGE. Purified intermediate molecules and were then mixed for approximately 18 hours at 4 °C to undergo ligation, forming a bsFpF molecule . bsFpFs were then purified using cation exchange chromatography followed by SEC. The purity of the bsFpF was assessed using silver staining and the concentration of the purified bsFpF was calculated using a micro-BCA assay.
and FabIL6R–PEG Tz ) were purified using a Macrocap SP cation exchange column (Macrocap SP, 5 mL). Fractions (1.0 mL) were analysed by SDS–PAGE. Purified intermediate molecules and were then mixed for approximately 18 hours at 4 °C to undergo ligation, forming a bsFpF molecule . bsFpFs were then purified using cation exchange chromatography followed by SEC. The purity of the bsFpF was assessed using silver staining and the concentration of the purified bsFpF was calculated using a micro-BCA assay.
The products are denoted using subscript ‘n’ on PEGn to indicate the PEG molar weight, e.g. FabVEGF–PEG15–FabTNFα is derived from the FabVEGF from bevacizumab, FabTNFα from infliximab, the 10 kDa for the PEG10 reagent and the 5 kDa for the PEG5 reagent .
:1 binding model. The sensorgram was fitted globally over the association and dissociation phases. Equilibrium dissociation constants (affinity) were calculated from the rate constants (KD = koff/kon).
Results
bsFpFs prepared by conjugation
The di(bis-sulfone) reagent derived from PEG with a molecular weight of 10 kDa was used to prepare a bsFpF from Fabs targeted to VEGF and HER2 (Scheme 2). Fabs can be obtained by thiol protease digestion of IgGs using either immobilised or soluble papain.39 Bevacizumab was the IgG that was proteolytically digested to give the Fab targeted to VEGF and trastuzumab was the IgG source for the Fab targeted to HER2. The accessible disulfide of FabVEGF was reduced with dithiothreitol (DTT) (Fig. 1A, lanes 2 and 3).
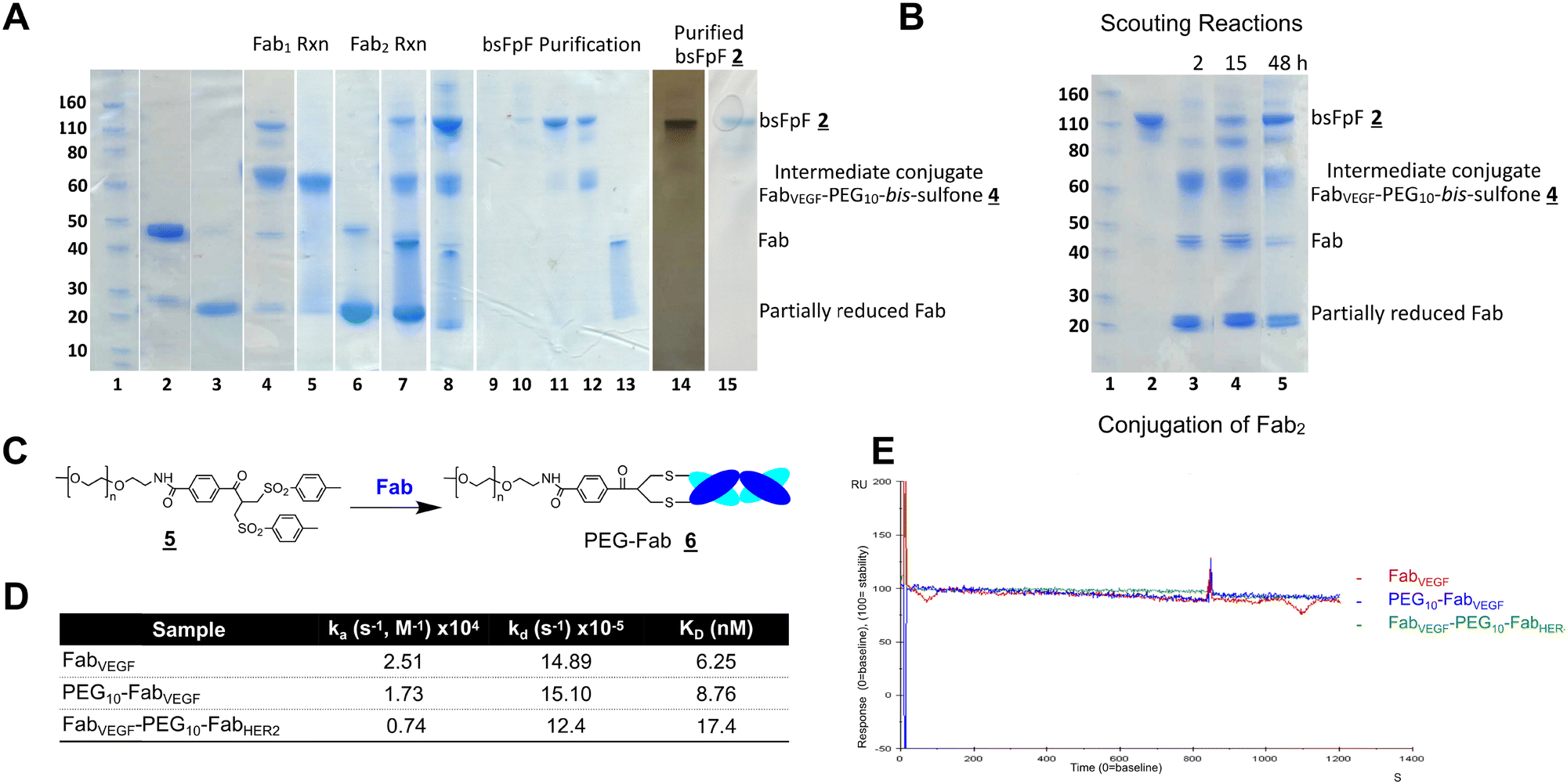 | ||
| Fig. 1 (A) Representative SDS–PAGE analysis for preparation of bsFpF using the di(bis-sulfone) reagent and the SDS-PAGE was stained with instant blue for protein staining (lanes 1–15) and silver staining for detecting any trace of impurity (lane 14), lane 1: Novex pre-stained protein marker, lane 2: FabVEGF (no DTT), lane 3: FabVEGF + DTT, lane 4: reaction mixture between PEG reagent (4–5 eq.) and reduced-FabVEGF (1 eq.) after 1 h incubation at ambient temperature, lane 5: FabVEGF–PEG–bis sulfone was obtained using ion-exchange chromatography, lane 6: FabHER-2 + DTT, lane 7: reaction mixture between FabVEGF–PEG–bis sulfone and reduced FabHER-2 (crude PEG reagent was used for first conjugation), lane 8: reaction mixture between PEG–FabVEGF and reduced FabHER-2 (HPLC purified PEG reagent was used for the conjugation), lanes 9–13, purification fraction using size-exclusion chromatography, lane 14: purified FabVEGF–PEG10–FabHER-2 and lane 15: purified FabVEGF–PEG10–FabTNFa appeared at the 110 kDa MW band. (B) SDS–PAGE analysis for scouting reactions at 2, 15 and 48 h for the conjugation of the second Fab (FabHER2) to the intermediate conjugate (FabVEGF–PEG–bis sulfone). (C) Synthesis of PEG–Fab . (D) Average kinetic rate constants and parameters achieved for FabVEGF, PEG10–FabVEGF and bispecific FabVEGF–PEG10–FabHER-2 using a CM3 chip immobilised with VEGF (55 RU). The numbers of replicates were 2 for bispecific conjugate and 3 for other samples. (E) Dissociation profiles overlayed for FabVEGF, PEG10–FabVEGF and FabVEGF–PEG10–FabHER-2. | ||
Fig. 1 After removal of DTT using a PD-10 column,1,7 the reduced Fab (1.0 mg in 3.3 mL) was allowed to incubate with an excess of the di(bis-sulfone) reagent (5 equivalents, 1 mg) in the PEGylation buffer with pH 7.8 for 1 hour at ambient temperature (Fig. 1A, lane 4). The reaction solution essentially comprised a mixture of the desired intermediate conjugate FabVEGF–PEG10–bis-sulfone , unreacted di(bis)-sulfone reagent , trace starting FabVEGF and the undesired homodimer FpF, FabVEGF–PEG10–FabVEGF. The excess di(bis-sulfone) reagent was used to minimise the formation of the homodimer, FabVEGF–PEG10–FabVEGF (Fig. 1A, lane 4). The reaction mixture was then eluted over an ion exchange column (a MacroCap SP) to give predominantly a band at 70 kDa, thought to be the desired intermediate conjugate FabVEGF–PEG10–bis-sulfone (Fig. 1A, lane 5). The excess di(bis-sulfone) reagent was removed from the reaction mixture preferably by ion exchange chromatography or by centrifugal filtration to prevent suppression of bsFpF formation.
In a separate vial, FabHER2 (1.0 mg in 1.0 mL of the PEGylation buffer) was incubated with DTT (1.0 mg), then DTT was removed and reduced-FabHER2 (Fig. 1A, lane 6) was incubated with the intermediate conjugate, FabVEGF–PEG10–bis-sulfone for 12 hours to give the desired bsFpF (FabVEGF–PEG10–FabHER2). Scouting reactions (Fig. 1B) indicated that the conjugation of FabHER2 to the intermediate FabVEGF–PEG10–bis-sulfone required a longer incubation time than conjugation of the first Fab (FabVEGF) to the starting di(bis-sulfone) reagent .
Purification of the di(bis-sulfone) reagent by HPLC (Fig. S1, ESI†) resulted in better conversion to the desired FabVEGF–PEG10–FabHER2 (Fig. 1A, lanes 7 and 8) which was purified by size exclusion chromatography (SEC) (Fig. 1A, lanes 9–13). The purity of the bsFpF , FabVEGF–PEG10–FabHER2, was confirmed by silver staining (Fig. 1A, lane 14).
Binding of FabVEGF–PEG10–FabHER2 was evaluated by surface plasmon resonance (SPR) with each ligand immobilised on separate CM3 chips at low response units to allow kinetic studies to be conducted (VEGF (55 RU) and HER2 (65 RU)). Ligand binding was first confirmed with the parent antibodies and bevacizumab for VEGF and trastuzumab for HER2. Additionally, there was no non-specific binding observed when bevacizumab was incubated with the immobilised HER2 chip and when trastuzumab was incubated with the immobilised VEGF chip (Fig. S2A and B, ESI†). The bsFpF , FabVEGF–PEG10–FabHER2, displayed concentration-dependent binding to each immobilised ligand (Fig. S2C and D, ESI†). Both VEGF and HER2 were immobilised to a single CM3 chip and binding of bevacizumab and trastuzumab was observed (control) as well as the concentration-dependent binding of bsFpF, FabVEGF–PEG20–FabHER2 (Fig. S2E and F, ESI†).
SPR kinetic studies were performed with FabVEGF, PEG10–FabVEGF and the bsFpF , FabVEGF–PEG10–FabHER2 using VEGF immobilised to a CM3 chip. PEG10–FabVEGF was prepared from the PEG10 bis-sulfone reagent ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) used for protein PEGylation as previously described (Fig. 1C).1,2,40 The SPR data indicated that FabVEGF exhibited a faster association rate constant (ka) compared to both PEG10–FabVEGF and FabVEGF–PEG10–FabHER2. This is likely attributed to the smaller molecular weight of FabVEGF. However, no discernible difference was observed in the dissociation rate constant (kd) between FabVEGF, PEG10–FabVEGF and FabVEGF–PEG10–FabHER2 upon dissociation from immobilised VEGF (Fig. 1D and E).
used for protein PEGylation as previously described (Fig. 1C).1,2,40 The SPR data indicated that FabVEGF exhibited a faster association rate constant (ka) compared to both PEG10–FabVEGF and FabVEGF–PEG10–FabHER2. This is likely attributed to the smaller molecular weight of FabVEGF. However, no discernible difference was observed in the dissociation rate constant (kd) between FabVEGF, PEG10–FabVEGF and FabVEGF–PEG10–FabHER2 upon dissociation from immobilised VEGF (Fig. 1D and E).
A second bsFpF (FabVEGF–PEG10–FabTNFα) derived from FabVEGF and a Fab targeted to tumour necrosis factor alpha (TNF-α) was prepared using the di(bis-sulfone) reagent (Fig. 1A, lane 15). FabVEGF–PEG10–FabTNFα also displayed concentration-dependent binding as observed by SPR to both VEGF and TNF-α (Fig. S3, ESI†).
During the preparation of bsFpFs using the di(bis-sulfone) reagent , it was thought that a reagent with more different conjugation reactivity at each PEG terminus would allow more efficient preparation of a bsFpF by a conjugation only approach. It was thought that a reagent with reduced conjugation reactivity on one terminus would require a lower excess of the FpF reagent for the first Fab conjugation while producing a less homodimer.
The bis-sulfone conjugation moiety functions by a sequence of addition–elimination reactions (Scheme S1, ESI†). Initial elimination of one equivalent of the toluene sulfinic acid leaving group is necessary to generate the α,β-unsaturated carbonyl moiety (e.g. structure , Scheme 3A). The initial elimination reaction is driven by the pKa of the α-proton to the carbonyl electron-withdrawing group in the bis-sulfone conjugating moiety ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) (Scheme 3A). If the pKa value of the α-proton was increased slightly as in the bis-sulfide precursor (Scheme 3A), this would reduce the rate of the initial elimination step to potentially slow conjugation compared to the bis-sulfone moiety.
(Scheme 3A). If the pKa value of the α-proton was increased slightly as in the bis-sulfide precursor (Scheme 3A), this would reduce the rate of the initial elimination step to potentially slow conjugation compared to the bis-sulfone moiety.
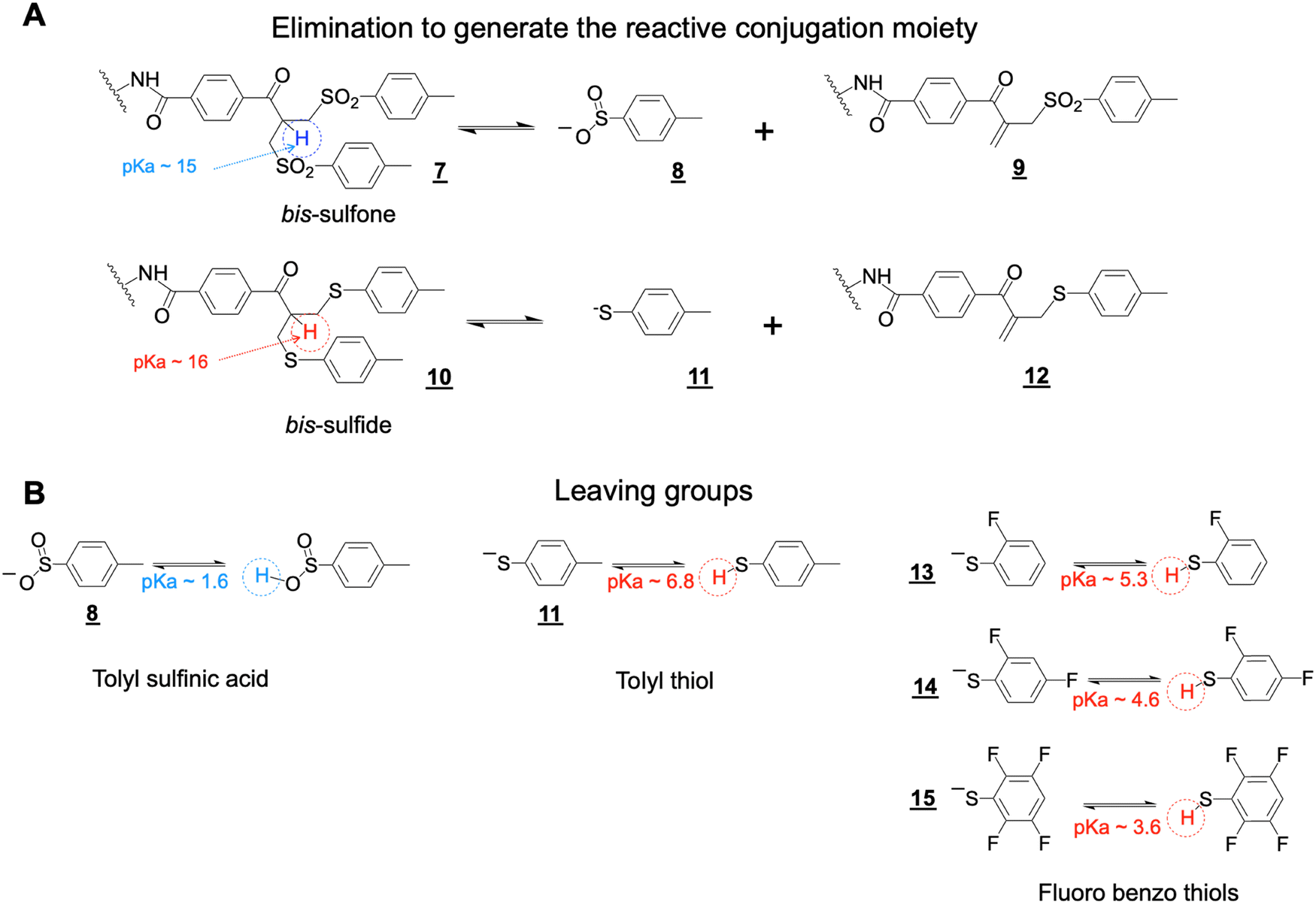 | ||
| Scheme 3 (A) Elimination reactions of the bis-sulfone and bis-sulfide conjugation moieties to yield enones capable of alkylation by a thiolate from a reduced disulfide. The pKa value of the α-proton in bis-sulfone is thought to be slightly less than that of the α-proton in bis-sulfide . (B) Estimates of leaving group pKas. | ||
The conjugation reactivity of the bis-sulfone moiety is also due to the pKa of the toluene sulfinic acid leaving group (Scheme 3B) which we estimate to be about ∼1.6 and is much lower than that for the cysteine thiol (pKa ∼ 10) to drive conjugation. Increasing the pKa of the leaving groups (e.g. structures and –, Scheme 3) relative to toluene sulfinic acid would potentially contribute to reduced conjugation reactivity. We therefore sought to examine the asymmetric conjugation reagent (Scheme 4). A bsFpF could potentially be made by conjugation to the bis-sulfone moiety with the first Fab (Fab1) and then conjugation of the second Fab (Fab2) to the less reactive bis-sulfide moiety in the intermediate Fab–PEG–bis sulfide intermediate (Scheme 4).
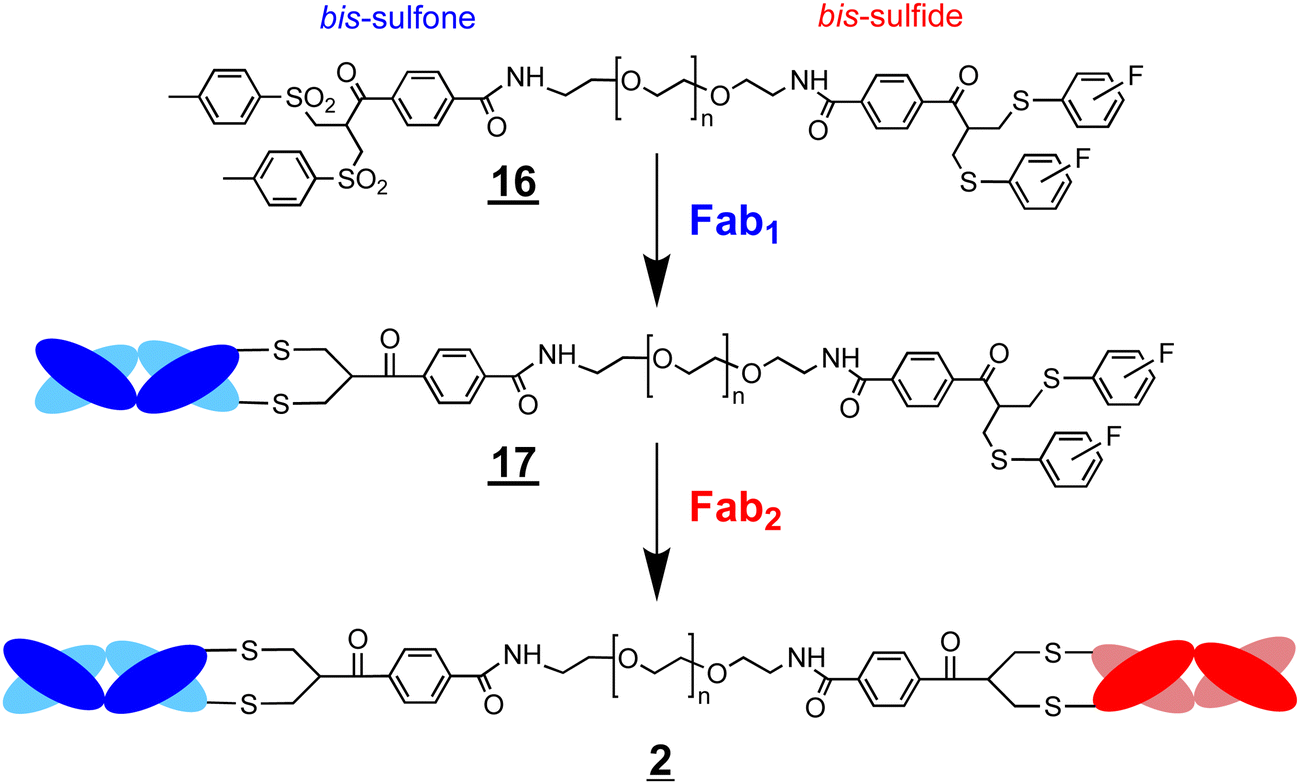 | ||
| Scheme 4 Preparation of a bsFpF by the conjugation of Fab1 with the bis-sulfone bis-sulfide reagent . Conjugation is intended to occur on the bis-sulfone moiety of reagent 16 to give the intermediate conjugate can be purified by IEX or by spin filtration to remove the excess starting reagent and unreacted Fab1. Conjugation of Fab2 to the intermediate conjugate gives the bsFpF . | ||
We prepared the bis-sulfide PEGylation reagents , , , and (Structures 2) to examine relative conjugation reactivities with the PEG–bis sulfone (Fig. 1C). PEG10–bis-sulfide with the unsubstituted tolyl thiol leaving group was not reactive enough under the mild conditions normally employed for Fab conjugation (Figures S4A and B, ESI†). In contrast, the aryl tetra-fluoro thiol leaving group in PEG10–bis sulfide appeared to have comparable conjugation reactivity with the bis-sulfone moiety in PEG–bis sulfone (Figure S4A, ESI†).
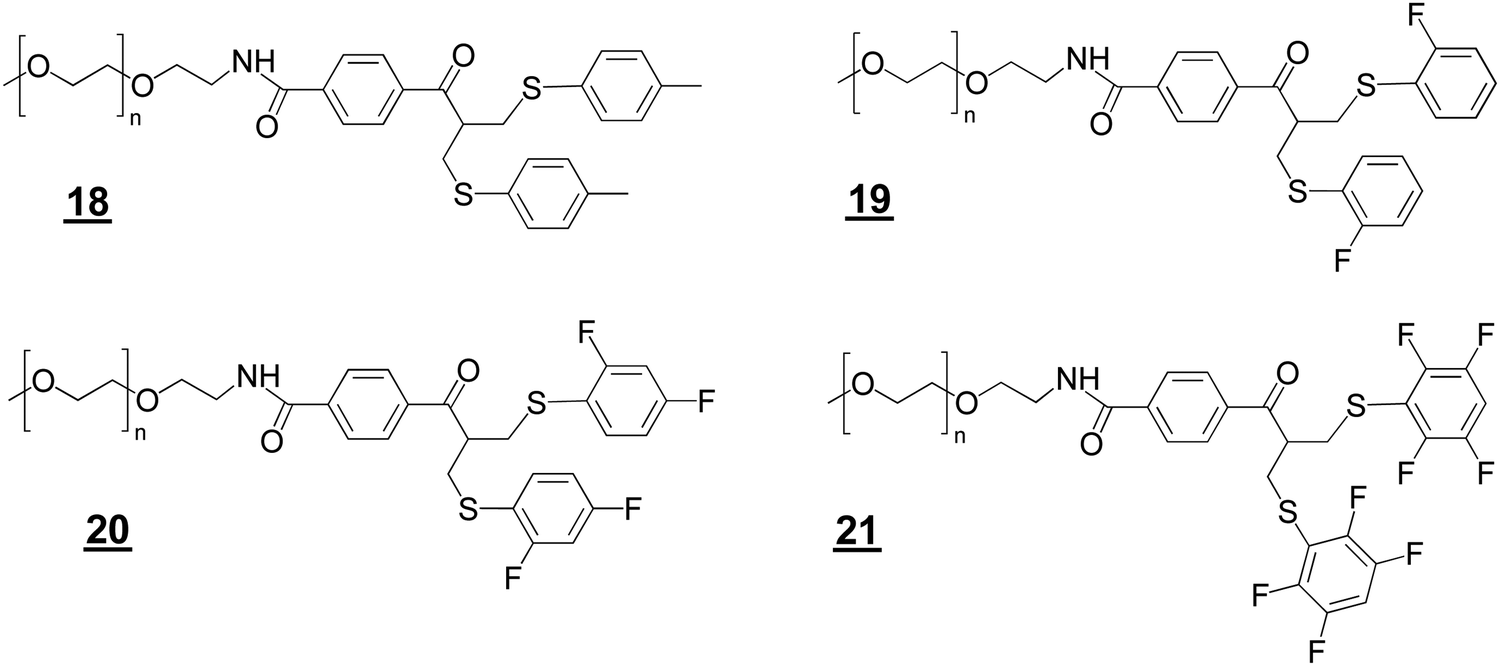
The ortho-fluoro and di-fluoro PEG10–bis sulfides and underwent Fab conjugation (Fig. 2A and B). Reagents and appeared to have sufficient conjugation reactivity while being less reactive than that of the bis-sulfone moiety. An experiment to examine the sequential conjugation reactions of FabVEGF to ortho-fluoro bis-sulfide bis-sulfone reagent did appear to give a good conversion to homodimeric FpF (Fig. 2C). An analogous conjugation was conducted with the di-fluoro bis-sulfide bis-sulfone reagent where formation to the desired FpF (Fig. S4C, ESI†) was also observed. These preliminary efforts indicated that considerable Fab2 remained which made the IEX purification difficult necessitating SEC purification. Fab2 conjugation also appeared to be slower with the intermediate conjugate Fab1–PEG–bis sulfide intermediate than Fab conjugation with PEG–fluoro-bis-sulfone (e.g.).
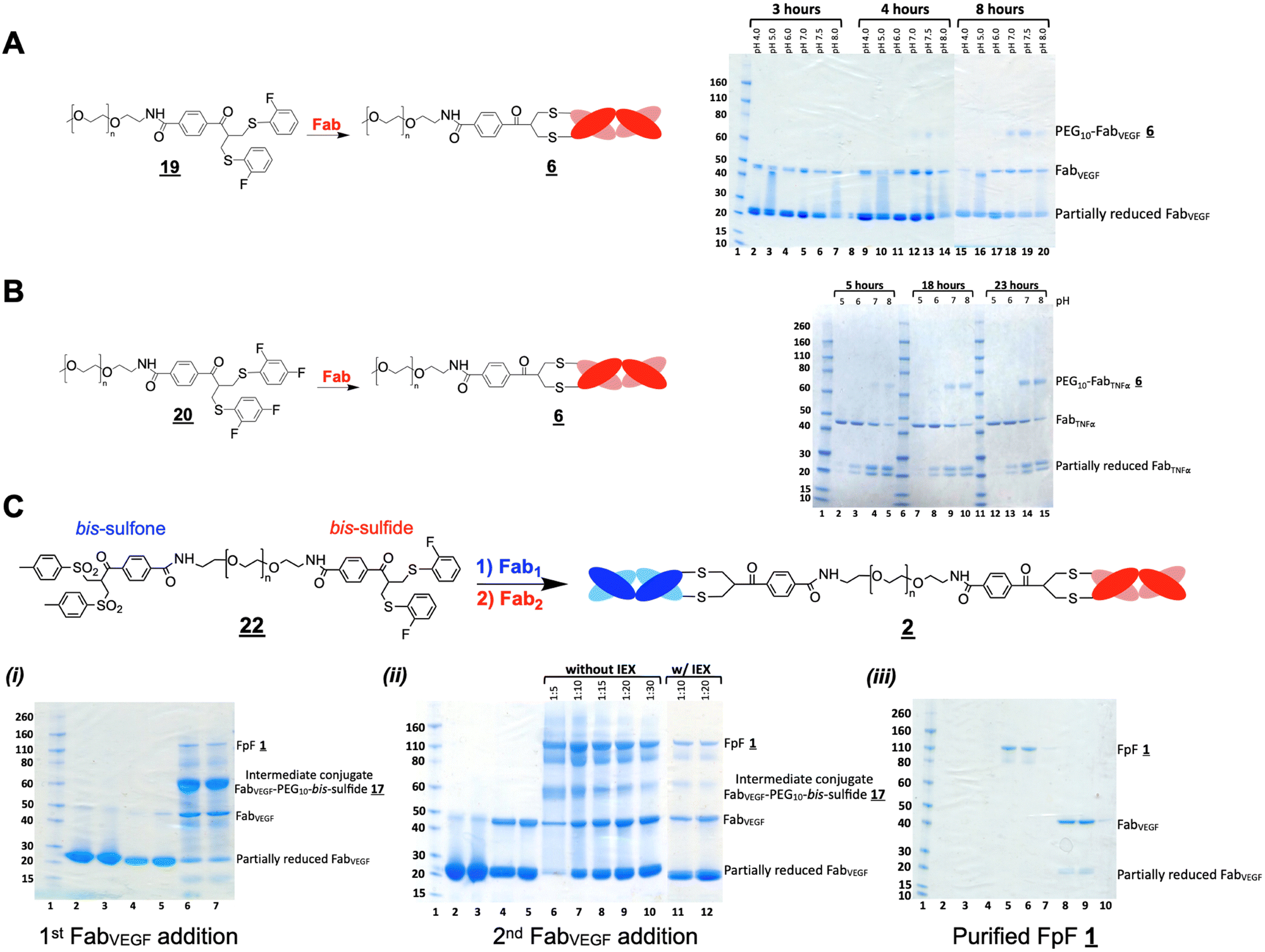 | ||
| Fig. 2 Conjugation of fluorodisulfide reagents (novex BisTris 4–12% gel stained with oomassie blue). (A) Scouting conjugation of FabVEGF to PEG10–bis-sulfide ortho-fluoride (2 eq.) from pH 4 to 8 at different incubation times. Lane 1: protein markers, lanes 2–7: 3-hour incubation time, lane 8: blank, lanes 9–14: 18-hour incubation time, lanes 15–20: 23-hour incubation time. (B) Scouting conjugation of FabTNFα to PEG10–bis-sulfide-difluoride (2 eq.) from pH 5 to 8 at different incubation times. Lane 1: protein markers, lanes 2–5: 5-hour incubation time, lane 6: protein markers, lanes 7–10: 18-hour incubation time, lane 11: protein markers, lanes 12–15: 23-hour incubation time. (C) Scouting reactions to prepare a FpF in two steps using the ortho-fluoro bis-sulfide bis-sulfone reagent . The same FabVEGF was used in both conjugation steps, so FpF was prepared rather than a bsFpF : (i) first FabVEGF conjugation reaction, lane 1: protein markers, lanes 2–3: 2 batches of reduced FabVEGF before PD10 elution, lanes 4 and 5: reduced FabVEGF after PD10 elution, lanes 6 and 7: first conjugation of FabVEGF to the reagent . (ii) Second FabVEGF conjugation reaction, lanes 2 and 3: 2 batches of reduced FabVEGF before PD10 elution, lanes 4 and 5: reduced FabVEGF after PD10 elution, lanes 6–10: second conjugation of increasing stoichiometries of FabVEGF with the corresponding intermediate conjugate, FabVEGF–PEG10–ortho bis sulfone , which had not been eluted over an IEX column, lanes 11 and 12: second conjugation of increasing stoichiometries of FabVEGF with the corresponding intermediate conjugate, FabVEGF–PEG10–ortho bis sulfone , which had been eluted over an IEX column. (iii) Fractions from SEC purification of the FpF , FabVEGF–PEG10–FabVEGF after a first step of IEX elution. | ||
bsFpFs prepared by conjugation–ligation
Considering the encouraging binding properties of FabVEGF–PEG10–FabHER2, we wanted to evaluate a larger number of bsFpF molecules. The di(bis-sulfone) and the asymmetric conjugation bis-sulfide bis-sulfone reagents are good reagents and can be used to prepare a bsFpF or other heterodimeric protein conjugates. To prepare several bsFpFs that could be used in early preclinical research, we decided to examine preparing these molecules by a conjugation–ligation strategy (Scheme 5). Conjugation–ligation strategies to prepare protein–protein and protein–drug conjugates are well known.41–43 In principle, conjugation ligation reagents and are first used to site-specifically modify separate proteins by disulfide rebridging conjugation to give intermediate conjugates and that can be ligated to give the desired bsFpF . Different Fabs can be used to provide stock solutions of the intermediate conjugates and that can be ligated to form a family of bsFpFs. Such an approach minimises the number of protein conjugation reactions needed and the need to purify bsFpFs from the unmodified protein.
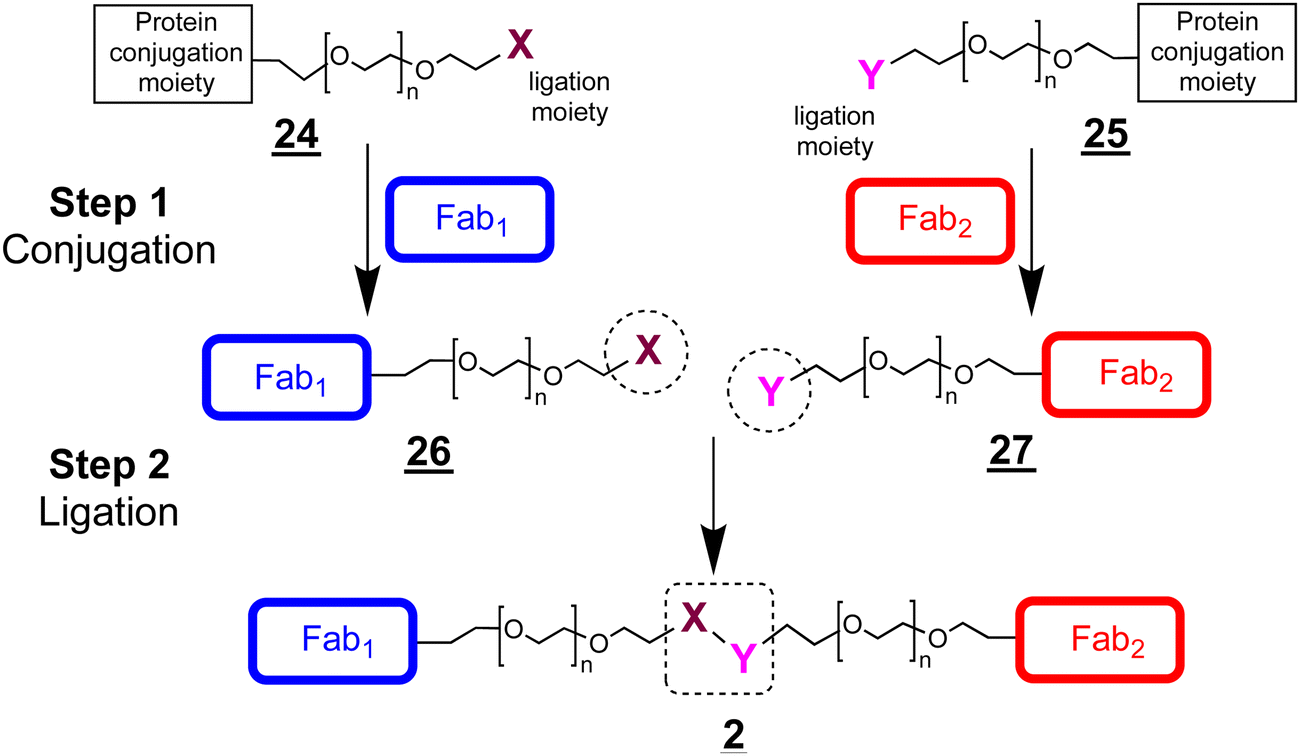 | ||
| Scheme 5 A conjugation–ligation strategy to prepare bsFpFs . The conjugate intermediates (e.g. and ) can be used with different Fabs in different combinations to prepare families of bsFpFs . | ||
The ligation step requires reactive moieties that will not undergo a reaction with the conjugated protein. Many ligation strategies and chemical moieties have been described, and we focus here on ligation moieties capable of undergoing cycloaddition.41,43 To prepare bsFpFs by a conjugation–ligation approach, we examined the bis-sulfone trans-cyclooctene (TCO) and tetrazine (Tz) reagents and (Scheme 6). The bis-sulfone–PEG–TCO reagent was derived from a mono-Boc protected PEG di-amine precursor with a molecular weight of either 5 and 10 kDa and the bis-sulfone–PEG–Tz reagent was derived from a PEG precursor of 5 and 10 kDa (ESI†). The TCO and Tz bis-sulfone reagents and readily underwent ligation at pH values ranging from 5 to 9 (ESI,† Fig. S5). Several Fabs were conjugated to the TCO and Tz bis-sulfone reagents and using our standard conjugation conditions with 1.5 to 2.0 equivalents of the conjugation reagent40,44 to produce intermediate Fab conjugates and for ligation (Scheme 6).
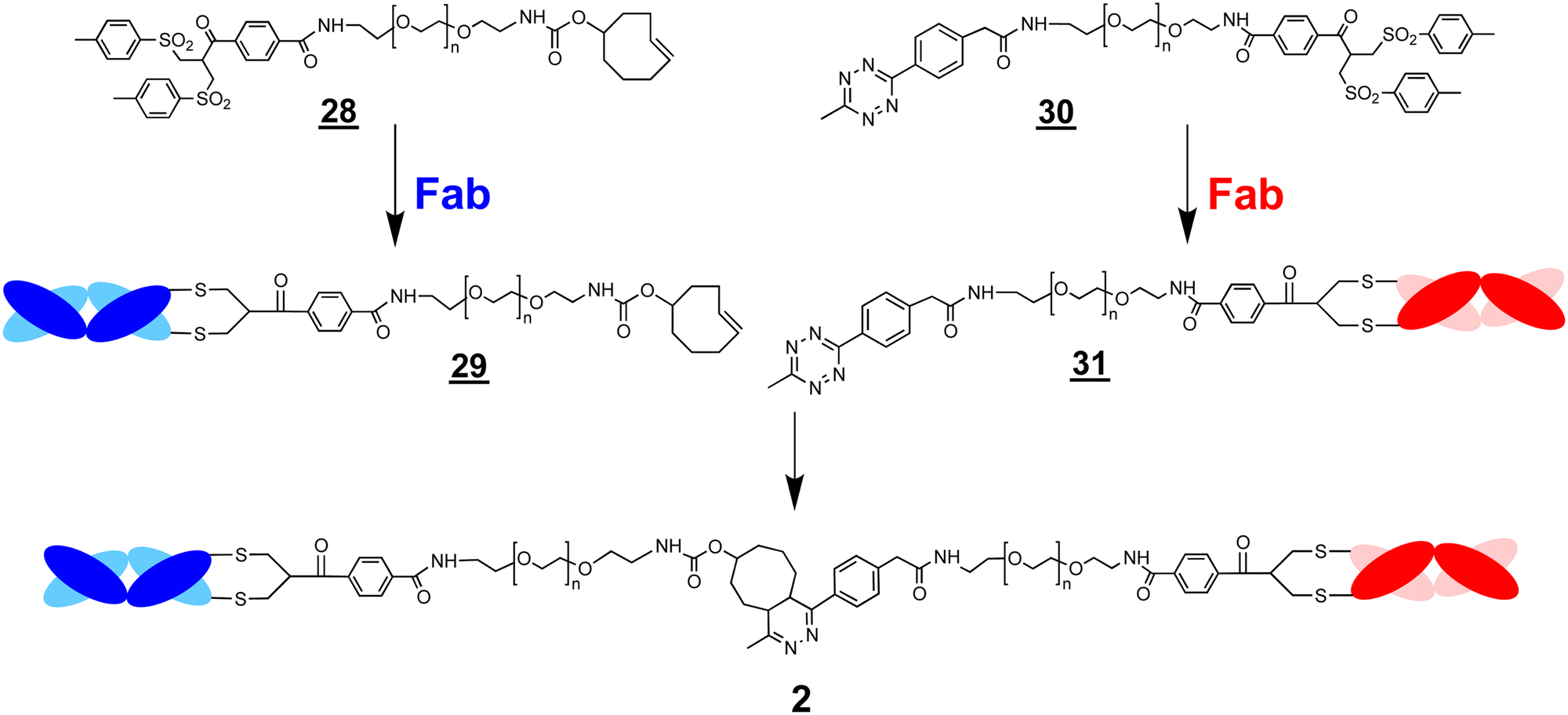 | ||
| Scheme 6 A conjugation–ligation strategy using the reagents, bis-sulfone–PEG–TCO and bis-sulfone–PEG–Tz , to prepare bsFpFs . The PEG–Fab conjugate intermediates and are prepared by site-specific conjugation of two different Fabs through the bis-sulfone functional group. Ligation of these conjugate intermediates results in the preparation of the bsFpF . | ||
A representative SDS–PAGE gel is shown in Fig. 3 displaying the intermediate conjugates and (lanes 4 and 5). Ligation to produce the bsFpF (or the homodimeric FpF ) was accomplished by first eluting the individual conjugation reaction mixtures over an ion exchange column to remove any un-conjugated TCO and TZ bis-sulfone reagents and . It was also possible to remove unconjugated reagents by viva spin. A representative ligation reaction mixture is shown in the gel in Fig. 3 (lane 6). Purification of the final bsFpFs was achieved by IEX and SEC with examples shown in lanes 7 to 12 (silver staining was used for detection in lanes 9–12) with the bsFpFs displaying a band at about 115 kDa (Fig. 3).
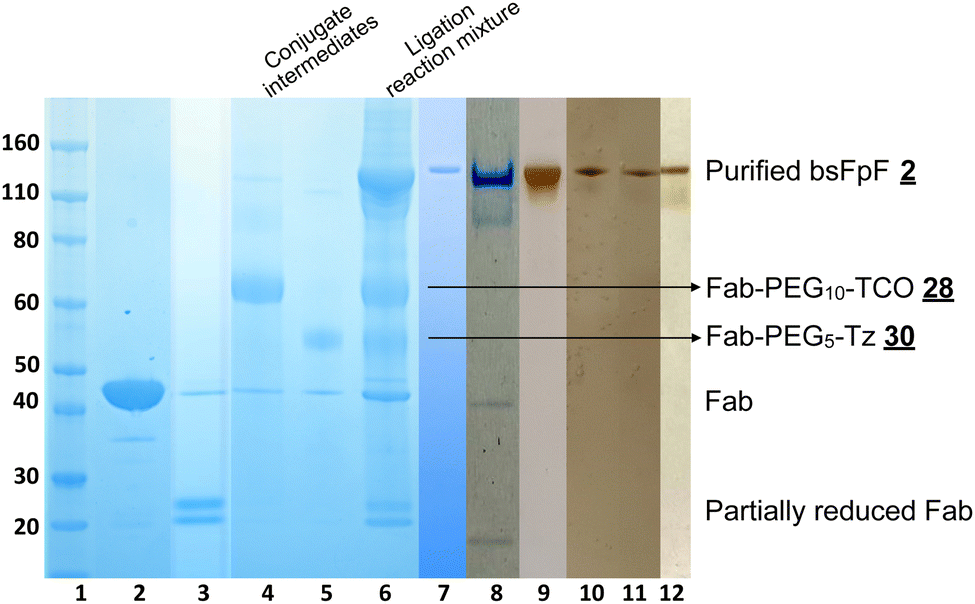 | ||
| Fig. 3 Representative SDS–PAGE analysis for preparation of bsFpF using the bis-sulfone conjugation–ligation reagents and , and the SDS–PAGE was stained with instant blue for protein staining (lanes 1–8) and silver staining for detecting any trace of impurity (lanes 9 to 12), lane 1: protein markers, lane 2: FabVEGF, lane 3: reduced FabVEGF, lane 4: FabVEGF conjugated to the bis-sulfone–PEG10–TCO reagent , lane 5: FabVEGF conjugated to the bis-sulfone–PEG5–Tz reagent , lane 6: reaction mixture for the ligation reaction between conjugate intermediates FabVEGF–PEG10–TCO and FabVEGF–PEG5–Tz , lanes 7–12: purified bsFpFs (Fab1–PEG15–Fab2, silver stain used for lanes 9–12 to assess purity). Different Fabs are used for conjugation; lane 7: homodimer FabVEGF–PEG15–FabVEGF, lane 8: FabVEGF–PEG15–FabHA, lane 9: FabVEGF–PEG15–FabIL6R, lane 10: FabVEGF–PEG15–FabTNFα, lane 11: FabIL6R–PEG15–FabTNFα, and lane 12: FabVEGF–PEG15–FabCOL2. | ||
Fig. 3 Several clinically approved IgGs including bevacizumab (for anti-VEGF Fab), tocilizumab (for anti-IL6R Fab), infliximab (for anti-TNFα Fab) and secukinumab (for anti-IL17 Fab) were digested to provide the Fabs that were used. We have previously described scaling of the papain digestion process to accommodate 100 mg of IgG resulting in the isolation of 50 mg of pure and stable Fab.39Table 1 lists the bsFpFs that were prepared by conjugation–ligation using the TCO and Tz bis-sulfone reagents and . To facilitate binding comparisons, the homodimer FpFVEGF was also prepared using the same reagents and , with FabVEGF sourced from digestion of bevacizumab. Additionally, another homodimer FpFVEGF construct was prepared using the di(bis-sulfone) reagent . The concentration of both FpFs and bsFpFs was determined by the micro-BCA-assay. Isolated yields varied between 15 and 20% at the reaction scales used and typically about 0.2 mg of purified bsFpF could be obtained starting from 1 mg of each Fab.
and targeting pro-inflammatory targets (TNFα, IL6R and IL17) and pro-angiogenic target (VEGF) and affinity targets (HA and collagen-2) along with their isolation yields
| Fab1 | Fab2 | bsFpF Isolated yields % |
|---|---|---|
| FabVEGF | FabIL6R | 20 |
| FabVEGF | FabTNFα | 16 |
| FabTNFα | FabIL6R | 14 |
| FabVEGF | FabIL17A | 11 |
| FabTNFα | FabIL17A | 11 |
| FabVEGF | FabCOL2 | 13 |
| FabVEGF | FabHA | 12 |
A conjugation–ligation–conjugation strategy was also examined (Scheme 7). The FabVEGF–PEG10–TCO conjugate was ligated with the Tz–PEG5–bis-sulfone reagent (1.5 equivalents) to give the ligation intermediate, FabVEGF–PEG15–bis-sulfone (Fig. 4, lane 6). The Fab targeted to an interleukin-6 receptor (FabIL6R) was then conjugated to intermediate to give the final bsFpF (FabVEGF–PEG15–FabIL6R) (Fig. 4, lane 7). The conversion to give this bsFpF, FabVEGF–PEG15–FabILR6 appeared to be about the same as when prepared by ligation of the separate conjugate intermediates and (Scheme 6).
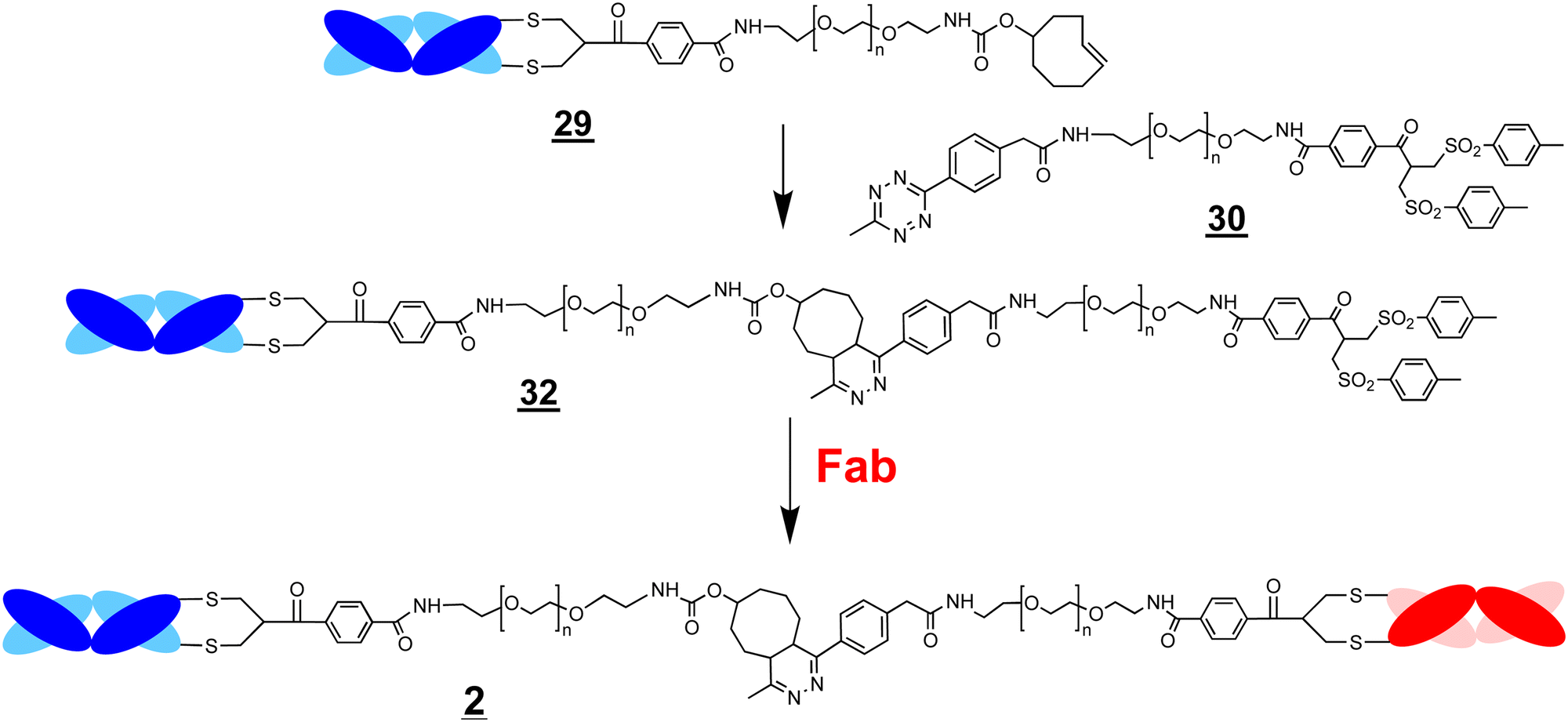 | ||
| Scheme 7 A conjugation–ligation–conjugation strategy to prepare the bsFpF . The Fab–PEG–TCO conjugate intermediate undergoes ligation with the bis-sulfone–PEG–Tz reagent to give the intermediate Fab–PEG–TCO . A second Fab is then conjugated to the intermediate to give the final bsFpF . | ||
 | ||
| Fig. 4 The SDS–PAGE gel displaying preparation of a bsFpF by conjugation–ligation–conjugation; Fab is first conjugated to bis-sulfone–PEG10–TCO to give the Fab–PEG10–TCO conjugate intermediate which is then ligated to bis-sulfone–PEG5–Tz to form the bis-sulfone terminated ligation intermediate (note Scheme 7) which can undergo conjugation with another Fab to give the bsFpF. Lane 1: protein markers, lane 2: FabVEGF, lane 3: FabVEGF + DTT, lanes 4 and 5: conjugation of reduced FabVEGF to bis-sulfone–PEG10–TCO , and then purified, lane 6: FabVEGF–PEG10–TCO ligation with bis-sulfone–PEG5–Tz to give the ligation intermediate which then underwent conjugation with reduced FabIL6R in lane 7: resulting in the formation of the FabVEGF–PEG15–PEGIL6R bispecific molecule. | ||
We also prepared bis-sulfone–PEG3-N3 and bis-sulfone–PEG5–DBCO (ESI,† Fig. S6) to examine the well-known strain-promoted alkyne–azide cycloaddition which uses azide (N3) and dibenzocyclooctyene (DBCO) moieties for ligation.42,45,46 Good conjugation conversion with FabTNFα was achieved with 1.5 to 2.0 equivalents of the bis-sulfone reagents and after a 5-hour incubation time at pH 8 to give the respective conjugate intermediates and (ESI,† Fig. S6A and B). The conjugation solutions were centrifuged using a viva spin column to remove excess starting reagents. Upon mixing the intermediate conjugates and (ESI,† Fig. S6C), the desired homodimeric FpF was formed but at apparently lower conversion than with the corresponding conjugate intermediates derived from the TCO and Tz bis-sulfone reagents and . Similar ligation results were observed for preparation of the homodimeric FpF derived from FabVEGF (ESI,† Fig. S7A) and ligation could not be improved when an excess reagent was removed by IEX chromatography instead of viva spin. Commercially available methoxy PEG10–DBCO and bis-sulfone PEG3-N3 did undergo ligation as would be expected without the presence of the conjugated Fabs (ESI,† Fig. S7B, lane 4).
The presence of the conjugated protein in each ligation intermediate (e.g. Fab–PEG-N3 and Fab–PEG–DBCO , ESI,† Fig. S6) may have resulted in conformational masking of the hydrophobic ligation moiety in the relatively large PEG linker element when one terminus of the PEG is conjugated to a 50 kDa protein (i.e. Fab). The TCO and Tz moieties are known to undergo faster ligation than the azide and DBCO moieties43 and this may allow more efficient ligation of the Fab conjugate intermediates. Shorter PEG linkers would be expected to allow more facile ligation.
The ELISA was first used to evaluate the binding affinity of FabVEGF–PEG15–FabIL6R. Separate plates were coated with VEGF and IL6R. There was no non-specific binding observed when infliximab (anti-TNFα IgG) was incubated with the VEGF or IL6R coated plates. The binding affinities (KD) determined by the ELISA for the parent anti-IL6R antibody (tocilizumab) and the associated FabIL6R obtained by proteolytic digestion were 0.13 nM for tocilizumab and 1.50 nM for FabIL6R (ESI,† Fig. S8). As expected, tocilizumab exhibited a lower KD and higher binding affinity due to its bivalent nature as an IgG compared to the monovalent FabIL6R. ELISA affinities represent average values from two replicates. The ELISA binding affinity of the bispecific FabVEGF–PEG15–FabIL6R to VEGF (KD 1.80 nM) and IL6R (KD 2.55 nM) is shown in Fig. 5. For comparison, the ELISA derived binding of PEG10–FabVEGF (KD 2.25 nM) and PEG5–FabIL6R (KD 3.20 nM) was determined (Table 2).
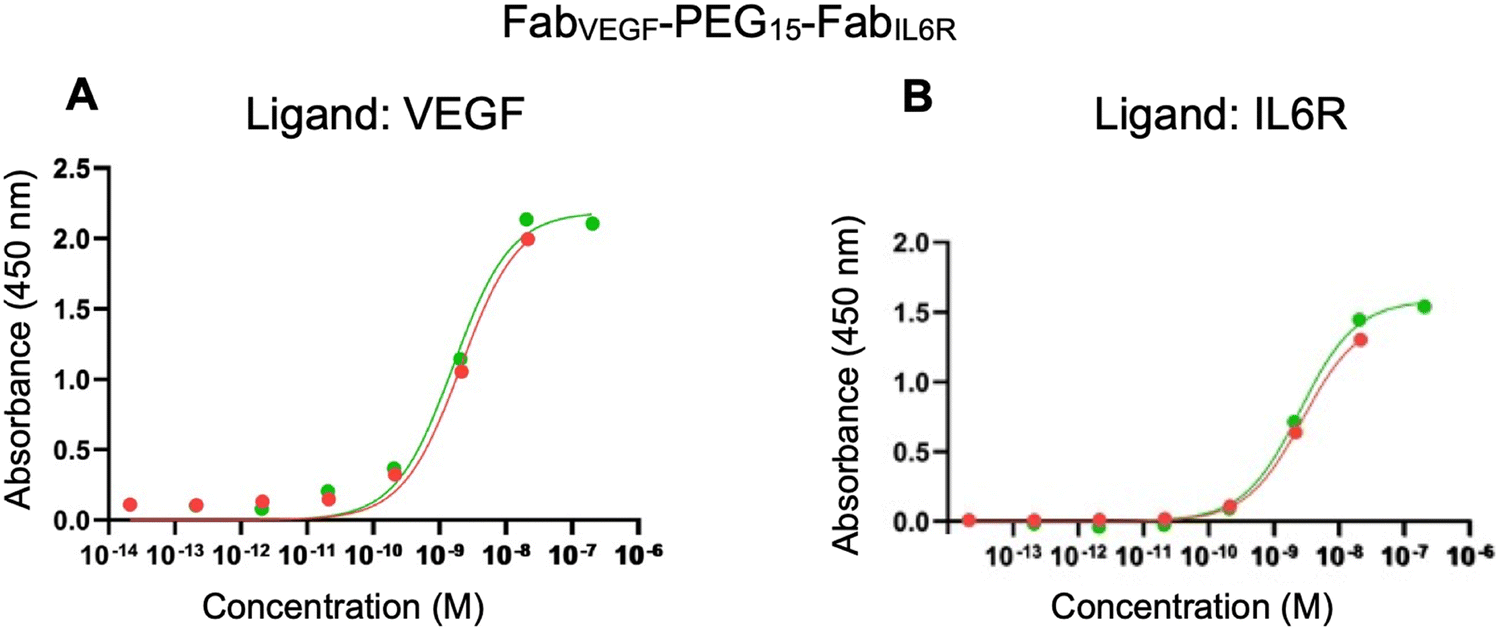 | ||
| Fig. 5 ELISA results for (A) FabVEGF–PEG15–FabIL6R (green data point) and mono PEG–FabVEGF (red data point) with a concentration range of 1.3 × 10−7 to 1.3 × 10−13 M over wells coated with VEGF (0.1 μg). (B) FabVEGF–PEG15–FabIL6R (green data point) and mono PEG–FabIL6R (red data point) with a concentration range of 1.3 × 10−7 to 1.3 × 10−13 M over wells coated with IL6R (0.1 μg). | ||
| Conjugates | Ligand | K D (nM) |
|---|---|---|
| PEG10–FabVEGF | VEGF | 2.25 |
| FabVEGF–PEG15–FabIL6R | VEGF | 1.80 |
| PEG5–FabIL6R | IL6R | 3.20 |
| FabVEGF–PEG15–FabIL6R | IL6R | 2.55 |
The binding affinity of FabVEGF–PEG15–FabIL6R was also evaluated by SPR. VEGF165 was immobilised to a CM3 chip (95 RU) and his-Tag IL6R was immobilised to a nitrilotriacetic acid (NTA) chip. The precursor Fabs and PEG–Fab conjugates were used for comparison (i.e. FabVEGF, FabIL6R, PEG10–FabVEGF and PEG5–FabIL6R). The concentration-dependent binding of the bispecific FabVEGF–PEG15–FabIL6R was observed for both VEGF and IL6R (ESI,† Fig. S9). The kinetic rate constants and affinities were calculated (Table 3) using a 1:1 binding model. ESI,† Table S1 summarises the SPR derived kinetic rate constants and affinities that were obtained for bsFpFs prepared by conjugation–ligation using the TCO and Tz bis-sulfone reagents and .
:1 binding model. Chi2 values, a fitting quantitative measure, were 0.12 for FabVEGF–PEG15–FabIL6R and 0.52 for mono-PEG–Fabs. The optimal Chi2 value is within a 10% range of the Rmax value
| Conjugates | Ligand | k a (1/Ms) × 104 | SD ka × 104 | k d (1/s) × 10−4 | SD kd × 10−4 | K D (nM) | SD KD |
|---|---|---|---|---|---|---|---|
| PEG10–FabVEGF | VEGF | 1.30 | 0.06 | 1.4 | 0.35 | 10.9 | 3.0 |
| FabVEGF–PEG15–FabIL6R | VEGF | 0.84 | 0.31 | 1.0 | 0.4 | 12 | 7.8 |
| PEG5–FabIL6R | IL6R | 3.90 | 1.83 | 12.5 | 4.94 | 38.1 | 29.6 |
| FabVEGF–PEG15–FabIL6R | IL6R | 2.80 | 1.30 | 5.7 | 0.56 | 23.3 | 14.4 |
The homodimer FpFVEGF prepared by the di(bis-sulfone) PEG reagent appeared to have lower KD due to a faster association rate (ka) constant and a slower dissociation rate (kd) compared to the homodimer FpFVEGF synthesised by a conjugation–ligation (reagents and ) approach, studied by both ELISA and SPR (Fig. 6).
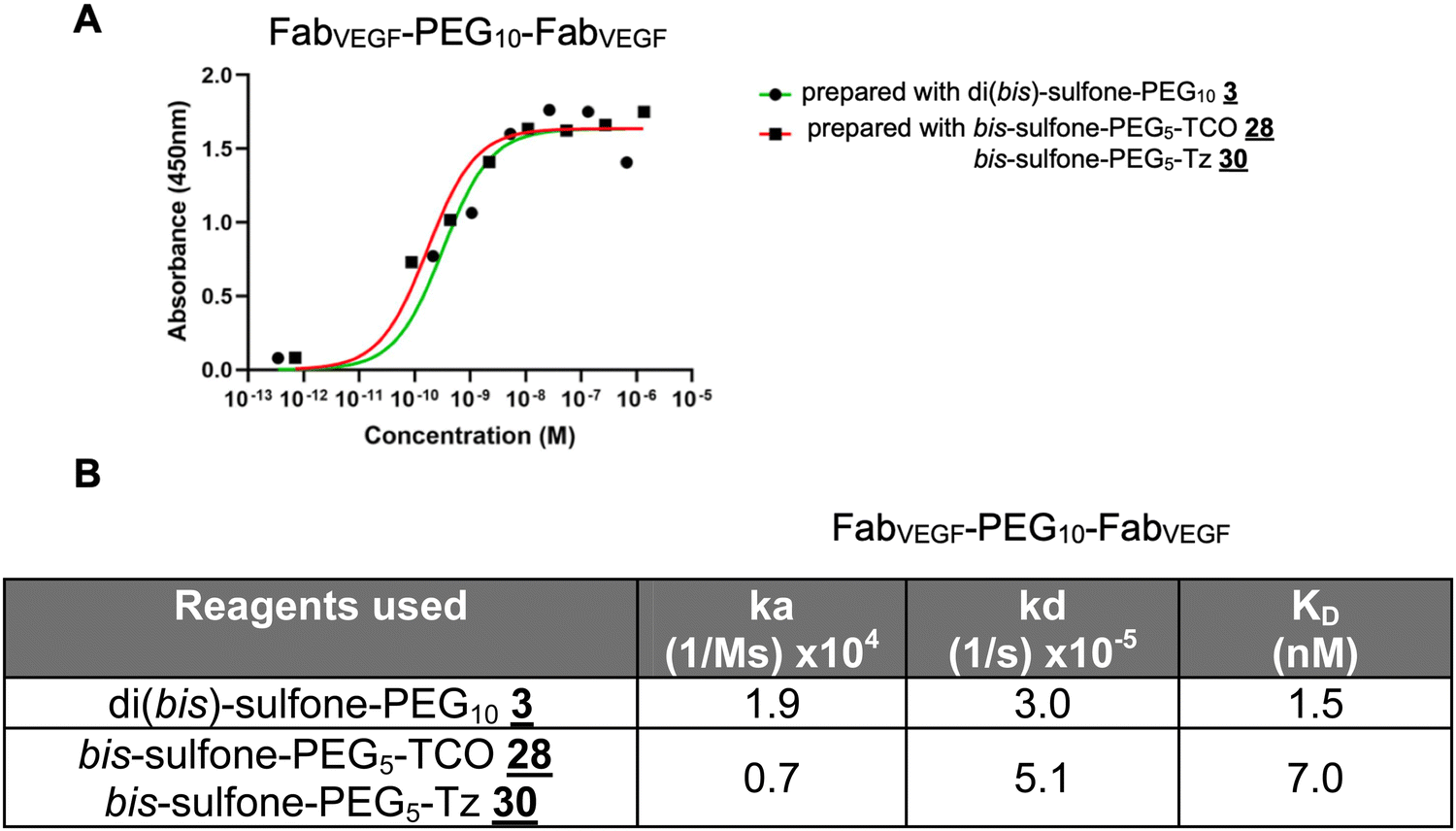 | ||
| Fig. 6 Binding analysis of the homodimer FpFVEGF prepared using the conjugation reagent and conjugation–ligation reagents and using (A) ELISA, plate coated with VEGF165 (0.1 μg) and (B) SPR technique, and the CM3 chip immobilised with VEGF (95 RU). | ||
Immunoblotting (dot blot assays) is also useful for qualitative assessment of binding interactions; for example, FabVEGF–PEG15–FabCOL2 displayed binding to its respective antigens using a dot blot assay (Fig. S10, ESI†). A limitation of ELISA, SPR and dot blot assays is that these techniques require immobilisation of one of the binding partners which may not fully represent the dynamic interactions of the binding partners in solution.47 Microscale thermophoresis (MST) enables the practical measurement of binding interactions in solution. Using MST, the binding analysis of bispecific FabVEGF–PEG15–FabIL6R was performed on VEGF, IL6R and a mixture of VEGF/IL6R (Fig. S11, ESI†). The binding traces revealed that the bispecific FabVEGF–PEG15–FabIL6R maintained its binding towards both VEGF and IL6R, both as individual targets and when presented as a mixed target (VEGF/IL6R) in solution.
Discussion
There is a rich history of investigating dimeric low molecular weight molecules and proteins linked by PEG, e.g.48–51 As a dimeric antibody-based molecule, the FpF motif is a good IgG binding mimetic.1,2 Each Fab is covalently bound to a terminus of a linear, flexible PEG in the region where the Fab is naturally anchored in an IgG. The PEG linker appears to mimic some of the conformational properties of the IgG hinge to allow flexible Fab–epitope interactions during binding, which are important for antibody functions.3 FpFs evaluated to date appear to have comparable binding thermodynamics to the corresponding IgG antibody with the same Fabs.1,2 FpF affinity is achieved by a slower association rate (ka) that is then compensated by a correspondingly slower dissociation rate (kd). A slower kd may have important therapeutic potential to help maintain localised therapeutic concentrations in tissues.52–54FpFs do not have the extended solution structure of a PEG–Fab conjugate where there is a protein only on one terminus of the PEG molecule.1 FpFs appear to have a similar solution size to IgGs broadly independent on the PEG linker size in the range of 5–20 kDa.1 It is thought that the Fab moieties in a FpF may self-associate to reduce the size of PEG in solution compared to what is observed for PEGylated proteins with a protein conjugated at only one PEG terminus. FpFs in solution may possess some of the conformational properties associated with A–B–A block copolymers55,56 where the Fabs (A block) can self-associate. Since bsFpFs have a Fab at each terminus of a PEG molecule like the homo-dimeric FpFs, it would be expected that the bsFpFs will have a similar size to the homodimeric FpFs.
It will be important to examine the differences in the solution structures for the FpFs made by the conjugation-only approach and by the conjugation–ligation approach where there is a ligation element located within the PEG linker. Although binding appears broadly similar, the ligation elements may exert conformational influences (Fig. 6). It is also clear from other studies that the PEG linker length in dimeric protein conjugates can influence binding, especially below a threshold length that is shorter than the ligand or the epitope distance.49,50,57 We recognise that there is also potential to optimise the bsFpF linker length depending on the specific application.
Since the IgG hinge region is susceptible to degradation, the use of a PEG linker and site-specific bis-alkylation conjugation at the accessible Fab disulfide contribute to the stability and reduced propensity for aggregation of FpFs.1 The thiol–ether bonds conjugating each Fab to the flexible PEG linker acting as the surrogate hinge are more stable than the unmodified accessible disulfide in the Fab. Much effort remains focused on developing antibody-based molecules with optimal physicochemical properties,58 some of which FpFs may have the potential to display.
Multifunctional protein conjugates including bispecific antibodies can be made using proteins that are readily accessible by recombinant means (e.g. Fabs and single chain fragments) which are then conjugated by selective chemical strategies. Outstanding studies have been published exemplifying this approach,35,59–61 including strategies to conjugate proteins or peptides at each terminus of a functionalised PEG linker analogous to what we have described for FpFs.9,10,62 FpFs comprise elements (e.g. Fabs and PEG) which separately have been clinically proven viable for use.
The di(bis-sulfone) reagent was examined to prepare the heterodimeric bsFpFs because this is an effective reagent to prepare the homodimeric FpFs . We anticipated that conjugation of the first Fab to the di(bis-sulfone) would be faster than the second Fab due to possible hydrophilic steric shielding effects of the second bis-sulfone conjugation moiety after conjugation of Fab1. Modification of the leaving groups in the bis-sulfone conjugation moiety to increase hydrophilicity was considered,63 although an excess reagent would still be required for the first conjugation step in a conjugation-only approach with a di(bis-sulfone) reagent. The addition of excess di(bis-sulfone) during the conjugation of Fab1 predominantly gave the desired Fab1–PEG–bis sulfone intermediate (e.g. FabVEGF–PEG10–bis-sulfone , Fig. 1A). The use of excess di(bis-sulfone) necessitates its removal prior to the conjugation of Fab2. Ion exchange chromatography (IEX) effectively removed the excess reagent along with trace remaining Fab1 and the homodimer (i.e. Fab1–PEG–Fab1).
Conjugation of Fab2 required longer incubation times (Fig. 1B), which may not be ideal for maintaining Fab2 stability and preventing reoxidation of the Fab2 accessible disulfide. The di(bis)-sulfone was purified by precipitation,64 which works well for reagents designed to undergo a single conjugation reaction, e.g. bis-sulfone PEGylation reagents (Fig. 1C).65 The use of pure PEG precursors and the chromatographic purification of the di(bis-sulfone) does give a reagent with less possible dead chain ends (Fig. S1, ESI†)9 and does appear more effective for the second conjugation step (Fig. 1A, lane 8). Although we are keen to utilise reagents that could be prepared without a tedious purification to isolate the reagent, it is important to acknowledge that minimisation of dead chain ends in all conjugation reagents is generally preferred.
The asymmetric FpF reagents (Schemes 3 and 4) indicate that we could reduce the excess of the reagent needed for the conjugation of Fab1 and reduce the formation of the undesired homodimer. The fluoro-substituted bis-sulfide conjugation moieties are less reactive than the bis-sulfone conjugation moiety; however, all these bis-alkylation moieties yield the same conjugate product (e.g. PEG–Fab , compare Fig. 1C and 2A). There are few if any di-conjugation reagents that undergo the same site-specific conjugation reaction at each moiety but with varied reactivity.
Our preliminary experiments indicated that the combination of reduced reactivity for the bis-sulfide conjugation moiety for Fab2 and the reduced reaction rate for the second conjugation to the Fab–PEG–bis-sulfide intermediate (Scheme 4) meant that there was often remaining Fab2 present with the conditions that were examined. Considering the advantages of a cleaner reaction for Fab1, more work is required to optimise the conjugation of the second protein (e.g. Fab2).
The ligation approach to prepare bsFpFs is to utilise bis-alkylation conjugation that site-specifically rebridges the two cysteine thiols from the accessible native disulfide of a Fab to give conjugate intermediates that are then ligated via an orthogonal cycloaddition reaction. Many ligation strategies have been described (e.g. ref. 66–68). Ligations are often accomplished by orthogonal reactions (e.g. ref. 69), with cycloaddition reactions dominating in recent years (e.g. ref. 45, 46 and 68) to make multifunctional proteins including bispecific antibody mimetics.35,45 Protein modification strategies involving conjugation and/or ligation will inevitably evolve in the context of engineering protein structure recombinantly to best match a given modification strategy. Although our use of reagents with PEG molecular weights greater than 3 kDa is designed to yield bsFpFs that can be optimised to exploit spatial–temporal relationships that exist with IgGs, the use of these relatively long PEG linkers may also result in conjugation intermediates (e.g. Fab–PEG10–TCO, Fab–PEG5–Tz) that have reduced ligation reactivity due to conformational masking of the ligation moiety.
Conjugation–ligation (Scheme 6) allows for a combinatorial approach to prepare bsFpFs . For example, one conjugate intermediate (e.g.) could undergo ligation with many different versions of its partner conjugate intermediate (e.g.) to give a small family of bsFpFs . Reaction orthogonality to the protein in the ligation step also reduces the potential for non-selective protein conjugation which is possible by the conjugation only approach when longer incubation times are used (e.g. >1 day). A conjugation–ligation approach also avoids generation of homo-dimeric protein conjugates that can result from a conjugation only approach, minimising the number of protein conjugation reactions needed and the need to purify bsFpFs from the unmodified protein.
Ligation using trans-cyclooctene (TCO) and tetrazine (Tz) moieties is faster than the cycloaddition between azide and DBCO moieties.42,43,70 Bis sulfone–PEG–TCO and bis sulfone–PEG–Tz were prepared and used to make several bsFpFs (Scheme 6) that were isolated (Fig. 3 and Table 1). We found that the isolated yields of purified proteins modified by different thiol specific conjugation strategies at a small scale (∼0.5 to 2.0 mg) often give moderate to low yields due to the loss of protein during purification.71,72 It is possible with high conversion that disulfide rebridging PEGylation reactions at these or slightly higher scales give isolated yields of 45–65% of the modified protein (e.g. ref. 40). Although the conjugation–ligation reagents and were precipitated 3–4 times during isolation (ESI†), additional chromatographic purification of the reagent (e.g. Fig. S1, ESI†) would be expected to increase overall conversion and isolated yields of the bsFpFs at the scales we examined.
Challenges exist in the assay development of dual-targeting molecules.73–76 Using FabVEGF–PEG15–FabIL6R as a representative example, we examined the binding affinity by dot blot, ELISA, SPR and MST (Tables 2 and 3). The KD value obtained from the binding assays varies across these different experiments (compare the KD value for the same bsFpF molecule between Tables 2 and 3). ELISA experiments indicate each Fab in FabVEGF–PEG15–FabIL6R has a similar affinity (KD) as the corresponding PEG–Fab (Table 2). PEG conjugation reduces protein activity generally, and certainly Fab affinity compared to the unmodified Fab due to steric shielding effects of PEG. Disulfide-rebridging conjugation at the accessible Fab disulfide is near the region of the Fab that is connected to the hinge in a native IgG and is maximally distal to the complementarity-determining region (CDR) responsible for Fab binding to its respective target. Conjugation at this accessible disulfide is thought to cause less reduction in Fab affinity than conjugation elsewhere in the Fab.1,40
The binding kinetics to give the association (ka) and dissociation (kd) rate constants for three bsFpFs determined by SPR are summarised in Table 3 and ESI,† Table S1. Low-density ligand immobilisation and a high flow rate (30 μL min−1) were used to minimise mass transfer limitations and re-binding effects to the immobilised ligand.77 Considering binding to immobilised VEGF165, the apparent KD and kd values for mono-PEG10–FabVEGF (KD 10.9 nM; kd 1.4 × 10−4 1/s) were similar to the bsFpFs that with one FabVEGF; (i) FabVEGF–PEG15–FabIL6R (KD 12 nM and kd 1.0 × 10−4 1/s) (Table 3) and (ii) FabVEGF–PEG15–FabTNFα (KD 10 nM and kd 0.81 × 10−4 1/s) (ESI,† Table S1).
We and others have previously shown that the dissociation rate constant of PEG modified Fabs remains broadly similar compared to the unmodified Fab.40,78 This trend was also observed for the dissociation of Fabbeva in PEG10–Fabbeva, and FabVEGF–PEG15–FabIL6R from VEGF and FabIL6R in PEG5–FabIL6R and FabVEGF–PEG15–FabIL6R from IL6R. Similar trends in the dissociation rate constants were observed for FabVEGF–PEG10–FabHER2 prepared using the PEG di(bis-sulfone) reagent (Fig. 1D). The data indicate that there was a little difference between the dissociation rate constants for FabVEGF, PEG10–FabVEGF and FabVEGF–PEG10–FabHer-2 conjugates (Fig. 1D). There is potential that the preparation of PEG–Fab1 will be a good surrogate for the binding of Fab1 in the corresponding bsFpF (i.e. Fab1–PEG–Fab2).
The association rate constants (ka) were smaller for the FabVEGF–PEG15–FabIL6R products to both immobilised VEGF and IL6R compared to the corresponding mono-PEG–Fab constructs (Table 3). The association rate constant of FabVEGF–PEG15–FabIL6R is slower probably because the bsFpF is larger in molecular weight than a PEG–Fab which results in a slower rate of transport from bulk solution to the chip surface. A similar trend was observed for FabVEGF–PEG10–FabHER2 to have a slower association rate constant compared to PEG10–FabVEGF. Reduction in binding affinities (KD) for Fab conjugates compared to the corresponding unmodified Fab tends to be due to reductions in the association rate constants (ka).1,40,78 All bsFpFs molecules have a larger mass than PEG10–Fab conjugates which results in a slower rate of transport over the chip surface.
ELISA, SPR and dot blot techniques are useful to confirm the binding of each Fab element in a bsFpF but immobilisation of binding targets is required on a chip or the surface of a plate. Such assays do not fully replicate the dynamic nature of in vivo interactions where the binding partners interact in solution while moving freely.79,80 Isothermal calorimetry can be used to evaluate binding properties in solution, but MST is also a real-time, solution-based method to measure binding interactions. MST requires that one protein be non-specifically labelled with a fluorescent dye. We elected to label the bsFpF in these experiments to utilise the same labelled molecule to evaluate both binding moieties in the bsFpF. Using MST, the binding analysis of bispecific FabVEGF–PEG15–FabIL6R was performed on VEGF, IL6R and a mixture of VEGF/IL6R (Fig. S11A–C, ESI†). MST binding curves can appear in both directions as observed in Fig. S11A–C (ESI†) depending on the diffusion coefficients of the labelled protein and complex.81 The binding curves indicate that the bispecific FabVEGF–PEG15–FabIL6R maintained its binding towards both VEGF and IL6R separately and as a mixture.
To consider the presence of the ligation element in the PEG linker, we evaluated a homodimer FpFVEGF prepared by PEG di(bis)-sulfone and homodimer FpFVEGF prepared by conjugation–ligation (reagents and ). While the dissociation rate constant (kd) appeared broadly similar, the binding affinity (KD) was smaller for the FpFVEGF prepared by the ligation approach (Fig. 6). The lower binding affinity for the ligated FpFVEGF appears to be due to a slightly slower association rate constant (ka). The ligation element may slightly restrict the freedom for the association of the second Fab to the immobilised ligand. It will be worthwhile to explore the conformational influences of the presence and absence of the ligation element, specifically with the PEG linker molecular weight.
A range of bispecific FpFs targeting different antigen epitopes with therapeutic properties for pro-inflammatory targets (tumour necrosis factor alpha or TNF-α, interleukins 6R, and interleukins17) and a pro-angiogenic target (vascular endothelial growth factor or VEGF) were prepared. We also explored the preparation of 2 bsFpFs, FabVEGF–PEG–FabHA and FabVEGF–PEG–FabCOL2 where one Fab functions to bind to a specific non-therapeutic tissue (affinity targeting) and the other Fab functions to bind to a therapeutic target. Hyaluronic acid (HA) and collagen-II (COL2) are two endogenous tissue targets that are envisaged for increasing the biological residence time of a molecule within the vitreous cavity of the eye.82–85 If the relevant Fab can be sourced, then bsFpFs may have utility as bispecific antibody mimetics for early preclinical studies designed to explore new therapeutic strategies.
Conclusions
FpFs are IgG mimetics with the potential to act as IgG surrogates in a range of applications.1 Bispecific FpFs were prepared as possible mimetics of bispecific IgGs. Although each element of the bsFpF (e.g. Fab and PEG linker) has been used clinically or can be sourced (e.g. FabCOL2 and FabHA), the potential of bsFpFs may be in preclinical research to develop strategies for (i) drug delivery where one Fab targets a specific anchor point (e.g. hyaluronic acid or collagen II in the vitreous cavity of the eye) to achieve the enhanced residence time in the tissue or organ of interest, (ii) tissue regeneration where one moiety would bind to a scaffold86–88 while the other moiety would provide a required biological function or for cell immobilisation89 and (iii) drug target development to optimise the spatial–temporal aspects90,91 of binding kinetics of a bispecific antibody and where it is not possible to use multiple antibodies in a single dosage form (e.g. intraocular indications).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. C. is thankful for the PhD funding from the University of East London. N. I. is grateful for the PhD funding from the Santen Pharmaceutical Co., Ltd. W. R. G is thankful for the VC PhD scholarship from the University of West London. S. A., P. T. K., and S. B. are grateful for the funding from the National Institute of Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, the Helen Hamlyn Trust (in memory of Paul Hamlyn), Moorfields Eye Charity, Fight for Sight, and the Michael and Ilse Katz foundation.References
- H. Khalili, P. Khaw, R. Lever, A. Godwin and S. Brocchini, Fab-PEG-Fab as a potential antibody mimetic, Bioconjugate Chem., 2013, 24(11), 1870–1882 CrossRef CAS PubMed.
- H. Khalili, R. Lee, P. Khaw, S. Brocchini and A. Dick, An anti-TNF alpha antibody mimetic to treat ocular inflammation, Sci. Rep., 2016, 6, 36905 CrossRef CAS PubMed.
- R. Nezlin, Dynamic Aspects of the Immunoglobulin Structure, Immunol. Invest., 2019, 48(8), 771–780 CrossRef CAS PubMed.
- L. Harris, S. Larson, E. Skaletsky and A. McPherson, Comparison of the conformations of two intact monoclonal antibodies with hinges, Immunol. Rev., 1998, 163, 35–43 CrossRef CAS PubMed.
- I. R. Correia, Stability of IgG isotopes in serum, mAbs, 2010, 2, 221–232 CrossRef PubMed.
- H. Khalili, Using different proteolytic enzymes to digest antibody and its impact on stability of antibody mimetics, J. Immunol. Methods, 2021, 489, 112933 CrossRef CAS PubMed.
- S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, PEGylation of native disulfide bonds in proteins, Nat. Protoc., 2006, 1(5), 2241–2252 CrossRef CAS PubMed.
- S. Balan, J. W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Site-specific PEGylation of protein disulfide bonds using a three-carbon bridge, Bioconjugate Chem., 2007, 18(1), 61–76 CrossRef CAS PubMed.
- A. Herrington-Symes, J. Choi and S. Brocchini, Interferon dimers: IFN-PEG-IFN, J. Drug Targeting, 2017, 25(9–10), 881–890 CrossRef CAS PubMed.
- H. Khalili, P. Khaw and S. Brocchini, Fc-fusion mimetics, Biomater. Sci., 2016, 4(6), 943–947 RSC.
- S. E. Sedykh, V. V. Prinz, V. N. Buneva and G. A. Nevinsky, Bispecific antibodies: design, therapy, perspectives, Drug Des., Dev. Ther., 2018, 12, 195–208 CrossRef CAS PubMed.
- U. Brinkmann and R. E. Kontermann, The making of bispecific antibodies, mAbs, 2017, 9(2), 182–212 CrossRef CAS PubMed.
- D. Nagorsen, P. Kufer, P. A. Baeuerle and R. Bargou, Blinatumomab: A historical perspective, Pharmacol. Ther., 2012, 136(3), 334–342 CrossRef CAS PubMed.
- E. Wolf, R. Hofmeister, P. Kufer, B. Schlereth and P. A. Baeuerle, BiTEs: bispecific antibody constructs with unique anti-tumor activity, Drug Discovery Today, 2005, 10(18), 1237–1244 CrossRef CAS PubMed.
- H. Shim, Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations, Biomolecules, 2020, 10(3), 360 CrossRef CAS PubMed.
- S. W. Pipe, M. Shima, M. Lehle, A. Shapiro, S. Chebon, K. Fukutake, N. S. Key, A. Portron, C. Schmitt, M. Podolak-Dawidziak, N. S. Bienz, C. Hermans, A. Campinha-Bacote, A. Kiialainen, K. Peerlinck, G. G. Levy and V. Jiménez-Yuste, Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study, Lancet Haematology, 2019, 6(6), E295–E305 CrossRef PubMed.
- M. Collins, S. Awwad, N. Ibeanu, P. T. Khaw, D. Guiliano, S. Brocchini and H. Khalili, Dual-acting therapeutic proteins for intraocular use, Drug Discovery Today, 2021, 26(1), 44–55 CrossRef CAS PubMed.
- T. Hansel, H. Kropshofer, T. Singer, J. Mitchell and A. George, The safety and side effects of monoclonal antibodies, Nat. Rev. Drug Discovery, 2010, 9, 325–338 CrossRef CAS PubMed.
- P. Mayes, K. Hance and A. Hoos, The promise and challenges of immune agonist antibody development in cancer, Nat. Rev. Drug Discovery, 2018, 17, 509–527 CrossRef CAS PubMed.
- G. Riethmuller, Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on, Cancer Immun., 2012, 12, 12 Search PubMed.
- M. Brennan, P. F. Davison and H. Paulus, Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin-G1 fragments, Science, 1985, 229(4708), 81–83 CrossRef CAS PubMed.
- U. D. Staerz, O. Kanagawa and M. J. Bevan, Hybrid antibodies can target sites for attack by t-cells, Nature, 1985, 314(6012), 628–631 CrossRef CAS PubMed.
- M. J. Glennie, H. M. McBride, A. T. Worth and G. T. Stevenson, Preparation and performance of bispecific F(ab’-gamma)2 antibody containing thioether-linked Fab’-gamma fragments, J. Immunol., 1987, 139(7), 2367–2375 CrossRef CAS.
- M. Surowka, W. Schaefer and C. Klein, Ten years in the making: application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins, mAbs, 2021, 13(1), 1967714 CrossRef PubMed.
- S. Dickopf, G. J. Georges and U. Brinkmann, Format and geometries matter: Structure-based design defines the functionality of bispecific antibodies, Comput. Struct. Biotechnol. J., 2020, 18, 1221–1227 CrossRef CAS PubMed.
- T. Hofmann, S. Krah, C. Sellmann, S. Zielonka and A. Doerner, Greatest Hits-Innovative Technologies for High Throughput Identification of Bispecific Antibodies, Int. J. Mol. Sci., 2020, 21(18), 6551 CrossRef CAS PubMed.
- J. Steinhardt, Y. Wu, R. Fleming, B. T. Ruddle, P. Patel, H. Wu, C. Gao and N. Dimasi, Fab-Arm Exchange Combined with Selective Protein A Purification Results in a Platform for Rapid Preparation of Monovalent Bispecific Antibodies Directly from Culture Media, Pharmaceutics, 2020, 12, 1 Search PubMed.
- L. Qian, X. Lin, X. Gao, R. U. Khan, J.-Y. Liao, S. Du, J. Ge, S. Zeng and S. Q. Q. Yao, The Dawn of a New Era: Targeting the “Undruggables” with Antibody-Based Therapeutics, Chem. Rev., 2023, 123(12), 7782–7853 CrossRef CAS PubMed.
- O. Harel and M. Jbara, Chemical Synthesis of Bioactive Proteins, Angew. Chem., Int. Ed., 2023, 62(13), e202217716 CrossRef CAS PubMed.
- L. Xu, S. L. Kuan and T. Weil, Contemporary Approaches for Site-Selective Dual Functionalization of Proteins, Angew. Chem., Int. Ed., 2021, 60(25), 13757–13777 CrossRef CAS PubMed.
- R. J. Taylor, M. B. Geeson, T. Journeaux and G. J. L. Bernardes, Chemical and Enzymatic Methods for Post-Translational Protein-Protein Conjugation, J. Am. Chem. Soc., 2022, 144(32), 14404–14419 CrossRef CAS PubMed.
- S. C. Lee, J. S. Y. Ma, M. S. Kim, E. Laborda, S. Choi, E. N. Hampton, H. Yun, V. Nunez, M. T. Muldong, C. N. Wu, W. Ma, A. A. Kulidjian, C. J. Kane, V. Klyushnichenko, A. K. Woods, S. B. Joseph, M. Petrassi, J. Wisler, J. Li, C. A. M. Jamieson, P. G. Schultz, C. H. Kim and T. S. Young, A PSMA-targeted bispecific antibody for prostate cancer driven by a small-molecule targeting ligand, Sci. Adv., 2021, 7(33), eabi8193 CrossRef CAS PubMed.
- B. M. Hutchins, S. A. Kazane, K. Staflin, J. S. Forsyth, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Selective Formation of Covalent Protein Heterodimers with an Unnatural Amino Acid, Chem. Biol., 2011, 18(3), 299–303 CrossRef CAS PubMed.
- H. Y. Liu, P. Zrazhevskiy and X. Gao, Solid-Phase Bioconjugation of Heterobifunctional Adaptors for Versatile Assembly of Bispecific Targeting Ligands, Bioconjugate Chem., 2014, 25(8), 1511–1516 CrossRef CAS PubMed.
- F. Thoreau, P. A. Szijj, M. K. Greene, L. N. C. Rochet, I. A. Thanasi, J. K. Blayney, A. Maruani, J. R. Baker, C. J. Scott and V. Chudasama, Modular Chemical Construction of IgG-like Mono- and Bispecific Synthetic Antibodies (SynAbs), ACS Cent. Sci., 2023, 9(3), 476–487 CrossRef CAS PubMed.
- S. Kishimoto, Y. Nakashimada, R. Yokota, T. Hatanaka, M. Adachi and Y. Ito, Site-Specific Chemical Conjugation of Antibodies by Using Affinity Peptide for the Development of Therapeutic Antibody Format, Bioconjugate Chem., 2019, 30(3), 698–702 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Site-selective modification strategies in antibody-drug conjugates, Chem. Soc. Rev., 2021, 50(2), 1305–1353 RSC.
- E. A. Hoyt, P. Cal, B. L. Oliveira and G. J. L. Bernardes, Contemporary approaches to site-selective protein modification. Nature Reviews, Chemistry, 2019, 3(3), 147–171 CAS.
- M. Collins and H. Khalili, Soluble Papain to Digest Monoclonal Antibodies; Time and Cost-Effective Method to Obtain Fab Fragment, Bioengineering, 2022, 9(5), 209 CrossRef CAS PubMed.
- H. Khalili, A. Godwin, J.-W. Choi, R. Lever and S. Brocchini, Comparative Binding of Disulfide-Bridged PEG-Fabs., Bioconjugate Chem., 2012, 23(11), 2262–2277 CrossRef CAS PubMed.
- M. Handula, K.-T. Chen and Y. Seimbille, IEDDA: An Attractive Bioorthogonal Reaction for Biomedical Applications, Molecules, 2021, 26(15), 4640 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Cu-free click cycloaddition reactions in chemical biology, Chem. Soc. Rev., 2010, 39(4), 1272–1279 RSC.
- K. Johann, D. Svatunek, C. Seidl, S. Rizzelli, T. A. Bauer, L. Braun, K. Koynov, H. Mikula and M. Barz, Tetrazine- and trans-cyclooctene-functionatised potypept(o)ides for fast bioorthogonal tetrazine ligation, Polym. Chem., 2020, 11(27), 4396–4407 RSC.
- C. Ginn, J. Choi and S. Brocchini, Disulfide-bridging PEGylation during refolding for the more efficient production of modified proteins, Biotechnol. J., 2016, 11(8), 1088–1099 CrossRef CAS PubMed.
- L. Bartels, H. L. Ploegh, H. Spits and K. Wagner, Preparation of bispecific antibody-protein adducts by site-specific chemo-enzymatic conjugation, Methods, 2019, 154, 93–101 CrossRef CAS PubMed.
- A. Battigelli, B. Almeida and A. Shukla, Recent Advances in Bioorthogonal Click Chemistry for Biomedical Applications, Bioconjugate Chem., 2022, 33(2), 263–271 CrossRef CAS PubMed.
- Y. Tang, X. Zeng and J. Liang, Surface Plasmon Resonance: An Introduction to a Surface Spectroscopy Technique, J. Chem. Educ., 2010, 87(7), 742–746 CrossRef CAS PubMed.
- V. M. Krishnamurthy, V. Semetey, P. J. Bracher, N. Shen and G. M. Whitesides, Dependence of effective molarity on linker length for an intramolecular protein-ligand system, J. Am. Chem. Soc., 2007, 129(5), 1312–1320 CrossRef CAS PubMed.
- R. Das, E. Baird, S. Allen, B. Baird, D. Holowka and B. Goldstein, Binding mechanisms of PEGylated ligands reveal multiple effects of the PEG scaffold, Biochemistry, 2008, 47(3), 1017–1030 CrossRef CAS PubMed.
- R. H. Kramer and J. W. Karpen, Spanning binding sites on allosteric proteins with polymer-linked ligand dimers, Nature, 1998, 395(6703), 710–713 CrossRef CAS PubMed.
- S. Kubetzko, E. Balic, R. Waibel, U. Z. Wittke and A. Pluckthun, PEGylation and multimeriztion of the anti-p185 HER-2 single chain Fv fragment 4D5, J. Biol. Chem., 2006, 281, 35186–35201 CrossRef CAS PubMed.
- G. Vauquelin and S. J. Charlton, Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action, Br. J. Pharmacol., 2010, 161(3), 488–508 CrossRef CAS PubMed.
- T. Ren, X. Zhu, N. M. Jusko, W. Krzyzanski and W. J. Jusko, Pharmacodynamic model of slow reversible binding and its applications in pharmacokinetic/pharmacodynamic modeling: review and tutorial, J. Pharmacokinet. Pharmacodyn., 2022, 49(5), 493–510 CrossRef PubMed.
- J. Gabrielsson and S. Hjorth, Turn On, Tune In, Turnover! Target Biology Impacts In Vivo Potency, Efficacy, and Clearance, Pharmacol. Rev., 2023, 75(3), 416–462 CrossRef CAS PubMed.
- A. Halperin, Polymeric vs. monomeric amphiphiles: Design parameters, Polym. Rev., 2006, 46(2), 173–214 CAS.
- H. Jung, S.-E. Gang, J.-M. Kim, T.-Y. Heo, S. Lee, E. Shin, B.-S. Kim and S.-H. Choi, Regulating Dynamics of Polyether-Based Triblock Copolymer Hydrogels by End-Block Hydrophobicity, Macromolecules, 2020, 53(23), 10339–10348 CrossRef CAS.
- P. Pan, T. Geng, Z. Li, X. Ding, M. Shi, Y. Li, Y. Wang, Y. Shi, J. Wu, L. Zhong, D. Ji, Z. Li and X. Meng, Design, Synthesis, and Biological Evaluation of Proteolysis-Targeting Chimeras as Highly Selective and Efficient Degraders of Extracellular Signal-Regulated Kinase 5, J. Med. Chem., 2023, 66(19), 13568–13586 CrossRef CAS PubMed.
- H. L. Svilenov, P. Arosio, T. Menzen, P. Tessier and P. Sormanni, Approaches to expand the conventional toolbox for discovery and selection of antibodies with drug-like physicochemical properties, mAbs, 2023, 15(1), 2164459 CrossRef PubMed.
- R. J. Taylor, M. A. Rangel, M. B. Geeson, P. Sormanni, M. Vendruscolo and G. J. L. Bernardes, p-Clamp-Mediated Homo- and Heterodimerization of Single-Domain Antibodies via Site-Specific Homobifunctional Conjugation, J. Am. Chem. Soc., 2022, 144(29), 13026–13031 CrossRef CAS PubMed.
- W.-C. Lo, S. K. Kawade, W.-H. Kuo, A. K. Adak and C.-C. Lin, Site-selective heterodimerization of antibody fragments for bispecific antibody complexes enabled by divinylpyrimidine reagent, J. Chin. Chem. Soc., 2023, 70(12), 2177 CrossRef CAS.
- P. Szijj and V. Chudasama, The renaissance of chemically generated bispecific antibodies, Nat. Rev. Chem., 2021, 5(2), 78–92 CrossRef CAS PubMed.
- C. Sun, J. L. Trevaskis, C. M. Jodka, S. Neravetla, P. Griffin, K. Xu, Y. Wang, D. G. Parkes, B. Forood and S. S. Ghosh, Bifunctional PEGylated Exenatide-Amylinomimetic Hybrids to Treat Metabolic Disorders: An Example of Long-Acting Dual Hormonal Therapeutics, J. Med. Chem., 2013, 56(22), 9328–9341 CrossRef CAS PubMed.
- A. Godwin, G. Badescu, M. Bird, P. Bryant, D. Morris and M. Frigerio, Process for the conjugation of a peptide or protein with a reagent comprising a leaving group including a portion of PEG, 2016, p. 188.
- Y. Cong, E. Pawlisz, P. Bryant, S. Balan, E. Laurine, R. Tommasi, R. Singh, S. Dubey, K. Peciak, M. Bird, A. Sivasankar, J. Swierkosz, M. Muroni, S. Heidelberger, M. Farys, F. Khayrzad, J. Edwards, G. Badescu, I. Hodgson, C. Heise, S. Somavarapu, J. Liddell, K. Powell, M. Zloh, J.-W. Choi, A. Godwin and S. Brocchini, Site-Specific PEGylation at Histidine Tags., Bioconjugate Chem., 2012, 23(2), 248–263 CrossRef CAS PubMed.
- S. Brocchini, A. Godwin, S. Balan, J.-W. Choi, M. Zloh and S. Shaunak, Disulfide bridge based PEGylation of proteins., Adv. Drug Delivery Rev., 2008, 60(1), 3–12 CrossRef CAS PubMed.
- R. Pihl, Q. Zheng and Y. David, Nature-inspired protein ligation and its applications, Nat. Rev. Chem., 2023, 7(4), 234–255 CrossRef CAS PubMed.
- A. H. Keeble and M. Howarth, Power to the protein: enhancing and combining activities using the Spy toolbox, Chem. Sci., 2020, 11(28), 7281–7291 RSC.
- C. Schindler, C. Faust, H. Sjuts, C. Lange, J. Kuehn, W. Dittrich, W. D. Leuschner, W. Schiebler, J. Hofmann, E. Rao and T. Langer, A multivalent antibody assembled from different building blocks using tag/catcher systems: a case study, Protein Eng., Des. Sel., 2022, 35, gzac014 CrossRef PubMed.
- E. M. Sletten and C. R. Bertozzi, Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality, Angew. Chem., Int. Ed., 2009, 48(38), 6974–6998 CrossRef CAS PubMed.
- T. Reiner and B. M. Zeglis, The inverse electron demand Diels-Alder click reaction in radiochemistry, J. Labelled Compd. Radiopharm., 2014, 57(4), 285–290 CrossRef CAS PubMed.
- K. Muneeruddin, C. E. Bobst, R. Frenkel, D. Houde, I. Turyan, Z. Sosic and I. A. Kaltashov, Characterization of a PEGylated protein therapeutic by ion exchange chromatography with on-line detection by native ESI MS and MS/MS, Analyst, 2017, 142(2), 336–344 RSC.
- J. E. Seely and C. W. Richey, Use of ion-exchange chromatography and hydrophobic interaction chromatography in the preparation and recovery of polyethylene glycol-linked proteins, J. Chromatogr. A, 2001, 908(1–2), 235–241 CrossRef CAS PubMed.
- A. C. Register, S. S. Tarighat and H. Y. Lee, Bioassay Development for Bispecific Antibodies-Challenges and Opportunities, Int. J. Mol. Sci., 2021, 22(10), 5350 CrossRef CAS PubMed.
- M. Pei, Y. Wang, L. Tang, W. Wu, C. Wang and Y.-L. Chen, Dual-target Bridging ELISA for Bispecific Antibodies, Bio-Protoc., 2022, 12(19), e4522 CAS.
- S. Mak, A. Marszal, N. Matscheko and U. Rant, Kinetic analysis of ternary and binary binding modes of the bispecific antibody emicizumab, mAbs, 2023, 15(1), 2149053 CrossRef PubMed.
- A. V. Madsen, O. Mejias-Gomez, L. E. Pedersen, K. Skovgaard, P. Kristensen and S. Goletz, Immobilization-Free Binding and Affinity Characterization of Higher Order Bispecific Antibody Complexes Using Size-Based Microfluidics, Anal. Chem., 2022, 94(40), 13652–13658 CrossRef CAS PubMed.
- A. Erbas and F. Inci, The Role of Ligand Rebinding and Facilitated Dissociation on the Characterization of Dissociation Rates by Surface Plasmon Resonance (SPR) and Benchmarking Performance Metrics, Methods Mol. Biol., 2022, 2385, 237–253 CrossRef PubMed.
- S. Kubetzko, C. A. Sarkar and A. Plückthun, Protein PEGylation decreases observed target association rates via a dual blocking mechanism, Mol. Pharmacol., 2005, 68(5), 1439–1454 CrossRef CAS PubMed.
- A. Erbas, M. Olvera de la Cruz and J. F. Marko, Receptor-Ligand Rebinding Kinetics in Confinement, Biophys. J., 2019, 116(9), 1609–1624 CrossRef CAS PubMed.
- M. A. Cooper, Label-free screening of bio-molecular interactions, Anal. Bioanal. Chem., 2003, 377(5), 834–842 CrossRef CAS PubMed.
- M. Jerabek-Willemsen, T. Andre, R. Wanner, H. M. Roth, S. Duhr, P. Baaske and D. Breitsprecher, MicroScale Thermophoresis: Interaction analysis and beyond, J. Mol. Struct., 2014, 1077, 101–113 CrossRef CAS.
- J. G. Ghosh, A. A. Nguyen, C. E. Bigelow, S. Poor, Y. B. Qiu, N. Rangaswamy, R. Ornberg, B. Jackson, H. Mak, T. Ezell, V. Kenanova, E. de la Cruz, A. Carrion, B. Etemad-Gilbertson, R. G. Caro, K. Zhu, V. George, J. R. Bai, R. Sharma-Nahar, S. Y. Shen, Y. Q. Wang, K. K. Subramanian, E. Fassbender, M. Maker, S. Hanks, J. Vrouvlianis, B. Leehy, D. Long, M. Prentiss, V. Kansara, B. Jaffee, T. P. Dryja and M. Roguska, Long-acting protein drugs for the treatment of ocular diseases, Nat. Commun., 2017, 8, 14837 CrossRef CAS PubMed.
- M. Huelsmann and E. Kopetzki, Fusion proteins for ophthalmology with increased eye retention, 2017, p. 67.
- F. E. Di Padova, J. Ghosh, T. Huber and J. R. Rondeau, Compositions and methods for long-acting antibodies targeting IL-17, 2015, p. 218.
- M. Dang and M. S. Shoichet, Long-Acting Ocular Injectables: Are We Looking In The Right Direction?, Adv. Sci., 2024, 11(8), 2306463 CrossRef CAS PubMed.
- K. Vulic and M. S. Shoichet, Affinity-Based Drug Delivery Systems for Tissue Repair and Regeneration, Biomacromolecules, 2014, 15(11), 3867–3880 CrossRef CAS PubMed.
- M. Cai, L. Chen, T. Wang, Y. Liang, J. Zhao, X. Zhang, Z. Li and H. Wu, Hydrogel scaffolds in the treatment of spinal cord injury: a review, Front. Neurosci., 2023, 17, 1211066 CrossRef PubMed.
- M. J. Kratochvil, A. J. Seymour, T. L. Li, S. P. Pasca, C. J. Kuo and S. C. Heilshorn, Engineered materials for organoid systems, Nat. Rev. Mater., 2019, 4(9), 606–622 CrossRef CAS PubMed.
- M. A. Kinney and T. C. McDevitt, Emerging strategies for spatiotemporal control of stem cell fate and morphogenesis, Trends Biotechnol., 2013, 31(2), 78–84 CrossRef CAS PubMed.
- J. W. Andrejecsk, W. G. Chang, J. S. Pober and W. M. Saltzman, Controlled protein delivery in the generation of microvascular networks, Drug Delivery Transl. Res., 2015, 5(2), 75–88 CrossRef CAS PubMed.
- J. E. Samorezov and E. Alsberg, Spatial regulation of controlled bioactive factor delivery for bone tissue engineering, Adv. Drug Delivery Rev., 2015, 84, 45–67 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00130c |
| ‡ Equal first authorship |
| This journal is © The Royal Society of Chemistry 2024 |