
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrocapsule fabrication by ATRP at the interface of non-aqueous emulsions†
Nicholas C.
Starvaggi
 a,
Chase B.
Somodi
b,
Eliandreina Cruz
Barrios
b,
Patrick J.
Shamberger
b and
Emily B.
Pentzer
*ab
a,
Chase B.
Somodi
b,
Eliandreina Cruz
Barrios
b,
Patrick J.
Shamberger
b and
Emily B.
Pentzer
*ab
aDept. of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: emilypentzer@tamu.edu
bDept. of Materials Science & Engineering, College Station, TX 77843, USA
First published on 28th March 2024
Abstract
We present soft-template encapsulation of salt hydrate phase change materials (PCMs) using modified silica particles to both stabilize emulsions and serve as initiators for organocatalyzed photoredox ATRP. The resulting core–shell structures have high core loading and are robust to thermal cycling. Critically, this strategy eliminates the need for a reagent in the core phase, thus preserving purity, and offers the ability to tailor shell composition for desired applications.
Encapsulation of solids, liquids, and gases finds application in biomedicine,1 personal hygiene,2 energy management,3 and foodstuffs4 among other diverse industries. Commonly spherical and several microns in diameter, microcapsules typically comprise a solid or fluid core wrapped by an organic5 or metallic shell.6 Encapsulation of sensitive materials offers advantages over their bulk counterparts, such as preventing chemical changes, ensuring containment for transport, and facilitating controlled release in response to stimuli.7,8 Encapsulation also resolves intrinsic challenges with bulk liquid processing (e.g., high viscosity) and low mass transfer rates, due to increased surface area to volume ratio.9 By tuning properties such as size, morphology, and shell composition, microcapsules can be implemented in advanced applications such as CO2 capture,10 contaminant removal,11 and targeted drug delivery.12
Common techniques to prepare microcapsules include microfluidics, hard-template approach, and soft-template method. Perhaps the most widely used, soft-templating typically leverages interfacial polymerization in emulsions between monomers in the continuous and discontinuous phases.13 Co et. al. synthesized hexadecane-containing microcapsules from oil-in-water emulsions stabilized by dodecyltrimethylammonium bromide; maleate monomer in the oil and divinyl ether monomer in the water underwent free-radical alternating copolymerization at the interface to yield a polymer shell around the hydrocarbon core.14 Our group and others have used graphene oxide (GO) nanosheets to stabilize Pickering emulsions and coupled these with interfacial polymerization.15–17 As prepared GO stabilizes oil-in-water emulsions whereas “alkylated” (hydrophobized) GO stabilizes non-aqueous emulsions and broadens the scope of fluids that can be encapsulated to include water-sensitive, water-miscible, and hygroscopic components.18
Microcapsule formation by interfacial polymerization limits the purity of the core, as it is tainted with monomer. Thus, this approach is not suitable for composition-sensitive cores such as salt hydrate phase change materials (PCMs), as slight changes in composition alter properties and performance in thermal energy management. Prior approaches to encapsulate salt hydrates include the use of water-in-oil in situ miniemulsion polymerization, as reported by Shchukin et al.; the authors prepared nanocapsules with a core of magnesium nitrate hexahydrate (MNH) and shell of poly(ethyl-2-cyanoacrylate), but required as much as 80 wt% extra water to maintain hydration, which decreased the loading of MNH.19 The same group also reported an inverse silica-stabilized Pickering emulsion to encapsulate MNH by acid-catalyzed interfacial condensation of tetraethyl orthosilicate, in this case using 50 wt% extra water.20 More recently, Lak and colleagues used alkylated GO to stabilize droplets of MNH or zinc nitrate hexahydrate (ZNH) in toluene, forming the emulsion above the PCM melting point; polymer was precipitated onto the droplets, forming a core–shell structure. The ionic liquid (IL) 1-ethyl-3-methylimidazolium hexafluorophosphate ([EMIM][PF6]) could also be encapsulated, and different commodity polymers could be used (i.e., polystyrene, poly(methyl methacrylate), and polyethylene).21 Although this approach overcame the need for excess water, shell composition was limited.
Herein, we use non-aqueous emulsions stabilized by reactive particle surfactants and atom transfer radical polymerization (ATRP) to prepare microcapsules with high loading and purity of hygroscopic core components. Our hypothesis is that modified silica can be used to (i) stabilize PCM droplets in oil and (ii) bear initiators for in situ shell formation. Silica particles were modified with α-bromo ester groups, which served as ATRP initiators (SiO2–Br), as confirmed by Fourier-transform infrared (FTIR) spectroscopy and thermogravimetric analysis (TGA). These particles were dispersible in nonpolar solvents (e.g., toluene) and showed interfacial activity at PCM-oil interface. Organocatalyzed photoredox ATRP of hydrophobic monomers in the continuous phase from SiO2–Br particles at the interface gave core–shell structure of microcapsules that can undergo thermal cycling without leakage (Fig. 1). This work, to the best of our knowledge, is the first report of encapsulating PCMs by reactions of modified, interfacially assembled particle surfactants to aid in core–shell formation. Critically, this strategy offers the ability to tailor composition while maintaining a pristine core, critical to application-focused properties.
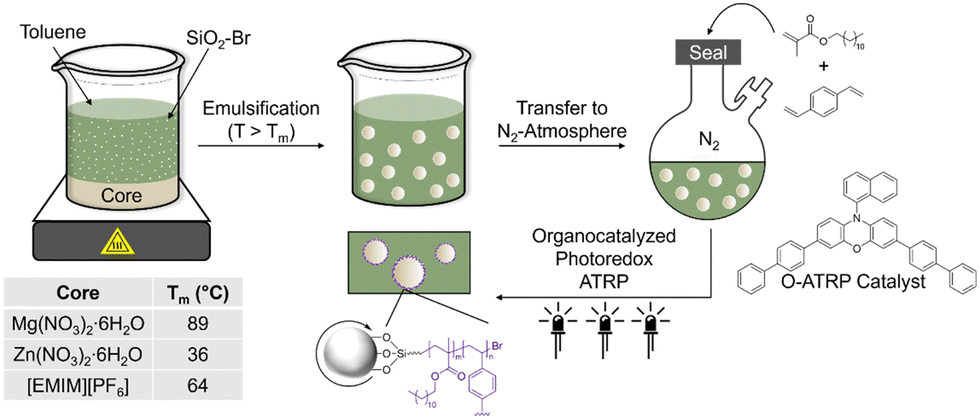 | ||
| Fig. 1 Overview of microcapsule fabrication method using silica particles modified with ATRP initiators as the surfactant. | ||
Particle surfactants with surface-immobilized ATRP initiators were prepared by functionalizing SiO2 particles (20 nm in diameter) in a two-step procedure22 (Fig. 2A). Pristine silica was dried under vacuum, dispersed in toluene, and reacted with (3-aminopropyl)triethoxysilane, yielding SiO2–NH2. Silanes readily undergo hetero-condensation with surface silanol (Si–OH) groups, resulting in formation of covalent siloxane linkages.23 The pendant amines of SiO2–NH2 were amidated24 with 2-bromopropionyl bromide to yield SiO2–Br.
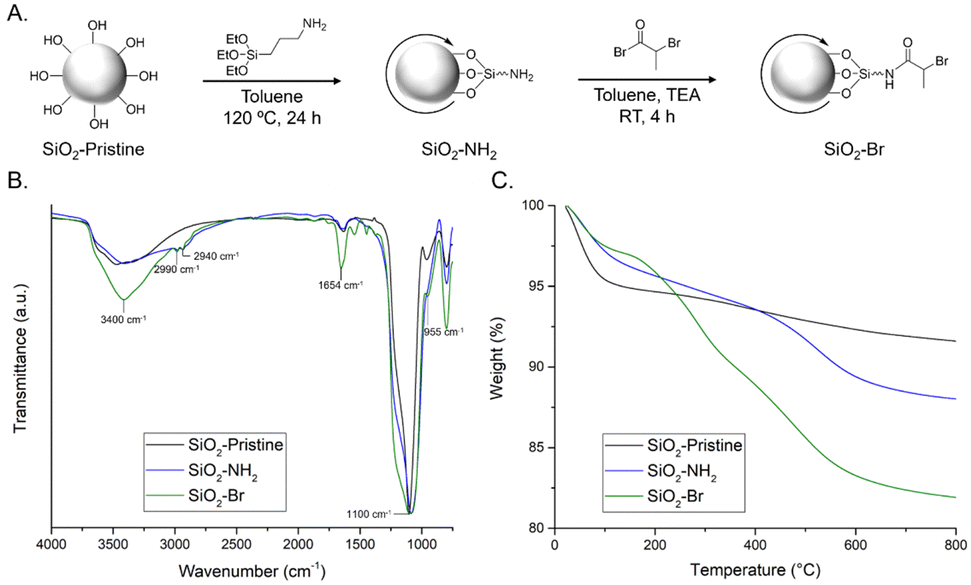 | ||
| Fig. 2 (A) Schematic of two-step synthesis of SiO2–Br from pristine SiO2; (B) normalized FTIR spectra; and (C) TGA weight loss profiles of SiO2–pristine, SiO2–NH2, and SiO2–Br particles. | ||
Modified silica particles were characterized using field emission scanning electron microscopy (FE-SEM), FTIR spectroscopy, TGA, and elemental analysis. Individual particle size was not expected to change upon functionalization; FE-SEM images reveal that pristine SiO2 is ∼22 nm and both SiO2–NH2 and SiO2–Br are ∼24 nm in diameter (Fig. S4, ESI†). Particle modification was confirmed via FTIR spectroscopy (Fig. 2B). All samples show three characteristic stretching frequencies: Si–O–Si (∼1100 cm−1), Si–OH (∼955 cm−1), and broad O–H (∼3400 cm−1). The spectrum of SiO2–NH2 also contains a stretching frequency at ∼2900 cm−1 attributed to the sp3 C–H groups. As expected, intensity of this C–H peak increases for SiO2–Br; additionally, the appearance of a peak ∼1650 cm−1 is attributed to the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. TGA qualitatively characterized particle functionalization (Fig. 2C). Pristine silica particles are hydroscopic and show significant mass from water.25 By contrast, the primary mass loss events for SiO2–NH2 and SiO2–Br occur >200 °C, and can be attributed to decomposition of organic moieties.26 Elemental analysis of SiO2–NH2 and SiO2–Br confirms the presence of N and Br, respectively (Table S1, ESI†).
O. TGA qualitatively characterized particle functionalization (Fig. 2C). Pristine silica particles are hydroscopic and show significant mass from water.25 By contrast, the primary mass loss events for SiO2–NH2 and SiO2–Br occur >200 °C, and can be attributed to decomposition of organic moieties.26 Elemental analysis of SiO2–NH2 and SiO2–Br confirms the presence of N and Br, respectively (Table S1, ESI†).
Modification of the silica colloids alters particle wettability, as well as interfacial assembly. Consistent with prior reports, we found that pristine silica is dispersible in water, but not interfacially active, whereas SiO2–NH2 is both dispersible in water and can stabilize oil-in-water emulsions.27,28 In contrast, SiO2–Br is more hydrophobic, readily dispersible in nonpolar solvents, and exhibits interfacial activity for PCM/toluene.
Emulsions for soft-template microcapsule fabrication were prepared by melting the desired core (MNH, ZNH, or [EMIM][PF6]), then adding a toluene dispersion of SiO2–Br at the same temperature. This biphasic mixture was emulsified via probe sonication at elevated temperatures and then cooled to ambient temperature resulting in solid, spherical droplets. These particles (i.e., pre-polymerized) were characterized via optical microscopy and laser scattering at room temperature; salt hydrate and ionic liquid droplets ranged from 10–15 μm in diameter (Fig. S5 and Table S2, ESI†). In all, a clear continuous phase supports SiO2–Br particles are associated with the interface. Notably, if toluene and the core are treated with the same conditions in the absence of SiO2–Br, emulsion droplets rapidly coalesce, further supporting the interfacial activity of SiO2–Br.
Microcapsules were fabricated via organocatalyzed photoredox ATRP in a grafting from polymerization on the interfacially assembled SiO2–Br, as shown in Fig. 1. Initial attempts to leverage conventional ATRP conditions, as previously reported for oil-in-water systems,29 were unsuccessful; the copper catalyst system showed poor solubility in toluene and migrated to the core (Fig. S6, ESI†). Thus, organocatalyzed photoredox ATRP was used; phenoxazine photocatalyst was added to a vessel prior to sealing under nitrogen, then lauryl methacrylate and divinyl benzene were added to the continuous phase, serving as the monomer and crosslinker respectively. Upon irradiation with UV, ATRP proceeded from SiO2–Br at the interface to give crosslinked polymer and a composite shell around the solid core. Critically, neither catalyst nor monomer is soluble in the core, allowing fabrication of microcapsules with a pristine core. The reaction mixture was exposed to air and microcapsules were isolated via centrifugation and washing to remove residual monomer and catalyst, then dried under reduced pressure.
The structure of the microcapsules was characterized by microscopy and composition was determined by FTIR and TGA, as well as extraction of the core. As shown in Fig. 3A, microcapsules with an MNH core exhibit a rough surface morphology and a nonuniform size, typical of soft-template encapsulation methods. Table S2 (ESI†) summarizes microcapsule size for all samples, indicating a reasonable correlation with emulsion droplet diameter. Remarkably, inter-microcapsule crosslinking is not observed, which we attribute to the dispersion of the droplets and localization of propagating chain ends at the interface and in the growing shell. Post-polymerization GC-MS analysis of the supernatant (containing unreacted monomers, solvent, and catalyst) indicates 61.2% conversion of lauryl methacrylate and 69.4% conversion of divinyl benzene (Fig. S7, ESI†). The core weight percent was determined by extraction of the salt hydrate (into water) or IL (into deuterated acetone) and characterization by UV-vis or 1H NMR, respectively. The loading of MNH, ZNH, and [EMIM][PF6] was found to be 94.6 ± 1.5, 95.6 ± 2.4, and 95.2 ± 0.01 wt%, respectively (Table S2, ESI†). This high core loading indicates a thin composite shell.
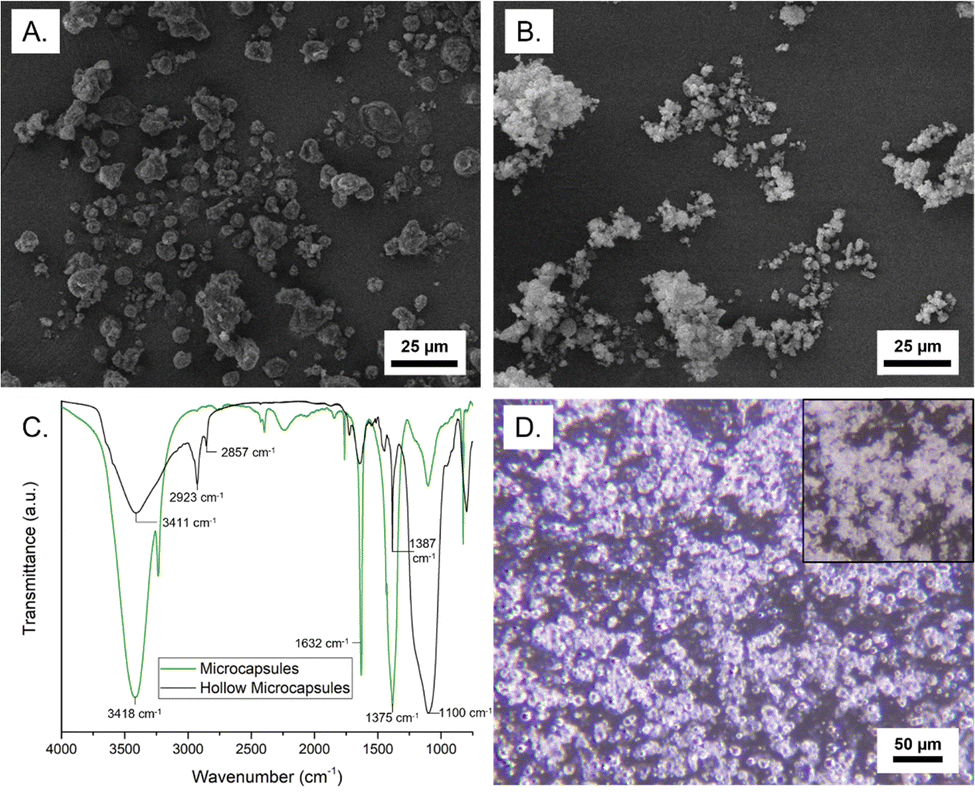 | ||
| Fig. 3 SEM images of MNH microcapsules (A) before and (B) after core extraction; (C) FTIR spectra of MNH microcapsules (green) and hollow shells (black); (D) optical microscopy images of MNH microcapsules after heating to 100 °C for 10 min with inset depicting microcapsules at room temperature. | ||
Hollow shells were isolated by centrifugation after core extraction, then drying under reduced pressure. Shells of the MNH microcapsules were semi-spherical and had a rough surface, similar to the filled microcapsules (Fig. 3B). The FTIR spectrum of the MNH microcapsules supports the presence of MNH (O–H at ∼3400 cm−1 and N–O at ∼1600 cm−1 and ∼1375 cm−1);30 the spectrum of the silica/polymer composite shell does not have these peaks but instead shows an intense signal at ∼2900 cm−1 (sp3 C–H) and peak at ∼1400 cm−1 (CC aromatic), as shown in Fig. 3C. In both spectra, stretching frequencies at ∼3400 cm−1 and ∼1100 cm−1 arise from the silica. Notably, the FTIR spectrum of pre-polymerization emulsion droplets after extraction is consistent with SiO2–Br, as expected (Fig. S8, ESI†). TGA weight loss profiles of bulk MNH and MNH microcapsules show 15–20% weight loss at ∼100 °C (loss of water) and major weight loss between 340 °C and 500 °C (Fig. S9, ESI†), indicating decomposition of MNH. Interestingly, the microcapsules show an additional weight loss at ∼320 °C, which we attribute to polymer decomposition. For both, ∼20% residual mass at 600 °C is due to the formation of MgO219 and, in the case of the microcapsules, residual silica.
To determine stability to thermal cycling, microcapsules were imaged via optical microscopy upon heating above the Tm of the core. The core will remain contained in the shell upon melting and solidification if encapsulation is robust. For MNH microcapsules, above 89 °C, the refractive index of the microcapsules changes (Movie 1), which suggests the MNH liquifies, yet the microcapsules retain their spherical shape with no apparent leakage of liquid (Fig. 3D). By contrast, the pre-polymerization particles rapidly coalesce above 89 °C.
Fig. 4 depicts the DSC thermograms of bulk MNH and MNH microcapsules. Bulk MNH shows an undercooling of 24.4 °C, suggesting a nucleation-limited process in which freezing occurs below the equilibrium melting temperature.31 Undercooling is determined by subtracting the peak temperature of the crystallization exotherm (Tcr) from the peak temperature of the melting exotherm (Tm). For the microcapsules, both exothermic and endothermic transformations are sharp, consistent with a uniform volume undergoing nucleation and growth, but undercooling remains (undercooling of 78.6 °C, Fig. 4). This dramatic shift in undercooling compared to bulk MNH is due to different configurations; bulk MNH is pre-melted into a uniform volume so only one nucleation event is required but each microcapsule must nucleate and grow individually, which can take significantly longer given the volumetric and surface energy dependence associated with nucleation.
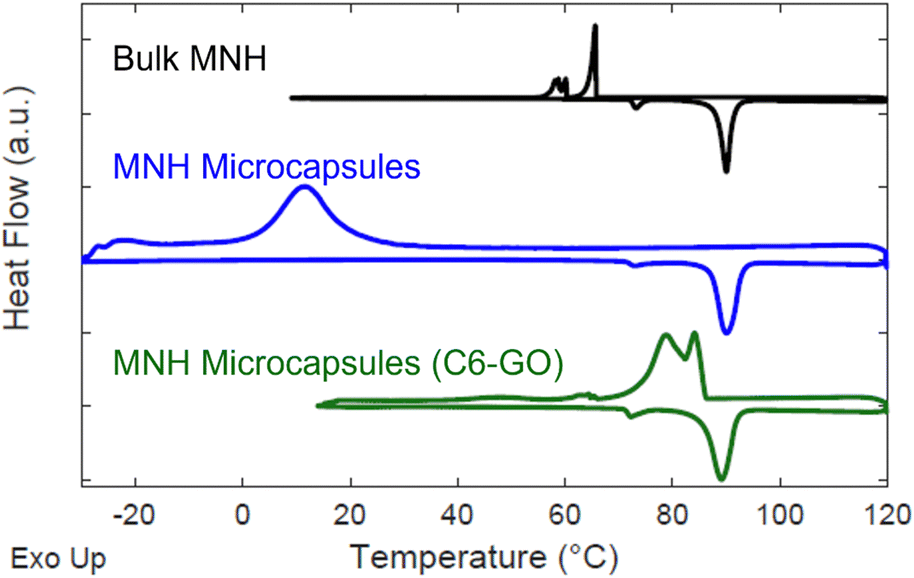 | ||
| Fig. 4 Offset DSC data for bulk MNH (black), MNH microcapsules (blue), and MNH microcapsules with C6-GO nanosheets (green). On heating of the bulk sample, melting is observed at ∼81 °C, and a lower temperature endotherm is observed at 72 °C, reported to correspond with a solid–solid transition.32,33 | ||
To circumvent undercooling, nucleating agents are commonly added in the encapsulation process.34 Based on the prior report by Lak and colleagues, alkylated GO nanosheets can serve as non-specific nucleating agents for MNH, as well as particle surfactants.21 Remarkably, we discovered that using C6-GO as a co-surfactant with SiO2–Br significantly reduces undercooling of MNH. To this end, the concentration of C6-GO in the emulsion was evaluated and 0.2 mg mL−1 was determined to reduce undercooling in comparison to bulk MNH (i.e., by 19.4 °C with ΔTMicrocapsules ≈ 5.0 °C and ΔTBulk ≈24.4 °C, Fig. 4).
To demonstrate applicability of this non-aqueous emulsion ATRP platform, we also demonstrated the encapsulation of the salt hydrate ZNH (Tm = 36 °C) and the IL [EMIM][PF6] (Tm = 64 °C). As shown in Fig. S10 (ESI†), microcapsules of ZNH exhibit similar surface morphology to MNH microcapsules whereas [EMIM][PF6] microcapsules are slightly larger, but in agreement with the [EMIM][PF6] droplet diameter. FTIR spectra of ZNH and [EMIM][PF6] microcapsules are dominated by the encapsulated core; consequently, signals from the shell are only observed in the spectra of the hollow shells (Fig. S11, ESI†). TGA weight loss profiles of these microcapsules show reasonable correlation to the respective bulk weight loss profiles, as expected (Fig. S12, ESI†). Both ZNH and [EMIM][PF6] microcapsules are stable upon heating above their Tm, suggesting robust encapsulation.
In summary, we report a new soft-template encapsulation method for water-sensitive materials with high core loading (> 90%) via organocatalyzed photoredox ATRP using silica particle surfactants with surface-immobilized initiators. Silica particles were modified with ATRP initiators, and used to stabilize emulsions formed above the melting point of the desired core. ATRP of hydrophobic monomers in the continuous phase from the interfacially assembled particles produced microcapsules with rough surface morphology as visualized by SEM. FTIR analysis of the microcapsules pre- and post-core extraction indicates a PCM or IL core and silica/polymer composite shell. The core can undergo multiple melting-solidification cycles without leakage, and undercooling of the salt hydrate PCM was mitigated by addition of a co-surfactant, which served as a nucleating agent. Critically, this approach eliminates the need for monomer in the core and lays the foundation for tuning of the microcapsule shell composition. Our ongoing work addresses endowing the shell with specific properties for bespoke performance-related properties.
Conflicts of interest
There are no conflicts to declare.References
- S. De Koker, R. Hoogenboom and B. G. De Geest, Chem. Soc. Rev., 2012, 41, 2867 RSC.
- F. Casanova and L. Santos, J. Microencapsulation, 2016, 33, 1–17 CrossRef CAS PubMed.
- A. Palacios, M. E. Navarro-Rivero, B. Zou, Z. Jiang, M. T. Harrison and Y. Ding, J. Energy Storage, 2023, 72, 108597 CrossRef.
- V. Nedovic, A. Kalusevic, V. Manojlovic, S. Levic and B. Bugarski, Procedia Food Sci., 2011, 1, 1806–1815 CrossRef CAS.
- K. C. Bentz and D. A. Savin, Polym. Chem., 2018, 9, 2059–2081 RSC.
- J. Hitchcock, A. L. White, N. Hondow, T. A. Hughes, H. Dupont, S. Biggs and O. J. Cayre, J. Colloid Interface Sci., 2020, 567, 171–180 CrossRef CAS PubMed.
- V. Eriksson, M. Andersson Trojer, S. Vavra, M. Hulander and L. Nordstierna, J. Colloid Interface Sci., 2020, 579, 645–653 CrossRef CAS PubMed.
- Y. Wang, N. Starvaggi and E. Pentzer, Polym. Chem., 2023, 14, 4033–4047 RSC.
- Q. Luo and E. Pentzer, ACS Appl. Mater. Interfaces, 2020, 12, 5169–5176 CrossRef CAS PubMed.
- Y.-Y. Lee, K. Edgehouse, A. Klemm, H. Mao, E. Pentzer and B. Gurkan, ACS Appl. Mater. Interfaces, 2020, 12, 19184–19193 CrossRef CAS PubMed.
- Q. Luo, Y. Wang, Z. Chen, P. Wei, E. Yoo and E. Pentzer, ACS Appl. Mater. Interfaces, 2019, 11, 9612–9620 CrossRef CAS PubMed.
- W. Ye, N. Wang, K. Hu, L. Zhang, A. Liu, C. Pan, T. Gong, T. Liu and H. Ding, RSC Adv., 2018, 8, 27253–27259 RSC.
- X. Wang, A.-C. Amason, R. T. Miceli, P. He, Y. Lei, R. Gabbard, J. A. Wieland, R. J. Linhardt, D. T. Corr and R. A. Gross, Colloids Surf., A, 2022, 648, 129243 CrossRef CAS.
- C. Scott, D. Wu, C.-C. Ho and C. C. Co, J. Am. Chem. Soc., 2005, 127, 4160–4161 CrossRef CAS PubMed.
- P. Wei, Q. Luo, K. J. Edgehouse, C. M. Hemmingsen, B. J. Rodier and E. B. Pentzer, ACS Appl. Mater. Interfaces, 2018, 10, 21765–21781 CrossRef CAS PubMed.
- E. Pentzer, E. Cruz Barrios and N. Starvaggi, Acc. Mater. Res., 2023, 4, 641–647 CrossRef CAS.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- B. Rodier, A. De Leon, C. Hemmingsen and E. Pentzer, ACS Macro Lett., 2017, 6, 1201–1206 CrossRef CAS PubMed.
- M. Graham, E. Shchukina, P. F. De Castro and D. Shchukin, J. Mater. Chem. A, 2016, 4, 16906–16912 RSC.
- M. Graham, J. Smith, M. Bilton, E. Shchukina, A. A. Novikov, V. Vinokurov and D. G. Shchukin, ACS Nano, 2020, 14, 8894–8901 CrossRef CAS PubMed.
- S. N. Lak, S. Ahmed, P. J. Shamberger and E. B. Pentzer, J. Colloid Interface Sci., 2022, 628, 605–613 CrossRef CAS PubMed.
- L. Zhou, W. Yuan, J. Yuan and X. Hong, Mater. Lett., 2008, 62, 1372–1375 CrossRef CAS.
- Y. Millot, A. Hervier, J. Ayari, N. Hmili, J. Blanchard and S. Boujday, J. Am. Chem. Soc., 2023, 145, 6671–6681 CrossRef CAS PubMed.
- J. A. Howarter and J. P. Youngblood, Langmuir, 2006, 22, 11142–11147 CrossRef CAS PubMed.
- F. Kunc, V. Balhara, Y. Sun, M. Daroszewska, Z. J. Jakubek, M. Hill, A. Brinkmann and L. J. Johnston, Analyst, 2019, 144, 5589–5599 RSC.
- J. McElwee, R. Helmy and A. Y. Fadeev, J. Colloid Interface Sci., 2005, 285, 551–556 CrossRef CAS PubMed.
- E. Ruckenstein, Langmuir, 1996, 12, 6351–6353 CrossRef CAS.
- N. C. Starvaggi, B. J. Bradford, C. D. L. Taylor and E. B. Pentzer, Soft Matter, 2023, 19, 7635–7643 RSC.
- Y. Chen, C. Wang, J. Chen, X. Liu and Z. Tong, J. Polym. Sci. A Polym. Chem., 2009, 47, 1354–1367 CrossRef CAS.
- N. Gupta, A. Kumar, S. K. Dhawan, H. Dhasmana, A. Kumar, V. Kumar, A. Verma and V. K. Jain, Mater. Today: Proc., 2020, 32, 463–467 CAS.
- P. Honcova, R. Pilar, V. Danielik, P. Soska, G. Sadovska and D. Honc, J. Therm. Anal. Calorim., 2017, 129, 1573–1581 CrossRef CAS.
- G. Sádovská, P. Honcová, R. Pilař, L. Oravová and D. Honc, J. Therm. Anal. Calorim., 2016, 124, 539–546 CrossRef.
- P. Honcova, R. Pilar, V. Danielik, P. Soska, G. Sadovska and D. Honc, J. Therm. Anal. Calorim., 2017, 129, 1573–1581 CrossRef CAS.
- P. J. Shamberger and M. J. O’Malley, Acta Mater., 2015, 84, 265–274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00736k |
| This journal is © The Royal Society of Chemistry 2024 |