
Cascade alkyl migration in 2-alkynylanilines for the synthesis of benzenoid ring multi-functionalized indoles†
Jiwen
He‡
,
Xiaohua
Li‡
,
Qiuqin
He
* and
Renhua
Fan
 *
*
Department of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, China. E-mail: rhfan@fudan.edu.cn
First published on 16th November 2023
Abstract
Cascade alkyl migration of 2-alkynylanilines via an aromaticity destruction and reconstruction process is reported. The first alkyl migration is triggered by generation of a dearomatized arenium species via oxidation and cyclization, and the second is driven by the force to restore the aromaticity of rearrangement products. The reaction gave rise to a range of multi-functionalized indoles.
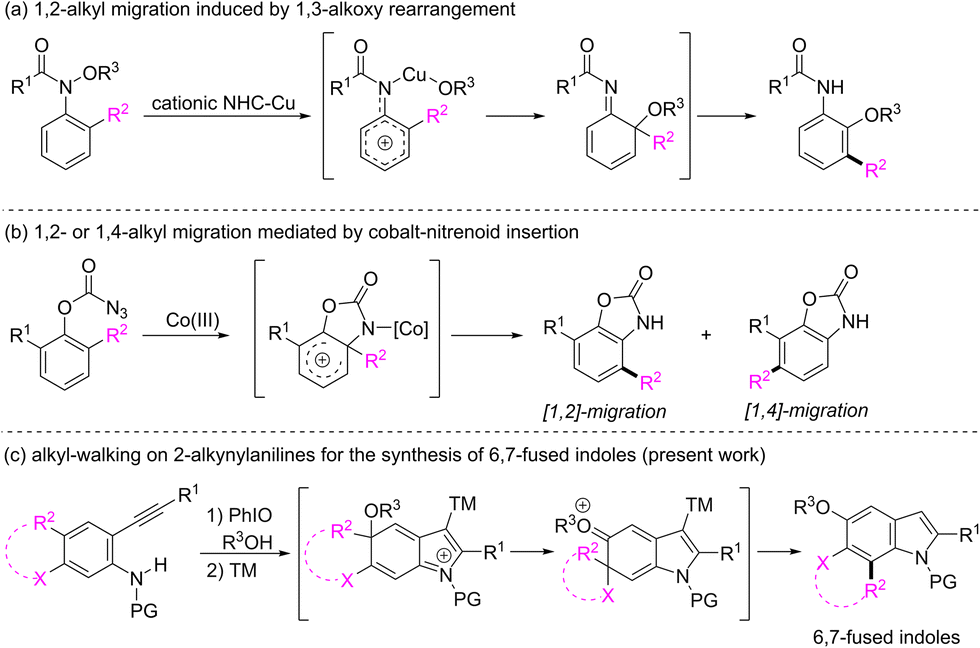
Alkyl migration remains a transformation of central importance in synthetic chemistry because it provides numerous possibilities for the construction of molecular complexity.1 Since the observations of the 1,2-alkyl migration in Friedel–Crafts alkylation reactions2 and the 1,4-methyl migration during the Fischer indole synthesis,3 alkyl migration in the arene frameworks has drawn particular attention because it affords a promising option for the synthesis of multi-functionalized aromatic molecules that are difficult to prepare by electrophilic substitution reactions.4 For example, recently, Nakamura and Terada demonstrated a cationic N-heterocyclic carbene and copper-catalyzed domino 1,3-alkoxy rearrangement and 1,2-alkyl migration to convert N-methoxyanilines to multisubstituted ortho-anisidines (Scheme 1a).5 Chang and co-workers reported a cobalt-nitrenoid insertion-mediated amidative carbon rearrangement of 2,6-disubstituted phenyl azidoformates leading to the formation of multisubstituted benzoxazolones (Scheme 1b).6 In these elegant tactics, dearomatized arenium species have been regarded as key intermediates for alkyl migrations.7 In connection with our recent works on the synthesis of functionalized indoles from 2-alkynylanilines via a dearomatization process,8 we are interested in the alkyl migration of the dearomatized intermediate of 2-alkynyl-4-alkylanilines. In the presence of a transition-metal catalyst, the 5-endo cyclization of the dearomatized intermediate might lead to the in situ generation of an arenium intermediate containing a quaternary carbon. The clockwise or counter-clockwise 1,2-alkyl migration might give rise to 4-alkyl or 6-alkyl substituted indoles, respectively.9 Accordingly, we envisioned that the counter-clockwise alkyl migration might occur if the C4 alkyl group is linked with the C5 carbon of substrates (Scheme 1c). Although the counter-clockwise migration might generate a spiro intermediate, aromatization to restore the system's aromaticity would drive a consequent alkyl migration leading to the formation of 6,7-fused indoles, which are highly valuable synthetic targets10 owing to their existence as core structures in many bioactive indole alkaloids such as teleocidins,11 trikentrins12 and herbindoles.13
 | ||
| Scheme 1 Synthesis of multi-functionalized aromatic molecules via alkyl migration. | ||
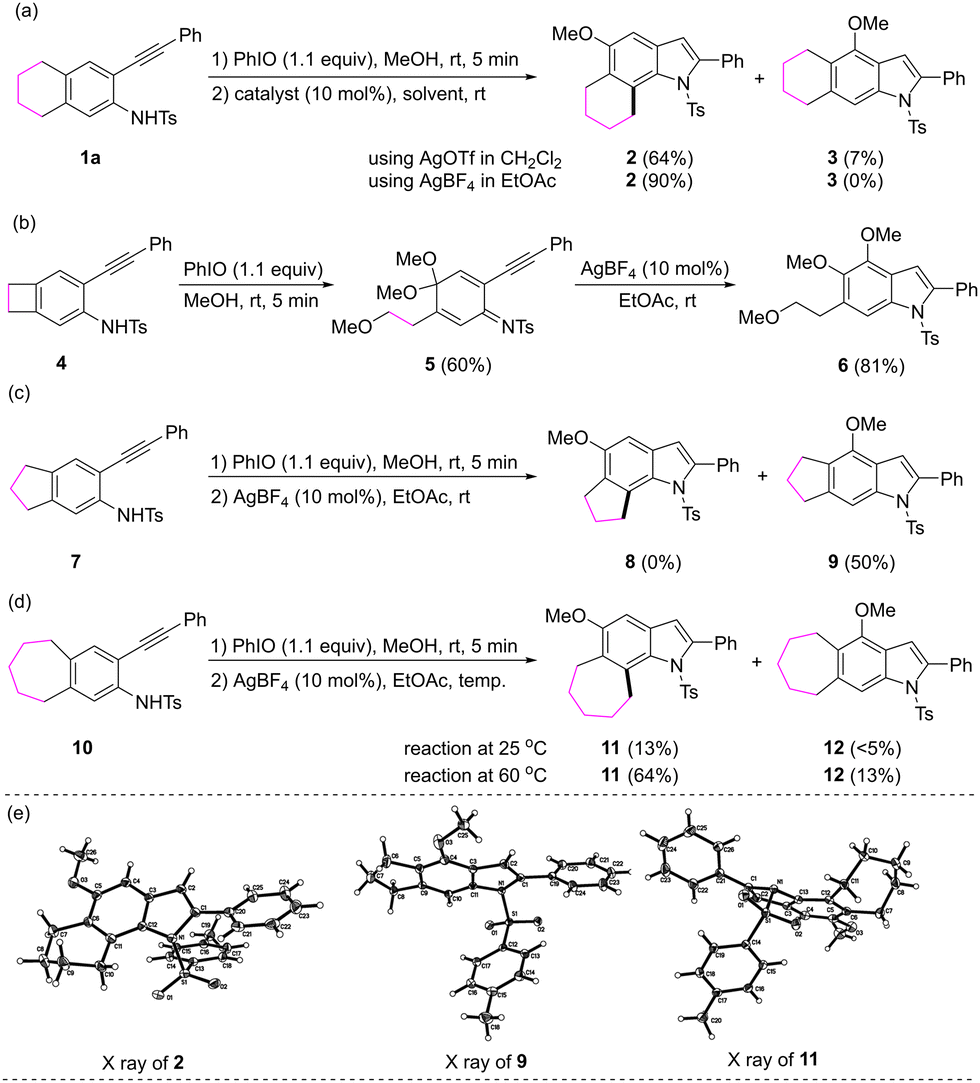
Initially, compound 1a, which possesses a four-carbon linkage at the C4 and the C5 carbons of 2-alkynylaniline, was treated with 1.1 equivalents of iodosobenzene in methanol for 5 minutes. After the removal of methanol under vacuum, catalytic amounts of AgOTf and CH2Cl2 were added. The reaction afforded the expected 6,7-fused indole derivative 2 in a 64% yield, along with a small amount of compound 3 (7%), which might be derived from the 1,2-methoxy clockwise migration or from the Michael addition by the residual methanol to the dearomatized intermediate. A set of reaction variables including metal catalyst, solvent and temperature were investigated to set up the optimized conditions. The reaction in the presence of 0.1 equivalent of AgBF4 in EtOAc at room temperature gave rise to 2 in a 90% yield (Scheme 2a). We next sought to evaluate the effect of the length of the linkages between the C4 and the C5 carbons. For compound 4, which possesses a two-carbon linkage, the oxidative dearomatization led to the ring opening of the four-membered ring and the treatment of the dearomatized product with AgBF4 afforded 4,5-dimethoxy-6-(2-methoxyethyl) substituted indole 6 (Scheme 2b). For compound 7, which possesses a three-carbon linkage, the alkyl migration was not observed, the dearomatization proceeded well but the alkyl migration did not occur, and methoxy rearrangement product 9 was isolated (Scheme 2c). For compound 10, which possesses a five-carbon linkage, the alkyl migration reaction at room temperature proceeded sluggishly, and the reaction at 60 °C afforded corresponding 6,7-fused indole 11 as the major product (Scheme 2d). The structures of compounds 2, 9 and 11 were confirmed by their X-ray crystallography (Scheme 2e).
 | ||
| Scheme 2 Initial test and evaluation of the effect of the length of the linkages. | ||
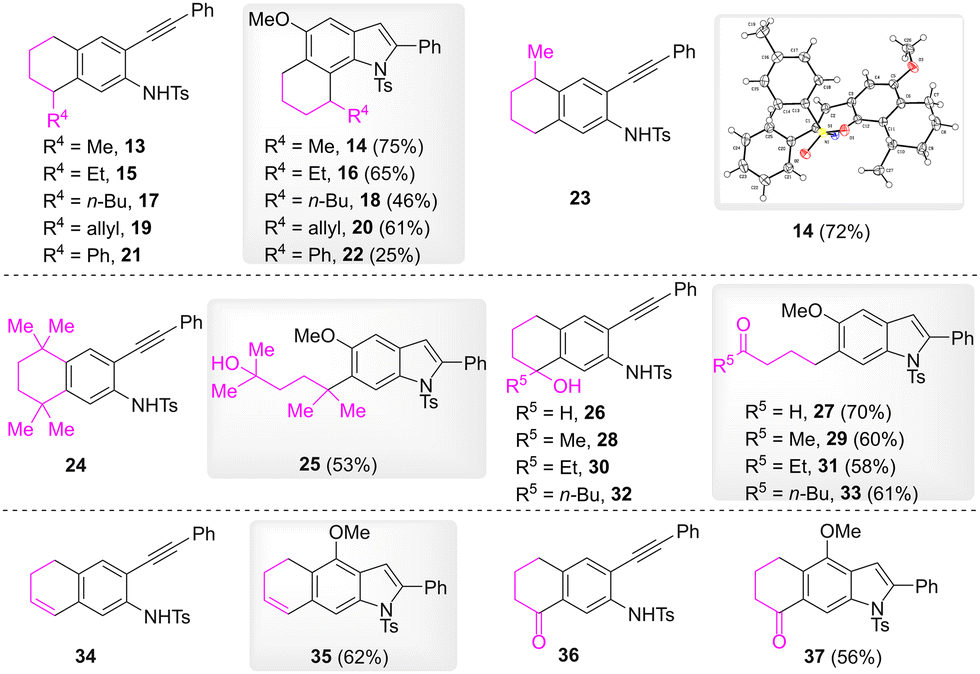
Subsequently, substrates possessing different four-carbon linkages were examined (Scheme 3). Compounds 13, 15, 17, and 19, which bear an alkyl or an allylic substitution at the meta benzyl position, were effectively converted to 6,7-fused indoles 14, 16, 18, and 20, respectively, in which the substitutions located at the C7 benzyl position. When the substitution was a phenyl group, 6,7-fused indole 22 was obtained in a 25% yield. It is noteworthy that reaction of substrate 23, which bears a methyl substitution at the para benzyl position, gave rise to the same product 14 with the reaction of substrate 13. The structure of 14 was confirmed by its X-ray crystallography. When the meta and para benzyl positions were substituted by four methyl groups, the formation of 6,7-fused indole was not observed, and 6-(5-hydroxy-2,5-dimethylhexan-2-yl)-indole 25 was formed instead. In the cases of compounds 26, 28, 30, and 32, which bear a hydroxyl substitution at the meta benzyl position, the reaction gave rise to 6-(4-oxoalkyl) substituted indoles 27, 29, 31, and 33 as products. When the meta group of the substrates was an alkenyl or a carbonyl group, the corresponding 1,2-methoxy rearrangement products were obtained.
 | ||
| Scheme 3 Evaluation of the scope of four-carbon linkages. | ||
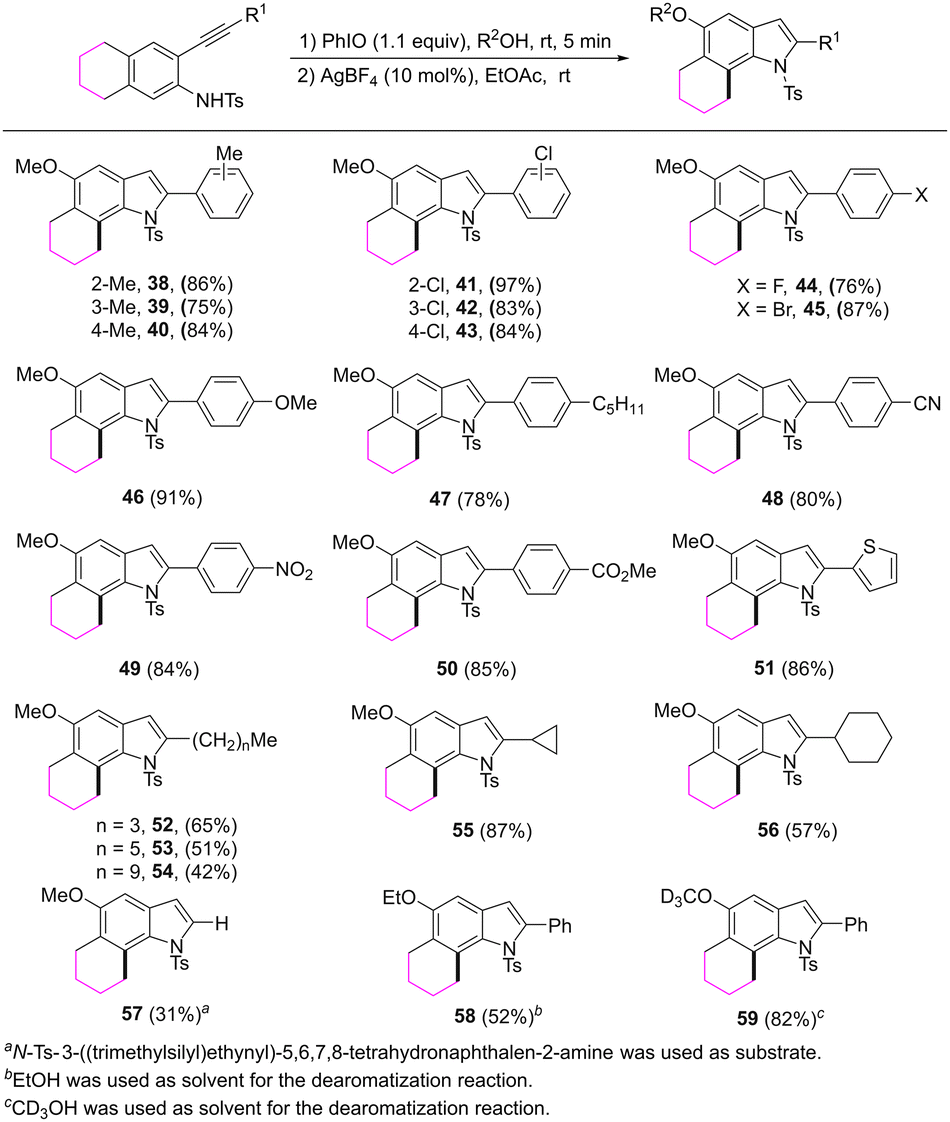
The reaction scope was further investigated by varying the substituents at the 2-alkynyl moiety of the substrates (Scheme 4). The reaction showed broad substrate compatibility and a range of functional groups could be tolerated. For example, no matter whether the substituted group on the aryl ring of the alkynyl moiety is an electron-donating or an electron-withdrawing group, the corresponding 6,7-fused indoles 38–50 were obtained in good yields. 2-Thienyl substituted 6,7-fused indole was also formed in an 86% yield. The length of the linear alkyl group of the alkynyl moiety had some influence on the transformation. 2-Butyl substituted product 52 was isolated in a higher yield compared with 2-hexyl or 2-decyl substituted products 53 and 54. Reactions of substrates bearing a cyclopropyl or cyclohexyl group proceeded well and afforded 55 and 56 in 87% and 57% yields, respectively. When a substrate, which possesses a trimethylsilylethynyl, was employed, the reaction provided 2-unsubstituted 6,7-fused indole 57 in a 31% yield. The 5-OMe group in the products could be replaced by OEt or OCD3 groups by varying the dearomatization solvents.
 | ||
| Scheme 4 Scope investigation of the 2-alkynyl moiety of the substrates. | ||
When the substituent at the 2-alkynyl moiety is a tert-butyl group, the reaction at room temperature gave rise to a mixture of compounds 61 and 62, in which the tosyl group at the nitrogen atom migrates to the C3 position of indole. Reaction temperature has an effect on the selectivity of the formation of these two compounds (Scheme 5a). The reaction at 0 °C gave rise to N-tosyl 2-tert-butyl substituted product 61, while the reaction at 70 °C gave rise to 3-tosyl 2-tert-butyl substituted product 62. The sulfonyl migration was also observed when various arylsulfonyl groups were used as protecting groups of the nitrogen atom in the substrates (Scheme 5b).
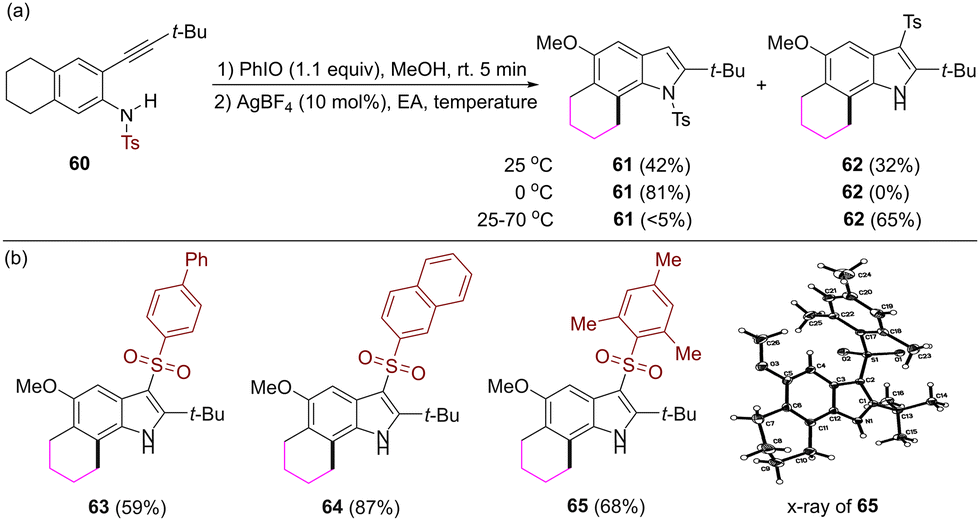 | ||
| Scheme 5 Formation of 3-sulfonyl substituted 6,7-fused indoles. | ||
Some control experiments were conducted to get clues on the reaction pathway. The treatment of 61 in EtOAc at 70 °C for 12 hours gave rise to compound 62 in a 47% yield. When 10 mol% AgBF4 was added, the reaction under the same conditions produced 62 in a 93% yield (Scheme 6a). These results indicated that 3-tosylindole might be formed from the isomerization of N-tosylindole, and the silver catalyst can promote the migration process Additionally, the oxidative dearomatization reactions of substrates 60 and 66 bearing a naphthalenesulfonyl group were conducted in MeOH and EtOH, respectively, and the resulting crude products were mixed together to be treated with AgBF4 under the standard conditions. The reaction only provided 62 and 5-ethyloxy-3-naphthalenesulfonyl indole 67 as products. This result indicated that the arylsulfonyl migration might occur in an intramolecular manner (Scheme 6b). To clarify whether the cyclic structure is essential for the cascade alkyl migration, the reaction of a substrate with two methyl groups both at the C4 and C5 positions was investigated. The reaction gave rise to 4,6-dimethyl-5-methoxy indole 71, which was derived from the clockwise migration of the C4 methyl group in the substrate as the major product (Scheme 6c).
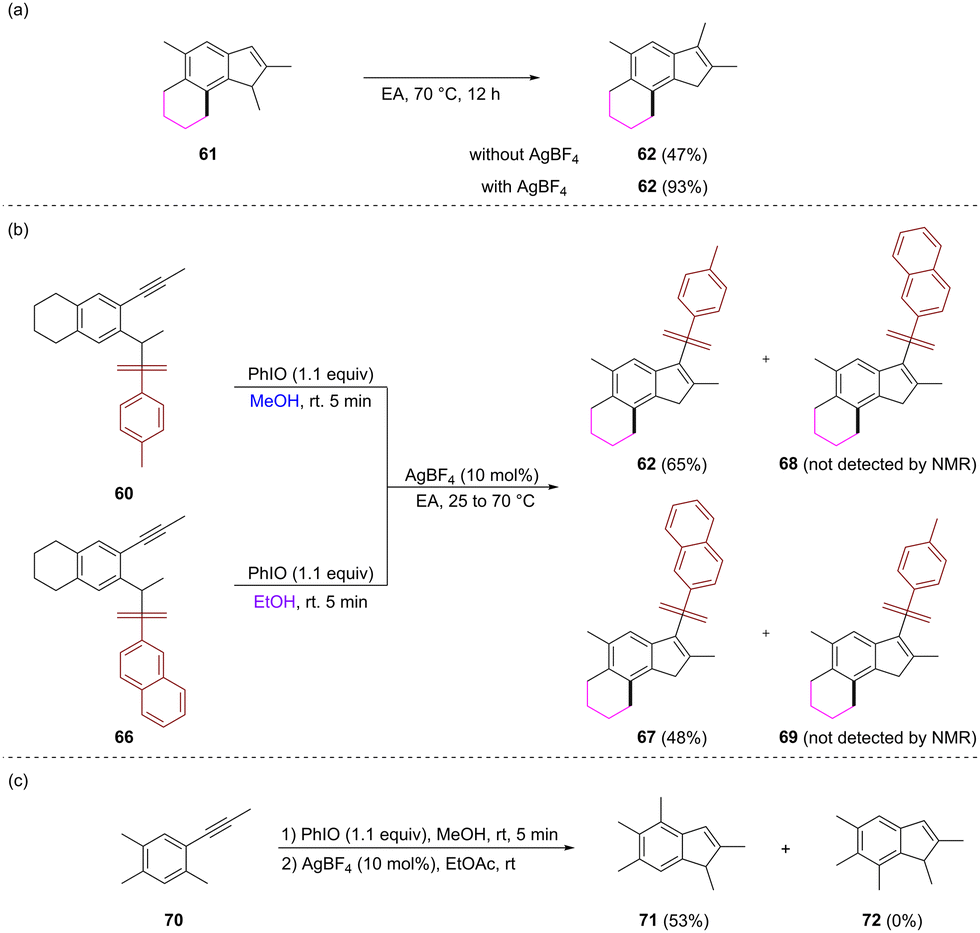 | ||
| Scheme 6 Control experiments. | ||
A plausible pathway of the alkyl migration reaction is depicted in Scheme 7. In the presence of a silver catalyst, the 5-endo cyclization of 2-alkynylcyclohexadienimines lead to the formation of dearomatized arenium species, such as iminium intermediate A and carbon cation intermediate B. The counter-clockwise 1,2-alkyl migration of intermediate B affords a five-or six-member-ring spiro intermediate C (path a). The generation of the spiro intermediate can be supported by the isolation of the same product 14 from the reactions of 13 and 23. To restore the aromaticity, one alkyl group of the spiro intermediate migrates to the C7 position to provide 6,7-fused indoles. When the linkage was a three-carbon unit, the alkyl migration to form the corresponding four-membered-ring spiro intermediate is difficult; clockwise methoxy migration occurs (path b). In the case of substrates that bear a hydroxyl substitution at the meta benzyl position, because the positive charge can be stabilized by the oxygen atom of the hydroxyl group in intermediate G, the cleavage of the carbon–carbon bond of the spiro intermediate gives rise to 6-(4-oxoalkyl) substituted indoles (path c).
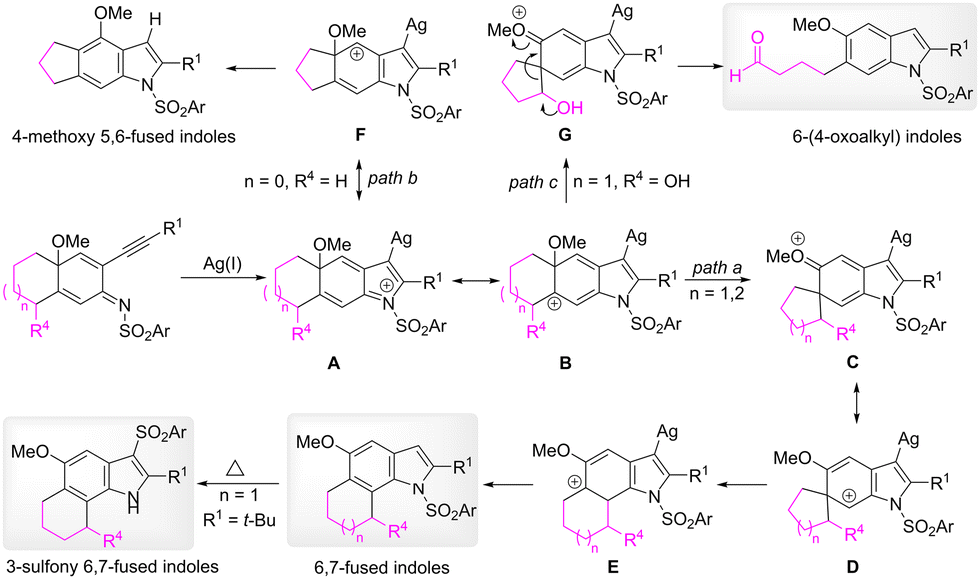 | ||
| Scheme 7 Proposed pathway for the alkyl migration reaction. | ||
In summary, we reported a cascade alkyl migration reaction of 2-alkynylanilines via an aromaticity destruction and reconstruction process. The first alkyl migration is triggered by the generation of a dearomatized arenium species via silver-catalyzed cyclization, and the second alkyl migration of the spiro intermediate is driven by aromatization to restore the aromaticity. The reaction showed broad substrate compatibility and gave rise to a range of functionalized 6,7-fused indoles. The use of this method in the synthesis of natural products is currently underway in our laboratory.
We are grateful to the National Natural Science Foundation of China (21971043, 22371051) for support of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. A. Steele, L. A. Cohen and E. Mosettig, J. Am. Chem. Soc., 1963, 85, 1134–1138 CrossRef CAS; (b) M. Alekseychuk and P. Heretsch, J. Am. Chem. Soc., 2022, 144, 21867–21871 CrossRef CAS; (c) L. Liu, F.-F. Duan, Y. Gao, X.-G. Peng, J.-L. Chang, J. Chen and H.-L. Ran, Org. Lett., 2021, 23, 9620–9624 CrossRef CAS PubMed; (d) R. B. Woodward and T. Singh, J. Am. Chem. Soc., 1950, 72, 494–500 CrossRef CAS; (e) C. P. Whittle, Tetrahedron Lett., 1968, 9, 3689–3692 CrossRef; (f) B. E. Arney, R. C. White, A. Ramanathan, L. Barham, S. Sherrod, P. McCall, P. Livanec, K. Mangus and K. White, Photochem. Photobiol. Sci., 2004, 3, 851–853 CrossRef CAS.
- R. M. Roberts and D. Shiengthong, J. Am. Chem. Soc., 1964, 86, 2851–2857 CrossRef CAS.
- (a) B. Miller and E. R. Matjeka, J. Am. Chem. Soc., 1980, 102, 4772–4780 CrossRef CAS; (b) R. B. Carlin and M. S. Moores, J. Am. Chem. Soc., 1959, 81, 1259 CrossRef CAS.
- (a) A. Suárez, S. Suárez-Pantiga, O. Nieto-Faza and R. Sanz, Org. Lett., 2017, 19, 5074–5077 CrossRef PubMed; (b) Y.-A. Qiu, D. Ma, W. Kong, C. Fu and S. Ma, Org. Chem. Front., 2014, 1, 62–67 RSC; (c) J. Wang, H.-T. Zhu, Y.-F. Qiu, Y. Niu, S. Chen, Y.-X. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2015, 17, 3186–3189 CrossRef CAS PubMed; (d) S. Yaragorla, D. Bag, R. Dada and K. V. J. Jose, ACS Omega, 2018, 3, 15024–15034 CrossRef CAS; (e) S. Maiti and P. Mal, Org. Lett., 2017, 19, 2454–2457 CrossRef CAS PubMed; (f) Z. Zhang, X. Tang, Q. Xu and M. Shi, Chem. – Eur. J., 2013, 19, 10625–10631 CrossRef CAS.
- Y. Ishida, I. Nakamura and M. Terada, J. Am. Chem. Soc., 2018, 140, 8629–8633 CrossRef CAS PubMed.
- J. Lee, B. Kang, D. Kim, J. Lee and S. Chang, J. Am. Chem. Soc., 2021, 143, 18406–18412 CrossRef CAS.
- (a) P. G. Gassman and J. E. Granrud, J. Am. Chem. Soc., 1984, 106, 2448–2449 CrossRef CAS; (b) H. Takeuchi, M. Maeda, M. Mitani and K. Koyama, J. Chem. Soc., Chem. Commun., 1985, 287–289 RSC; (c) M. Luo, X. Zhu, R. Liu, S. Yu and W. Wei, J. Org. Chem., 2020, 85, 3638–3654 CrossRef CAS; (d) Q. Zhou and Y. Li, J. Am. Chem. Soc., 2014, 136, 1505–1513 CrossRef CAS PubMed; (e) S. Wang, G. W. Morrow and J. S. Swenton, J. Org. Chem., 1989, 54, 5364–5371 CrossRef CAS; (f) B. Miller and A. K. Bhattacharya, J. Am. Chem. Soc., 1980, 102, 2450–2451 CrossRef CAS; (g) L. Li, M. Yang, Q. He and R. Fan, Nat. Commun., 2020, 11, 4805 CrossRef CAS PubMed; (h) J. J. Kulagowski, C. J. Moody and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1985, 2725–2732 RSC; (i) H. Takeuchi and K. Takano, J. Chem. Soc., Perkin Trans. 1, 1986, 611–618 RSC.
- (a) J. He, X. Zhang, Q. He, H. Guo and R. Fan, Chem. Commun., 2021, 57, 5442–5445 RSC; (b) T. Xu, Q. He and R. Fan, Org. Lett., 2022, 24, 2665–2669 CrossRef CAS PubMed; (c) Z. Chen, Q. He, H. Guo and R. Fan, Chem. Commun., 2022, 58, 6797–6800 RSC.
- L. Li, X. Li, W. Wang, Q. He and R. Fan, Org. Lett., 2021, 23, 2130–2134 CrossRef CAS PubMed.
- (a) P. D. Thornton, N. Brown, D. Hill, B. Neuenswander, G. H. Lushington, C. Santini and K. R. Buszek, ACS Comb. Sci., 2011, 13, 443–448 CrossRef CAS PubMed; (b) N. Brown, D. Luo, J. A. Decapo and K. R. Buszek, Tetrahedron Lett., 2009, 50, 7113–7115 CrossRef CAS PubMed; (c) A. N. Garr, D. Luo, N. Brown, C. J. Cramer, K. R. Buszek and D. VanderVelde, Org. Lett., 2010, 12, 96–99 CrossRef CAS.
- (a) R. Annunziata, M. Cinquini, F. Cozzi and L. Raimondi, J. Chem. Soc., Chem. Commun., 1986, 366–367 RSC; (b) R. R. Webb II, M. C. Venuti and C. Eigenbrot, J. Org. Chem., 1991, 56, 4706–4713 CrossRef.
- (a) W. Liu, H. J. Lim and T. V. Rajanbabu, J. Am. Chem. Soc., 2012, 134, 5496–5499 CrossRef CAS PubMed; (b) K. R. Buszek, N. Brown and D. Luo, Org. Lett., 2009, 11, 201–204 CrossRef CAS; (c) R. J. Huntley and R. L. Funk, Org. Lett., 2006, 8, 3403–3406 CrossRef CAS.
- (a) R. A. Leal, C. Bischof, Y. V. Lee, S. Sawano, C. C. McAtee, L. N. Latimer, Z. N. Russ, J. E. Dueber, J.-Q. Yu and R. Sarpong, Angew. Chem., Int. Ed., 2016, 55, 11824–11828 CrossRef CAS; (b) S. K. Jackson, S. C. Banfield and M. A. Kerr, Org. Lett., 2005, 7, 1215–1218 CrossRef CAS PubMed; (c) H. Muratake, A. Mikawa and M. Natsume, Tetrahedron Lett., 1992, 33, 4595–4598 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures, characterization data, and 1H and 13C NMR spectra of new compounds. Crystallographic data for compounds 2, 9, 11, 14 and 65. CCDC 2294393–2294397. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04815b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |