
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved access to polythioesters by heterobimetallic aluminium catalysis†
Bhargav R.
Manjunatha
 a,
Merlin R.
Stühler
b,
Luise
Quick
b and
Alex J.
Plajer
*a
a,
Merlin R.
Stühler
b,
Luise
Quick
b and
Alex J.
Plajer
*a
aMakromolekulare Chemie 1, Universität Bayreuth, Universitätsstraße 30, 95447, Bayreuth, Germany. E-mail: alex.plajer@uni-bayreuth.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, Berlin 14195, Germany
First published on 13th March 2024
Abstract
Bimetallic Al(III) catalysis mediates thioanhydride/epoxide copolymerisation at greatly improved rates and monomer tolerance than analogous Cr(III) catalysis. Moving to sulfurated monomers furthermore generally improves rates and selectivites.
Heteroatom containing polymers are more susceptible to degradation than current commodity polymers comprising an all-carbon backbone, due to the presence of reactive centres in the polymer main-chain.1–4 In this regard, moving from oxygenated polymers like polyester and polycarbonates to sulfur containing analogues imparts additional desirable thermal and optical properties onto the polymers while further enhancing their degradability.5–14 In this regard the ring-opening copolymerisation (ROCOP) of cyclic thioanhydrides with epoxides results in poly(ester-thioesters) from alternating enchainment of the two monomers.15–23 Bicomponent catalysts featuring a metal or main group based Lewis acid in combination with soluble Lewis bases or onium salts (which have also been combined within the same scaffold) can mediate this process, yet these suffer from distinct challenges: as many rely on the use of toxic and expensive chromium or [(Ph3P)2N]+Cl−; are not air and moisture tolerant; and are synthetically non-trivial to access. Tackling some of these problems, our group established the use of methoxy-substituted bisphenoxyimine based chromium catalysts in the ROCOP of phthalic thioanhydride (PTA) or CS2 with cyclohexene oxide (CHO) to yield sulfurated polymers.24–26 These could solubilise alkali metal acetates AMOAc (AM = Li, Na, K, Rb, Cs) by means of coordination to an all-oxygen donor site in the ligand sufficiently, to form heterobimetallic complexes that also form in situ during catalysis, and these then facilitate PTA/CHO or CS2/CHO ROCOP. Viewed differently these complexes coordinatively enable the use of cheap AMOAc salts as cocatalysts in place of onium salts. Our previous work on Cr(III) catalysts revealed that both ligand modifications and alkali metal choice affect activity and selectivity and that there are parallelling trends regarding this for CS2/CHO and CO2/CHO ROCOP. Unfortunately though, these complexes still relied on the use of toxic and expensive Chromium, which is undesirable as catalysts traces in the produced polymers rule out many applications (as e.g. in a biological context). Hence, we explore in this report the use of the Al(III) variants featuring an earth abundant and non-toxic metal and not only find these to exhibit improved rates but also offer insight into how monomer sulfuration affects catalysis while improving the monomer scope to yield unexplored co and terpolymers from thioanhydrides (Fig. 1).
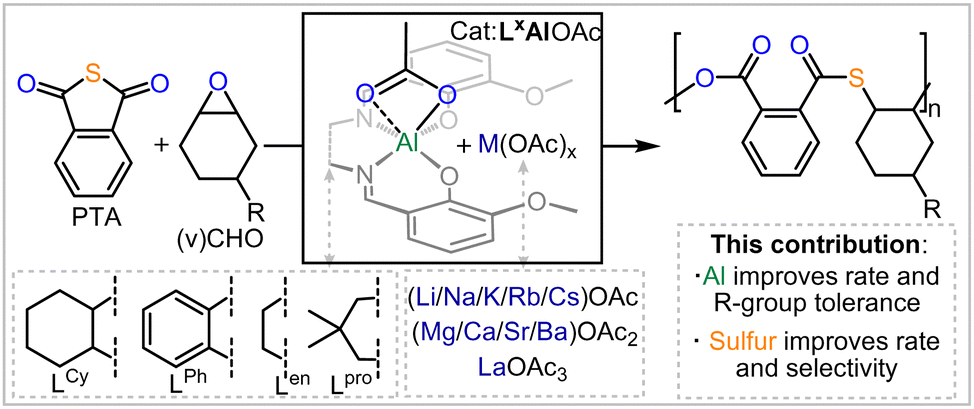 | ||
Fig. 1 Outline of the current work; R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H (CHO), CHCH2(vCHO). H (CHO), CHCH2(vCHO). | ||
We initially prepared LCyAlOAc, i.e. the Al(III) version of our previous Cr(III) catalyst via the literature reported procedure.27,28 Testing LCyAlOAc in PTA/CHO ROCOP at a loading of 1 eq. LCyAlOAc: 250 eq. PTA: 1000 eq. CHO, with one equivalent of added RbOAc for in situ complexation, results in 97% PTA conversion in 25 min at 80 °C. The same activity was found when the preformed adduct was employed. To do this, LCyAlOAc and RbOAc were combined in methanol, followed by removal of volatiles in vacuum, and the adduct formation could be confirmed by 1H NMR spectroscopy (see ESI,† Fig. S6). Due to the greater practicality of this method, we employed all following catalysis with preformed adducts. We followed the monomer consumption via aliquot analysis by 1H NMR which showed a maximum TOF of 638 h−1 for the initial linear increase of the PTA conversion versus time plots. In comparison, the previously investigated Cr(III) version showed a maximum TOF of 50 h−1 meaning that, moving to non-toxic and abundant Al(III) improves activity by at least one order of magnitude.‡ The formed polymer shows an apparent Mn of 11.3 kg mol−1 with a polydispersity of Đ = 1.4 (see ESI,† Fig. S37). The GPC curve is bimodal indicating two co-occuring initiation processes which are explored in more detail below. No (thio)ether links are formed indicating alternating insertion of CHO and PTA which would result in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 distribution of thioester:ester links, i.e. 50% thioester. Yet, only 45% thioester links are formed, and this can be rationalised by an oxygen enrichment mechanism (see ESI,† Scheme S2) producing thiirane by-products which could be identified in the crude reaction mixture (see ESI,† Fig. S11). Note that LCyAl itself without RbOAc also catalyses PTA/CHO ROCOP, albeit with the formation of (thio)ether links from epoxide or thiirane homopropagation as well as a low TOF < 20 h−1, furthermore supporting the cooperative catalysis between LCyAlOAc and cocatalytic RbOAc.29 As we previously found the ligand backbone on the bisphenoxyimine spectator ligand to influence the catalysis we proceeded to investigate the phenylene variant LPhAlOAc, the ethylene variant LenAlOAc and the dimethylpropylene variant LproAlOAc in PTA/CHO ROCOP under the same conditions. Whilst all aliphatic variants exhibited the same activity (see ESI,† Fig. S10), the aromatic LPhAlOAc shows a reduced maximum TOF of 11 h−1 and also forms (thio)ether links. This contrasts the behaviour of the Cr(III) case for which the aromatic backbone showed similar reactivity to other aliphatic ones. Furthermore, the Al(III) complexes were not all equally stable. LproAlOAc was found to decompose in methanol solution under ambient conditions (ESI,† Fig. S5), while LCyAlOAc, LenAlOAc and LPhAlOAc remained stable. Hence, we proceeded with LCyAlOAc to investigate how the choice of acetate salt affects the catalytic activity.
:1 distribution of thioester:ester links, i.e. 50% thioester. Yet, only 45% thioester links are formed, and this can be rationalised by an oxygen enrichment mechanism (see ESI,† Scheme S2) producing thiirane by-products which could be identified in the crude reaction mixture (see ESI,† Fig. S11). Note that LCyAl itself without RbOAc also catalyses PTA/CHO ROCOP, albeit with the formation of (thio)ether links from epoxide or thiirane homopropagation as well as a low TOF < 20 h−1, furthermore supporting the cooperative catalysis between LCyAlOAc and cocatalytic RbOAc.29 As we previously found the ligand backbone on the bisphenoxyimine spectator ligand to influence the catalysis we proceeded to investigate the phenylene variant LPhAlOAc, the ethylene variant LenAlOAc and the dimethylpropylene variant LproAlOAc in PTA/CHO ROCOP under the same conditions. Whilst all aliphatic variants exhibited the same activity (see ESI,† Fig. S10), the aromatic LPhAlOAc shows a reduced maximum TOF of 11 h−1 and also forms (thio)ether links. This contrasts the behaviour of the Cr(III) case for which the aromatic backbone showed similar reactivity to other aliphatic ones. Furthermore, the Al(III) complexes were not all equally stable. LproAlOAc was found to decompose in methanol solution under ambient conditions (ESI,† Fig. S5), while LCyAlOAc, LenAlOAc and LPhAlOAc remained stable. Hence, we proceeded with LCyAlOAc to investigate how the choice of acetate salt affects the catalytic activity.
We employed the series of alkali metal acetates AMOAc (AM = Li, Na, K, Cs), alkaline earth metal acetates AEM(OAc)2 (AEM = Mg, Ca, Sr, Ba) as well as La(OAc)3. For all cases, but LiOAc and Mg(OAc)2, adduct formation could be confirmed via1H NMR, (see ESI,† Fig. S7–S9) suggesting that the lighter metals do not seem to be a good size match for the oxygenated binding pocket of LCyAlOAc. Again, kinetic studies were carried out through 1H NMR aliquot analysis following the consumption of PTA. It should be noted that although trying to minimise systematic errors associated with the aliquoting method, the precise value of the maximum TOF presented in Table 1 should viewed with caution! Yet our results do allow us to draw conclusions on how metal acetate choice comparatively affects PTA/CHO and PA/CHO ROCOP, which will be discussed in the following. Surprisingly, all added acetate salts led to an improvement of the PTA/CHO ROCOP activity compared to stand-alone LCyAl yielding poly(ester-thioesters) without (thio)ether links with 39–47% thioester links. The alkali metals Cs, Rb and K are the most active exhibiting very similar kinetic traces (Fig. 2) while Li and Na are substantially less active. The alkaline earth metals Mg, Ca and Ba show similar performances to Li and Na, while an initiation period is observed in the case of Ba. La shows activity ranging between the best alkali and alkaline earth metals while giving the best thioester content of 47%. Next, we were wondering how this compared to the all-oxygen case, as one open question in ROCOP catalysis is, how catalyst selection criteria change when moving down the periodic table from O to S. Therefore PA/CHO ROCOP was investigated under identical conditions with all catalyst combinations and analysed by 1H NMR aliquot analysis. As can be seen, from the kinetic results presented in Table 1, PTA conversion in PTA/CHO ROCOP is faster than PA conversion in PA/CHO ROCOP for almost all the catalysts, and we attribute this to the increased nucleophilicity of thiocarboxylate intermediates formed by PTA insertion, compared to carboxylate intermediates from PA insertion. Most interestingly though for LCyAlOAc with LiOAc, Ca(OAc)2, Sr(OAc)2, Ba(OAc)2 and La(OAc)3 ether links from CHO homopropagation are formed during the polymerisation process. This is most extreme with Mg(OAc)2, where only polyether and no polyester is formed. Ether formation is associated with broadened polydispersities, in that, while, LCyAlOAc with RbOAc yields a perfect polyester with Mn = 6.7 kg mol−1 and Đ = 1.2, the polyether formed with Mg(OAc)2 shows a Mn = 18.8 kg mol−1 and Đ = 1.9 and the poly(ether-ester) with Ca(OAc)2 a Mn = 7.0 kg mol−1 and Đ = 2.0 (see Fig. S37, ESI†). In contrast, the same catalysts for PTA/CHO ROCOP produce no ether links whatsoever and this implies that PTA incorporation might occur more facilely than PA incorporation. To investigate this, we performed PTA/PA/CHO terpolymerisation employing LCyAlOAc with RbOAc at a loading of 1 eq. Cat.: 250 eq. PTA: 250 eq. PA: 1000 eq. CHO, but observed preferential PA incorporation by 1H aliquot analysis (see ESI,† Fig. S36). Only once PA is consumed PTA/CHO ROCOP occurs to eventually form a polymer with a Mn = 11.1 kg mol−1 (Đ = 1.6) after full PA and 64% PTA consumption. Hence, we infer that the linkage selectivity improvement when moving from PA to PTA ROCOP cannot stem from more facile PTA over PA insertion, but much rather the suppression of epoxide homopropagation by the sulfurated species, which needs to be the subject of further investigation.
| Alkali metal | PTA conv.a [%] | Time [h] | Max. TOFb [h−1] | Ester:thioester:etherc |
PA conv.a [%] | Time [h] | Max. TOFb [h−1] | Ester:etherc |
|---|---|---|---|---|---|---|---|---|
| ROCOPs conducted with 1 eq. LCyAlOAc: 1 eq. M(OAc)x: 250 eq. P(T)A: 1000 eq. CHO at 80 °C.a Relative integral of aromatic resonances from residual PTA/PA/CHO versus polymer in the normalised 1H NMR spectrum of final aliquot.b TOF (turnover frequency) determined by linear fitting of PTA/PA conversion vs. time. Data points are excluded once conversion versus time plots deviate from linearity due to depleting monomer concentration.c Relative integral of ester, thioester vs. ether resonances in normalised 1H NMR spectrum of the crude polymer. | ||||||||
| Li(I) | 89 | 6 | 52 | 61:39:0 |
26 | 4 | 19 | 9:91 |
| Na(I) | 94 | 6 | 54 | 59:41:0 |
94 | 10 | 43 | 100:0 |
| K(I) | 94 | 0.75 | 491 | 55:45:0 |
98 | 3 | 79 | 100:0 |
| Rb(I) | 99 | 0.42 | 638 | 55:45:0 |
99 | 2.5 | 107 | 100:0 |
| Cs(I) | 98 | 1.25 | 248 | 56:44:0 |
98 | 2.5 | 96 | 100:0 |
| Mg(II) | 91 | 8 | 39 | 56:44:0 |
0 | 2 | 0 | 0:100 |
| Ca(II) | 97 | 5.5 | 39 | 59:41:0 |
53 | 13 | 9 | 26:74 |
| Sr(II) | 83 | 5.5 | 35 | 56:44:0 |
54 | 4.5 | 31 | 20:80 |
| Ba(II) | 85 | 10 | 25 | 54:46:0 |
26 | 10 | 8 | 67:33 |
| La(III) | 98 | 2.5 | 105 | 53:47:0 |
22 | 10 | 2 | 13:87 |
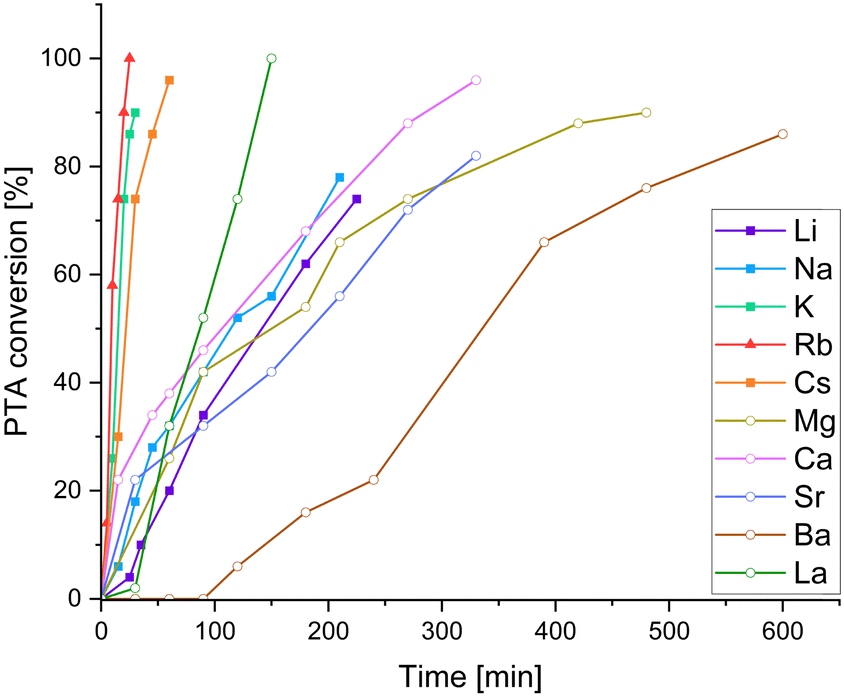 | ||
| Fig. 2 PTA/CHO ROCOP conversion versus time plots corresponding to Table 1. For PA/CHO ROCOP see ESI,† Fig. S25. | ||
Having established that moving from Cr(III) to Al(III) accelerates heterobimetallic PTA/CHO ROCOP, we wondered whether this also enables the use of epoxides with functional vinyl substituents, as the chromium analogues cannot achieve their copolymerisation with PTA at all (see ESI,† Fig. S38). Hence, we attempted the ROCOP of 4-vinyl cyclohexene oxide (vCHO) with PTA employing LCyAlOAc with RbOAc at a loading of 1 Cat.: 250 PTA: 1000 vCHO at 80 °C, which resulted in 99% PTA conversion after 45 min to yield a poly(ester-thioester) with Mn = 15.8 kg mol−1 (Đ = 1.4) (Table 2 run #1). This represents an approximately 40% improvement of Mn over the analogous PTA/CHO ROCOP. Employment of vCHO rather than CHO to improve molecular weights has been used before in PA ROCOP with related catalysts.30 As vCHO also apparently results in improved molecular weights in PTA ROCOP, we thereafter decreased the catalyst loading to investigate the maximum obtainable molecular weights employing LCyAlOAc with RbOAc. Reducing the loading to 1 eq. Cat.: 1000 eq. PTA: 2000 eq. vCHO yields a copolymer with Mn = 45.6 kg mol−1 (Đ = 1.6) at 80% PTA conversion while moving to 1 eq. Cat.: 2000 eq. PTA: 4000 eq. vCHO further improves the Mn to 64.6 kg mol−1 (Đ = 1.7) at 92% PTA conversion (Table 1 run #2 and #3); a further reduction of loading only resulted in slight improvement of Mn which can also be attributed to the decreased maximum PTA conversion of 70% (Table 1 run #4). Although the molecular weights range within the highest achieved for PTA/epoxide ROCOP, they are somewhat lower than the theoretical weights of Mn,theo. = 36.1–265.3 kg mol−1 (Mn,GPC = 15.8–67.2 kg mol−1). We attribute this to side reactions and protic diol impurities in the vCHO monomer operating as bifunctional initiators that act alongside acetate initiation by the catalyst coligands. This also helps explain the bimodal molecular weight distributions observed as reported previously. Seeking to confirm this notion we deliberately added 20 eq. of a diol chain transfer agent in the form of 1,4-benzendimethanol to a PTA/vCHO ROCOP conducted at 1 eq. Cat.: 250 eq. PTA: 1000 eq. vCHO. This yields a copolymer with a narrowed monomodal molecular weight distribution with a Mn of 3.7 kg mol−1 (Đ = 1.2) at ca. one third of the molecular weight than without diol chain transfer agent (see ESI,† Fig. S43). This supports that protic impurities are a primary factor for the deviation of molecular weights and broadened bimodal distributions. Nevertheless PTA/vCHO ROCOP could be combined with other ROCOP processes such as CO2/vCHO ROCOP producing some polycarbonate links in PTA/CO2/vCHO ring-opening terpolymerisation (ROTERP, see ESI,† Section S5 and Fig. 3). Terpolymerisation at a loading of 1 Cat.: 300 PTA: 300 vCHO under 4 bar CO2 pressure at 80 °C for 1.75 h yields a terpolymer with Mn = 10.8 kg mol−1 (Đ = 1.3) at 58% PTA conversion in which approximately 6% of links are carbonates that result from CO2 insertion and 94% of links are poly(thioester-esters) that result from PTA insertion.§ Increasing the CO2 pressure to 40 bar yields a terpolymer with Mn = 11.8 kg mol−1 (Đ = 1.4) at 80% PTA conversion in which approximately 16% of links resulting from CO2 insertion.
| Cat:PTA:vCHO |
Time (h) | PTA conv.a (%) | M n (kg mol−1) | Đ |
|---|---|---|---|---|
| T = 80 °C.a Relative integral in the normalised 1H NMR spectrum of aromatic resonances from residual PTA versus polymer.b Determined by GPC (gel permeation chromatography) measurements conducted in THF, using narrow polystyrene standards to calibrate the instrument. | ||||
| 1:250:1000 |
0.75 | >99 | 15.8 | 1.4 |
| 1:1000:2000 |
3 | 80 | 45.6 | 1.6 |
| 1:2000:4000 |
16 | 92 | 64.5 | 1.7 |
| 1:4000:6000 |
24 | 70 | 67.2 | 1.8 |
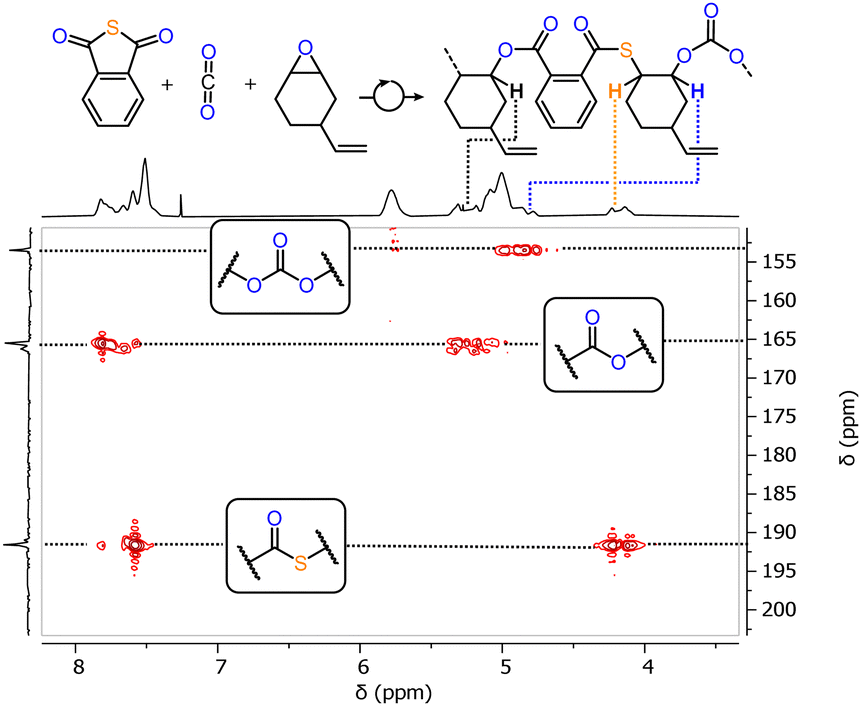 | ||
| Fig. 3 1H–13C HMBC spectrum (CDCl3) of PTA/CO2/vCHO terpolymer. | ||
Finally, we sought to demonstrate the utility of the vinyl substituents on the vCHO copolymers that the aluminium catalysis enabled us to incorporate. Hence vulcanisation with elemental sulfur, a waste product of the petrochemical industry, was attempted to crosslink the vinyl functional groups, an underexplored method in the context of ROCOP copolymers (see ESI,† Section S6). Therefore, a powdered blend of 90 wt% vCHO/PTA copolymer (Table 2 run #3) and 10 wt% sulfur was hot processed at 200 °C for 20 minutes and a vulcanized polymer film was obtained as a red, brittle sheet. FTIR analysis shows the stark reduction of the vinylene double bonds upon vulcanisation. In contrast to the parent linear polymer which shows good solubility in organic solvents like DCM, no solubility (95% gel fraction) is observed for the vulcanised material. Furthermore, while the parent linear thioester could be degraded by aminolysis in methanolic ammonia,26 no degradation was observed for the vulcanised materials further substantiating crosslinking.
In conclusion, we report that moving from chromium to aluminium based heterobimetallic catalysts leads to a drastic improvement of reaction rate in thioanhydride/epoxide ROCOP. Compared to the performance of aluminium catalysis in the all-oxygenated anhydride/epoxide ROCOP a general improvement of rate and linkage selectivity was also found. This allowed for the introduction of functional vinyl substituents, the synthesis of high Mn materials and novel thioanhydride based terpolymers. Our report highlights clear performance advantages of catalysts featuring sustainable metals in the synthesis of sulfurated polymer which could inspire future catalyst development.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed
.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mat., 2020, 5, 501–516 CrossRef CAS
- B. von Vacano, H. Mangold, G. W. M. Vandermeulen, G. Battagliarin, M. Hofmann, J. Bean and A. Künkel, Angew. Chem., Int. Ed., 2023, 135, e202210823 CrossRef
-
S. Kernbichl and B. Rieger, Engineering Solutions for CO2 Conversion, John Wiley & Sons, Ltd, 2021, pp. 385–406 Search PubMed
- T.-J. Yue, W.-M. Ren and X.-B. Lu, Chem. Rev., 2023, 24, 14038–14083 CrossRef PubMed
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed
- Y. Wang, M. Li, J. Chen, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 22547–22553 CrossRef CAS PubMed
- Y. Xia, X. Yue, Y. Sun, C. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202219251 CrossRef CAS PubMed
- Y.-L. Su, L. Yue, H. Tran, M. Xu, A. Engler, R. Ramprasad, H. J. Qi and W. R. Gutekunst, J. Am. Chem. Soc., 2023, 145, 13950–13956 CrossRef CAS PubMed
- E. F. Clark, G. Kociok-Köhn, M. G. Davidson and A. Buchard, Polym. Chem., 2023, 14, 2838–2847 RSC
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 145, 18560–18567 CrossRef CAS PubMed
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 CrossRef CAS PubMed
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS PubMed
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Macromolecules, 2019, 52, 2439–2445 CrossRef CAS
- T.-J. Yue, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Eur. Polym. J., 2023, 190, 111985 CrossRef CAS
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC
- C. Fornacon-Wood, M. R. Stühler, C. Gallizioli, B. R. Manjunatha, V. Wachtendorf, B. Schartel and A. J. Plajer, Chem. Commun., 2023, 59, 11353–11356 RSC
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CrossRef CAS PubMed
- X.-F. Zhu, R. Xie, G.-W. Yang, X.-Y. Lu and G.-P. Wu, ACS Macro Lett., 2021, 10, 135–140 CrossRef CAS PubMed
- J. Stephan, M. R. Stühler, S. M. Rupf, S. Neale and A. J. Plajer, Cell Rep. Phys. Sci., 2023, 101510 CrossRef CAS
- C. Fornacon-Wood, B. R. Manjunatha, M. R. Stühler, C. Gallizioli, C. Müller, P. Pröhm and A. J. Plajer, Nat. Commun., 2023, 14, 4525 CrossRef CAS PubMed
- M. R. Stühler, C. Gallizioli, S. M. Rupf and A. J. Plajer, Polym. Chem., 2023, 14, 4848–4855 RSC
- W. T. Diment, G. L. Gregory, R. W. F. Kerr, A. Phanopoulos, A. Buchard and C. K. Williams, ACS Catal., 2021, 11, 12532–12542 CrossRef CAS
- E. J. K. Shellard, W. T. Diment, D. A. Resendiz-Lara, F. Fiorentini, G. L. Gregory and C. K. Williams, ACS Catal., 2024, 14, 1363–1374 CrossRef CAS PubMed
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS PubMed
- W. T. Diment and C. K. Williams, Chem. Sci., 2022, 13, 8543–8549 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00811a |
| ‡ For Cr(III), polymerisation were conducted at 100 °C, though at these temperature, we found the Al(III) catalyst to be too fast for the TOF to be determined by the same experimental technique implying that one order of magnitude improvement is the lower limit of the performance improvement. |
| § This particularly loading was chosen to avoid CO2/CHO ROCOP after PTA consumption allowing the determination of the relative CO2 incorporation during the ROTERP process. |
| This journal is © The Royal Society of Chemistry 2024 |