
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoredox-catalyzed carbonylative acylation of styrenes with Hantzsch esters†
Qiangwei
Li
ab,
Le-Cheng
Wang
ab,
Zhi-Peng
Bao
ab and
Xiao-Feng
Wu
 *abc
*abc
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 3rd April 2024
Abstract
Ketones exist widely in naturally occurring products and are indispensable building blocks in organic synthesis. Carbonylation represents one of the most straightforward methods for ketone preparation and has become an attractive field in modern organic chemistry as well. Among the strategies, photocatalytic carbonylation is also worthy of further exploration. Herein, we developed a three-component carbonylation that provides a new method for the synthesis of ketones from Hantzsch esters, CO and styrenes. The reaction was performed under a blue light environment and yields a series of ketones with moderate to good yields.
Ketones exist widely in naturally occurring products and are also important building blocks in modern organic synthesis.1 Numerous synthetic procedures use ketones as their starting materials. Therefore, the development of new and alternative procedures for the construction of carbonyl-containing compounds plays an important role in modern chemistry. Among the various possibilities, transition-metal catalyzed carbonylation has been greatly developed for ketone synthesis.2 Meanwhile, carbonylation via radical intermediates is also noteworthy.3 Based on these achievements and attracted by photo-chemistry, new carbonylation procedures under light irradiation conditions are becoming attractive.4
Among the various types of ketones, alkyl ketones are attractive which are traditionally produced by the direct reaction of carboxylic acids or their derivatives with organometallic reagents.5 However, these traditional synthetic routes towards alkyl ketones often suffer from various limitations including pre-synthesis of acyl functional groups, use of reactive organometallic reagents, and limited functional group tolerance (Scheme 1a).6 In recent years, transition metal-catalyzed carbonylation has become one of the most effective methods for constructing carbonyl functional groups.7 Compared to the synthesis of amides and esters, the carbonylation method for synthesizing ketones is mainly limited to the well-established aryl ketones, benzylic ketones and alkynyl ketones.8 In addition, these carbonylation strategies still rely on using stoichiometric amounts of main group organometallic reagents and activated organic halides (Scheme 1b).9 Among the various achievements, very recently, Chen and co-workers described a novel nickel-catalyzed carbonylation of unactivated alkyl halides with organozinc reagents to yield a variety of alkyl ketones in good yields in general.10
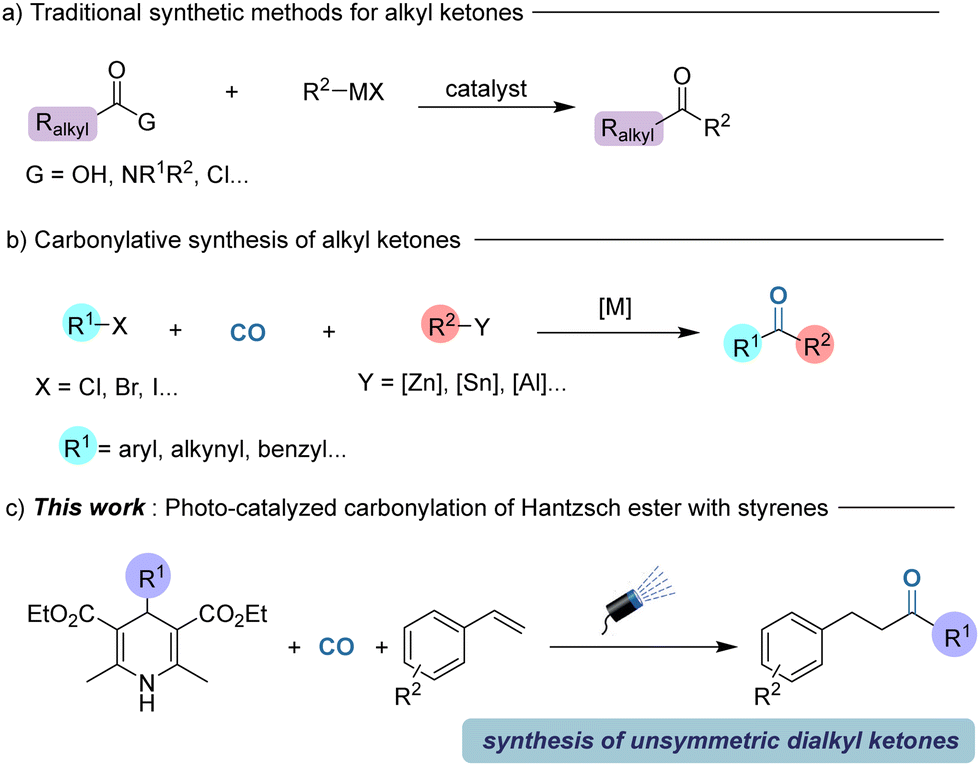 | ||
| Scheme 1 Strategies in carbonylation of Hantzsch esters. | ||
In recent years, photocatalytic carbonylation has developed rapidly. In the field of photoredox-catalyzed organic synthesis, Hantzsch esters have attracted much attention due to their unique light-absorbing properties.11 Hantzsch esters, which were replaced by other groups at the 4-position, transfer an electron and a hydrogen atom to participate in the reduction reaction. Our group reported a Mn-mediated double carbonylative reaction to synthesize dicarbonyl compounds by employing Hantzsch esters as a precursor, but excess loading of catalyst was required in this reaction.12 Herein, we developed a three-component reaction of the radical precursors produced by Hantzsch esters, CO and styrenes under a blue light environment in the presence of a photo-catalyst without any other additives (Scheme 1c).
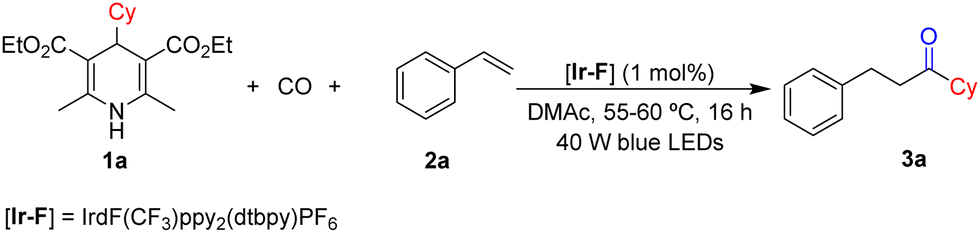
In order to realize this transformation, we chose 4-cyclohexyl substituted Hantzsch ester 1a and styrene 2a as the model substrates to figure out the best conditions for this reaction (Table 1). After testing various solvents, we found that N,N-dimethylacetamide (DMAc) was a better choice of solvent with good solubility than other tried solvents (for details, see the ESI†). We finally can obtain the desired product 3a in 74% yield when the reaction was stirred under 60 bar CO with 1 mol% of IrdF(CF3)ppy2(dtbpy)PF6 ([Ir-F]) in DMAc (2 mL) for 16 h (Table 1, entry 1). When we use organic photosensitizer 4CzIPN or eosin Y to carry out the reaction, the yield of the targeted product was 45% and 0%, respectively (Table 1, entries 2 and 3). Then we used 1.0 equiv. 1a and 2.0 equiv. 2a to carry out the reaction, and the target product was produced in 66% yield, which was lower than expected (Table 1, entry 7). With the addition of a higher amount of catalyst, the yield of product 3a dropped (Table 1, entry 9). A similar yield was obtained when the pressure of CO was decreased to 30 bar, but cannot be further decreased (Table 1, entry 10). Moreover, the control experiment showed that light was crucial for this transformation (Table 1, entry 11). Notably, compound formed due to non-carbonylation was detected as the main by-product.
|
|
||||
|---|---|---|---|---|
| Entrya | Pc |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Solvent | Yield (%) |
| a Reaction conditions: 1a (0.15 mmol), 2a (0.1 mmol), Pc (1 mol%), and DMAc (2 mL) for 16 h under CO (60 bar). Yields were determined by GC-FID analysis using dodecane as the internal standard. b Isolated yield. c 1 mL of solvent. d 2 mol% of [Ir-F]. e CO (30 bar). f In the dark. | ||||
| 1 | [Ir-F] | 1.5:1.0 |
DMAc | 74 (71)b |
| 2 | 4CzIPN | 1.5:1.0 |
DMAc | 45 |
| 3 | Eosin Y | 1.5:1.0 |
DMAc | 0 |
| 4 | [Ir-F] | 1.5:1.0 |
DCM | 48 |
| 5 | [Ir-F] | 1.5:1.0 |
THF | 51 |
| 6 | [Ir-F] | 1.5:1.0 |
DMF | 57 |
| 7 | [Ir-F] | 1.0:2.0 |
DMAc | 66 |
| 8 | [Ir-F]c | 1.5:1.0 |
DMAc | 61 |
| 9 | [Ir-F]d | 1.5:1.0 |
DMAc | 61 |
| 10 | [Ir-F]e | 1.5:1.0 |
DMAc | 66 |
| 11 | [Ir-F]f | 1.5:1.0 |
DMAc | 0 |
In view of the above results, we then investigated the substrate scope for this reaction (Scheme 2). We first tested various group substituted styrenes such as –Me (3b), –OMe (3e), –tBu (3f)), –Cl (3g), and –Br (3k), and the results showed that when substituents gradually change from the para-position to ortho-position, irrespective of the electron-donating group or electron-withdrawing group, the yields increased. When the para-position of styrene has a stronger electron-withdrawing group such as –CN and –NO2, the desired reaction did not occur. When α-methyl or β-methyl substituted styrene was tested as the substrate, the yield of the desired ketone can be still be obtained in a moderate rate (3n and 3o). When styrene was replaced with bistyrene, the yield of the corresponding product was 37% (3p). However, when tested with naphthene or α-methyl styrene, the yields of the desired products were isolated in 66% in both cases (3q and 3r). Aliphatic alkenes and ethylene were also tested with 1a under our standard conditions, but desired product was detectable. Then we continued to test the reactivity of different substituted Hantzsch esters, and moderate yields can be obtained in general (3s–3v).
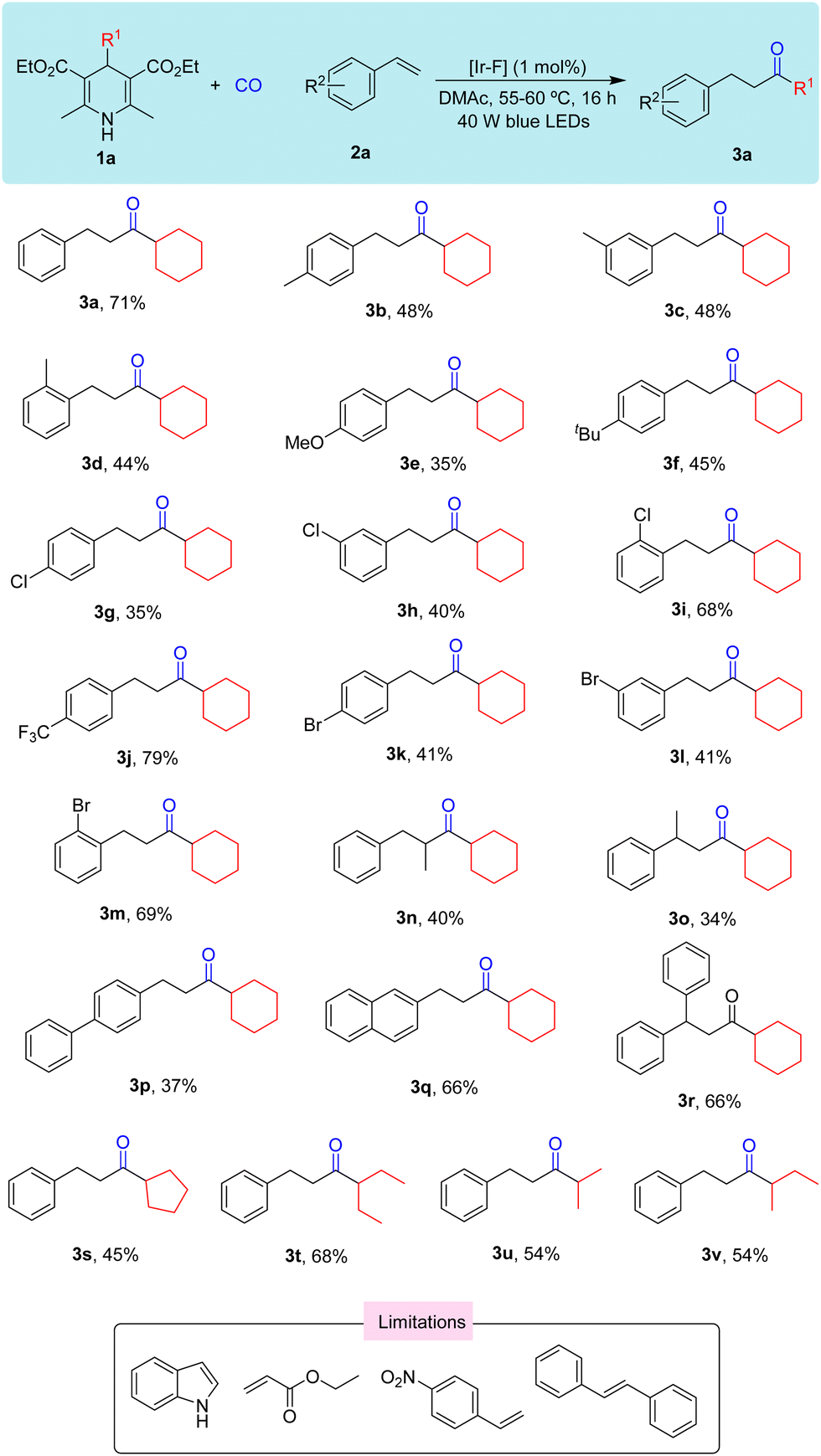 | ||
| Scheme 2 Substrate testing. Reaction conditions: 1a (0.15 mmol), 2a (0.1 mmol), [Ir-F] (1 mol%), and DMAc (2 mL) for 16 h under CO (60 bar). | ||
In order to verify the reaction mechanism, we conducted control experiments. No reaction occurred when the reaction was performed under dark conditions (Table 1, entry 11). The model reaction was fully inhibited when TEMPO (2,2,6,6-tetramethylpiperidinooxy) was added as the radical inhibitor (Scheme 3), which implies the existence of the radical intermediate. The pyridine derivative from Hantzsch esters can be obtained after the reaction.
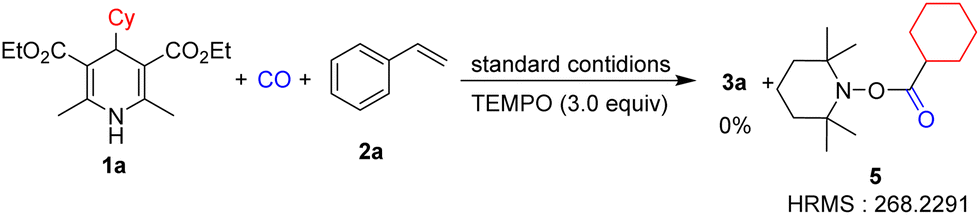 | ||
| Scheme 3 Radical capture experiment. | ||
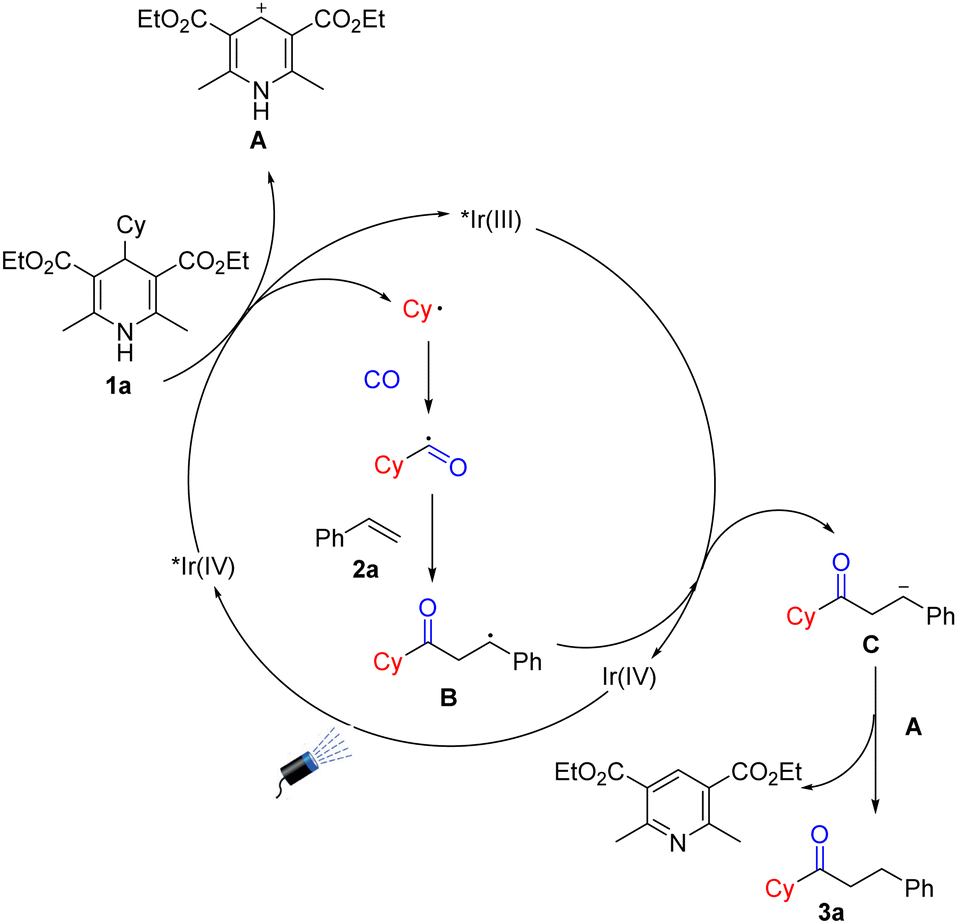
Based on the above mechanism studies and the literature, we propose a possible mechanism (Scheme 4). First, the photosensitizer was excited under light irradiation conditions, and Hantzsch esters were oxidized by the activated photosensitizer to produce cyclohexyl radicals together with the release of intermediate A. The cyclohexyl radical was then captured by CO to obtain acyl radicals which were then added to styrene to produce intermediate B. Subsequently, intermediate B will be reduced by Ir(IV) to obtain intermediate C. The removal of H+ from the intermediate A promotes the protonation of C to generate the final product along with the formation of a molecule of pyridine derivative.
 | ||
| Scheme 4 Proposed mechanism. | ||
In summary, we have established photoredox-catalyzed carbonylation of Hantzsch esters and styrenes. The reaction took place under blue light irradiation to give a series of alkyl ketones with moderate to good yields under mild conditions.
We thank the financial support from National Key R&D Program of China (2023YFA1507500).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. Sribalan, A. Sangili, G. Banuppriya and V. Padmini, New J. Chem., 2017, 41, 3414–3421 RSC; (b) R. O. Owor, K. G. Bedane, S. Zühlke, S. Derese, G. O. Ong’amo, A. Ndakala and M. Spiteller, J. Nat. Prod., 2020, 83, 996–1004 CrossRef CAS PubMed; (c) H. H. Otto and T. Schirmeister, Chem. Rev., 1997, 97, 133–172 CrossRef CAS PubMed; (d) M. Knaack, P. Emig, J. Bats, M. Kiesel, M. Arndt and E. Günther, Eur. J. Org. Chem., 2001, 3843–3847 CrossRef CAS; (e) S.-L. Jang, S.-L. Jang, K.-J. Kim, H.-J. Kim, H.-H. Yu, R. Park, H.-M. Kim and Y.-O. You, Planta Med., 2003, 69, 1057–1059 CrossRef CAS PubMed; (f) G.-B. Bettolo, C.-G. Casinovi and C. Galeffi, Tetrahedron Lett., 1965, 52, 4857–4864 CrossRef.
- (a) C. Shen and X.-F. Wu, Synlett, 2016, 1269–1273 CAS; (b) C. Shen, A. Spannenberg and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 5067–5070 CrossRef CAS PubMed; (c) Y. Li, Z. Wang and X.-F. Wu, ACS Catal., 2018, 8, 738–741 CrossRef CAS; (d) F.-P. Wu, J.-B. Peng, X. Qi and X.-F. Wu, Catal. Sci. Technol., 2017, 7, 4924–4928 RSC; (e) Y. Li, Z. Wang and X.-F. Wu, Green Chem., 2018, 20, 969–972 RSC.
- (a) L.-C. Wang, B. Chen and X.-F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202203797 CrossRef CAS PubMed; (b) L.-C. Wang, B. Chen, Y. Zhang and X.-F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207970 CrossRef CAS PubMed; (c) L.-C. Wang, Y. Yuan, Y. Zhang and X.-F. Wu, Nat. Commun., 2023, 14, 7439 CrossRef PubMed; (d) Z.-P. Bao and X.-F. Wu, Angew. Chem., Int. Ed., 2023, 62, e202301671 CrossRef CAS PubMed; (e) Y. Zhang, B.-H. Teng and X.-F. Wu, Chem. Sci., 2024, 15, 1418–1423 RSC; (f) G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318–323 CrossRef CAS PubMed; (g) Y. Liu, C. Zhou, M. Jiang and B. A. Arndtsen, J. Am. Chem. Soc., 2022, 144, 9413–9420 CrossRef CAS PubMed.
- (a) B. Lu, M. Xu, X. Qi, M. Jiang, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed; (b) I. Ryu and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050–1066 CrossRef; (c) I. Ryu, Chem. Soc. Rev., 2001, 30, 16–25 RSC.
- (a) C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040–4043 CrossRef CAS PubMed; (b) Y. Li, Z. Ma, X. Liu, Z. Li, F. Zhao and J. Wu, Org. Chem. Front., 2023, 10, 1710–1714 RSC; (c) A. R. Sahoo, G. Lalitha, V. Murugesh, C. Bruneau, G. V. M. Sharma, S. Suresh and M. Achard, J. Org. Chem., 2017, 82, 10727–10731 CrossRef CAS PubMed.
- D. Wang, K. Zhao, P. Ma, C. Xu and Y. Ding, Tetrahedron Lett., 2014, 55, 7233–7235 CrossRef CAS.
- (a) Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC; (b) J. B. Peng, F. P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed; (c) W. Li, D. Jiang, C. Wang and L.-J. Cheng, Chin. J. Chem., 2023, 41, 3419–3432 CrossRef CAS.
- (a) X. Chen, G. Chen and Z. Lian, Chin. J. Chem., 2024, 42, 177–189 CrossRef CAS; (b) Y. Wang, B. Wang, Z. Ren and Z. H. Guan, Asian J. Org. Chem., 2023, 12, e202300021 CrossRef CAS.
- (a) L. J. Cheng and N. P. Mankad, Acc. Chem. Res., 2021, 54, 2261–2274 CrossRef CAS PubMed; (b) P. Bung and N. P. Mankad, J. Am. Chem. Soc., 2023, 145, 9423–9427 CrossRef PubMed; (c) L. J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2017, 139, 10200–10203 CrossRef CAS PubMed.
- Y. Zhang, Q. Cao, Y. Xi, X. Wu, J. Qu and Y. Chen, J. Am. Chem. Soc., 2024, 146, 7971–7978 CrossRef CAS PubMed.
- P.-Z. Wang, J.-A. Chen and W.-J. Xia, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- B. Chen, C.-S. Kuai, J.-X. Xu and X.-F. Wu, Adv. Synth. Catal., 2022, 364, 487–492 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytical data, and NMR spectra. See DOI: https://doi.org/10.1039/d4cc01293c |
| This journal is © The Royal Society of Chemistry 2024 |