
Iodine-promoted transfer of dihydrogen from ketones to alkenes, triphenylmethyl, and diphenylmethyl derivatives†
Yiping
Duan
b,
Wenyi
Zhong
a,
Zhaolan
Zeng
b,
Jiajie
Feng
b,
Jinyi
Xu
 *b,
Fulai
Yang
*a and
Jie
Liu
*a
*b,
Fulai
Yang
*a and
Jie
Liu
*a
aDepartment of Organic Chemistry, School of Science, China Pharmaceutical University, Nanjing, 210009, P. R. China. E-mail: flycpu@cpu.edu.cn
bState Key Laboratory of Natural Medicines and Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing, 210009, P. R. China
First published on 31st October 2023
Abstract
Herein, a novel class of transfer hydrogenation agent, cycloheptanone, was successfully employed in metal-free hydrogenation facilitated by iodine. A series of alkenes, triphenylmethyl derivatives, and diphenylmethyl derivatives were reduced to the desired compounds in moderate to excellent yields. The transfer hydrodeuteration of alkenes using α-deuterated cyclododecanone exhibited high regioselectivity. Preliminary mechanism studies confirmed the origins of the two hydrogen atoms involved in the reduction of alkenes. The current study paves the way for the use of ketones as unique transfer hydrogenation agents in chemical synthesis.
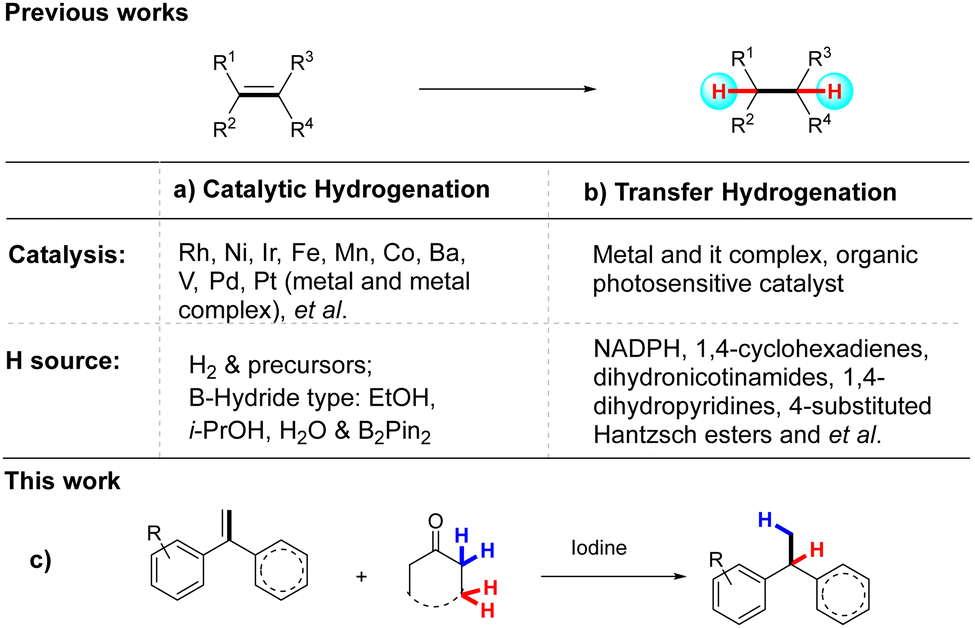
The catalytic hydrogenation reaction is one of the most fundamental transformations in organic synthesis with a long history.1 Typical methods often use more than 1 atmosphere of molecular H2 as the source of hydrogen, most of which is currently supplied from processes reforming fossil resources, such as natural gas and oil.2 However, the ongoing escalation of global concerns about environmental issues strongly demands the replacement of any fossil resource-based technologies with more sustainable schemes. Moreover, most of the catalytic hydrogenation reactions require the use of precious metal catalysts,3 such as ruthenium (Ru), rhodium (Rh), palladium (Pd), platinum (Pt), and iridium (Ir).4 (Scheme 1a). The toxicity, environmental unfriendliness, and use of precious metals hinder the application of hydrogenation reactions. Especially in the pharmaceutical industry, due to the high standard of heavy metals in the food and drug industry that limited the application of precious metal catalysts.5
 | ||
| Scheme 1 The method of the hydrogenation reaction. | ||
Transfer hydrogenation (TH), which refers to the addition of hydrogen to a compound from a source other than molecular H2, usually under mild temperature and ambient pressure conditions, is a convenient and powerful method for accessing various hydrogenated compounds.6 It is an attractive alternative to catalytic hydrogenation, and it has recently become the center of research in hydrogenation science. Transfer hydrogenation does not require potentially hazardous pressurised H2 gas or elaborate experimental set-ups, and the hydrogen donors are readily available, inexpensive, and easy to handle. Traditionally, it uses hydrogen donor compounds, such as formic acid,7a alcohols,7b–e water7f–h or dihydroanthracene,7i which also act as solvents for the reaction. Most of these reactions have now been developed using ruthenium and rhodium complexes.7j,k Excitingly, as a well-studied alternative to transition metal catalysts, organocatalytic transfer hydrogenations are now undergoing explosive development1a,e,8 and one of the main trends is focused on the exploration of more effective transfer hydrogenation agents, which mainly include 1,4-cyclo-hexadienes,9a–c dihydronicotinamides,9d 1,4-dihydropyridines,9e–k and 4-substituted Hantzsch esters9l–n (Scheme 1b). Most of these transfer hydrogenation agents are not commercially available and require preparation. To address these issues, it is highly desirable to explore new transfer hydrogenation agents under metal-free reaction conditions.
Ketones are not only essential functionalities of various biologically active compounds10a,b but also important reagents in modern organic synthesis.10c,d For example, well-known name reactions, such as the Baeyer–Villiger reaction, Robinson reaction, Witting reaction, and Favorski reaction, are widely used in laboratories and industry, which can reduce ketones to the corresponding alcohols or alkanes, or oxidise to esters. In sharp contrast, no report has been published on the use of ketone as a transfer hydrogenation agent. In the course of exploring new reactions of ketones,11 we found that alkenes could be reduced using ketone as a transfer hydrogenation agent (Scheme 1c). We considered the system that consisted of two separate steps. In the first step, the alkenes are protonated and the alpha-hydrogen of the ketone separates as a proton source. In the second step, the more substituted carbon atom abstracts another hydrogen to give the reduction product. To test this idea, the treatment of alkenes with α-deuterated ketone could yield the hydrodeuteration product with excellent regioisomeric ratios, with the deuterium attached to the less substituted carbon atom. This method could achieve highly regioselective incorporation of deuterium and hydrogen at the desired positions of the starting material. Site-selective monodeuteration of alkenes is practised to provide access to isotopically labelled compounds with broad utility in chemical research, such as organic synthesis,12 mechanistic investigations,13 and spectrometric/scopic analysis14 as well as in the pharmaceutical industry.15 Although a number of impressive deuteration methods have been reported for alkenes, the extension to metal-free regioselective hydrodeuteration of alkenes is less developed. The challenge is that chemically similar hydrogen and deuterium must be selectively inserted via a π bond.
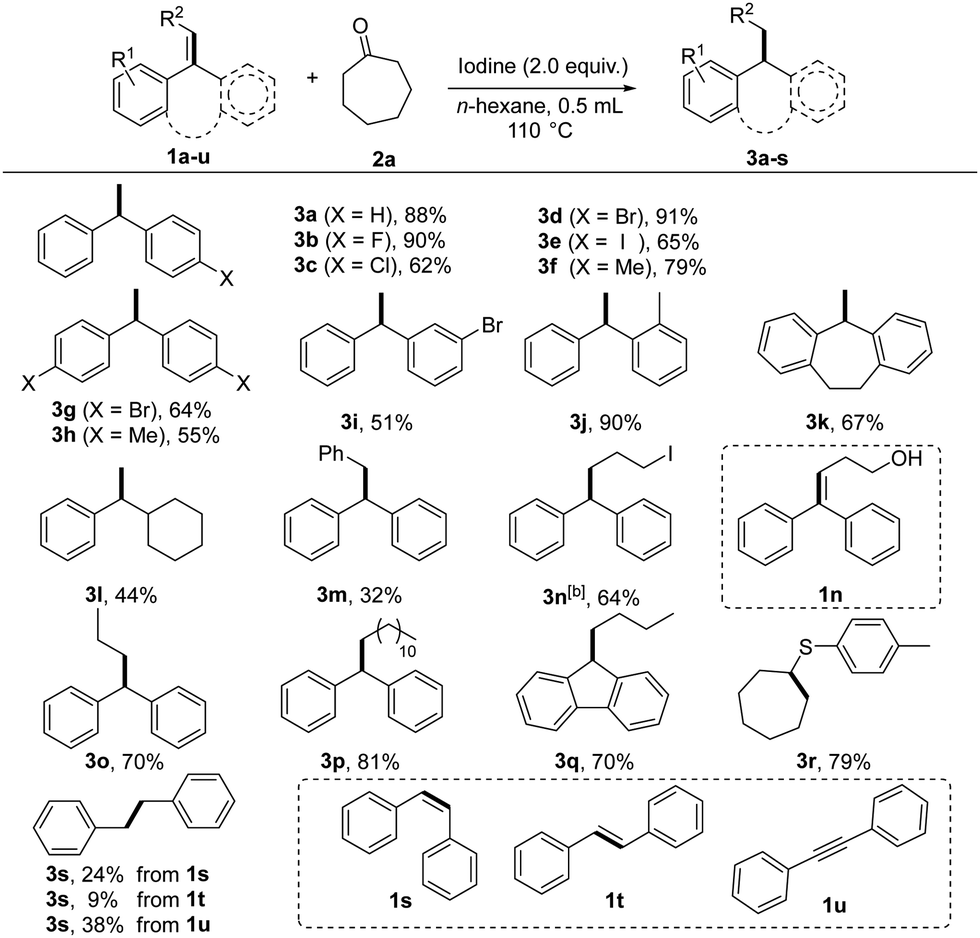
The reduction of 1,1-diphenylethylene 1a with cycloheptanone 2a was selected as the model reaction. Initially, the reaction did not occur in n-hexane in the presence of iodine and open air at room temperature. To our delight, heating the mixture at 110 °C in a Shrek tube led to the formation of product 3a in 88% isolated yield (Scheme S1, ESI†). The dimerization of 1,1-diphenylethylene afforded the by-product. To confirm the role of cycloheptanone and iodine in the reaction, treatment of 1,1-diphenylethylene 1a with iodine gave the dimerization compound 4 in almost quantitative yield, indicating that the ketone is essential in the reaction. Encouraged by this result, various ketones were examined. Alicyclic ketones could be used as hydrogen sources to provide reduction products in good yields, such as cycloheptanone (2a), cyclohexanone (2c), and cyclododecanone (2g). For comparison, no desired product was obtained when replacing alicyclic ketones with chain aliphatic ketones and aromatic ketones. Finally, considering price, the manoeuvrability, and hydrogenation efficiency, cycloheptanone was chosen as the transfer hydrogenation reagent.
Next, various solvents were examined, but no better yield was obtained. Lowering the reaction temperature led to a lower yield. Furthermore, replacing iodine with N-iodosuccinimide (NIS) and HI reduced the yield to 29% and 37%, respectively, and no desired product was obtained when replacing iodine with Bu4NI. We know that elemental iodine itself may also provide electrons to serve as a reductant. To rule out this possibility, treatment of 1,1-diphenylethylene 1a and cycloheptanone 2a with non-reducing trifluoromethanesulfonic acid gave the product 3a in 62% yield, which suggests that the ketone acted as a transfer hydrogenation reagent rather than iodine as a reductant in the reaction. Thus, the optimal reaction conditions were 1.0 equiv. of 1a, 2.5 equiv. of cycloheptanone, and 2.0 equiv. of I2, in n-hexane at 110 °C. (Table S1, ESI†).
Under the optimized reaction conditions, a series of alkenes were investigated for this reaction (Table 1). 1,1-Diarylalkenes (1a–1k) smoothly underwent hydrogenation with a ketone in ethanol to produce structurally diverse products (3a–3k) in moderate to excellent yields. Notably, the reaction tolerated a variety of functional groups, such as fluoro, chloro, bromo, iodo and methyl with a minor steric effect. However, replacing one of the aryl groups with an alkyl group afforded 3l in moderate yields. Moreover, trisubstituted alkenes (1m–1q) could also perform under the reaction conditions and afforded the desired products 3m–3q in moderate to good yields. To our delight, when alkenyl sulfide (1r) was used as a substrate, sulfide (3r) was obtained in acceptable yield (79%). Furthermore, diphenylacetylene 1s–1t (cis and trans) could also react and generate 3s under these conditions, albeit with a lower yield. Diphenylacetylene (1u) could also be reduced to produce the corresponding alkane product (3s).
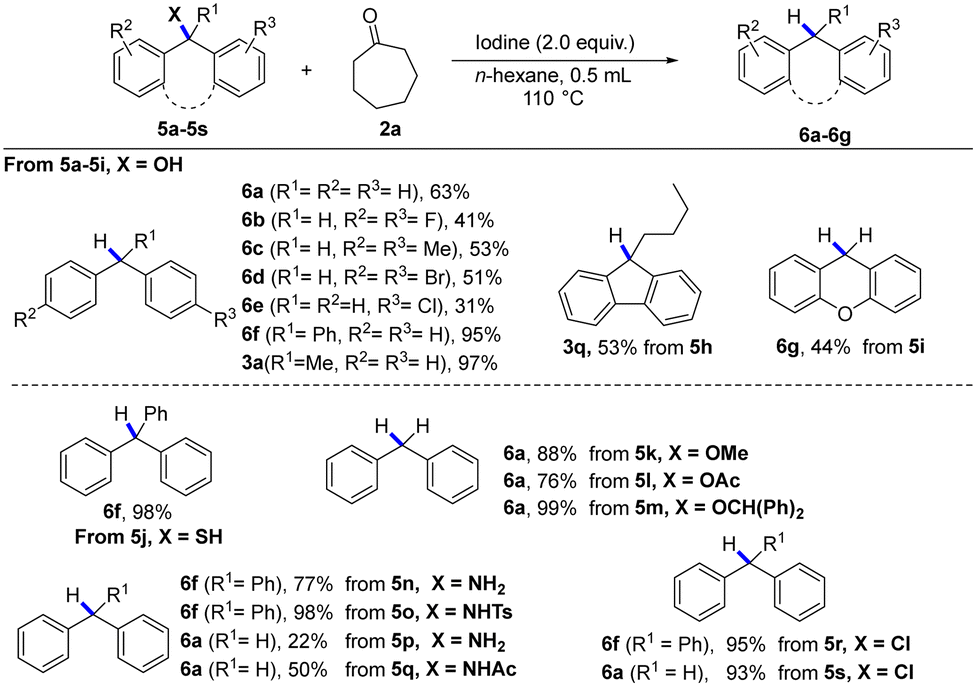
Subsequently, other reduction reactions, such as deoxidation, desulfurization, dehalogenation, and deamination reactions, were also tested under the standard conditions (Table 2). Gratefully, large steric hindrance diphenylmethyl derivatives and triphenylmethyl derivatives (5a–5s) could be reduced to afford the corresponding alkane products (6a–g) under standard conditions in good yields. It is worth noting that reductive deamination is a useful and important method to achieve functional group transformation, but little has been published on the cleavage of the carbon–nitrogen bonds of 10 amines for the hydrogenation reaction.16 Interestingly, reductive deamination (5n–5q) could be efficiently implemented under this metal-free condition, producing the desired products (6f, 6a). Moreover, the deoxidation of substituted benzyl alcohols (5k–5m) also reacted smoothly and gave the desired product (6a) in moderate yields.
| a Reaction conditions: 1a (0.3 mmol), 2a (0.75 mmol), and iodine (0.6 mmol) in n-hexane (0.5 mL), at 110 °C (oil bath), for 4 h. |
|---|
|
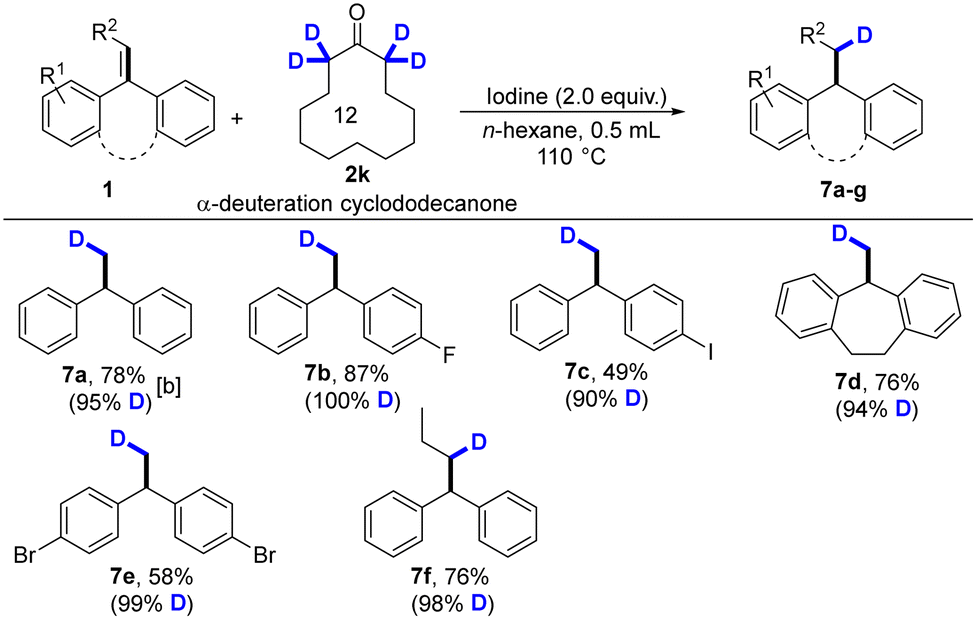
The regioselective transfer hydrodeuteration of alkenes (1a, 1b, 1e, 1g, 1k, and 1o) with α-deuterated ketone was performed under the standard conditions. This hydrodeuteration reaction showed a wide scope and functional-group tolerance and generated deuterated compound 7a–7f in good yields with high regioselectivity, and selected examples are presented in Table 3.
To gain insight into the reaction mechanism, a series of control experiments with possible intermediates were performed. (Scheme 2) First, we carried out electron-spray ionization (ESI) mass spectrometric analysis of the reaction mixture of 1a and cycloheptanone, and tentatively identified one minor product of compound 8, according to the high-resolution mass data (Scheme 2, eqn (1)). To further confirm the formation of compound 8, cycloheptanone was treated with iodine to give the compound 8 in 19% yield (Scheme 2, eqn (2)). The formation of compound 8 may proceed as follows: aldol condensation of cycloheptanone gives β-hydroxyketones, followed by intramolecular ring closure and aromatization reaction. To test this, treatment of compound 9 with iodine or TfOH gave the desired product in 79% and 76% yields, respectively (Scheme 2, eqn (3)). In addition, treatment of compound 1a with 9-1 under optimized conditions gave compound 3a in 91% yield and the minor compound 8 was identified using MS (Scheme 2, eqn (4)). These pieces of evidence suggest that β-hydroxyketones may act as a reducing agent. Furthermore, to clarify the hydrogen source, deuterium-labeling experiments were conducted. We selected α-deuterated cyclopentanone as the transfer hydrogenation reagent because it has only two types of hydrogens (Scheme 2, eqn (5)). Deuterium incorporation at the C-1 position and no deuterium incorporation at the C-2 position were observed for the reaction of α-deuterated cyclopentanone with 1a in CDCl3, which implies that C-1 hydrogen is from α-position of cyclopentanone and C-2 hydrogen is from β-position of cyclopentanone.
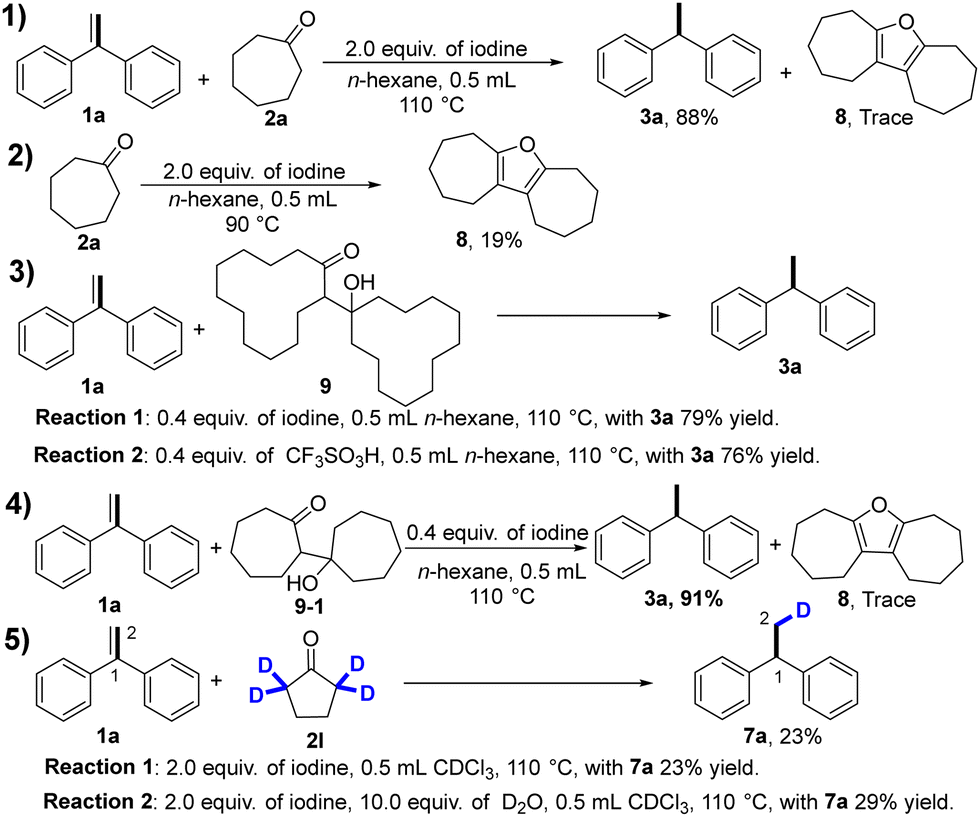 | ||
| Scheme 2 The key study explored the reaction mechanism. | ||
According to the above experimental results, we propose the following reaction pathways, as shown in Scheme 3. First, in the presence of iodine, ketone undergoes an aldol condensation to produce intermediate III and releases an α-H+, and then intermediate III takes an enol tautomerization to generate intermediate IV. At the same time, the alkene receives the α-H+ to generate the carbonium ion intermediate VIII. Furthermore, intermediate IV undergoes a 1,5-hydrogen shift to generate the intermediate VI,17a–c which undergoes an aromatization reaction to produce intermediate VII and β-H−. Intermediate VIII captures β-H− to afford the desired product 3a. Finally, intermediate VII is transferred to compounds 8X. The reduction of diphenylmethyl and triphenylmethyl derivatives may follow a similar process.
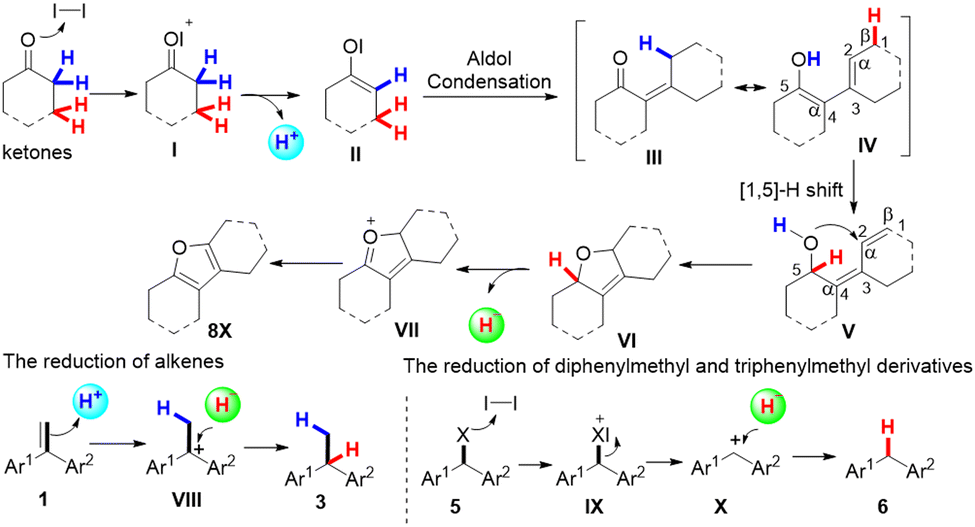 | ||
| Scheme 3 Possible mechanism of the hydrogenation reaction. | ||
In summary, we have developed an iodine-promoted metal-free hydrogenation reaction using ketones as a new transfer hydrogenation agents. A series of alkenes, triphenylmethyl derivatives, and diphenylmethyl derivatives were reduced to the desired compounds in moderate to excellent yields. In particular, reductive deamination could be efficiently implemented under this transition-metal-free conditions. Furthermore, the transfer hydrodeuteration of alkenes with α-deuterated ketone was performed with high regioselectivity. A preliminary mechanism study confirmed the presence of two hydrogen sources of alkene reduction products. The current study paves the way for the use of ketones as the unique transfer hydrogenation agents in chemical synthesis.
We are grateful for the financial support from the National Natural Science Foundation of China (21502182) and the Fundamental Research Funds for the Central Universities (2632019ZD12 and 2632020ZD03).
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211 RSC; (b) P. Oswal, A. Arora, S. Singh, D. Nautiyal, S. Kumar and G. K. Rao, Dalton Trans., 2020, 49, 12503 RSC; (c) Z. An and J. Li, Green Chem., 2022, 24, 1780 RSC; (d) J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592 RSC; (e) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681 CrossRef CAS.
- B. C. Tashie-Lewis and S. G. Nnabuife, Chem. Eng. J. Adv., 2021, 8, 100172 CrossRef CAS.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459 RSC.
- (a) Q.-A. Chen, Z.-S. Ye, Y. Duan and Y.-G. Zhou, Chem. Soc. Rev., 2013, 42, 497 RSC; (b) W.-H. Lai, L.-F. Zhang, W.-B. Hua, S. Indris, Z.-C. Yan, Z. Hu and S.-X. Dou, Angew. Chem., Int. Ed., 2019, 58, 11868 CrossRef CAS PubMed.
- T. K. Das and A. Poater, Int. J. Mol. Sci., 2021, 22, 13383 CrossRef CAS PubMed.
- (a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179 CrossRef CAS; (b) A. Fürstner, J. Am. Chem. Soc., 2019, 141, 11 CrossRef; (c) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC; (d) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed; (e) N. Garg, A. Sarkar and B. Sundararaju, Coord. Chem. Rev., 2021, 433, 213728 Search PubMed.
- (a) R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang and C. C. Xu, ACS Catal., 2021, 11, 1071 CrossRef CAS; (b) Y. Wang, P. Prinsen, K. S. Triantafyllidis, S. A. Karakoulia, A. Yepez, C. Len and R. Luque, ChemCatChem, 2018, 10, 3459 CrossRef CAS; (c) R. Wang, Y. Tang, M. Xu, C. Meng and F. Li, J. Org. Chem., 2018, 83, 2274 CrossRef CAS; (d) A. N. Ajjou and J. L. Pinet, J. Mol. Catal. A: Chem., 2004, 214, 203 CrossRef CAS; (e) F. Yang, J. Chen, G. Shen, X. Zhang and B. Fan, Chem. Commun., 2018, 54, 4963 RSC; (f) G. Shen, J. Chen, D. Xu, X. Zhang, Y. Zhou and B. Fan, Org. Lett., 2019, 21, 1364 CrossRef CAS PubMed; (g) Y. Dai, J. Chen, Z. Wang, T. Wang, L. Wang, Y. Yang, X. Qiao and B. Fan, J. Org. Chem., 2021, 86, 7141 CrossRef CAS; (h) W. Wu, H. Zhao, J. Chen, F. Zhang and B. Fan, Chem. – Eur. J., 2022, 28, e202202460 CrossRef CAS PubMed; (i) Q. Lu, G. Zhang, Z.-X. Zhang, B. Hu and K. Li, J. Anal. Appl. Pyrol., 2020, 150, 105103 CrossRef; (j) V. Ritleng and C. Michon, Molcules., 2022, 27, 4703 CrossRef CAS; (k) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588 CrossRef CAS PubMed.
- J. Wang and Y.-G. Zhou, Organocatalytic Transfer Hydrogenation. In Homogeneous Hydrogenation with Non-Precious Catalysts, ed. J. F. Teichert, Wiley, 2019, p. 261 Search PubMed.
- (a) L. Li, S. Kail, S. M. Weber and G. Hilt, Angew. Chem., Int. Ed., 2021, 60, 23661 CrossRef CAS; (b) S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688 RSC; (c) I. Chatterjee and M. Oestreich, Org. Lett., 2016, 18, 2463 CrossRef CAS; (d) A. McSkimming and S. B. Colbran, Chem. Soc. Rev., 2013, 42, 5439 RSC; (e) H.-M. Jiang, J.-H. Qin, Q. Sun, D. Zhang, J.-P. Jiang, X.-H. Ouyang and J.-H. Li, Org. Chem. Front., 2022, 9, 4070 RSC; (f) W. Li, H. Yang, R. Li, H. Lv and X. Zhang, ACS Catal., 2020, 10, 2603 CrossRef CAS; (g) M. Pang, L.-L. Shi, Y. Xie, T. Geng, L. Liu, R.-Z. Liao and W. Wang, ACS Catal., 2022, 12, 5013 CrossRef CAS; (h) Q. He, Z. Xu, D. Jiang, W. Ai, R. Shi, S. Qian and Z. Wang, RSC Adv., 2014, 4, 8671 RSC; (i) L. Bernardi and M. Fochi, Molecules, 2016, 21, 1000 CrossRef PubMed; (j) J. Wang, Z.-H. Zhu, M.-W. Chen, Q.-A. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2019, 58, 1813 CrossRef CAS PubMed; (k) Y.-L. Su, G.-X. Liu, J.-W. Liu, L. Tram, H. Qiu and M. P. Doyle, J. Am. Chem. Soc., 2020, 142, 13846 CrossRef CAS; (l) G.-B. Shen, L. Xie, H.-Y. Yu, J. Liu, Y.-H. Fu and M. Yan, RSC Adv., 2020, 10, 31425 RSC; (m) X. Wang, H. Li, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 2062 RSC; (n) S. Ye and J. Wu, Acta. Chim. Sin., 2019, 77, 814 CrossRef CAS.
- (a) J. Wang, Y. Lv, Y. Shang, Z. Cui, K.-H. Wang, D. Huang and Y. Hu, Chin. J. Org. Chem., 2022, 42, 2300 CrossRef CAS; (b) N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209 CrossRef CAS; (c) Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592 CrossRef CAS PubMed; (d) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876 CrossRef CAS.
- B. Chaudhary, N. Kulkarni, N. Saiyed, M. Chaurasia, S. Desai, S. Potkule and S. Sharma, Adv. Synth. Catal., 2020, 362, 4794 CrossRef CAS.
- (a) M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495 CrossRef CAS; (b) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396 CrossRef CAS PubMed.
- (a) K. B. Wiberg, Chem. Rev., 1955, 55, 713 CrossRef CAS; (b) F. H. Westheimer, Chem. Rev., 1961, 61, 265 CrossRef CAS; (c) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef; (d) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS; (e) E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2006, pp. 421–477 Search PubMed.
- (a) S. E. Scheppele, Chem. Rev., 1972, 72, 511 CrossRef; (b) P. Liuni, E. Olkhov-Mitsel, A. Orellana and D. J. Wilson, Anal. Chem., 2013, 85, 3758 CrossRef CAS PubMed.
- (a) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219 CrossRef CAS PubMed; (b) A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS PubMed; (c) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276 CrossRef CAS PubMed.
- (a) Z.-B. Zhang and J.-B. Xia, Org. Lett., 2020, 22, 9609 CrossRef CAS PubMed; (b) Y.-Q.-Q. Yi, W.-C. Yang, D.-D. Zhai, X.-Y. Zhang, S.-Q. Li and B.-T. Guan, Chem. Commun., 2016, 52, 10894 RSC.
- (a) M. Rehan, S. Maity and L. K. Morya, Angew. Chem., Int. Ed., 2016, 55, 7728 CrossRef CAS; (b) Y.-J. Wang, T.-T. Wang, L. Yao, Q.-L. Wang and L.-M. Zhao, J. Org. Chem., 2020, 85, 9514 CrossRef CAS; (c) V. Nagaraju, C. E. Raju, D. Purnachandar, V. J. Rao and G. V. Karunakar, ChemistrySelect, 2019, 4, 2053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03409g |
| This journal is © The Royal Society of Chemistry 2024 |