
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing expansion of spirocyclopropanes with stabilized sulfonium ylides: highly diastereoselective synthesis of cyclobutanes†
Hisanori
Nambu
 *ab,
Yuta
Onuki
a,
Kana
Aso
a,
Momoka
Kanamori
a,
Keisuke
Tomohara
b,
Kiyoshi
Tsuge
c and
Takayuki
Yakura
*a
*ab,
Yuta
Onuki
a,
Kana
Aso
a,
Momoka
Kanamori
a,
Keisuke
Tomohara
b,
Kiyoshi
Tsuge
c and
Takayuki
Yakura
*a
aFaculty of Pharmaceutical Sciences, University of Toyama, Sugitani, Toyama 930-0194, Japan. E-mail: yakura@pha.u-toyama.ac.jp
bLaboratory of Pharmaceutical Manufacturing Chemistry, Kyoto Pharmaceutical University, Kyoto 607-8412, Japan. E-mail: hnambu@mb.kyoto-phu.ac.jp
cGraduate School of Science and Engineering, University of Toyama, Gofuku, Toyama 930-8555, Japan
First published on 14th March 2024
Abstract
A novel method was devised for regioselective ring expansion of Meldrum's acid-derived spirocyclopropanes to spirocyclobutanes with stabilized sulfonium ylides, affording 1,2-trans-disubstituted 6,8-dioxaspiro[3.5]nonane-5,9-diones in up to 87% yields without the formation of any isomers. The aforementioned reaction was also applied to the barbituric acid-derived spirocyclopropane, resulting in the formation of the corresponding cyclobutanes.
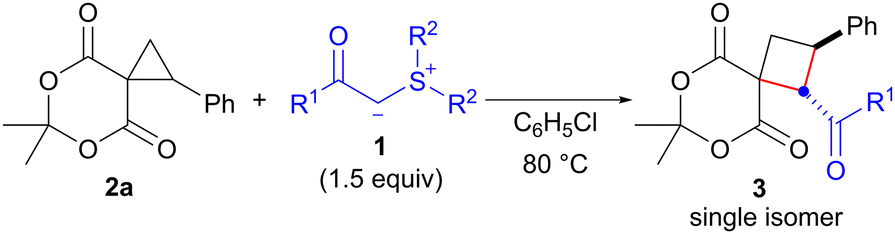
Sulfonium ylides stabilized by electron-withdrawing groups (EWG) have been used as a versatile methylene synthon in the synthesis of a variety of carbo- and heterocyclic compounds.1,2 As a pioneering work, Payne reported that the reaction of α,β-unsaturated diethylmalonate with EWG-stabilized sulfonium ylide 1 (EWG = CO2Et) afforded cyclopropane in 90% yield (Scheme 1A).3 In this reaction, the Michael addition of 1 followed by SN2-type cyclization of the carbanion (C-cyclization) proceeded with the concomitant release of the sulfide. In contrast, the reaction of the corresponding 1,3-diketone with stabilized sulfonium ylide 1 unexpectedly produced dihydrofuran in 83% yield through enolate cyclization (O-cyclization, Scheme 1A).3 The regioselectivity of these reactions may be attributed to the inherent difference between esters and ketones. Recently, we reported the ring-opening cyclization of spirocyclopropanes4 with EWG-stabilized sulfonium ylides 1 to afford hexahydrobenzopyranone as a single isomer via the regioselective ring-opening of cyclopropane with sulfonium ylide 1 and subsequent SN2-type O-cyclization (Scheme 1B, eqn (1)).4f Considering the similar reactivity of cyclopropane and carbon–carbon double bonds, we expected that the reaction of ester-derived spirocyclopropane 2 with stabilized sulfonium ylide 1 would provide spirocyclobutane 3 through C-cyclization (Scheme 1C). Because cyclobutane is a useful scaffold found in several biologically active natural products and pharmaceutically active compounds,5 the development of a synthetic method for cyclobutane is currently the subject of intense research.6 Although several instances of cyclopropane to cyclobutane ring expansion have been documented thus far,7,8 to the best of our knowledge, there have been no examples of sulfonium ylide-mediated ring expansion (C-cyclization).9 Herein, we describe the ring expansion of Meldrum's acid-derived spirocyclopropanes 2 to spirocyclobutanes 3 using EWG-stabilized sulfonium ylides 1 (Scheme 1C).
 | ||
| Scheme 1 Reactions of various carbonyl compounds with stabilized sulfonium ylides 1 as nucleophiles. | ||
Initially, we examined the reaction of 6,6-dimethyl-1-phenyl-5,7-dioxaspiro[2.5]octane-4,8-dione (2a)10 with dimethylsulfonium benzoylmethylide (1a) as an EWG-stabilized sulfonium ylide (Table 1). The ring expansion of 1a proceeded under the reaction conditions previously reported by our group (1.5 equiv. of 1a in refluxing CH2Cl2),4f affording 1-benzoyl-7,7-dimethyl-2-phenyl-6,8-dioxaspiro[3.5]nonane-5,9-dione (3a) after 24 h in 75% yield (entry 1). Notably, no isomer formation was observed during this process. The structure of 3a including its stereochemistry was confirmed by a single-crystal X-ray diffraction analysis. This analysis revealed that the structure corresponds to that of cyclobutane with a 1,2-trans configuration (see ESI† for details). Screening of the solvents at reflux revealed that benzene and halogenated solvents, such as dichloromethane and 1,2-dichloroethane, were suitable for this reaction (entry 1 vs. entries 2–5). Finally, we found that chlorobenzene at 80 °C was the most effective and afforded 3a in 86% yield after 6 h (entry 6).

After determining the optimal conditions, we investigated the reaction of spirocyclopropane 2a using a range of sulfonium ylides 1 that are stabilized by carbonyl functional groups (Table 2). The reaction with 1.5 equiv. of p-methoxybenzoyl sulfonium ylide 1b in chlorobenzene at 80 °C afforded the corresponding spirocyclobutane 3b as the sole product after 6 h in 74% yield (entry 2). The use of m- and o-methoxybenzoyl sulfonium ylides 1c11 and 1d12 provided the corresponding products 3c and 3d in 86% and 87% yields, respectively (entries 3 and 4). The reaction with sulfonium ylide 1e bearing a p-nitro group as a strong EWG decreased the product yield, and a significantly longer reaction time was required to achieve full conversion (61% yield, 24 h, entry 5 vs. entry 1). In contrast, the reaction with p-chlorobenzoyl sulfonium ylide 1f under the optimized conditions proceeded smoothly to completion within 5 h, furnishing 3f in 83% yield (entry 6). We also investigated the suitability of an acetyl sulfonium ylide for this reaction. To this end, we used tetrahydrothiophenium acetylmethylide (1g) because of the difficulty in preparing dimethylsulfonium acetylmethylide. The reaction of 2a with 1g afforded the desired product 3g as a single isomer, albeit with a prolonged reaction time and lower yield (24 h, 36% yield, entry 7 vs. entry 1). Moreover, ethoxycarbonyl group-substituted sulfonium ylide 1h was used in the present protocol, and the corresponding cyclobutane 3h was obtained in 53% yield after 23 h (entry 8).
Next, we examined the scope of the reaction with the spirocyclopropane substrates 2 using benzoyl-substituted sulfonium ylide 1a (Table 3). Treatment of spirocyclopropanes 2b, 2c and 2d, which possess p-acetoxy-, p-methyl-, and p-bromophenyl groups on the cyclopropane, respectively, with 1a under the optimized conditions (chlorobenzene at 80 °C), afforded the corresponding products 3i, 3j, and 3k in 64%–80% yields with perfect diastereoselectivities (entries 1–3). The reaction of m-methoxyphenyl-substituted spirocyclopropane 2e for 12 h provided cyclobutane 3l in 69% yield (entry 4). Although spirocyclopropane 2f, which possesses a 2-naphthyl group, required a relatively long reaction time (48 h), 3m was obtained in a good yield (80%, entry 5). There was a concern that the use of vinyl-substituted spirocyclopropane 2g would compete with the conjugate addition, but the reaction of 2g proceeded uneventfully and afforded the desired product 3n in 68% yield (entry 6). Finally, the reaction of the simple spirocyclopropane 2h (R = H)13 was investigated (entry 7). A 2′,3′-nonsubstituted spirocyclopropane was found to be less reactive than an aryl-substituted one,4e which resulted in a lower yield of product 3o (26% yield).

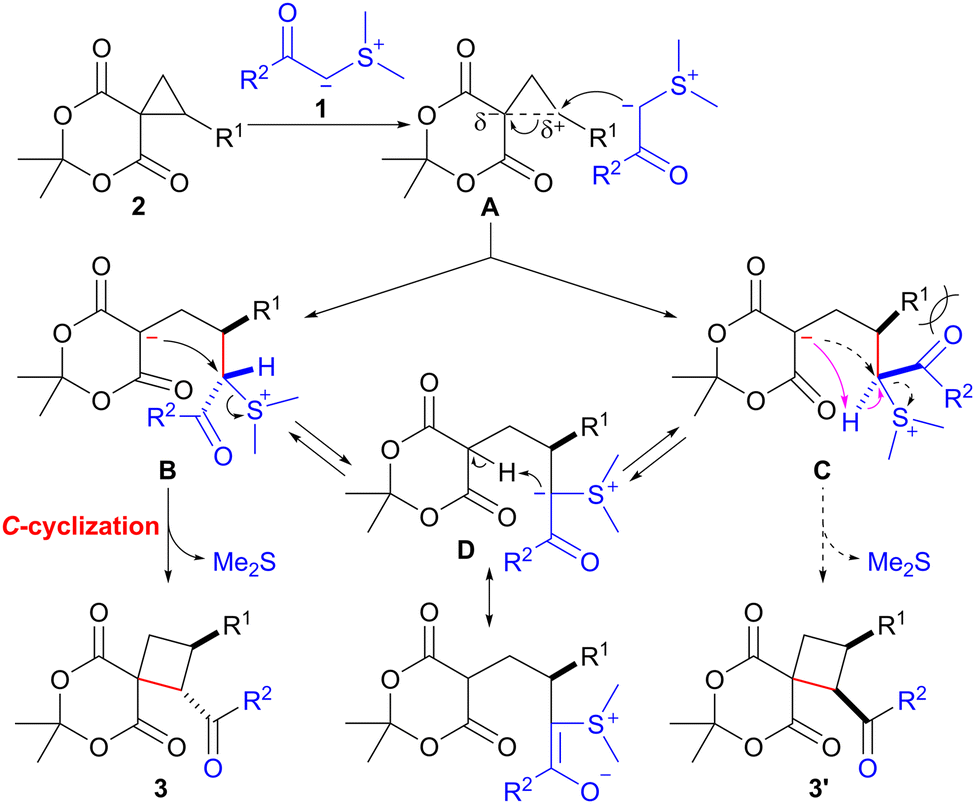
A plausible mechanism for the ring expansion of spirocyclopropane 2 with sulfonium ylide 1, stabilized by an acyl group, is shown in Scheme 2. The ring opening of spirocyclopropane 2 would proceed through the nucleophilic attack of the carbanion in 1 on the electrophilic cyclopropane carbon possessing an R1 substituent in A. This reaction would lead to the formation of betaine intermediates B and C. SN2-type C-cyclization of the carbanion in B would occur smoothly to afford trans-product 3 with the concomitant release of dimethyl sulfide. In contrast, the C-cyclization of C would hardly proceed owing to the severe steric repulsion between the acyl group (R2CO) and substituent R1 in C. Consequently, intermediate C could be converted into cyclization precursor B through reversible intramolecular proton transfer via the stabilized sulfonium ylide D,14,15 finally providing trans-isomer 3.
 | ||
| Scheme 2 Plausible reaction mechanism. | ||
To demonstrate the utility of the present protocol, we examined the conversion of spirocyclobutane 3a into highly substituted non-spiro cyclobutane 4 (Scheme 3). The treatment of 3a with sulfuric acid in methanol/diethyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 50 °C led to a transesterification process, resulting in the formation of dimethyl ester. The reaction yielded the corresponding cyclobutane 4 in 82% yield. Since the reaction of dimethyl 2-phenylcyclopropane-1,1-dicarboxylate (5) with sulfonium ylide 1a did not proceed,4f,16 spiro form 3a was required for the synthesis of diester 4. This ring-expansion reaction of spirocyclopropanes could be a useful method for the preparation of substituted cyclobutanes.
:1) at 50 °C led to a transesterification process, resulting in the formation of dimethyl ester. The reaction yielded the corresponding cyclobutane 4 in 82% yield. Since the reaction of dimethyl 2-phenylcyclopropane-1,1-dicarboxylate (5) with sulfonium ylide 1a did not proceed,4f,16 spiro form 3a was required for the synthesis of diester 4. This ring-expansion reaction of spirocyclopropanes could be a useful method for the preparation of substituted cyclobutanes.
 | ||
| Scheme 3 Conversion of spirocyclobutane 3a into cyclobutane 4. | ||
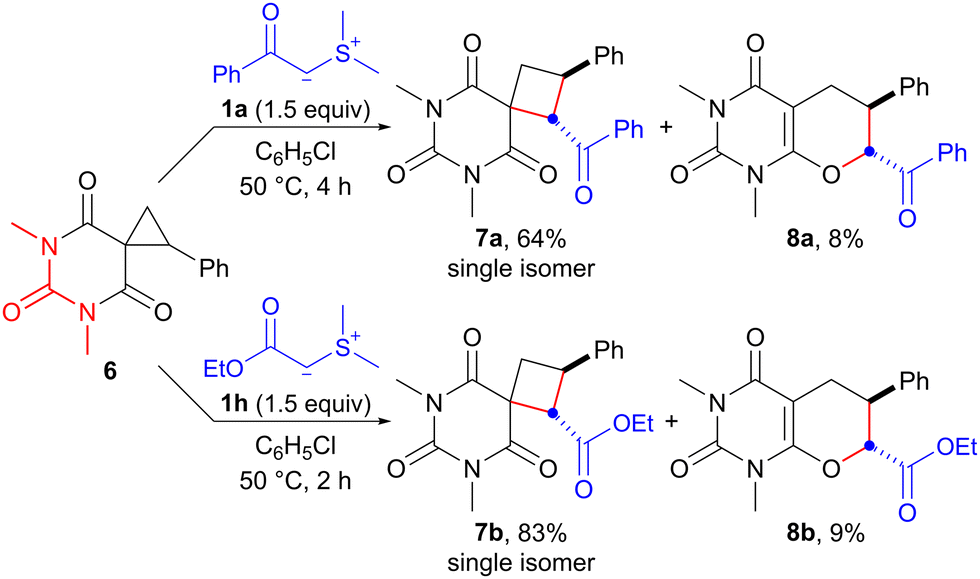
Having achieved ring expansion of ester-derived spirocyclopropanes, we further investigated the reaction of an amide-derived spirocyclopropane with an EWG-stabilized sulfonium ylide. The reaction of spirocyclopropane 6,17 derived from barbituric acid, with sulfonium ylides 1a and 1h in chlorobenzene proceeded smoothly at 50 °C to provide the corresponding spirocyclobutanes 7a and 7b in 64% and 83% yields, respectively (Scheme 4). Interestingly, unexpected products 8a and 8b, which indicated that SN2-type O-cyclization of the enolate ion instead of the carbanion would occur, were also obtained in 8% and 9% yields, respectively. Although the results are still preliminary, the reaction of barbituric acid-derived spirocyclopropane with sulfonium ylide exhibits promise as a synthetic method of spirobarbiturate cyclobutane analogs. These compounds have potential as pharmaceutical agents.18
 | ||
| Scheme 4 Ring expansion of barbituric acid-derived spirocyclopropane 6 with sulfonium ylides 1a and 1h. | ||
In conclusion, we devised a novel method for regioselective ring expansion of cyclopropanes to cyclobutanes using stabilized sulfonium ylides. Meldrum's acid-derived spirocyclobutanes with EWG-stabilized sulfonium ylides afforded the corresponding spirocyclobutanes as single diastereomers in yields of up to 87%. The present reaction provides an efficient route to highly substituted cyclobutanes. To the best of our knowledge, this is the first example of a ring expansion of cyclopropanes with sulfonium ylides. This reaction may be envisaged as a formal [3+1] cycloaddition, facilitating the construction of the four-membered ring system.19 The expansion reaction could be applied to the transformation of barbituric acid-derived spirocyclopropane into the corresponding spirocyclobutane. Ongoing efforts are being made to apply the present method to the synthesis of a variety of cyclobutane derivatives.
This research was supported, in part, by a Grant-in-Aid for Scientific Research (C) (Grant No. 21K05048) from the Ministry of Education, Culture, Sports, Science and Technology, Japan and by a Grant from Hoansha Foundation (Osaka, Japan).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews, see: (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (b) G. D. Bisag, S. Ruggieri, M. Fochi and L. Bernardi, Org. Biomol. Chem., 2020, 18, 8793–8809 RSC; (c) P. Y. Ushakov, S. L. Ioffe and A. Y. Sukhorukov, Org. Chem. Front., 2022, 9, 5358–5382 RSC.
- For recent examples, see: (a) C. Wang, L. Fang, L. Zhang, Y. Wang, F. Gao and Z. Wang, Org. Chem. Front., 2022, 9, 2204–2208 RSC; (b) X. Wang, J. Yu, M. Xu, H. Mao, Y. Shan, X. Lv and L. Zhou, Org. Lett., 2022, 24, 5896–5901 CrossRef CAS PubMed; (c) W. Yu, C.-H. Tung and Z. Xu, Adv. Synth. Catal., 2022, 364, 3749–3753 CrossRef CAS; (d) J. Yu, X. Wang, M. Xu, B. Zhang, Z. Xiong, H. Mao, X. Lv and L. Zhou, Org. Chem. Front., 2023, 10, 916–922 RSC; (e) M. Xu, M. You, Y. Su, B. R. Lu, L. Liu, X. Lv, S. Wang, H. Mao and L. Zhou, Org. Chem. Front., 2023, 10, 1521–1526 RSC.
- G. B. Payne, J. Org. Chem., 1967, 32, 3351–3355 CrossRef CAS.
- (a) H. Nambu, M. Fukumoto, W. Hirota and T. Yakura, Org. Lett., 2014, 16, 4012–4015 CrossRef CAS PubMed; (b) H. Nambu, M. Fukumoto, W. Hirota, N. Ono and T. Yakura, Tetrahedron Lett., 2015, 56, 4312–4315 CrossRef CAS; (c) H. Nambu, N. Ono and T. Yakura, Synthesis, 2016, 1892–1901 CrossRef; (d) H. Nambu, W. Hirota, M. Fukumoto, T. Tamura and T. Yakura, Chem. – Eur. J., 2017, 23, 16799–16805 CrossRef CAS PubMed; (e) H. Nambu, Y. Onuki, N. Ono and T. Yakura, Adv. Synth. Catal., 2018, 360, 2938–2944 CrossRef CAS; (f) H. Nambu, Y. Onuki, N. Ono, K. Tsuge and T. Yakura, Chem. Commun., 2019, 55, 6539–6542 RSC; (g) H. Nambu, T. Tamura and T. Yakura, J. Org. Chem., 2019, 84, 15990–15996 CrossRef CAS PubMed; (h) Y. Onuki, H. Nambu and T. Yakura, Chem. Pharm. Bull., 2020, 68, 479–486 CrossRef CAS PubMed; (i) H. Nambu, Y. Onuki, K. Yamazaki and T. Yakura, Heterocycles, 2021, 103, 1099–1107 CrossRef PubMed; (j) Y. Onuki, K. Yamazaki, N. Ono, Y. Masuda, T. Yakura and H. Nambu, Adv. Synth. Catal., 2023, 365, 2536–2544 CrossRef CAS.
- (a) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559–1581 CrossRef CAS PubMed; (b) M. R. Bauer, P. D. Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC; (c) M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and S. Blanco-Ania, ChemMedChem, 2022, 17, e202200020 CrossRef CAS PubMed; (d) P. Yang, Q. Jia, S. Song and X. Huang, Nat. Prod. Rep., 2023, 40, 1094–1129 RSC.
- For selected reviews, see: (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed; (b) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (c) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC; (d) D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 1310–1326 CrossRef CAS; (e) J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed; (f) R. Guo and M. K. Brown, Acc. Chem. Res., 2023, 56, 2253–2264 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS; (b) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed; (c) B. Biletskyi, P. Colonna, K. Masson, J.-L. Parrain, L. Commeiras and G. Chouraqui, Chem. Soc. Rev., 2021, 50, 7513–7538 RSC.
- For selected examples, see: (a) H. Nemoto, H. Ishibashi, M. Nagamochi and K. Fukumoto, J. Org. Chem., 1992, 57, 1707–1712 CrossRef CAS; (b) B. M. Trost and T. Yasukata, J. Am. Chem. Soc., 2001, 123, 7162–7163 CrossRef CAS PubMed; (c) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed; (d) Z. Wu, D. Leboeuf, P. Retailleau, V. Gandon, A. Marinetti and A. Voituriez, Chem. Commun., 2017, 53, 7026–7029 RSC; (e) C.-G. Zhao, Z.-T. Feng, G.-Q. Xu, A. Gao, J.-W. Chen, Z.-Y. Wang and P.-F. Xu, Angew. Chem., Int. Ed., 2020, 59, 3058–3062 CrossRef CAS PubMed; (f) D. P. Hari, J. C. Abell, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 5515–5520 CrossRef CAS PubMed; (g) W. Ouyang, J. Huo and J. Wang, Synlett, 2023, 1507–1511 CAS; (h) M. Takatsuki, H. Aoyama, K. Murai, M. Arisawa and M. Sako, Chem. Commun., 2023, 59, 7467–7470 RSC.
- Chen and co-workers reported the ring expansion reaction of electron-deficient cyclopropanes with arsonium ylides. Y.-L. Chen, W.-G. Cao, W.-Y. Ding and X.-H. Sun, Chin. J. Chem., 2005, 23, 81–84 CrossRef CAS.
- Y. R. Lee and J. H. Choi, Bull. Korean Chem. Soc., 2006, 27, 503–507 CrossRef CAS.
- S. J. Sabounchei, A. Yousefi, M. Ahmadianpoor, A. Hashemi, M. Bayat, A. Sedghi, F. A. Bagherjeri and R. W. Gable, Polyhedron, 2016, 117, 273–282 CrossRef CAS.
- S. K. Pagire, N. Kumagai and M. Shibasaki, ACS Catal., 2021, 11, 11597–11606 CrossRef.
- H. Nambu, N. Ono, W. Hirota, M. Fukumoto and T. Yakura, Chem. Pharm. Bull., 2016, 64, 1763–1768 CrossRef CAS PubMed.
- Aggarwal and co-workers reported that a similar proton transfer event could intervene in sulfonium ylide-mediated cyclopropanations under certain conditions. S. L. Riches, C. Saha, N. F. Filgueira, E. Grange, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 7626–7630 CrossRef CAS PubMed.
- Crudden and co-workers also reported the deprotonation and reprotonation on the carbon atom of an α-hydroxy sulfonium ylide in the Corey–Chaykovsky epoxidation. D. R. Edwards, J. Du and C. M. Crudden, Org. Lett., 2007, 9, 2397–2400 CrossRef CAS PubMed.
- The spiro structure is crucial for the success of this ring expansion. (a) S. Danishefsky and R. K. Singh, J. Am. Chem. Soc., 1975, 97, 3239–3241 CrossRef CAS; (b) P. M. Jüstel, A. Stan, C. D. Pignot and A. R. Ofial, Chem. – Eur. J., 2021, 27, 15928–15935 CrossRef PubMed.
- (a) X. Wang and Y. R. Lee, Bull. Korean Chem. Soc., 2013, 34, 1735–1740 CrossRef CAS; (b) P. Qian, B. Du, R. Song, X. Wu, H. Mei, J. Han and Y. Pan, J. Org. Chem., 2016, 81, 6546–6553 CrossRef CAS PubMed.
- For a review, see: A. Bagherinejad and A. Alizadeh, Org. Biomol. Chem., 2022, 20, 7188–7215 RSC.
- Doyle and co-workers reported catalytic asymmetric [3+1]-cycloaddition with sulfonium ylides. Y. Deng, L. A. Massey, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2019, 58, 1955–1959 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, analytical data, and NMR spectra (PDF). CCDC 2312076 (3a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc06033k |
| This journal is © The Royal Society of Chemistry 2024 |