
A hetero-bimetallic Ru(II)–Ir(III) photosensitizer for effective cancer photodynamic therapy under hypoxia†
Mengsi
Zheng
a,
Xinlin
Lin
a,
Kai
Xiong
a,
Xiting
Zhang
*b,
Yu
Chen
 *a,
Liangnian
Ji
a and
Hui
Chao
*a
*a,
Liangnian
Ji
a and
Hui
Chao
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, Guangdong Basic Research Center of Excellence for Functional Molecular Engineering, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510006, P. R. China. E-mail: ceschh@mail.sysu.edu.cn; chenyu63@mail.sysu.edu.cn
bSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, 510006, P. R. China. E-mail: zhxt@gzhu.edu.cn
First published on 10th February 2024
Abstract
A hetero-bimetallic Ru(II)–Ir(III) photosensitizer was developed. Upon light exposure, contrary to the homogeneous Ru(II)–Ru(II) and Ir(III)–Ir(III) complexes that can only produce singlet oxygen, Ru(II)–Ir(III) can generate multiple reactive oxygen species and kill hypoxic tumors. This study presents the first example of a hetero-bimetallic type-I and type-II dual photosensitizer.
As a light-driven modality, photodynamic therapy (PDT) has the advantages of high tumor selectivity and low normal tissue damage and has been approved for cancer treatment worldwide.1 The photosensitizer is the critical component of PDT. When exposed to light, it is excited to the excited state, which eventually kills the tumor cells through two pathways. In type-I pathways, a radical intermediate is formed, which then reacts with oxygen-containing species to produce reactive oxygen species (ROS) such as superoxide anion radicals (O2•−) or hydroxyl radicals (•OH). In a type-II reaction, the excited photosensitizer directly transfers its energy to O2 to produce singlet oxygen (1O2).2 The FDA-approved photosensitizers PHOTOFRIN, Visudyne, and ALA are type-II photosensitizers.3 As oxygen-dependent photosensitizers, their effectiveness is limited by the hypoxic microenvironment of the tumor. Therefore, there is an urgent need to develop type-I photosensitizers to treat hypoxic tumors. However, in contrast to the well-established type-II photosensitizer design strategy, a design strategy for type-I photosensitizers is elusive.
The mainstream focus of photosensitizer development for the last half-century has been on organic molecules.4 However, this trend reversed in the past decade. Metal complexes, especially Ru(II) and Ir(III) complexes, have attracted emerging interest as photosensitizer candidates due to their ability to satisfy several needs for PDT.5 For example, a Ru(II)-based photosensitizer, TDL1433, is under phase II clinical trials, giving new hope to patients.6 The enormous potential of metal complexes for PDT has been summarized in many recent reviews.7 However, reports so far have focused on mononuclear complexes as type-II photosensitizers, which increase the excited state lifetimes and thus enhance their ability to transfer energy to oxygen by adjusting the ligands.8 By contrast, two metal centers of dinuclear complexes may exist in coupled interactions and exhibit unique excited states, photoinduced electrons, and energy transfer processes, giving access to peculiar photophysical and photochemical properties.9 Sun's group developed a series of dinuclear Ir(III) complexes showing a pronounced PDT effect via1O2 generation under visible light.10 Subsequently, they synthesized a dinuclear Ir(III) complex and established that it exhibited improved 1O2 generation ability compared to the corresponding mononuclear one.11 Bryce and Draper's work also confirmed this conclusion.12 Butera presented a series of dinuclear Ru(II)-pyrrolide complexes as both type-I and type-II photosensitizers by computational study, which needs experimental validation.13 Compared with the rigorous development of mononuclear photosensitizers, dinuclear metal complexes applied in PDT have been less well studied and mainly include dinuclear complexes with homogeneous metal centers. Reports on hetero-bimetallic photosensitizers are rare, some of which only verified their ability to cause cell-free DNA damage, while others are essentially mononuclear photosensitizers obtained through photodissociation.14 Additionally, as oxygen-dependent type-II photosensitizers, their ability against hypoxic tumors has not yet been evaluated.
Capitalizing on this, herein, the rational design, synthesis, theoretical calculation, and biochemical investigation of dinuclear metal complexes Ru-Ru, Ir-Ir, and Ru-Ir as photosensitizers are described. Upon irradiation, the two homogeneous complexes Ru-Ru and Ir-Ir efficiently produced 1O2 under normoxic conditions but they lost their photosensitizer ability under hypoxia. On the contrary, the heterometallic Ru-Ir generated multiple ROS in both normoxic (•OH, O2•−, and 1O2) and hypoxic (•OH, and O2•−) circumstances. After being encapsulated with DSPE-PEG2000 to form a nano drug to enhance cell uptake, the ability of Ru-Ir@PEG against hypoxic tumors was demonstrated (Scheme 1). To the best of our knowledge, Ru-Ir is the first heterometallic type-I and type-II dual photosensitizer.
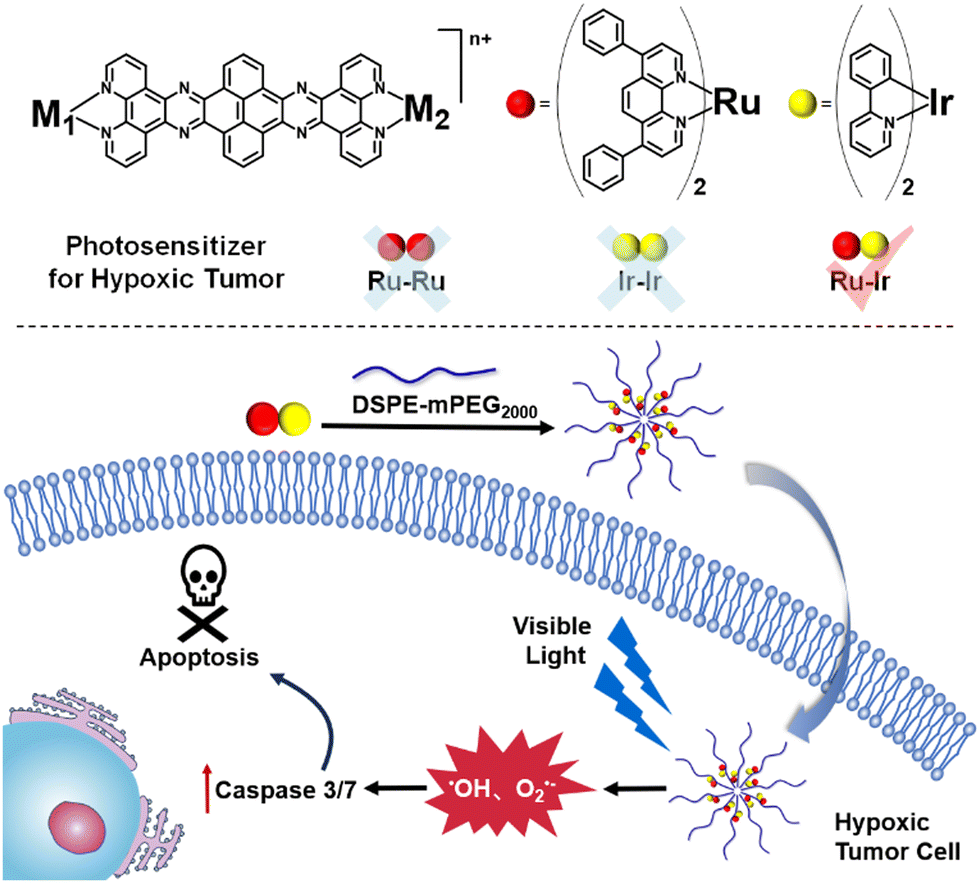 | ||
| Scheme 1 The chemical structures of the bimetallic photosensitizers and the illustration of Ru-Ir@PEG against hypoxic tumors. | ||
The synthetic routes of Ru-Ru, Ir-Ir, and Ru-Ir are presented in Scheme S1 (ESI†). The three complexes were characterized by 1H NMR spectroscopy and ESI-MS (Fig. S1–S6, ESI†). Ru-Ru showed characteristic absorption bands of intraligand charge transfer (ILCT) around 280 nm and broad metal-to-ligand charge transfer (MLCT) from 400 to 500 nm (Fig. S7A, ESI†). In contrast, Ir-Ir exhibited an additional ILCT band at 330 nm and sharp MLCT peaks at about 405 and 428 nm. The UV-vis spectrum of Ru-Ir included all the characteristic peaks mentioned above, indicating the absence of coupling between the two metal centers (Fig. S7A, ESI†).15 None of the three complexes could emit luminescence (Fig. S7B, ESI†).
In PDT, the ability and type of photoinduced ROS generation are the most important properties. The ROS productions of Ru-Ru, Ir-Ir, and Ru-Ir were investigated by electron paramagnetic resonance (EPR). As shown in Fig. 1A, upon irradiation and the presence of capture agent 2,2,6,6-tetramethyl-4-piperidone hydrochloride (TEMP), a triple peak (peak integral ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) was observed for Ru-Ru, Ir-Ir, and Ru-Ir, demonstrating that the three complexes efficiently produced 1O2 under normoxic conditions and could be applied as type-II photosensitizers.16 These EPR signals disappeared when adding histidine, a 1O2 scavenger, to the system (Fig. S8, ESI†). The 1O2 yields of the complexes were indirectly measured using 1,3-diphenylisobenzofuran (DPBF) as a probe (Fig. S9, ESI†). The 1O2 yields of Ru-Ru, Ir-Ir, and Ru-Ir were 0.78, 0.32, and 0.76, respectively, consistent with the EPR results. Note that due to the involvement of two MLCTs in Ru-Ir, its 1O2 yields under 405 and 450 nm irradiation were detected, with values of 0.65 and 0.76, respectively (Fig. S10, ESI†). Since 450 nm irradiation-induced higher 1O2 yield and the light damage of 450 nm is lower than 405 nm, 450 nm was used as the excitation wavelength for Ru-Ir in subsequent experiments.
:1:1) was observed for Ru-Ru, Ir-Ir, and Ru-Ir, demonstrating that the three complexes efficiently produced 1O2 under normoxic conditions and could be applied as type-II photosensitizers.16 These EPR signals disappeared when adding histidine, a 1O2 scavenger, to the system (Fig. S8, ESI†). The 1O2 yields of the complexes were indirectly measured using 1,3-diphenylisobenzofuran (DPBF) as a probe (Fig. S9, ESI†). The 1O2 yields of Ru-Ru, Ir-Ir, and Ru-Ir were 0.78, 0.32, and 0.76, respectively, consistent with the EPR results. Note that due to the involvement of two MLCTs in Ru-Ir, its 1O2 yields under 405 and 450 nm irradiation were detected, with values of 0.65 and 0.76, respectively (Fig. S10, ESI†). Since 450 nm irradiation-induced higher 1O2 yield and the light damage of 450 nm is lower than 405 nm, 450 nm was used as the excitation wavelength for Ru-Ir in subsequent experiments.
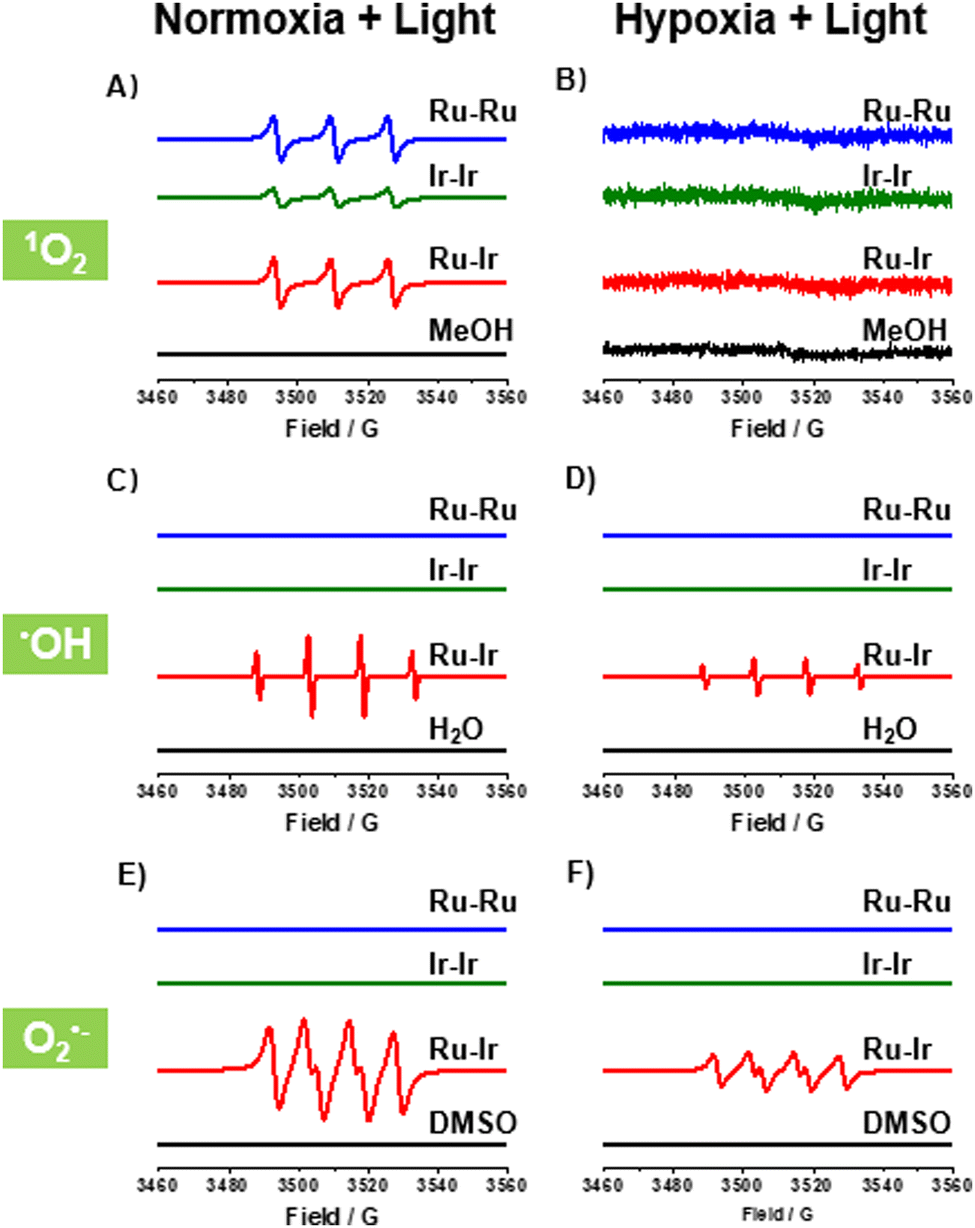 | ||
| Fig. 1 EPR spectra of Ru-Ru, Ir-Ir, and Ru-Ir (10 μM) trapped by (A) and (B) TEMP to detect the 1O2 signal in MeOH, (C) and (D) BMPO to detect the •OH signal in H2O, and (E) and (F) BMPO to detect the O2•− signal in DMSO. The testing environments were either under normoxic (21% O2) or hypoxic conditions (1% O2). Light: 450 nm LED for Ru-Ru and Ru-Ir; 405 nm LED for Ir-Ir. Light dose: 20 mW cm−2 for 5 min. | ||
Different from the effective 1O2 generation under normoxic conditions (21% O2), no 1O2 was detected as expected under hypoxia (1% O2, Fig. 1B), indicating that the efficiency of Ru-Ru, Ir-Ir, and Ru-Ir may be limited in hypoxic solid tumors. To overcome this problem, we examined the potential of the three complexes as type-I photosensitizers with the spin-trapping agent 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO).17 As shown in Fig. 1C and D, the characteristic BMPO-•OH adduct (peak integral ratio 1:2:2:1) was observed for heterometallic Ru-Ir in H2O, whether under normoxic or hypoxic conditions. The •OH generation was also evidenced by a typical colorimetric analysis based on 3,3′,5,5′-tetramethyl-benzidine (TMB). Upon light exposure, Ru-Ir oxidized TMB to yield blue-coloured ox-TMB with typical absorbances at 652 nm (Fig. S11, ESI†).16 Replacing the solution H2O with •OH quencher DMSO, a characteristic signal for the BMPO-O2•− adduct (peak integral ratio 1:1:1:1) was recorded (Fig. 1E and F), consistent with previous reports.17 Interestingly, there were no corresponding signals for the homogeneous complexes Ru-Ru and Ir-Ir in the same experimental conditions. These conclusions were further demonstrated in cells by cytotoxicity and commercial probe experiments which followed.
Encouraged by the performance of the three complexes for ROS production, the in vitro (photo)cytotoxicity was evaluated. Since Ru-Ru, Ir-Ir, and Ru-Ir are lipophilic with positive octanol/water partition coefficient (logPO/W) values of 1.13 ± 0.13, 1.08 ± 0.08, and 0.71 ± 0.05, respectively, Ru-Ru, Ir-Ir, and Ru-Ir were wrapped into nanoparticles with DSPE-PEG2000, named Ru-Ru@PEG, Ir-Ir@PEG, and Ru-Ir@PEG, to improve biocompatibility. These nanoparticles were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS), Zeta potential, and inductively coupled plasma mass spectrometry (ICP-MS). The three nanoparticles exhibited similar characteristics. Taking Ru-Ir@PEG as an example, the solution was characterized to be a monodisperse suspension with an average hydrodynamic diameter of 124.35 nm (Fig. S12A, ESI†). Similarly, the TEM study further demonstrated the spherical shape of Ru-Ir@PEG (Fig. S12B, ESI†). The Zeta potential of Ru-Ir@PEG was conducted to be −20.18 ± 0.63 mV (Fig. S13, ESI†), indicating its stability in solution. Using ICP-MS measurement, the encapsulation efficiency of Ru-Ir@PEG was 59.58%, consistent with previous reports.
The (photo)cytotoxicity of the three nanoparticles toward cisplatin-resistant cancer cells A549R was evaluated to study the potential anticancer properties. As shown in Table S1 (ESI†), under normoxic conditions, Ru-Ru@PEG, Ir-Ir@PEG, and Ru-Ir@PEG were found with cytotoxic values toward A549R in the moderate range (16.90–50.79 μM) in the dark, while being significantly more toxic (1.40–4.89 μM) with a mild light fluence (6 J cm−2). Ru-Ru@PEG and Ru-Ir@PEG showed similar PDT potency, with respective PI values of 20.4 and 21.9, much higher than Ir-Ir@PEG (3.46). This discriminative PDT potency can be attributed to their distinct cellular intake levels, organelle distribution, and 1O2 yield. As shown in Table S2 and Fig. S14 (ESI†), after 12 h incubation, Ru-Ru@PEG and Ru-Ir@PEG mainly localized in the cytoplasm with similar cellular content (12.21 ± 2.13 and 14.55 ± 2.94 ng/106 cell, Table S1, ESI†). In contrast, Ir-Ir@PEG accumulated in the mitochondria, leading to more toxicity in the dark, which synergistically contributed to its low PI value with poor singlet oxygen yield.
When reducing the oxygen concentration to 1%, Ru-Ru@PEG and Ir-Ir@PEG showed similar toxicity in the absence and presence of irradiation, revealing the limitation of type-II photosensitizers. Contrary to this, the heterometallic Ru-Ir@PEG kept excellent PDT lesions in A549R cells (IC50 = 3.48 + 0.66 μM), demonstrating its potential against hypoxic tumors. The commercial ROS probe staining also confirmed this observation. As shown in Fig. 2A, there was a significant increase in the enzymatic product 2′,7′-dichlorofluorescein (DCF) fluorescence intensity in A549R cells treated with Ru-Ir@PEG upon 450 nm irradiation (20 mW cm−2, 5 min), while cells of the control group showed no apparent fluorescence. Since 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) is not specific for any particular ROS, the O2•−-specific probe dihydroethidium (DHE) and •OH-specific detector hydroxyphenyl fluorescein (HPF) were further used to confirm the light-induced O2•− and •OH generated by Ru-Ir@PEG (Fig. 2A).18
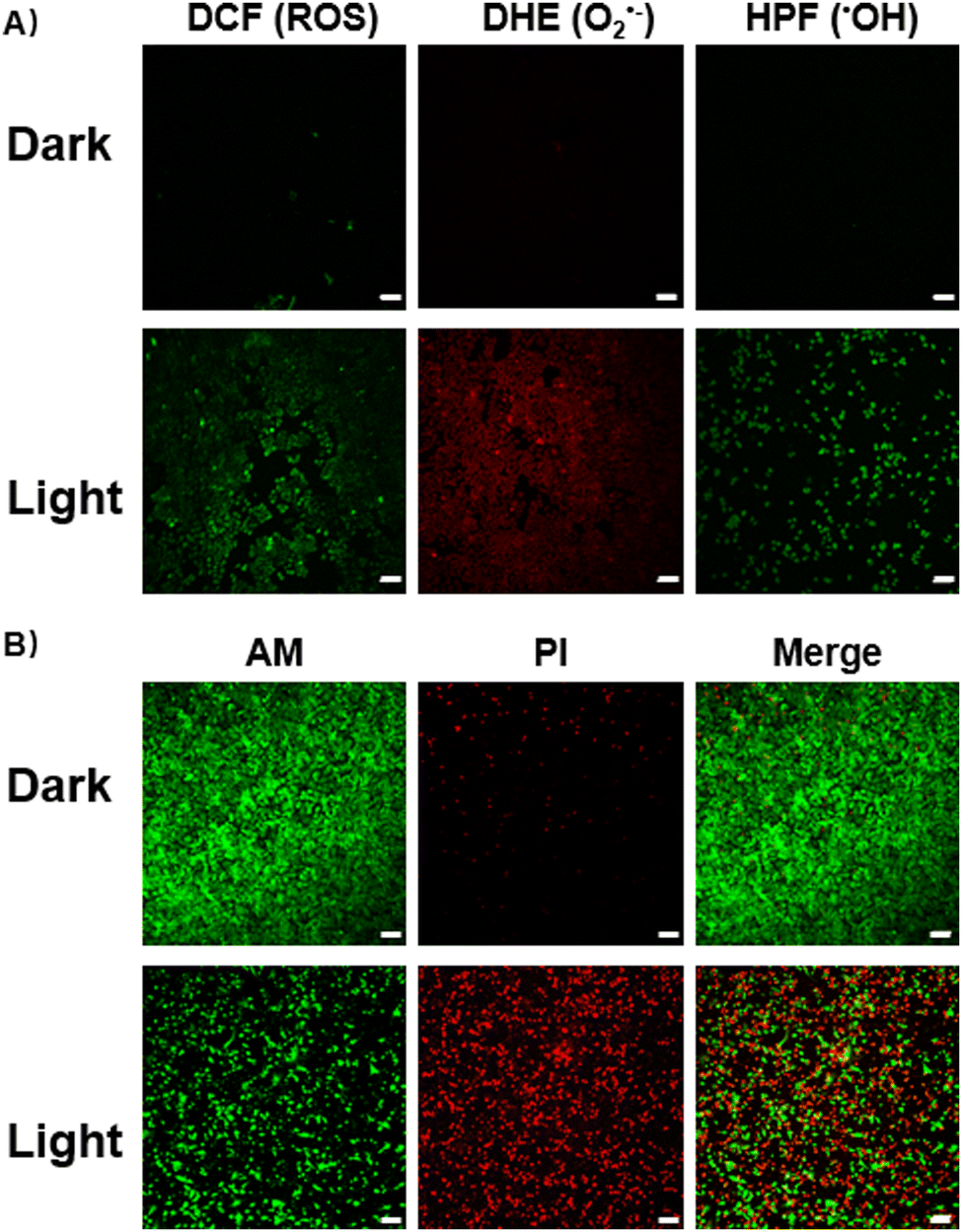 | ||
| Fig. 2 Confocal microscopy images of A549R cells with the corresponding dyes before and after Ru-Ir@PEG (5 μM) PDT at 450 nm (20 mW cm−2, 5 min). (A) Cellular ROS, •OH, and O2•− detection (DCF: λex = 488 nm, λem = 515–535 nm; DHE: λex = 510 nm, λem = 600–620 nm; HPF: λex = 490 nm, λem = 510–520 nm); (B) Ca-AM/PI co-staining (Ca-AM: λex = 490 nm, λem = 510–520 nm; PI: λex = 535 nm, λem = 610–630 nm). All testing environments were performed under hypoxic conditions (1% O2). Scale bars: 100 μm. | ||
To assess the phototherapeutic efficacy under hypoxia, A549R cells were co-stained with calcein acetoxymethyl ester (Ca-AM, green fluorescence, live cells)/propidium iodide (PI, red fluorescence, dead cells). After irradiation, for Ru-Ir@PEG-treated cells, a significant reduction of Ca-AM fluorescence accompanied by the appearance of PI signal was observed (Fig. 2B). The mechanism of cell death induced by Ru-Ir@PEG upon light exposition was evaluated next. For annexin V-FITC/PI double staining, the percentage of apoptotic cells was 62.8% for Ru-Ir@PEG and laser irradiation treatment. By contrast, the value for the control group was 5.91% (Fig. S15, ESI†). Moreover, the PDT process of Ru-Ir@PEG in A549R cells resulted in a 5.8-fold increase in the caspase 3/7 activation level, higher than classical apoptosis-inducer cisplatin (2.8-fold, Fig. S16, ESI†). The up-regulated protein expression level of cleaved-caspase 3 was also confirmed by Western Blot (Fig. S17, ESI†). These results indicated that Ru-Ir@PEG induced apoptotic cell death in A549R cells.16 This conclusion was further evidenced by cell death mode inhibitors. As shown in Fig. S18 (ESI†), after being co-incubated with apoptosis inhibitor Z-VAD-FMK, the cell viability of Ru-Ir@PEG increased from 67.94 ± 1.04% to 84.34 ± 0.79%, while the other cell death mode inhibitors, including autophagic inhibitor 3-methyladenine, necrosis/necroptosis inhibitor necrostatin-1, ferroptosis inhibitor ferrostatin-1, and lysosomal protease-mediated cell death inhibitor leupeptin, were ineffective.
In summary, three DSPE-PEG2000-encapsulating dinuclear photosensitizers were developed. When in normoxic conditions, both of them acted as type-II photosensitizers by generating singlet oxygen upon irradiation. However, the homometallic Ru-Ru@PEG and Ir-Ir@PEG lost their phototherapeutic efficacy under hypoxia. On the contrary, the heterometallic Ru-Ir@PEG was demonstrated to generate multiple ROS (•OH, and O2•−) and thus applied as a type-I photosensitizer against hypoxic tumor cells, indicating that the generated ROS types can be regulated by adjusting the metal center of the dinuclear complexes. To the best of our knowledge, Ru-Ir@PEG is the first hetero-bimetallic type-I and type-II dual photosensitizer. Since solid tumors are known to form a normoxic region and a hypoxic core, a dual photosensitizer will synergistically kill tumor cells by both type-I and type-II mechanisms in the normoxic region and achieve antitumor activity by a type-II mechanism in the hypoxic core region. We hope that this work will provide a feasible route for developing type-I and type-II dual photosensitizers and bring promising new PDT reagents from the lab to the clinic.
This work was supported by the National Natural Science Foundation of China (No. 21977126, 22120102002, 22207134) and the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (No. 2021B1515020102).
Conflicts of interest
There are no conflicts to declare.References
- (a) P. Wu, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC; (b) S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed; (c) L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- (a) Y. Zhang, B. T. Doan and G. Gasser, Chem. Rev., 2023, 123, 10135–10155 CrossRef CAS PubMed; (b) K. Xiong, F. Wei, Y. Chen, L. Ji and H. Chao, Small Methods, 2023, 7, 2201403 CrossRef CAS PubMed; (c) E. Ortega-Forte, A. Rovira, M. López-Corrales, A. Hernández-García, F. J. Ballester, E. Izquierdo-García, M. Jordà-Redondo, M. Bosch, S. Nonell, M. D. Santana, J. Ruiz, V. Marchán and G. Gasser, Chem. Sci., 2023, 14, 7170–7184 RSC.
- (a) X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185 RSC; (b) J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 CrossRef CAS PubMed.
- (a) B. K. Kundu, G. Han and Y. Sun, J. Am. Chem. Soc., 2023, 145, 3535 CrossRef CAS PubMed; (b) C. Han, B. K. Kundu, Y. Liang and Y. Sun, Adv. Mater., 2024, 36, 2307759 CrossRef CAS PubMed.
- (a) Z. Deng, H. Li, S. Chen, N. Wang, G. Liu, D. Liu, W. Ou, F. Xu, X. Wang, D. Lei, P. C. Lo, Y. Y. Li, J. Lu, M. Yang, M. L. He and G. Zhu, Nat. Chem., 2023, 15, 930–939 CrossRef CAS PubMed; (b) X. Zhou, P. Wang, V. Ramu, L. Zhang, S. Jiang, X. Li, S. Abyar, P. Papadopoulou, Y. Shao, L. Bretin, M. A. Siegler, F. Buda, A. Kros, J. Fan, X. Peng, W. Sun and S. Bonnet, Nat. Chem., 2023, 15, 980–987 CrossRef CAS PubMed; (c) D. Wei, Y. Huang, B. Wang, L. Ma, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2022, 61, e202201486 CrossRef CAS PubMed; (d) K. B. K. Pragti, S. N. Upadhyay, N. Sinha, R. Ganguly, I. Grabchev, S. Pakhira and S. Mukhopadhyay, Dalton Trans., 2022, 51, 3937 RSC; (e) B. K. K. Pragti, S. Singh, W. A. C. Ranjith, S. Sarkar, A. Sonawane and S. Mukhopadhyay, ACS Appl. Mater. Interfaces, 2023, 15(37), 43345–43358 CrossRef PubMed.
- M. A. Munegowda, A. Manalac, M. Weersink, S. A. McFarland and L. Lilge, Coord. Chem. Rev., 2022, 470, 214712 CrossRef PubMed.
- (a) B. K. K. Pragti and S. Mukhopadhyay, Coord. Chem. Rev., 2021, 448, 214169 CrossRef; (b) X. Li, X. Zhao, W. Wang, Z. Shi, Y. Zhang, Q. Tian, Y. Yao, C. He and C. Duan, Coord. Chem. Rev., 2023, 495, 215366 CrossRef CAS.
- A. Mani, T. Feng, A. Gandioso, R. Vinck, A. Notaro, L. Gourdon, P. Burckel, B. Saubaméa, O. Blacque, K. Cariou, J. E. Belgaied, H. Chao and G. Gasser, Angew. Chem., Int. Ed., 2023, 62, e202218347 CrossRef CAS PubMed.
- (a) X. Li, Z. Wang, X. Hao, J. Zhang, X. Zhao, Y. Yao, W. Wei, R. Cai, C. He, C. Duan, Z. Guo, J. Zhao and X. Wang, J. Am. Chem. Soc., 2023, 145, 14766–14775 CrossRef CAS PubMed; (b) P. S. Felder, S. Keller and G. Gasser, Adv. Therap., 2020, 3, 1900139 CrossRef; (c) M. F. Wang, R. Yang, S. J. Tang, Y. A. Deng, G. K. Li, D. Zhang, D. Chen, X. Ren and F. Gao, Angew. Chem., Int. Ed., 2022, 61, e202208721 CrossRef CAS PubMed; (d) J. Wang, Y. Lu, N. McGoldrick, C. Zhang, W. Yang, J. Zhao and S. M. Draper, J. Mater. Chem. C, 2016, 4, 6131–6139 RSC.
- B. Liu, S. Monro, L. Lystrom, C. G. Cameron, K. Colón, H. Yin, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2018, 57, 9859–9872 CrossRef CAS PubMed.
- C. Lu, W. Xu, H. Shah, B. Liu, W. Xu, L. Sun, S. Y. Qian and W. Sun, ACS Appl. Bio. Mater., 2020, 3, 6865–6875 CrossRef CAS PubMed.
- (a) L. Zhang, Y. Li, W. Che, D. Zhu, G. Li, Z. Xie, N. Song, S. Liu, B. Z. Tang, X. Liu, Z. Su and M. R. Bryce, Adv. Mater., 2019, 6, 1802050 Search PubMed; (b) L. Zhang, Y. Geng, L. Li, X. Tong, S. Liu, X. Liu, Z. Su, Z. Xie, D. Zhu and M. R. Bryce, Chem. Sci., 2021, 12, 5918–5925 RSC; (c) J. Wang, Y. Lu, W. McCarthy, R. Conway-Kenny, B. Twamley, J. Zhao and S. M. Draper, Chem. Commun., 2018, 54, 1073–1076 RSC.
- V. Butera, G. Mazzone and H. Detz, ChemPhotoChem, 2022, 6, e202200094 CrossRef CAS.
- (a) J. Wang, J. Newman Jr., S. L. H. Higgins, K. M. Brewer, B. S. J. Winkel and K. J. Brewer, Angew. Chem., Int. Ed., 2013, 52, 1262–1265 CrossRef CAS PubMed; (b) C. Zhang, R. Guan, X. Liao, C. Ouyang, T. W. Rees, J. Liu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2019, 55, 12547 RSC.
- (a) J. Zhou, J. Li, K. Y. Zhang, S. Liu and Q. Zhao, Coord. Chem. Rev., 2022, 453, 214334 CrossRef CAS; (b) Q. Chen, J. Cuello-Garibo, L. Bretin, L. Zhang, V. Ramu, Y. Aydar, Y. Batsiun, S. Bronkhorst, Y. Husiev, N. Beztsinna, L. Chen, X. Zhou, C. Schmidt, I. Ott, M. J. Jager, A. M. Brouwer, B. W. Snaar-Jagalska and S. Bonnet, Chem. Sci., 2022, 13, 6899–6919 RSC; (c) H. Qin, C. Zhao, Y. Sun, J. Ren and X. Qu, J. Am. Chem. Soc., 2017, 139, 16201–16209 CrossRef CAS PubMed.
- F. Wei, J. Karges, J. Shen, L. Xie, K. Xiong, X. Zhang, L. Ji and H. Chao, Nano Today, 2022, 44, 101509 CrossRef CAS.
- (a) H. Zhang, C. Hao, A. Qu, M. Sun, L. Xu, C. Xu and H. Kuang, Angew. Chem., Int. Ed., 2020, 59, 7131–7138 CrossRef CAS PubMed; (b) M. Chang, Z. Hou, M. Wang, D. Wen, C. Li, Y. Liu, Y. Zhao and J. Lin, Angew. Chem., Int. Ed., 2022, 61, e202209245 CrossRef CAS PubMed.
- M. P. Murphy, H. Bayir, V. Belousov, C. J. Chang, K. J. A. Davies, M. J. Davies, T. P. Dick, T. Finkel, H. J. Forman, Y. Hanssen-Heininger, D. Gems, V. E. Kagan, B. Kalyanaraman, N. G. Larsson, G. L. Milne., T. Nyström, H. E. Poulsen, R. Radi, H. V. Remmen, P. T. Schumacker, P. J. Thornalley, S. Toyokuni, C. C. Winterbourn, H. Yin and B. Halliwell, Nat. Metab., 2022, 4, 651–662 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00072b |
| This journal is © The Royal Society of Chemistry 2024 |