
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dissipative sequential catalysis via six-component machinery†
Debabrata
Mondal‡
,
Emad
Elramadi‡
 ,
Sohom
Kundu
and
Michael
Schmittel
*
,
Sohom
Kundu
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, School of Science and Technology, University of Siegen, Adolf-Reichwein-Str. 2, Siegen D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 10th April 2024
Abstract
Triphenyl phosphane (TPP) and an epoxide as a fuel system transiently transformed a non-catalytic six-component turnstile into a four-component catalytic rotor releasing N-methyl pyrrolidine and a copper(I) complex. The two latter compounds acted synergistically as catalysts to perform first a Michael addition and then a 5-exo-dig cyclization, giving rise to dissipative sequential catalysis.
Biological machines1 that operate autonomously under dissipative conditions2 require continuous supply of chemical energy, e.g. for cargo transport across a cell membrane3 or catalytic processes.4 Inspired by such examples, researchers have focused on achieving dissipative control in catalysis.5 Nonetheless, to date there are only a few examples of dissipative catalysis executed by artificial molecular machinery.6 While multi-step catalysis is ubiquitous in multicellular organisms, the resulting challenge is now to drive sequential catalysis utilizing synthetic multi-component7 machinery under dissipative conditions. Establishing such a complex process with different components ((a) machinery, (b) catalyst system, (c) reaction system, and (d) fuel system) in a single environment demands an extremely high level of information handling8,9 and advanced self-sorting.10 The following prerequisites needed thus to be addressed: (i) selection of a multi-component system that is able to dissipatively activate multiple catalysts upon application of a chemical fuel, (ii) transient formation of catalysts that drive the sequential reaction of several reactants, and (iii) avoidance of detrimental cross-talk between any components, catalytic reactants/products, and fuel/fuel waste.11
Herein, we present the six-component turnstile [(T)·(4)2] = [CuZn(1)(2)(3)(4)2]3+ that as a catalytic OFF state deactivates two distinct catalysts, both of which are activated by chemical fuel (Fig. 1). The catalysts are N-methylpyrrolidine (4) and the copper(I) complex [Cu(3)(TPP)2]+ that cooperate in catalyzing two reactions, i.e., a Michael addition12 and 5-exo-dig cyclization13 in a sequential manner. The idea was the following: When the six-component turnstile, constructed from copper(I)-HETPHEN14 and zinc(II)-HETTAP15 complexation motifs, is exposed to triphenylphosphane16 (TPP) as a chemical fuel, then the copper(I) complex ought to be cleaved. Ideally, this should lead to complex [Cu(3)(TPP)2]+ and the rotor [(R)·(4)2], the latter now releasing the organocatalyst 4 due to fast motion.17 Gradually, TPP will be oxidized to triphenylphosphane oxide (TPPO) in the presence of the oxidant 5 so that the initial turnstile should reform.
The five-component turnstile18T = [ZnCu(1)(2)(3)]3+ was conceived on the basis of copper(I)-HETPHEN14 and zinc(II)-HETTAP linkages,15 where cleavage of the copper(I) complexation site should lead to rotor R = [Zn(1)(2)]2+. To prepare R, we chose stator 1, in which two degenerate zinc(II)-porphyrin (ZnPor) stations are connected to a terpyridine unit (Fig. 1). As a counterpart, ligand 2 was equipped with a 2,9-diarylphenanthroline binding site that is able to form a zinc(II)-based HETTAP15 complex with the terpyridine unit of 1. Furthermore, the triazole-pyrimidine side arm of 2 can connect via a Npym → ZnPor interaction to 1. Due to the complementary binding sites of 1 and 2, the three-component rotor R = [Zn(1)(2)]2+ was quantitatively obtained from 1, 2 and Zn2+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) in CD2Cl2, as validated by 1H NMR, ESI-MS and CHN analysis (ESI,† Fig. S11 and S22). In the 1H NMR, the 9-H proton signal of 2 appeared at 5.03 ppm whereas those of a-H and b-H of 1 showed up at 7.53 and 7.39 ppm, respectively, attesting the formation of a HETTAP complex. Moreover, the stator's proton signal r-H (of 1) appeared at 10.24 ppm, whereas the rotary arm 2 proton signal of the b′-H and c′-H proton appeared in the upfield region at 6.14 and 2.95 ppm, respectively (Fig. 2a–c). Those shifts confirm that the pyrimidine head of 2 is coordinated to the ZnPor unit of the stator 1.
:1:1) in CD2Cl2, as validated by 1H NMR, ESI-MS and CHN analysis (ESI,† Fig. S11 and S22). In the 1H NMR, the 9-H proton signal of 2 appeared at 5.03 ppm whereas those of a-H and b-H of 1 showed up at 7.53 and 7.39 ppm, respectively, attesting the formation of a HETTAP complex. Moreover, the stator's proton signal r-H (of 1) appeared at 10.24 ppm, whereas the rotary arm 2 proton signal of the b′-H and c′-H proton appeared in the upfield region at 6.14 and 2.95 ppm, respectively (Fig. 2a–c). Those shifts confirm that the pyrimidine head of 2 is coordinated to the ZnPor unit of the stator 1.
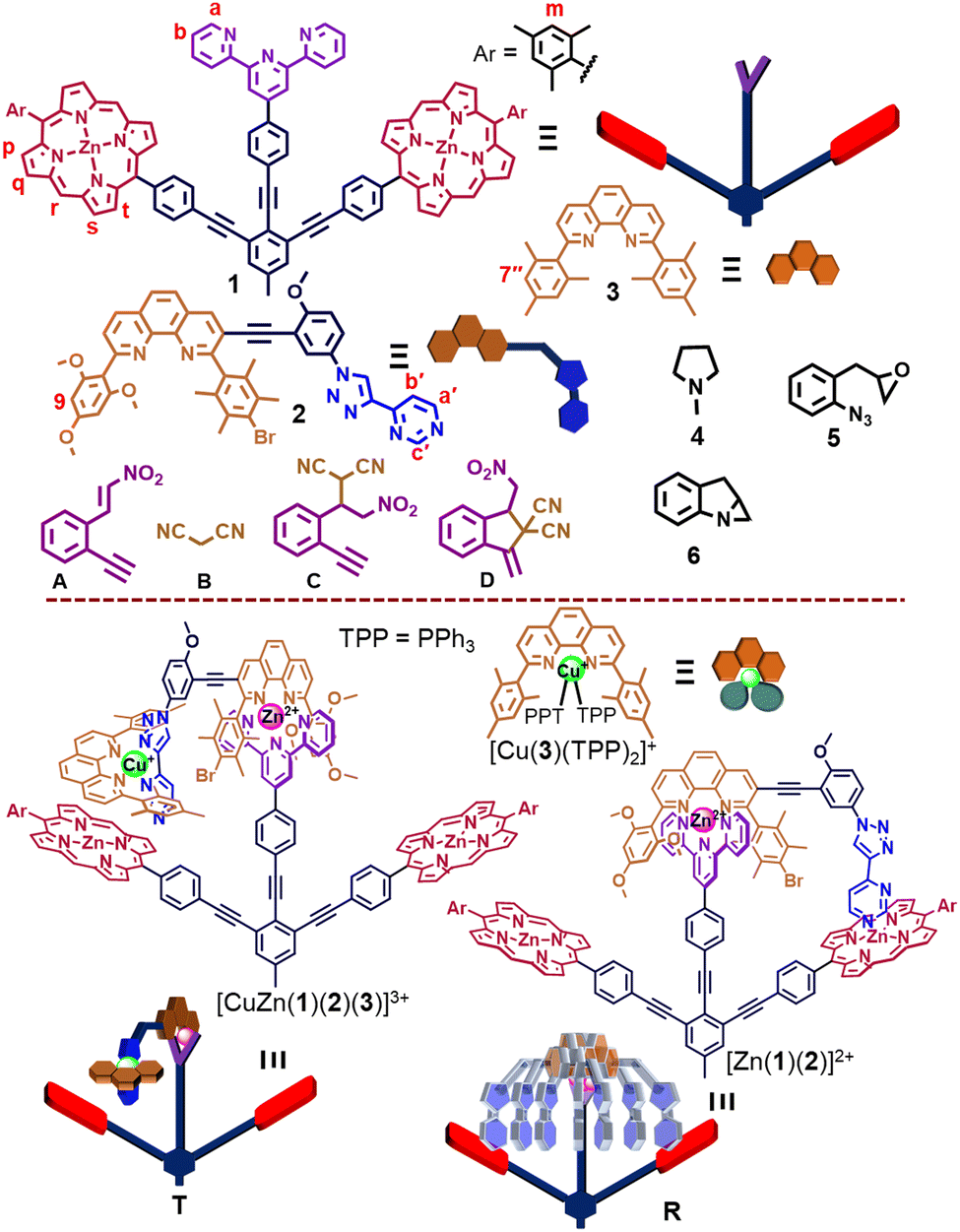 | ||
| Fig. 1 Chemical structure and their cartoon representations. | ||
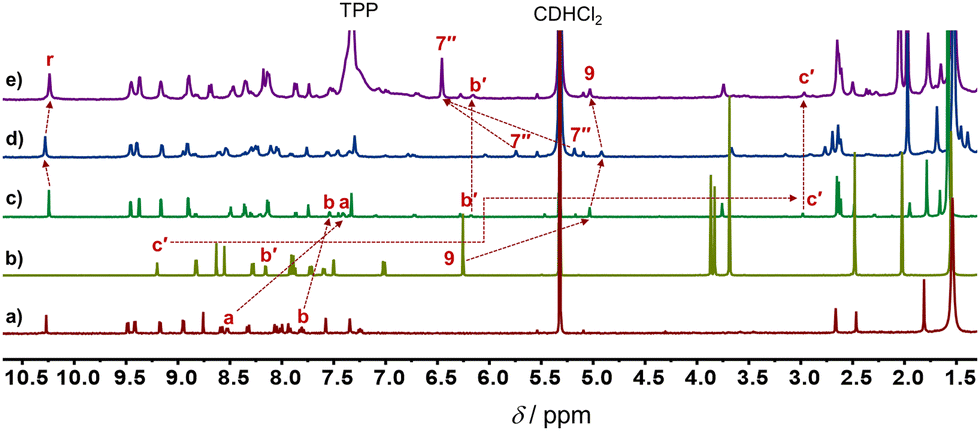 | ||
| Fig. 2 Partial 1H NMR (500 MHz, CD2Cl2, 298 K) of (a) ligand 1, (b) ligand 2, (c) rotor R = [Zn(1)(2)]2+, (d) turnstile T = [ZnCu(1)(2)(3)]3+, and (e) R + [Cu(3)(TPP)2]+. | ||
Since the mesityl protons m-H of both ZnPor stations in rotor R appeared as a singlet in the 1H NMR at room temperature, it suggested fast rotation of the rotary arm. Upon lowering the temperature in a variable temperature (VT) 1H NMR study, the m-H proton signals broadened and split into two sets of singlets at −75 °C in a 1:1 ratio at 7.27 and 7.31 ppm (Fig. 3). The rotational exchange speed at 298 K was determined as k298 = 3.1 × 105 s−1 using WINDNMR.19 The corresponding activation parameters were determined as ΔH‡ = 42.7 ± 0.3 kJ mol−1 and ΔS‡ = 3.0 ± 0.8 J mol−1 K−1, while the free activation energy barrier at 25 °C was obtained as ΔG‡298 = 41.8 ± 0.6 kJ mol−1 (ESI,† Fig. S16 and S17).
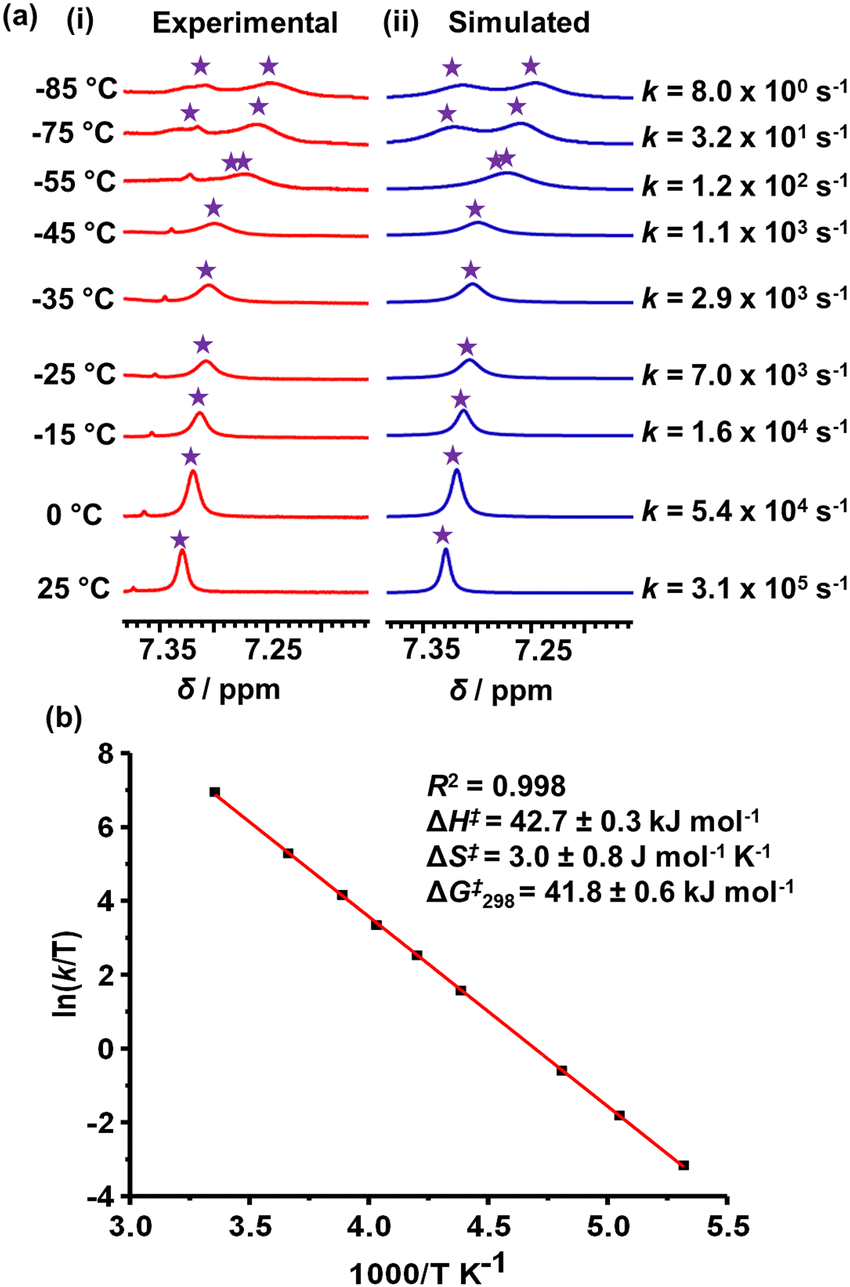 | ||
| Fig. 3 (a) VT 1H NMR (600 MHz, CD2Cl2) of the m-H proton signal of rotor R; (i) experimental and (ii) simulated NMR trace, and (b) Eyring-plot. | ||
Addition of 1 equiv. of the copper(I)-loaded phenanthroline [Cu(3)]+ to R = [Zn(1)(2)]2+ led to the instantaneous formation of a HETPHEN complex at the triazole-pyrimidine chelate site and loss of the Npym → ZnPor interaction. In the 1H NMR of the thus formed five-component turnstile T = [ZnCu(1)(2)(3)]3+, the mesityl 7′′-H signal of 3 showed up as two high-field shifted singlets (1:1) at 5.72 and 5.15 ppm, clearly advocating the formation of a HETPHEN complex. Furthermore, the proton signal r-H shifted from 10.24 ppm in R to 10.28 ppm in T, whereas those of b′-H and c′-H disappeared from their initial positions at 6.14 and 2.95 ppm in R confirming the loss of the Npym → ZnPor interaction in T (Fig. 2c and d). Importantly, upon addition of two equiv. of TTP to T, the rotor R was regenerated along with the formation of complex [Cu(3)(TPP)2]+, the latter being identified by independent synthesis (Fig. 2e).
After establishing the formation of rotor R and its reversible interconversion to turnstile T, we explored the chemical fuel that would trigger transient rotor formation. Therefore, ligands 1, 2 and 3, as well as [Zn(CF3SO3)2], [Cu(CH3CN)4]PF6 and co-fuel 5 were combined (1:1:1:1:1:9) in CD2Cl2, cleanly affording turnstile T. Presence of further added co-fuel 5 did not interfere with T, as advocated by 1H NMR (ESI,† Fig. S24). Instantaneously, right after the addition of two equiv. of TPP as chemical fuel, rotor R and complex [Cu(3)(TPP)2]+ formed (Fig. 4). Because the epoxide 5 slowly oxidized TPP to TPPO (generating waste 6), complex [Cu(3)(TPP)2]+ was destroyed and as a result turnstile T was restored after 10 h (ESI,† Fig. S24). Before starting the second dissipative cycle, the consumed amount of oxidant 5 (2 equiv. w.r.t. R) was added to the reaction mixture left after the first dissipative cycle. To restart the second dissipative cycle, 2 equiv. of TPP were charged to the mixture. As expected, we again noticed the transient formation of R and [Cu(3)(TPP)2]+ that reverted to the initial complex T after 10 h by producing two more equiv. of TPPO and waste 6 (ESI,† Fig. S25).
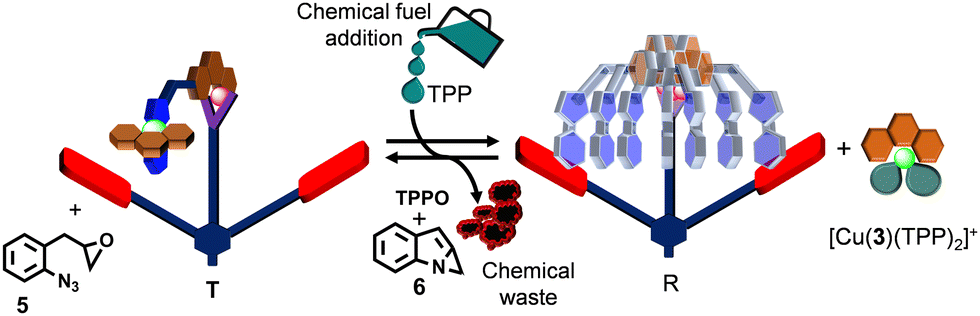 | ||
| Fig. 4 Cartoon representation of the fueled turnstile T to transient rotor R transformation. | ||
The next challenge was to choose suitable substrates and a reaction scheme that would make use of the transiently formed catalysts 4 and [Cu(3)(TPP)2]+. Furthermore, neither substrates nor products should interfere with the dissipative process. After an extensive screening, compounds A and B were chosen as reactants (Scheme 1) since in the presence of catalyst 4 they afforded intermediate C, which was further converted to product D with the help of both [Cu(3)(TPP)2]+ and 4 acting as a synergistic catalyst. Indeed, when substrates A (10 equiv. w.r.t. Cu+) and B (30 equiv. w.r.t. Cu+) were reacted at room temperature for 10 h in the presence of catalyst [Cu(3)(TPP)2]+ and base 4 (2 equiv. w.r.t. Cu+), the final product D was afforded in 62% yield with some amount of intermediate C (5% yield) remaining (ESI,† Fig. S27).
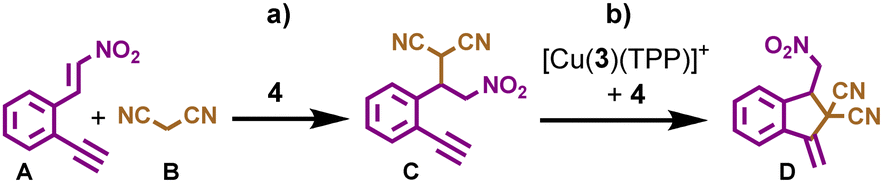 | ||
| Scheme 1 One-pot reaction with two catalytic steps. (a) Michael addition of A and B affords C using solely 4 as a catalyst, and (b) conversion of C to D using [Cu(3)(TPP)2]+ and base 4 as a synergistic catalyst pair. | ||
Next, we investigated the role of each catalyst in the sequential two-step reaction. Thus, substrates A (5 equiv. w.r.t. 4) and B (15 equiv. w.r.t. 4) were reacted in the presence of organo-catalyst 4 at room temperature for 10 h, furnishing the 1,4-addition product C in 64% yield (ESI,† Fig. S26). The ensuing cyclization of C (10 equiv. w.r.t. Cu+) employing both catalysts [Cu(3)(TPP)2]+ and 4 (2 equiv. w.r.t. Cu+) furnished D in 90% yield after 10 h at room temperature (ESI,† Fig. S31), whereas almost no conversion of C to D (only 4%) was observed in the absence of catalyst 4 using the same conditions (ESI,† Fig. S30). Obviously, both catalysts act in a synergistic manner accelerating the conversion of C to D.
After this insight into the role of each catalyst, we examined the catalytic activity of the multi-component mixture involving the highly dynamic catalyst-loaded rotor [(R)·(4)2] and complex [Cu(3)(TPP)2]+. Hereunto, turnstile T was prepared (as explained before) followed by the addition of catalyst 4 (2 equiv. w.r.t. T), and substrate A (10 equiv. w.r.t. T) and B (30 equiv. w.r.t. T). The reaction was followed by 1H NMR at room temperature for 10 h. Since, neither the formation of intermediate C nor of the final product D was validated from 1H NMR data, we denote this ensemble as the catalytic OFF state (ESI,† Fig. S32). In the OFF state, the organocatalyst 4 is tightly attached to the zinc(II)-porphyrin (ZnPor) units of T, while the copper(I) ion is deeply buried inside the HETPHEN complex site preventing any catalytic activity. Next, 2 equiv. of TPP were added to the catalytic OFF state, resulting in the instant formation of the catalytic rotor [(R)·(4)2] and [Cu(3)(TPP)2]+. The 1H NMR spectra after 10 h at room temperature revealed the formation of the final product D in 47% yield, whereas only 5% of intermediate C was obtained (ESI,† Fig. S33). As gauged by quantitative analysis, the combination of both the chelate effect and high rotational dynamics of rotor R released 1.67 equiv. of organocatalyst 4 into solution that initiated product formation (ESI,† Fig. S29). Notably, the formation of intermediate C reached a plateau within ca. 1.5 h (ESI,† Fig. S33) yielding 9% of C, which slowly decreased after 4.5 h to 5%. In the 1,4-addition, catalyst 4 showed a turnover number (TON) = 5.2, while for the cyclization both catalyst 4 and [Cu(3)(TPP)2]+ together displayed a TON = 4.7.
Finally, we addressed our final objective, the dissipative two-step sequential reaction using a multi-component ensemble. For this purpose, the catalytic OFF state was prepared in CD2Cl2, and subsequently 9 equiv. of 5 (w.r.t. T) were added and the mixture was kept at room temperature for 10 h (Fig. 5a). As expected, no product traces were observed (ESI,† Fig. S36). Thereafter, 2 equiv. of TPP were added which led to the immediate formation of the transient catalytic rotor [(R)·(4)2] along with complex [Cu(3)(TPP)2]+ (catalytic ON state) at room temperature (Fig. 5a). After 10 h, the 1H NMR proved that both the intermediate C and final product D had formed in 8% and 30% yield respectively (ESI,† Fig. S37) and that the initial catalytic OFF state was refurnished along with the generation of two equiv. TPPO and waste 6 (Fig. 5b). Under the dissipative conditions (use of chemical fuel), formation of the intermediate C reached a steady state after 2 h that continued for another 4 h. As expected, the final product D was afforded in lower yield under dissipative than non-dissipative conditions due to the lower overall concentrations of both catalysts 4 and [Cu(3)(TPP)2]+ in the reaction medium.
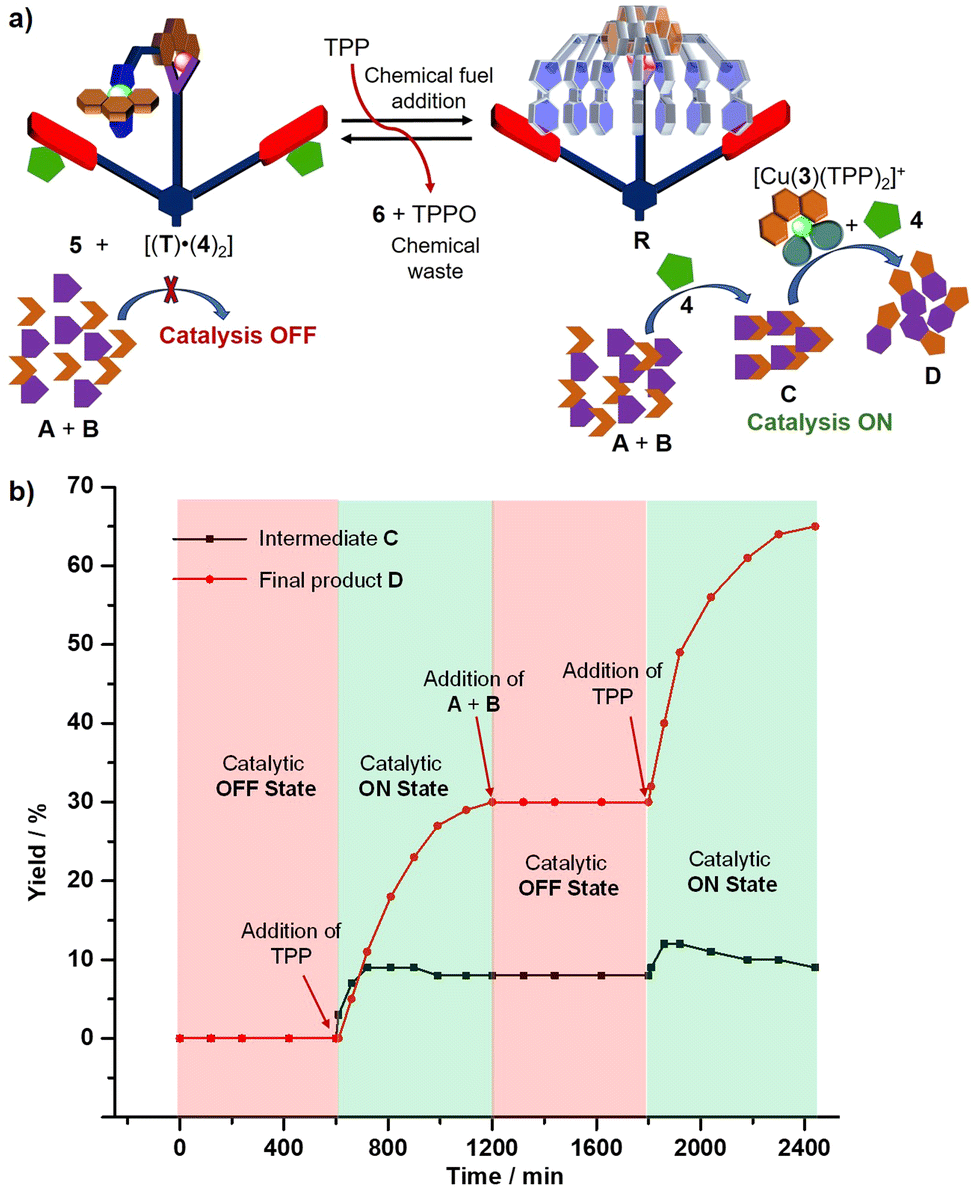 | ||
| Fig. 5 (a) Cartoon representation of dissipative sequential dual catalysis using TPP as a chemical fuel, and (b) formation of intermediate C and final product D over time. | ||
To reset the second dissipative cycle to the starting conditions, the used-up amounts of substrates A (3.8 equiv. w.r.t. T), B (3.8 equiv. w.r.t. T) and co-fuel 5 (2 equiv. w.r.t. T) were replenished (Fig. 5b) and the resultant mixture was allowed to react at room temperature. After 10 h, no further amounts of C or D than from the first cycle were detected (ESI,† Fig. S38). For turning on the second dissipative cycle, two equiv. of TPP were added thus affording the dissipative catalytic ON state over 10 h. The yield over two dissipative cycles amounted to 65% of D and 9% of C (ESI,† Fig. S39). In the interim, intermediate C reached a maximum amount of 12%, within ca. one hour. Two successive dissipative catalytic cycles were thus carried out in an interference-free manner supporting the robustness and resilience of the two-step sequential catalysis.
In summary, we present a multi-component catalytic machinery that reversibly toggles between a non-catalytic turnstile and catalytic rotor ensemble driven by a chemical fuel. Intriguingly, two catalysts are thereby simultaneously and transiently activated that catalyze a two-step reaction sequence. In detail, addition of TPP as the chemical fuel cleaves the copper(I)-HETPHEN complex unit in [(T)·(4)2] transiently furnishing catalyst [Cu(3)(TPP)2]+ and the catalytic rotor [(R)·(4)2]. The latter liberates the true catalyst 4 due to its high (k298 = 3.1 × 105 s−1) rotational dynamics. Both catalysts, i.e.4 and [Cu(3)(TPP)2]+, enable a Michael 1,4-addition followed by a 5-exo-dig cyclization. Thus, the dissipative double-catalyst activation employing a single chemical fuel endows a useful two-step reaction as the output. The present work impressively demonstrates that one can go beyond fueling machinery for single-step catalysis, which will facilitate to manage more complex reaction schemes (e.g. for self-repair,20 removal of undesired influx, smart materials properties) on the way to emulating advanced biological functions in artificial autonomous systems.
We thank the DFG (Schm 647/22-1) and the Univ. of Siegen for continued support. Moreover, we are indebted to Dr Paululat (Siegen) for the VT-1H NMR measurements. This work is dedicated to Prof. Manas K. Ghorai (IIT Kanpur) who has fully supported my research group over the last 20 years.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
D. S. Goodsell, The Machinery of Life, Springer, New York, 2010 Search PubMed
.
-
(a) D. Del Giudice and S. Di Stefano, Acc. Chem. Res., 2023, 56, 889–899 CrossRef CAS PubMed
- M. A. Watson and S. L. Cockroft, Angew. Chem., Int. Ed., 2016, 55, 1345–1349 CrossRef CAS PubMed
- J. E. Walker, Biochem. Soc. Trans., 2013, 41, 1–16 CrossRef CAS PubMed
-
(a) S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed
-
(a) I. Valiyev, A. Ghosh, I. Paul and M. Schmittel, Chem. Commun., 2022, 58, 1728–1731 RSC
-
(a) V. V. Rajasekaran, E. Elramadi, I. Valiyev, P. Howlader and M. Schmittel, Chem. Commun., 2023, 59, 3886–3889 RSC
- M. Schmittel, Isr. J. Chem., 2018, 59, 197–208 CrossRef
- P. Remón and U. Pischel, ChemPhysChem, 2017, 18, 1667–1677 CrossRef PubMed
-
(a) Q. Liu, B. Jin, Q. Li, H. Yang, Y. Luo and X. Li, Soft Matter, 2022, 18, 2484–2499 RSC
- A. Ghosh, I. Paul, M. Adlung, C. Wickleder and M. Schmittel, Org. Lett., 2018, 20, 1046–1049 CrossRef CAS PubMed
- T. Poon, B. P. Mundy and T. W. Shattuck, J. Chem. Educ., 2002, 79, 264 CrossRef CAS
- D. B. Ramachary, R. Mondal and C. Venkaiah, Eur. J. Org. Chem., 2010, 3205–3210 CrossRef CAS
- M. Schmittel, C. Michel, S.-X. Liu, D. Schildbach and D. Fenske, Eur. J. Inorg. Chem., 2001, 1155–1166 CrossRef CAS
- M. Schmittel, V. Kalsani, P. Mal and J. W. Bats, Inorg. Chem., 2006, 45, 6370–6377 CrossRef CAS PubMed
- C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS PubMed
-
(a) A. Goswami, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 15656–15663 CrossRef CAS PubMed
- For a two-component turnstile: T. Lang, A. Guenet, E. Graf, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2010, 46, 3508–3510 RSC
- H. J. Reich, J. Chem. Educ., Software, 1996, 3D, 2 Search PubMed
- D. Mondal, S. Kundu, E. Elramadi, I. Valiyev and M. Schmittel, Org. Lett., 2023, 25, 933–937 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, VT-NMR, and spectral data. See DOI: https://doi.org/10.1039/d4cc00786g |
| ‡ Debabrata Mondal and Emad Elramadi contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |