
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise design of copolymer-conjugated nanocatalysts for active electron transfer†
Reina
Hagiwara
,
Shun
Nishimura
 and
Kosuke
Okeyoshi
*
and
Kosuke
Okeyoshi
*
Graduate School of Advanced Science and Technology, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: okeyoshi@jaist.ac.jp
First published on 13th December 2023
Abstract
A copolymer-conjugated nanocatalytic system has been designed for active electron transfer. To enhance photoinduced H2 generation, we precisely synthesize ternary random copolymers capable of transferring electrons through phase transitions, extending and shrinking in response to viologen's redox changes within 2 nm distance from the surface of the catalytic nanoparticle.
In the field of photoinduced electron transfer, researchers have developed various precisely arranged molecular systems employing supramolecules or hybrid materials.1–14 Additionally, various organic/polymeric systems have emerged, serving roles in redox reactions and the broader context of photoinduced water splitting, such as artificial photosynthesis. For example, polymers have been used not only as protectors for stably dispersed nanoparticles (NPs)4,5 but also as rails for electron relays2 or molecular complexes with multiple components.13,14 These systems satisfied the theoretically estimated critical distance between the electron donor and acceptor of ∼2 nm.15 However, these systems have not evolved to encompass active molecular motion, such as electron transfer, particularly in fluidic aqueous environments. This gap hinders the development of highly efficient catalytic systems for key reactions. The motivation behind this study lies in the quest for highly efficient systems that can facilitate forward electron transfer by actively promoting electron transportation, thus minimizing side and reverse reactions. This active transportation strategy is a pivotal component in the development of advanced catalytic systems for sustainable energy generation and other applications. A prominent molecule serving as both an electron donor and acceptor is the viologen, which can reversibly transition between redox states and finds utility in various applications, including molecular machines and batteries.16–18
Polymer networks within aqueous environments have proven to be instrumental in various chemical reaction contexts, offering stable dispersions of catalytic NPs while inhibiting self-quenching.19 Photosensitizer-immobilized polymer networks provide an ideal platform for photoenergy conversion in H2- or O2-generating gel systems.20–25 By arranging photosensitizers and catalytic NPs in polymer networks with a combination of Ru(bpy)32+/3+/viologen2+/+/Pt,25–29 researchers have achieved efficient H2 generation driven by visible light energy, with a high overall quantum efficiency exceeding 13%, a superior result when compared to conventional solution-based approaches.19 Recently, we reported an electron transfer system using a copolymer, poly(N-isopropylacrylamide-co-viologen) (PNV), and surfactant-modified Pt NPs.30 Based on the thermosensitive phase transition of the main component of poly(N-isopropylacrylamide) (PNIPAAm), a conformational change occurred in accordance with the redox changes of the viologen. The greatest advantage of the PNV system lies in its capacity to accelerate cyclic electron transfer for H2 generation through the polymeric coil–globule transitions marked by hydrophilic/hydrophobic changes. However, the PNV system still has a notable disadvantage in that not only does the PNV in close proximity to the Pt NPs participate in electron transfer, but the free PNV situated farther from the NPs can also accept electrons. When the free PNV is in a reduced state, it contracts and readily coagulates with neighboring PNV molecules, thereby undergoing electron transfer in the forward reaction. To resolve this problem, a precisely designed polymeric system is required to remove the free PNV, a factor contributing to diffusion-limited reactions. The conjugation of such a polymer onto the Pt NPs offers a strategic solution, allowing for the precise solution of the active path for electron transfer and the creation of a novel catalytic system that functions efficiently.
In this study, we designed a copolymer-conjugated nanocatalytic system for active electron transfer during photoinduced H2 generation using a ternary random copolymer, poly(NIPAAm-co-acrylamide-co-viologen) (PNAV) (Fig. 1). Acrylamide (AAm) was used for conjugation onto the Pt NPs and arrangement of the copolymer only around the Pt NPs. When redox changes occur in the viologen unit, the polymeric coil–globule transitions, characterized by cyclic extending and shrinking dynamics, actively accelerate electron transport for H2 generation. In contrast to conventional solution systems, the polymeric system has the following advantages: (I) active electron transport by conformational changes in PNAV driven by redox changes in viologen, (II) distance control between viologen and the Pt NP surface by PNAV conjugation, and (III) inhibition of Pt NP aggregation at high salt concentrations and self-quenching among viologens. In addition, in comparison with the polymeric system using PNV and surfactant-Pt NPs, the system using PNAV-conjugated Pt NPs was precisely arranged on a single-nanometer scale for electron transfer. According to Marcus theory, electron transfer proves highly effective when the distance between an electron donor and an electron acceptor is less than ∼2 nm.15 In this system, the viologen unit located on the surface of the Pt NP lies within the reach of polymeric fluctuations. In the context of the photoinduced system, when a photoexcited sensitizer such as *Ru(bpy)32+ donates an electron to the viologen in the polymer, the reduced viologen triggers immediate shrinkage of the polymer, causing it to approach the surface of the Pt NPs. Subsequently, H2 is generated from the pooled electrons in the Pt NPs and protons. Following the pooling of electrons in the Pt NPs, the polymer undergoes expansion again, enabling the continuous expansion of the electronic transfer circuit. Throughout the redox cycles of the viologen, the polymer chain serves as a distance regulator, akin to tentacles.
 | ||
| Fig. 1 Photoinduced H2 generation system using poly(NIPAAm-co-AAm-co-viologen) (PNAV). Chemical structure and redox states of PNAV and electron transfer driven by coil–globule transitions of PNAV conjugated on Pt nanoparticles (NPs). The dot circle means the effective distance from the Pt NP surface. | ||
A semi-telechelic PNAV with a terminal amino group was synthesized through the radical telomerization30 of NIPAAm (87 mol%) and AAm (5 mol%), with a viologen monomer (8 mol%) using 2-aminoethanethiol (AESH) acting as a chain transfer agent (Fig. 2). Notably, the molecular structure of the viologen monomer was designed to introduce an acrylamide group, imparting characteristics akin to NIPAAm. The molecular weight of the polymer (Mw), as determined via gel permeation chromatography, was 5.02 × 103 g mol−1. The actually introduced NIPAAm, AAm, and viologen units were estimated as 85.6, 10.2, and 4.2 mol%, respectively, as calculated from the 1H-NMR spectra of PNAV (see ESI,† Fig. S1). As a rough estimation using the Mw value and introduced unit ratio, the number of each monomer unit in one PNAV chain was approximately calculated to be 36 NIPAAm, 4 AAm, and 2 viologen in 42 monomers on average. As presented in Fig. 2B, a schematic diagram of the PNAV linear chain is presented as a random ternary copolymer, incorporating these proportions. Considering that the calculated number of monomers and the C–C bond distance are 0.154 nm, the polymer length of PNV can be calculated to be approximately 13 nm on average. In this molecular arrangement, the AAm units were less than 2 nm from the viologen unit with an extremely high probability. Using the average number of viologen monomers in one PNAV chain, we calculated the distance between the viologen and the Pt NP surface to be 1.5 nm. This distance ensures that the electrons are capable of transferring from the donor to the acceptor, as estimated by Marcus theory. Furthermore, this calculation indicates that the viologen enters the reduced state, and the distance between the viologen and AAm can decrease because of the shrinking of the PNIPAAm parts. Thus, the precise design of the PNAV chain holds potential for controlling the distance when PNAV is conjugated to Pt NPs. Moreover, this precise molecular sequence contributes synergistically to the excluded volume effect, ensuring the stable dispersion of Pt NPs and active electron transport.
 | ||
| Fig. 2 Precise design of the ternary copolymer. (A) Preparation of poly(NIPAAm-co-AAm-co-viologen) by telomerization. (B) Schematic of PNAV as a linear chain, calculated with the values in polymer properties. | ||
As presented in Fig. 3A, the aqueous PNAV solution appears colorless in the oxidized state (PNAV2+) and takes on a violet hue in the reduced state (PNV+). The absorption spectra of PNAV in the oxidized and reduced states are illustrated in Fig. 3B. Maximum absorption in the reduced state occurred at a wavelength of approximately 550 nm, similar to that of the viologen monomer. Fig. 3C shows the changes in the transmittance of the PNAV solutions as a function of temperature. The redox states of viologen in the polymer were controlled using a reductant, Na2S2O4. Owing to the thermosensitivity of PNIPAAm, the transmittance suddenly decreased as the temperature increased in both states, indicative of the phase transition temperature (PTT) (see ESI,† Fig. S2). When the viologen unit is maintained in the reduced state (PNAV+), the PTT is lower than that in the oxidized state (PNAV2+). The decrease in the PTT for PNAV+ is attributed to an increase in hydrophobicity resulting from a reduction in charge within the unit. This behavior aligns with observations generally made for PNIPAAm-based polyelectrolytes when their ionic charges decrease. Furthermore, the influence of AAm on the polymer is evident as the PTT of PNAV+ is higher than that of homo-PNIPAAm (approximately 30 °C). This elevation in PTT is attributed to the greater hydrophilicity of PNAV+ compared to PNIPAAm and PNV+. In our prior report,30 PNV+ exhibited a lower PTT than PNIPAAm because of its increased hydrophobicity. Based on the deviations in the PTTs of PNAV2+ and PNAV+, it was expected that the polymer will undergo cyclic expansion and collapse as the viologen unit is cyclically oxidized and reduced, respectively (Fig. 3D). In the PNAV, the temperature range between 33 °C and 50 °C is of critical importance for changes in polymeric dispersion and flocculation, rendering this feature greatly advantageous for photoinduced electronic transfer circuits conducted at a constant temperature.
 | ||
| Fig. 3 Optical properties of poly(NIPAAm-co-AAm-co-viologen) (PNAV). (A) Photographs of PNAV solutions in oxidized (2+) and reduced (1+) states. (B) Absorption spectra of PNAV solutions (0.05 wt%) at 25 °C. (C) Temperature dependence of transmittances of PNAV solutions. PNAV: 0.05 wt%; Tris–HCl buffer solution (25 °C, pH 7.4, 500 mM); λ = 700 nm. Na2S2O4 (50 mM) for reducing viologen, PNAV+. (D) Schematic illustration of PNAV2+'s expansion for dispersion and PNAV+'s collapse for flocculation. | ||
Fig. 4A presents the preparation of PNAV-conjugated Pt NPs through reduction using PNAV, chloroplatinic acid (H2PtCl6) and sodium borohydride (NaBH4) under alkaline conditions. During the reduction process, the amine group of AAm within PNAV becomes conjugated to the Pt NPs, resulting in a change in the color of the sample from pale yellowish to dark brown, indicative of stable colloidal dispersion. The transmission electron microscopy (TEM) image in Fig. 4B clearly indicates that the NPs have an average diameter of 1.85 nm and are well dispersed without aggregation, even in the dried state. This dispersion stability suggests that the polymer chains are effectively conjugated to the Pt NPs without becoming entangled with each other. The elemental ratio between S and Pt was determined by measuring the elemental concentrations in the purified sample through dialysis using a membrane with a molecular weight cutoff (MWCO) of 100 kDa. Concentrations were measured using inductively coupled plasma mass spectrometry.
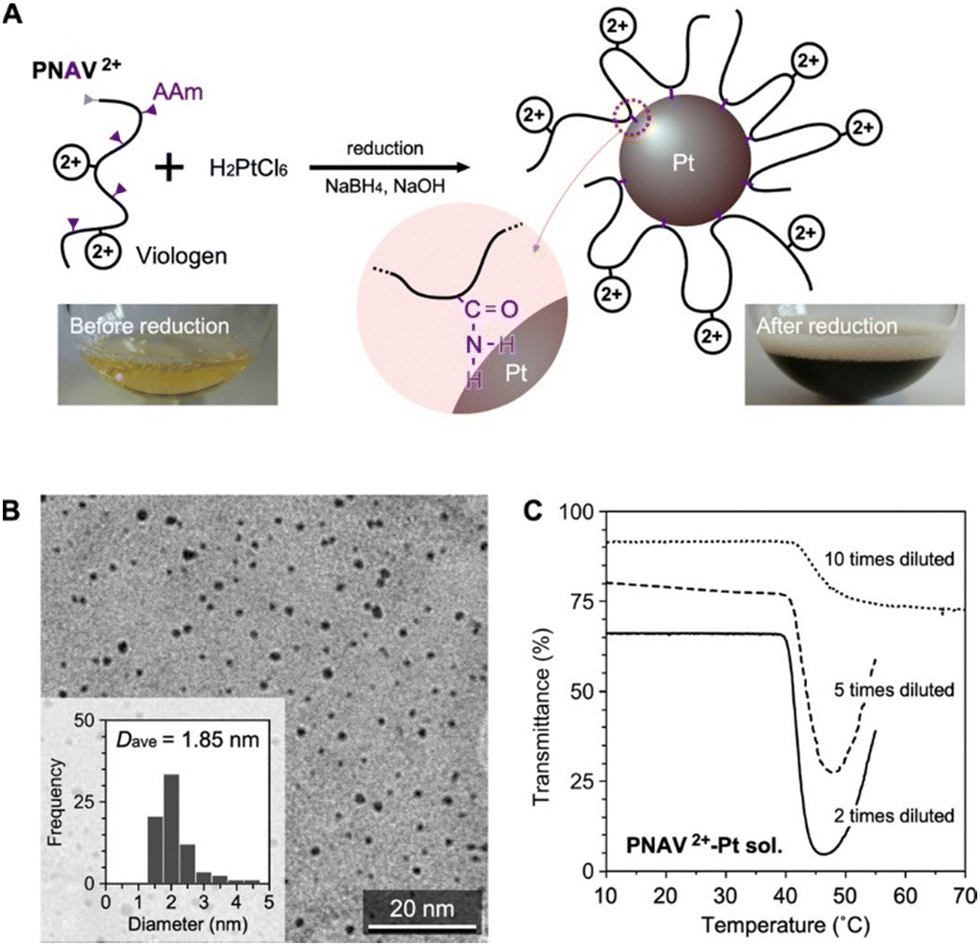 | ||
| Fig. 4 Preparation and properties of PNAV-conjugated Pt nanoparticles (NPs). (A) Synthetic scheme of PNAV-conjugated Pt NPs by the reduction method. Photos: before and after reduction of the aqueous mixture. (B) TEM image of PNAV-conjugated Pt NPs. Inset: Size distribution of the NPs. (C) Temperature dependence of the transmittances of the PNAV2+/+-conjugated Pt NP colloidal solution. Tris–HCl buffer solution (25 °C, pH 7.4, 500 mM); λ = 700 nm. | ||
To investigate the behavior of conjugated PNAV on the Pt NP, the temperature dependence of the transmittance was measured at various concentrations of the PNAV-Pt colloidal solution (Fig. 4C). Analogous to free PNAV solutions, the samples exhibited a substantial decrease in transmittance above 40 °C when the viologen was in its oxidized state. As the concentration increased, the degree of decrease in transmittance became more pronounced and substantial. These results indicate that the conjugated PNAV on the Pt NP is capable of inducing polymeric flocculation of the mass of PNAV-Pt NPs at higher temperatures. Considering that the transmittance of the non-conjugated PNAV2+ is drastically decreased at temperatures exceeding 50 °C (see Fig. 2C), the PTT (approximately 40 °C) of the PNAV-Pt colloidal solution is substantially influenced by the conjugation, a decrease possibly due to the influence of the AAm unit, which renders the PNV chain more hydrophobic compared to the PNAV chain. As for the reduced state of PNAV conjugated on Pt NP (PNAV+-Pt NP), there was a limit to maintain the stable dispersion during the measurement. Considering the irreversible reactions including the reducer, Na2S2O4 and the oxidation of PNAV+ on Pt, the alternative characterization for the equilibrium states is now under consideration.
In pursuit of active electron transfer within the realm of photoinduced H2 generation, we introduced a meticulously designed copolymer-conjugated nanocatalytic system. This innovation harnessed the advantages of a stimuli-responsive polymer chain to achieve active electron transfer. Specifically, a ternary random copolymer, poly(NIPAAm-co-AAm-co-viologen) (PNAV), was synthesized by precisely controlling the molecular weight and the introduction ratio of the units. One striking feature of PNAV was its temperature-responsive behavior, which manifested as a phase transition contingent on temperature. This unique copolymer displayed a distinct transition, transitioning between a swollen state in its oxidized form (PNAV2+) and a shrunken state in its reduced form (PNAV+). Furthermore, PNAV was conjugated to Pt NPs using a reduction method to control the distance between viologen and Pt. In particular, the swelling/shrinking of PNAV was confirmed on the Pt NPs, a factor critical to the success of the envisioned cyclic electron transfer process at a critical distance. Notably, this design aligns with Marcus theory. In the future, this copolymer-conjugated nanocatalytic system will be useful for active electron transfer in artificial photosynthetic reactions, such as water splitting.22,30 We envision that this polymeric design for distance control will facilitate not only photochemical reactions but also various electrochemical reactions31,32 and macromolecular recognitions.33,34 The sustainable cycle it enables in energy conversion systems offers the potential to drive innovation in diverse scientific domains, ultimately contributing to the broader landscape of sustainable energy technologies.
This study was supported by a Grant-in-Aid for Challenging Research (Exploratory) (Grant number: JP21K18998) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- M. Sykora, K. A. Maxwell, J. M. DeSimone and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7687–7691 CrossRef CAS PubMed.
- M. Kaneko, Prog. Polym. Sci., 2001, 26, 1101–1137 CrossRef CAS.
- N. Toshima, T. Takahashi and H. Hirai, J. Macromol. Sci., Part A: Pure Appl. Chem., 1988, 25, 669–686 CrossRef.
- T. Yonezawa and N. Toshima, J. Mol. Catal., 1993, 83, 167–181 CrossRef CAS.
- H. Inoue, S. Funyu, Y. Shimada and S. Takagi, Pure Appl. Chem., 2005, 77, 1019–1033 CrossRef CAS.
- Y. Zhong, K. Ueno, Y. Mori, X. Shi, T. Oshikiri, K. Murakami, H. Inoue and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 10350–10354 CrossRef CAS PubMed.
- S. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2014, 136, 586–589 CrossRef CAS PubMed.
- A. M. Sanders, T. J. Magnanelli, A. E. Bragg and J. D. Tovar, J. Am. Chem. Soc., 2016, 138, 3362–3370 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- T. Matsuo, T. Sakamoto, K. Takuma, K. Sakura and T. Ohsako, J. Phys. Chem., 1981, 85, 1277–1279 CrossRef CAS.
- K. Yamauchi, S. Masaoka and K. Sakai, J. Am. Chem. Soc., 2009, 131, 8404–8406 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Biochem. Biophys. Acta, 1985, 811, 265–322 CAS.
- N. L. Poul and B. Colasson, ChemElectroChem, 2015, 2, 475–496 CrossRef.
- M. Burgess, J. S. Moore and J. Rodríguez-López, Acc. Chem. Res., 2016, 49, 2649–2657 CrossRef CAS PubMed.
- V. Balzani, M. Clemente-Leo, A. Credi, B. Ferrer, M. Venturi, A. H. Flood and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1178–1183 CrossRef CAS PubMed.
- K. Okeyoshi and R. Yoshida, Chem. Commun., 2011, 47, 1527–1529 RSC.
- K. Okeyoshi and R. Yoshida, Soft Matter, 2009, 5, 4118–4123 RSC.
- K. Okeyoshi and R. Yoshida, Chem. Commun., 2009, 6400–6402 RSC.
- K. Okeyoshi and R. Yoshida, Adv. Funct. Mater., 2010, 20, 708–714 CrossRef CAS.
- K. Okeyoshi, D. Suzuki, A. Kishimura and R. Yoshida, Small, 2011, 7, 311–315 CrossRef CAS PubMed.
- K. Okeyoshi, R. Kawamura, R. Yoshida and Y. Osada, Chem. Commun., 2015, 51, 11607–11610 RSC.
- M. Grätzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 435–461 CrossRef CAS.
- E. Amouyal, Sol. Energy Mater. Sol. Cells, 1995, 38, 249–276 CrossRef CAS.
- B. Happ, A. Winter, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 2222–2255 RSC.
- K. Okeyoshi and R. Yoshida, Angew. Chem., Int. Ed., 2019, 58, 7304–7307 CrossRef CAS PubMed.
- S. Sen, J. Saraidaridis, S. Y. Kim and G. T. R. Palmore, ACS Appl. Mater. Interfaces, 2013, 5, 7825–7830 CrossRef CAS PubMed.
- M. Aizenberg, K. Okeyoshi and J. Aizenberg, Adv. Funct. Mater., 2018, 28, 1704205 CrossRef.
- A. E. Kaifer, Acc. Chem. Res., 1999, 32, 62–71 CrossRef CAS.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including preparation and characterization of materials. See DOI: https://doi.org/10.1039/d3cc05242g |
| This journal is © The Royal Society of Chemistry 2024 |